

Abstract
Genome variation studies in Plasmodium falciparum have focused on SNPs and, more recently, large-scale copy number polymorphisms and ectopic rearrangements. Here, we examine another source of variation: variable number tandem repeats (VNTRs). Interspersed low complexity features, including the well-studied P. falciparum microsatellite sequences, are commonly classified as VNTRs; however, this study is focused on longer coding VNTR polymorphisms, a small class of copy number variations. Selection against frameshift mutation is a main constraint on tandem repeats (TRs) in coding regions, while limited propagation of TRs longer than 975 nt total length is a minor restriction in coding regions. Comparative analysis of three P. falciparum genomes reveals that more than 9% of all P. falciparum ORFs harbor VNTRs, much more than has been reported for any other species. Moreover, genotyping of VNTR loci in a drug-selected line, progeny of a genetic cross, and 334 field isolates demonstrates broad variability in these sequences. Functional enrichment analysis of ORFs harboring VNTRs identifies stress and DNA damage responses along with chromatin modification activities, suggesting an influence on genome mutability and functional variation. Analysis of the repeat units and their flanking regions in both P. falciparum and Plasmodium reichenowi sequences implicates a replication slippage mechanism in the generation of TRs from an initially unrepeated sequence. VNTRs can contribute to rapid adaptation by localized sequence duplication. They also can confound SNP-typing microarrays or mapping short-sequence reads and therefore must be accounted for in such analyses.
Keywords: Malaria genomics, Polymorphisms, Plasmodium falciparum, Copy number, Variable number tandem repeats, Intragenic tandem repeats
Introduction
Plasmodium falciparum is the causative agent of the most severe and fatal form of human malaria; a disease that caused an estimated 243 million episodes and 863,000 deaths in 2008 (WHO 2009). This disease has persisted due in part to parasite-immune system evasion, and the development of antimalarial drug resistance. Studies of genome variation in this 81% AT rich (Gardner et al. 2002) parasite have focused predominantly on SNPs (Jeffares et al. 2007; Mu et al. 2007; Volkman et al. 2007b) or large structural variation (Carret et al. 2005; Cheeseman et al. 2009; Kidgell et al. 2006; Ribacke et al. 2007).
Tandem repeats (TRs), including simple sequence repeats, microsatellites, and minisatellites, are valuable as genetic markers due to their wide distribution, presumed neutrality, and relatively fast timescale of evolution (Ellegren 2004; Rando and Verstrepen 2007; Schlotterer 2004). Phenotypes affected by variable number TRs (VNTRs) include dog skull morphology (Fondon and Garner 2004), phase variation (van Belkum et al. 1998), and several human diseases and syndromes (Hancock and Simon 2005; Kenneson et al. 2001; Snell et al. 1993). VNTRs can produce homopeptide tracts (Fondon and Garner 2004), duplicate protein domains (Verstrepen et al. 2005), or affect gene expression levels (Vinces et al. 2009; Whetstine et al. 2002) and transcript splicing (Pagani et al. 2000). However, in a wide-variety of organisms TRs are rare in coding regions relative to non-coding regions (Edwards et al. 1998; Wang et al. 1994), indicating strong selection against repeats that cause frameshift mutations (Metzgar et al. 2000; Morgante et al. 2002; Toth et al. 2000).
Variable number tandem repeat polymorphisms are appreciated as a type of copy number variation (Conrad et al. 2009) and have been surveyed in the ORFs of Legionella pneumophila (Coil et al. 2008), Saccharomyces cerevisiae (Bowen et al. 2005; Verstrepen et al. 2005), Aspergillus fumigatus (Levdansky et al. 2007), Neisseria spp. (Jordan et al. 2003), and humans (O’Dushlaine et al. 2005). The reported percentage of ORFs with VNTRs varies in different species ranging from <0.01% in L. pneumophila to approximately 4.3% in S. cerevisiae. Most of these are likely to be underestimates due to limited sampling and/or conservative criteria. For example, based on their amount of sampling the human study estimates a true rate of 6% up from the 1.6% they empirically identified. However, the S. cerevisiae analysis identifying 4.3% of ORFs (Bowen et al. 2005) may be an overestimate because the authors assume all TRs in ORFs >4 nt in length are polymorphic. VNTR polymorphisms have not been examined genome-wide in P. falciparum.
Investigations of repeat structures in the P. falciparum genome include microsatellite sequences (Su et al. 1999; Su and Wellems 1996), repeats in genes that encode antigen or surface proteins (Cowman et al. 1984; Dame et al. 1984; Kemp et al. 1987; Stahl et al. 1985; Triglia et al. 1987), and subtelomeric repeats including the rep20 elements (Oquendo et al. 1986). Plasmodium TR arrays are likely to expand through slipped strand mispairing (Hughes 2004; Rich and Ayala 2000), and in some cases may play a role in immune evasion (Schofield 1991) or other particular functions (Kochan et al. 1986; O’Donnell et al. 2002). Other P. falciparum studies identified intragenic TRs of non-antigen encoding genes (Anderson et al. 2000; Vinayak et al. 2006) and an unusual abundance of nonglobular domain insertions at the amino acid level (Bowman et al. 1999; Gardner et al. 1998). However, these nonglobular domains were identified by the SEG algorithm (Wootton and Federhen 1996) which is commonly used to identify and mask low complexity amino acid sequences (Ye et al. 2006). More recent analysis concludes that <20% of these nonglobular domain insertions are unequivocally repetitious (Pizzi and Frontali 2001). The current work aims to comprehensively evaluate VNTRs in P. falciparum.
Here, we describe the genome-wide distribution for this class of repeat and its relationship to coding sequences. Repeat expansion in coding regions is restricted only for very long VNTRs or when frameshift mutations are introduced, leading to an unusually high repeat abundance and variability among P. falciparum strains. Gene ontology (GO) enrichment analysis of genes carrying VNTRs suggests components of rapid, adaptive variation. Structural features of the repeats and flanking regions indicate that a replication slippage mechanism can generate repeats in sequences that were originally unrepeated, and is supported by inter- and intraspecies comparative sequence analysis.
Materials and Methods
Genome-Wide TR Analysis
The 3D7 genome sequence was downloaded from PlasmoDB v5.3 (Bahl et al. 2003). The sequence for each chromosome was analyzed with Tandem Repeats Finder v4.0 (Benson 1999) with the maximum period size set to 2,000 and all other settings at default values. The results were parsed with custom perl scripts and stored in a database for querying. Data were analyzed with standard database tools and MATLAB software.
Telomeres and subtelomeric regions were excluded from any analyses that used the repeat categorization, “3× repeat” (a repeat where the motif length is a multiple of 3). Subtelomeric regions were defined by the location of the most proximal rep20 or telomere associated repetitive element (Figueiredo et al. 2000) that could be identified. Genes and repeats contained in or distal to this location were considered to be subtelomeric. Definitions for repeat array length, perfect repeats, and calculation of relative coding versus non-coding 3× repeat frequency are available in Supplementary materials and methods. The repeat distributions were compared with a Mann–Whitney–Wilcoxon test.
Tandem repeat density in a region was calculated as the total number of nucleotides comprising that region divided by the number of TRs found in that region to calculate a mean nucleotide spacing between TRs which we state as: 1 TR/X nt. The TR region was defined as the region where >50% of the repeat nucleotides was located.
Intragenic TR Location in Gene
Methods to assess spatial distribution were adapted from Huntley and Clark 2007 for all intragenic TRs identified in the 3D7 genome. In brief, each transcript sequence was divided into three segments of equal length: 5′ segment, midsegment, and 3′ segment. The TR midpoint was used to assign location into one of these groups. If the midpoint was located at the boundary of two segments, it was randomly assigned to one of them. Expected frequencies were calculated with the following formulas where L is transcript length and l is total intragenic TR length:
midsegment = (L/3)/(L − l)
5′ or 3′ segment = ((L/3) − (l/2))/(L − l).
A chi-square test was used to compare observed and expected frequencies.
Comparative Analysis of 3D7, HB3, and Dd2 Transcript Sequences
In order to identify VNTRs in coding regions, we analyzed 3D7, HB3, and Dd2 transcript sequences. We generated a genome-wide map informing which 3D7 transcript was homologous to which HB3 or Dd2 transcript using blast. Transcript sequences were analyzed with Tandem Repeats Finder. TRs in homologous transcripts from 3D7/HB3/Dd2 were mapped to each other using blast to compare the TR sequence, and the 30 flanking nucleotides up and down-stream. Only perfect repeats and near perfect repeats ≥12 nt in unit length were used in the TR mapping. If the repeat copy number varied between parasite strains, the TR was considered to be a VNTR. Further details are available in Supplementary materials and methods.
We used the GO Term Enrichment Tool available via GeneDB’s AmiGO interface to assess possible functional implications of VNTRs (Ashburner et al. 2000; Boyle et al. 2004; Hertz-Fowler et al. 2004). Plasmodium falciparum GO annotation v1.62, GOC validated 3/2009 was used.
VNTR Genotyping
Oligonucleotide primers were designed using VectorNTI (Invitrogen, Carlsbad, CA) to amplify and genotype PCR fragments (Supplemental Table S1). Fluorescently labeled primers were obtained from Sigma-Aldrich (St. Louis, MO). Polymerase chain reactions were setup with Phusion Flash High-Fidelity PCR Master Mix (Finnzymes, Inc., Woburn, MA) using two-step protocol cycling conditions. Samples were genotyped in triplicate on a CEQ8000 (Beckman Coulter, Fullerton, CA). Further details are available in Supplementary materials and methods.
Tandem Repeat Flanking Sequence Analysis
The Plasmodium knowlesi genome sequence (Pain et al. 2008) was downloaded from PlasmoDB v5.3. The ME49 Toxoplasma gondii genome sequence, an organism often used as a model for Plasmodium and other apicomplexans (Kim and Weiss 2004), was downloaded from ToxoDB v4.3 (Gajria et al. 2008). TR flanking regions were analyzed to identify sequences that were identical to the start of the repeat, a feature that we refer to as an “overhang.” Using custom perl scripts, we determined the overhang size in nucleotides. We used overhangs ≥4 nt to evaluate the possibility of a sequence repeat arising from slipped strand mispairing for both short and long repeats; using a longer overhang would be poorly suited when applied to short repeats because this would often indicate that the sequence was previously duplicated. Monte-Carlo sampling was used to estimate the genome-wide background incidence of overhangs using 1 million samples. Further details are available in Supplementary materials and methods. The observed number of overhangs present in the TR flanking sequences was compared to the estimated background rate using a chi-square test.
Homologous VNTR loci were identified in Plasmodium reichenowi using the blast interface at PlasmoDB. Sequences with a blast e-value of 0 were aligned to the 3D7 sequence through the EMBOSS Pairwise Alignment Algorithm (Rice et al. 2000): http://www.ebi.ac.uk/Tools/emboss/align/index.html.
Results
In the P. falciparum 3D7 reference genome, 49,798 TRs were identified (1 TR/467 nt). This number includes overlapping repeats such that compound repeats of different periods could be recognized. The distribution of TRs across the 14 chromosomes is shown in Fig. 1. In general, the largest repeats were found near the telomeres, however, a few large repeats are internal on the chromosomes. VNTRs were identified within 489 ORFs (Fig. 1, dark red circles).
Fig. 1.

Positions of TRs across the 3D7 genome. Each circle depicts one TR and its genomic location on the 14 chromosomes. Circle diameter indicates repeat unit size as shown in the legend. Red circles indicate VNTRs identified in gene coding regions
Tandem Repeats in Coding Regions
Tandem repeats with a motif size that is a multiple of three (3× repeats) do not cause a frameshift mutation when repeats are added or deleted. In non-coding regions, roughly 33% of TRs were 3× repeats (as would be expected from a random repeat size distribution); the same was true for introns (Fig. 2). However, in coding regions, nearly 94% of TRs were 3× repeats. The density of 3× repeats was very similar in coding (1 TR/889 nt) and non-coding regions (1 TR/965 nt). When comparing the repeat array length of 3× repeats in coding versus non-coding regions, the coding repeats have a greater mean and median length, and are not likely to be derived from the same distribution as non-coding repeats (mean: 97 vs. 71 and median: 69 vs. 48 in coding vs. non-coding, respectively; P < 0.0001). However, the largest repeats occur more frequently in non-coding regions (Supplemental Fig. S1). At ≥975 nt, the relative frequency of 3× repeats is the same in coding and non-coding regions. The relative frequencies become more non-coding biased at repeat lengths beyond 975 nt and repeat arrays ≥1,500 nt are approximately twice as frequent in non-coding than coding regions. Moreover, TR spatial distribution in genes is not likely to be drawn from a random distribution (P < 0.0001), with TRs observed more frequently in the transcript midsegment and 5′ segment, and less frequently in the 3′ segment than expected by chance (Fig. 3).
Fig. 2.
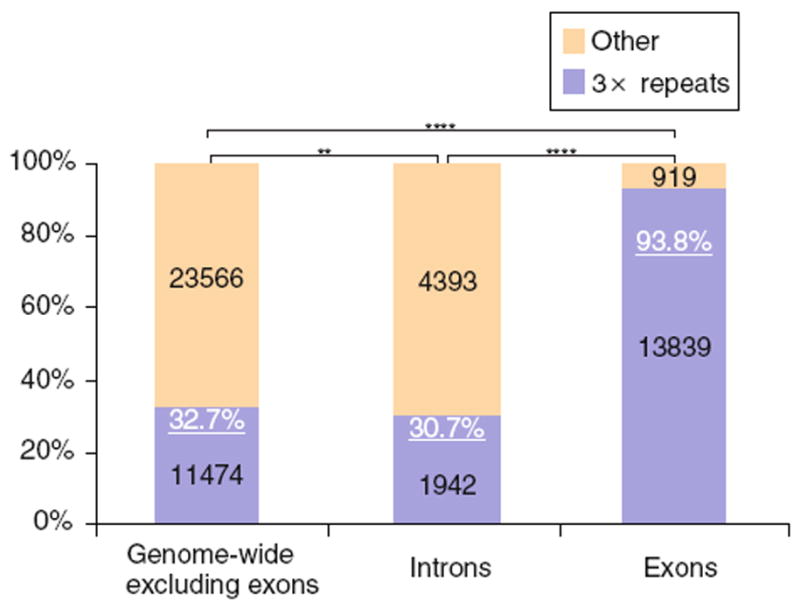
Selection against TRs that cause frameshift mutations. A very high percentage of TRs in exons were 3× repeats. The blue portions of the bars represent the proportion of TRs in each group that are 3× repeats. The black text indicates the number of TRs in each group; the white, underlined text indicates the percentage of 3× repeats for each group. A chi-square test was used to compare the categorical observations (** P < 0.01; **** P < 0.0001)
Fig. 3.
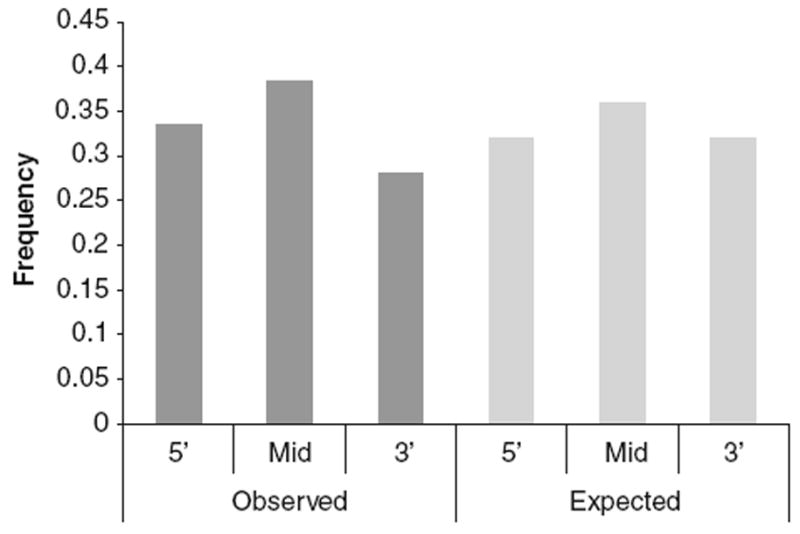
Spatial distribution of TRs in genes. The observed spatial distribution of TRs in genes compared to the expected frequency from a random distribution shows a greater frequency of TRs in the midsegment and 5′ segment than expected, and lower frequency observed in the 3′ segment than expected
Comparative Analysis of 3D7, HB3, and Dd2 Transcript Sequences for VNTRs
The set of predicted transcript sequences for 3D7, HB3, and Dd2 was analyzed for VNTRs in predicted coding regions. Overall, 4484 3D7 transcript sequences mapped to a homolog in HB3 and/or Dd2 and 489 of these harbored VNTRs, approximately 9% of all P. falciparum genes and 11% of 3D7 genes with a mapped homolog. The 489 VNTR genes were analyzed for GO term enrichment, identifying several terms associated with stress/DNA damage responses and chromatin modification (Supplemental Table S2).
Observations of VNTR Loci in Multiple Strains
As an initial exploration into variability and segregation at the population level, we genotyped six predicted VNTRs, chosen randomly, in several parasite lines to represent: the reference genome sequence (3D7); the two parents of a genetic cross (HB3, Dd2); and a drug-selected parasite (CF10) and its progenitor (106/1) (Table 1). In addition, we examined VNTR inheritance of one of these loci (PF14_0538) in progeny of the HB3 × Dd2 genetic cross (Wellems et al. 1990) and 334 Southeast Asian field isolates (Nair et al. 2007); we further validated this locus in 10 of these field isolates by capillary sequencing. Genotyping confirmed the predicted VNTR variability in lab strains and field isolates. Southeast Asian field isolates genotyped at the PF14_0538 locus had various alleles ranging from 1 to 2 repeat copies, to rare alleles with as many as six repeat copies (Supplemental Fig. S2). The flanking region around the repeats was identical in the sequenced field isolates and lab adapted parasites.
Table 1.
VNTR copy number in 3D7, HB3, Dd2, 106/1, and CF10 for six loci
| Repeat unit size (nt) | MAL7P1.145 | PFI1470c | PFE0575c | PF08_0114 | MAL13P1.39 | PF14_0538 |
|---|---|---|---|---|---|---|
| 42 | 54 | 30 | 24 | 42 | 36 | |
| Number of copies | ||||||
| 3D7 | 14 | 4 | 3 | 3 | 4 | 1 |
| HB3 | 8 | 2 | 5 | 6 | 5 | 4 |
| Dd2 | 11 | 2 | 2 | 8 | 6 | 1 |
| 106/1 | 11 | 2 | 4 | 5 | 7 | 1 |
| CF10 | 11 | 2 | 4 | 5 | 7 | 1 |
Six loci identified to harbor VNTRs through bioinformatics were genotyped. Repeat copy number was determined through sequence analysis and confirmed in genotyping experiments
Parasite clone CF10 was derived directly from 106/1 by in vitro drug selection without passage through meiotic stages (Cooper et al. 2002). Genotyping data revealed no differences between 106/1 and CF10, however, of 28 candidate 106/1 polymorphisms identified by microarray-based comparison to the P. falciparum reference genome, capillary sequencing demonstrated that 9 (32%) were due to polymorphic VNTRs.
Tandem Repeat Flanking Sequence Analysis
An analysis of the 33,576 TRs with ≥12 nt unit length revealed 73.4% of those TRs had a flanking sequence overhang at least 4 nt long; when this 73.4% overhang rate is compared to a Monte-Carlo based estimate for genome-wide overhang incidence (10.0%), a chi-square test finds a significant difference (P < 0.0001). Analysis of T. gondii and P. knowlesi genomes yielded similar results (Table 2); other apicomplexan genomes could not be properly analyzed because they were not assembled into chromosomes.
Table 2.
TR overhang analysis in three apicomplexan genomes
| TR density | TRs with overhangs (%) | Background overhang (%) | P value | |
|---|---|---|---|---|
| P. falciparum | 1 TR/467 nt | 73.4 | 10.0 | < 0.0001 |
| P. knowlesi | 1 TR/1,114 nt | 79.5 | 11.5 | < 0.0001 |
| T. gondii | 1 TR/3,578 nt | 73.6 | 2.9 | < 0.0001 |
TR density was calculated in three apicomplexan genomes. Overhang analysis was limited to repeats with unit length ≥ 12 nt to determine what proportion had an overhang at least 4 nt long for the three genomes. This was compared to an estimated background incidence with a chi-square test
These overhang features facilitate mispairing when DNA strands separate, causing the intervening sequence to become deleted or duplicated (Fig. 4). The ancestral state is assumed to be a single copy sequence flanked by a sequence overhang that undergoes subsequent expansion through slipped strand mispairing; in some cases, the sequence may become deleted. When compared to a state that has the repeated sequence, the unrepeated sequence can be considered to have “one copy” of the repeat unit, and the deleted sequence to have “zero copies” of the repeat unit. Polymorphisms in the flanking region will reduce sequence identity in the overhang region, diminishing the chance of mispairing. Several examples from P. falciparum and P. reichenowi sequence data demonstrate these points (Fig. 5); identification of homologous repeat regions through other species in the Plasmodium lineage is prevented by low sequence identity. Figure 5a, b depict VNTR loci in P. falciparum that are unrepeated in the P. reichenowi sequence where SNPs are present in the P. reichenowi overhang sequence reducing sequence identity to the start of the repeat units, eliminating the possibility of repeat generation. Figure 5c illustrates a locus that is deleted in P. reichenowi relative to P. falciparum, where only the overhang sequence remains. Through this slippage mechanism, a sequence that was not originally tandemly repeated can become duplicated to form a TR, or deleted.
Fig. 4.
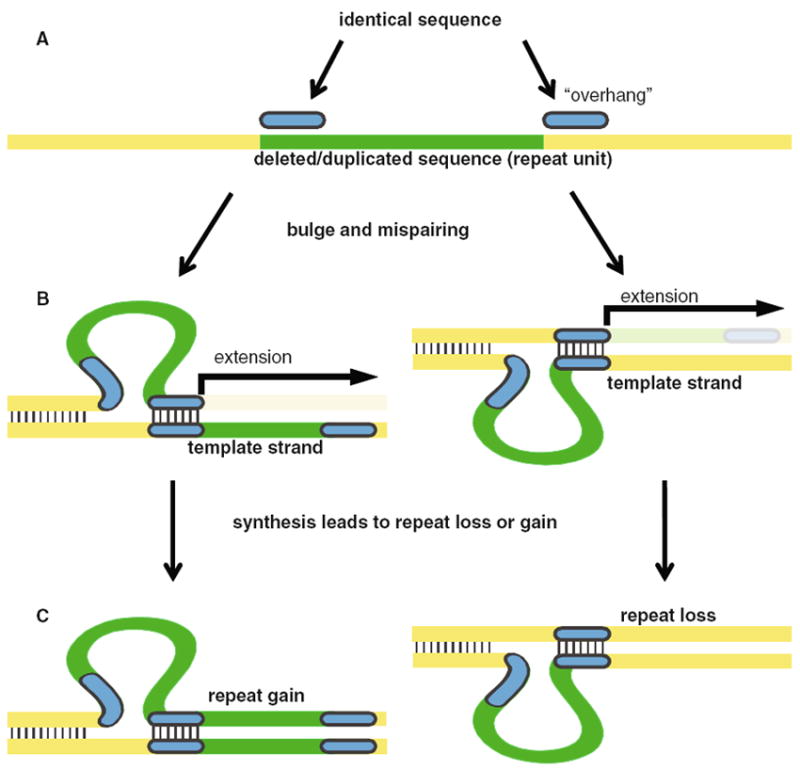
Slipped strand mispairing can generate sequence duplication. A sequence overhang may cause slipped strand mispairing to occur allowing sequence duplication. a No tandemly repeated sequence is present; however, two stretches of non-adjacent identical sequence are present (blue segments). b The overhang in the flanking region can become mispaired with the start region of the repeat unit. This can cause a bulge in the DNA in either the template or nascent strand. c DNA synthesis will lead to duplication or deletion. Duplication will facilitate further mispairing, increasing the likelihood of greater repeat variability
Fig. 5.
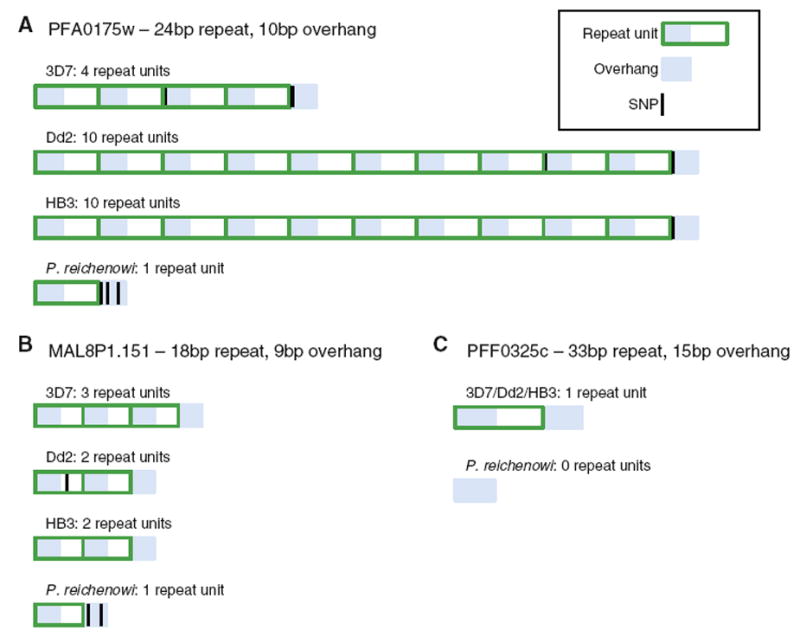
TRs and sequence overhangs in P. falciparum and P. reichenowi. Several VNTR loci from P. falciparum and P. reichenowi illustrate how sequence overhangs may initiate TR repeat generation or sequence deletion. a PFA0175w is a variable locus in P. falciparum where a 24 bp repeat with a 10 bp overhang in the flanking region is present. In P. reichenowi, the sequence is not repeated and the 10 bp overhang contains three SNPs that reduce sequence identity to the potential repeat motif, eliminating the chance for replication slippage to tandemly duplicate the sequence. b MAL8P1.151 contains an 18 bp repeat with a 9 bp overhang. In the homologous P. reichenowi sequence, the 18 bp sequence is not repeated and there are two SNPs in the overhang. c PFF0325c contains an unrepeated 33 bp sequence tduhat is flanked by a 15 bp sequence overhang. In P. reichenowi, the 33 bp sequence is absent, and the flanking 15 bp overhang is all that remains
Discussion
We identified a greater percentage of genes (9%) containing VNTR polymorphisms in the P. falciparum genome than has been reported for any other species to date (Bowen et al. 2005; Coil et al. 2008; Jordan et al. 2003; Levdansky et al. 2007; O’Dushlaine et al. 2005; Verstrepen et al. 2005). These current findings are expected to be an underestimate due to several factors: (1) This analysis compared three genomes and we expect variation in the genomes of other parasite strains; (2) 1,111 or 20% of the predicted 3D7 P. falciparum transcript sequences could not be mapped to a homolog; (3) TRs were bioinformatically recognized in each individual parasite sequence and subsequently compared for copy number, causing us to miss examples of one or zero TR “copies” (i.e., a sequence present zero or one times is not recognized as a TR in the initial bioinformatic step); (4) The VNTR analysis was limited to repeat sizes that would be considered minisatellites (≥12 nt repeat unit) to monitor how variable larger repeats are. Smaller TRs such as a 3 nt repeated unit in pfmdr1 that is variable between 3D7, HB3, and Dd2 were not tallied in this analysis. In addition to VNTRs, P. falciparum has the greatest intragenic TR content reported for any species, including fourfold higher than T. gondii, and 10- to 150-fold greater than nine other genomes surveyed (Goto et al. 2008).
Gene ontology enrichment analysis of the 489 genes containing VNTRs identified several overrepresented terms associated with stress or DNA damage responses, or chromatin modification—which influences transcription, DNA repair, and chromosome condensation (Kouzarides 2007). Copy number variation is important in P. falciparum drug resistance evolution (Carret et al. 2005; Cheeseman et al. 2009; Kidgell et al. 2006; Nair et al. 2007), and it could be a component of the accelerated resistance to multiple drugs (ARMD) phenotype reported by Rathod et al. 1997. Previous studies suggest defective DNA repair and nucleotide excision or mismatch repair in particular may be a potential mechanism for ARMD (Trotta et al. 2004) or generating genetic diversity (Bethke et al. 2007). We note that an association of VNTRs with stress or DNA damage response, or chromatin modification functions could suggest mechanisms that influence adaptive potential as has been described for other microorganisms (Chopra et al. 2003; Denamur and Matic 2006; Foster 2007). ARMD is a strain-specific phenotype, and it is plausible that mechanisms driving TR generation could also be strain specific.
We observed dense, fairly uniform TR distribution across the 14 P. falciparum chromosomes (Fig. 1). Concordant with yeast studies (Richard and Dujon 2006), we found no association between minisatellites and meiotic recombination hot spots. In coding regions, we observed strong selection against frameshift mutations as has been reported in other species (Metzgar et al. 2000; Morgante et al. 2002; Toth et al. 2000), and see no evidence for selection against in-frame TRs that effectively lengthen the amino acid sequence until the repeat array reaches lengths surpassing 975 nt.
Unique to P. falciparum, intragenic TRs are more frequent in the middle of transcript encoding regions. Studies in other species find repeat sequences overrepresented in the terminal segments (Alba and Guigo 2004; Huntley and Clark 2007; Siwach et al. 2006). However, due to the focus on homopeptide tracts of individual amino acids or low complexity regions, the studies may not be directly comparable with our nucleotide-level TR analysis. A more comparable nucleotide-level TR analysis of multiple Aspergillus genomes found TR spatial distributions that did not significantly deviate from a random distribution with the exception of Aspergillus niger (Gibbons and Rokas 2009). An underlying cause for spatial bias of repeats is currently unknown.
We genotyped six coding VNTRs through DNA fragment length analysis in different parasite populations. Each of these repeats was unique to one locus in the genome so they were not common low complexity regions. The VNTRs were variable in lab lines and field samples, but were stable over a 2-month period of drug selection and in vitro cultivation. Furthermore, these TRs were inherited in a Mendelian fashion in progeny of a genetic cross indicating short-term stability through meiosis and mitosis (data not shown). More extensive studies will be required to determine the stability of VNTRs and the rate at which these mutations occur, however, other studies indicate that indels/TRs provide at least as much polymorphism as SNPs in P. falciparum (Volkman et al. 2007a, b).
A common feature in the majority of TR flanking regions indicates TR generation through slipped strand mispairing. TRs with short unit lengths may occur by chance, but as repeat unit length increases, the chance of finding a tandemly repeated sequence decreases exponentially. Multiple examples of long, tandemly repeated sequences suggest a mechanism causes localized duplication of a single, originally unrepeated sequence to create a TR. Overhang sequences commonly found in TR flanking regions allow mispairing to occur (Fig. 4b), causing the intervening sequence to become deleted or duplicated (Fig. 4c). Through this slipped strand mispairing, a sequence that was not tandemly repeated can become duplicated locally to form a TR. Electron micrographs provide direct visualization of bulges and knots in DNA TR regions over 4 kb segments (Coggins and O’Prey 1989) demonstrating that dissociation and mispairing can occur over kilobase-sized regions. This predicts that two identical sequence patches can lead to duplication or deletion of the intervening sequence. P. reichenowi is the closest relative of P. falciparum (Escalante and Ayala 1994; Escalante et al. 1995) and the current understanding is that P. falciparum originated from P. reichenowi (Rich et al. 2009). Examples comparing P. falciparum and P. reichenowi sequence data exhibit behavior consistent with a role for sequence overhangs in TR generation (Fig. 5). The sequence overhang is necessary only for the initial TR formation after which TR units allow unequal crossover or further mispairing to alter the number of repeats (Chambers and MacAvoy 2000; Jeffreys et al. 1999; Levinson and Gutman 1987). Nearly, 75% of P. falciparum TRs demonstrate sequence overhangs and with more extensive genome sequence information from multiple parasite strains that will emerge from population surveys that use high-throughput sequencing, it will be possible to deduce the minimum overhang requirements for size, sequence identity, or maximum unit length.
Conclusions
Traditionally, investigations have focused on SNPs or copy number polymorphisms when investigating phenotypes of interest. If unaccounted for, these VNTR polymorphisms may cause complications in genomics studies such as in SNP-typing microarrays or when mapping short-sequence read data to a reference. However, they may also be incorporated into other genomics studies since VNTR polymorphisms are detectable through comparative genomic hybridization microarrays (Tan et al. 2009). TRs may affect primary protein sequence, transcript splicing, or gene expression levels but (excluding microsatellites) have not been extensively characterized in P. falciparum. It appears that there are no restrictions on TRs in coding regions other than selection against frameshift mutation, and selection against TRs with a total length above 975 nt. We find that TRs, which may play adaptive roles to selective pressures through DNA damage responses or immune evasion, are more abundant and variable than in other genomes. The interstrain variation caused by VNTRs is greater in P. falciparum than other organisms analyzed to date. Overhang sequences found in the majority of TR flanking regions provide support for repeat generation from a single source unit through slipped strand mispairing. This predicts that TRs may be generated at any sequence stretch flanked by a sequence overhang. We conclude that TRs are an underappreciated source of gene variation with previously unrecognized abundance and variability that must be accounted for in addition to SNPs and traditional copy number polymorphisms.
Supplementary Material
Acknowledgments
This study was supported by the National Institutes of Health (AI071121 and AI075145 to M.T.F), and an Arthur J. Schmitt Presidential Fellowship (to J.C.T.). We are grateful to F. Nosten, the Shoklo Malaria Research Unit, and T. Anderson for providing DNA samples, and the Broad Institute and Sanger Pathogen Sequencing Unit for making genome sequence data publicly available prior to publication. We acknowledge the Notre Dame Genomics Core Facility for excellent technical support.
Footnotes
Electronic supplementary material The online version of this article (doi:10.1007/s00239-010-9381-8) contains supplementary material, which is available to authorized users.
Contributor Information
John C. Tan, Email: jtan1@nd.edu.
Michael T. Ferdig, Email: ferdig.1@nd.edu.
References
- Alba MM, Guigo R. Comparative analysis of amino acid repeats in rodents and humans. Genome Res. 2004;14:549–554. doi: 10.1101/gr.1925704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson TJ, Su XZ, Roddam A, Day KP. Complex mutations in a high proportion of microsatellite loci from the protozoan parasite Plasmodium falciparum. Mol Ecol. 2000;9:1599–1608. doi: 10.1046/j.1365-294x.2000.01057.x. [DOI] [PubMed] [Google Scholar]
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahl A, Brunk B, Crabtree J, Fraunholz MJ, Gajria B, Grant GR, Ginsburg H, Gupta D, Kissinger JC, Labo P, Li L, Mailman MD, Milgram AJ, Pearson DS, Roos DS, Schug J, Stoeckert CJ, Jr, Whetzel P. PlasmoDB: the Plasmodium genome resource. A database integrating experimental and computational data. Nucleic Acids Res. 2003;31:212–215. doi: 10.1093/nar/gkg081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 1999;27:573–580. doi: 10.1093/nar/27.2.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bethke L, Thomas S, Walker K, Lakhia R, Rangarajan R, Wirth D. The role of DNA mismatch repair in generating genetic diversity and drug resistance in malaria parasites. Mol Biochem Parasitol. 2007;155:18–25. doi: 10.1016/j.molbiopara.2007.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowen S, Roberts C, Wheals AE. Patterns of polymorphism and divergence in stress-related yeast proteins. Yeast. 2005;22:659–668. doi: 10.1002/yea.1240. [DOI] [PubMed] [Google Scholar]
- Bowman S, Lawson D, Basham D, Brown D, Chillingworth T, Churcher CM, Craig A, Davies RM, Devlin K, Feltwell T, Gentles S, Gwilliam R, Hamlin N, Harris D, Holroyd S, Hornsby T, Horrocks P, Jagels K, Jassal B, Kyes S, McLean J, Moule S, Mungall K, Murphy L, Oliver K, Quail MA, Rajandream MA, Rutter S, Skelton J, Squares R, Squares S, Sulston JE, Whitehead S, Woodward JR, Newbold C, Barrell BG. The complete nucleotide sequence of chromosome 3 of Plasmodium falciparum. Nature. 1999;400:532–538. doi: 10.1038/22964. [DOI] [PubMed] [Google Scholar]
- Boyle EI, Weng S, Gollub J, Jin H, Botstein D, Cherry JM, Sherlock G. GO:TermFinder—open source software for accessing Gene Ontology information and finding significantly enriched Gene Ontology terms associated with a list of genes. Bioinformatics. 2004;20:3710–3715. doi: 10.1093/bioinformatics/bth456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carret CK, Horrocks P, Konfortov B, Winzeler E, Qureshi M, Newbold C, Ivens A. Microarray-based comparative genomic analyses of the human malaria parasite Plasmodium falciparum using Affymetrix arrays. Mol Biochem Parasitol. 2005;144:177–186. doi: 10.1016/j.molbiopara.2005.08.010. [DOI] [PubMed] [Google Scholar]
- Chambers GK, MacAvoy ES. Microsatellites: consensus and controversy. Comp Biochem Physiol B Biochem Mol Biol. 2000;126:455–476. doi: 10.1016/s0305-0491(00)00233-9. [DOI] [PubMed] [Google Scholar]
- Cheeseman IH, Gomez-Escobar N, Carret CK, Ivens A, Stewart LB, Tetteh KK, Conway DJ. Gene copy number variation throughout the Plasmodium falciparum genome. BMC Genomics. 2009;10:353. doi: 10.1186/1471-2164-10-353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chopra I, O’Neill AJ, Miller K. The role of mutators in the emergence of antibiotic-resistant bacteria. Drug Resist Updat. 2003;6:137–145. doi: 10.1016/s1368-7646(03)00041-4. [DOI] [PubMed] [Google Scholar]
- Coggins LW, O’Prey M. DNA tertiary structures formed in vitro by misaligned hybridization of multiple tandem repeat sequences. Nucleic Acids Res. 1989;17:7417–7426. doi: 10.1093/nar/17.18.7417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coil DA, Vandersmissen L, Ginevra C, Jarraud S, Lammertyn E, Anne J. Intragenic tandem repeat variation between Legionella pneumophila strains. BMC Microbiol. 2008;8:218. doi: 10.1186/1471-2180-8-218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad DF, Pinto D, Redon R, Feuk L, Gokcumen O, Zhang Y, Aerts J, Andrews TD, Barnes C, Campbell P, Fitzgerald T, Hu M, Ihm CH, Kristiansson K, Macarthur DG, Macdonald JR, Onyiah I, Pang AW, Robson S, Stirrups K, Valsesia A, Walter K, Wei J, Tyler-Smith C, Carter NP, Lee C, Scherer SW, Hurles ME. Origins and functional impact of copy number variation in the human genome. Nature. 2010;464(7289):704–712. doi: 10.1038/nature08516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper RA, Ferdig MT, Su XZ, Ursos LM, Mu J, Nomura T, Fujioka H, Fidock DA, Roepe PD, Wellems TE. Alternative mutations at position 76 of the vacuolar transmembrane protein PfCRT are associated with chloroquine resistance and unique stereospecific quinine and quinidine responses in Plasmodium falciparum. Mol Pharmacol. 2002;61:35–42. doi: 10.1124/mol.61.1.35. [DOI] [PubMed] [Google Scholar]
- Cowman AF, Coppel RL, Saint RB, Favaloro J, Crewther PE, Stahl HD, Bianco AE, Brown GV, Anders RF, Kemp DJ. The ring-infected erythrocyte surface antigen (RESA) polypeptide of Plasmodium falciparum contains two separate blocks of tandem repeats encoding antigenic epitopes that are naturally immunogenic in man. Mol Biol Med. 1984;2:207–221. [PubMed] [Google Scholar]
- Dame JB, Williams JL, McCutchan TF, Weber JL, Wirtz RA, Hockmeyer WT, Maloy WL, Haynes JD, Schneider I, Roberts D, et al. Structure of the gene encoding the immunodominant surface antigen on the sporozoite of the human malaria parasite Plasmodium falciparum. Science. 1984;225:593–599. doi: 10.1126/science.6204383. [DOI] [PubMed] [Google Scholar]
- Denamur E, Matic I. Evolution of mutation rates in bacteria. Mol Microbiol. 2006;60:820–827. doi: 10.1111/j.1365-2958.2006.05150.x. [DOI] [PubMed] [Google Scholar]
- Edwards YJ, Elgar G, Clark MS, Bishop MJ. The identification and characterization of microsatellites in the compact genome of the Japanese pufferfish, Fugu rubripes: perspectives in functional and comparative genomic analyses. J Mol Biol. 1998;278:843–854. doi: 10.1006/jmbi.1998.1752. [DOI] [PubMed] [Google Scholar]
- Ellegren H. Microsatellites: simple sequences with complex evolution. Nat Rev Genet. 2004;5:435–445. doi: 10.1038/nrg1348. [DOI] [PubMed] [Google Scholar]
- Escalante AA, Ayala FJ. Phylogeny of the malarial genus Plasmodium, derived from rRNA gene sequences. Proc Natl Acad Sci USA. 1994;91:11373–11377. doi: 10.1073/pnas.91.24.11373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escalante AA, Barrio E, Ayala FJ. Evolutionary origin of human and primate malarias: evidence from the circumsporozoite protein gene. Mol Biol Evol. 1995;12:616–626. doi: 10.1093/oxfordjournals.molbev.a040241. [DOI] [PubMed] [Google Scholar]
- Figueiredo LM, Pirrit LA, Scherf A. Genomic organisation and chromatin structure of Plasmodium falciparum chromosome ends. Mol Biochem Parasitol. 2000;106:169–174. doi: 10.1016/s0166-6851(99)00199-1. [DOI] [PubMed] [Google Scholar]
- Fondon JW, 3rd, Garner HR. Molecular origins of rapid and continuous morphological evolution. Proc Natl Acad Sci USA. 2004;101:18058–18063. doi: 10.1073/pnas.0408118101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster PL. Stress-induced mutagenesis in bacteria. Crit Rev Biochem Mol Biol. 2007;42:373–397. doi: 10.1080/10409230701648494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gajria B, Bahl A, Brestelli J, Dommer J, Fischer S, Gao X, Heiges M, Iodice J, Kissinger JC, Mackey AJ, Pinney DF, Roos DS, Stoeckert CJ, Jr, Wang H, Brunk BP. ToxoDB: an integrated Toxoplasma gondii database resource. Nucleic Acids Res. 2008;36:D553–D556. doi: 10.1093/nar/gkm981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner MJ, Tettelin H, Carucci DJ, Cummings LM, Aravind L, Koonin EV, Shallom S, Mason T, Yu K, Fujii C, Pederson J, Shen K, Jing J, Aston C, Lai Z, Schwartz DC, Pertea M, Salzberg S, Zhou L, Sutton GG, Clayton R, White O, Smith HO, Fraser CM, Adams MD, Venter JC, Hoffman SL. Chromosome 2 sequence of the human malaria parasite Plasmodium falciparum. Science. 1998;282:1126–1132. doi: 10.1126/science.282.5391.1126. [DOI] [PubMed] [Google Scholar]
- Gardner MJ, Hall N, Fung E, White O, Berriman M, Hyman RW, Carlton JM, Pain A, Nelson KE, Bowman S, Paulsen IT, James K, Eisen JA, Rutherford K, Salzberg SL, Craig A, Kyes S, Chan MS, Nene V, Shallom SJ, Suh B, Peterson J, Angiuoli S, Pertea M, Allen J, Selengut J, Haft D, Mather MW, Vaidya AB, Martin DM, Fairlamb AH, Fraunholz MJ, Roos DS, Ralph SA, McFadden GI, Cummings LM, Subramanian GM, Mungall C, Venter JC, Carucci DJ, Hoffman SL, Newbold C, Davis RW, Fraser CM, Barrell B. Genome sequence of the human malaria parasite Plasmodium falciparum. Nature. 2002;419:498–511. doi: 10.1038/nature01097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbons JG, Rokas A. Comparative and functional characterization of intragenic tandem repeats in 10 Aspergillus genomes. Mol Biol Evol. 2009;26:591–602. doi: 10.1093/molbev/msn277. [DOI] [PubMed] [Google Scholar]
- Goto Y, Carter D, Reed SG. Immunological dominance of Trypanosoma cruzi tandem repeat proteins. Infect Immun. 2008;76:3967–3974. doi: 10.1128/IAI.00604-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hancock JM, Simon M. Simple sequence repeats in proteins and their significance for network evolution. Gene. 2005;345:113–118. doi: 10.1016/j.gene.2004.11.023. [DOI] [PubMed] [Google Scholar]
- Hertz-Fowler C, Peacock CS, Wood V, Aslett M, Kerhornou A, Mooney P, Tivey A, Berriman M, Hall N, Rutherford K, Parkhill J, Ivens AC, Rajandream MA, Barrell B. GeneDB: a resource for prokaryotic and eukaryotic organisms. Nucleic Acids Res. 2004;32:D339–D343. doi: 10.1093/nar/gkh007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes AL. The evolution of amino acid repeat arrays in Plasmodium and other organisms. J Mol Evol. 2004;59:528–535. doi: 10.1007/s00239-004-2645-4. [DOI] [PubMed] [Google Scholar]
- Huntley MA, Clark AG. Evolutionary analysis of amino acid repeats across the genomes of 12 Drosophila species. Mol Biol Evol. 2007;24:2598–2609. doi: 10.1093/molbev/msm129. [DOI] [PubMed] [Google Scholar]
- Jeffares DC, Pain A, Berry A, Cox AV, Stalker J, Ingle CE, Thomas A, Quail MA, Siebenthall K, Uhlemann AC, Kyes S, Krishna S, Newbold C, Dermitzakis ET, Berriman M. Genome variation and evolution of the malaria parasite Plasmodium falciparum. Nat Genet. 2007;39:120–125. doi: 10.1038/ng1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffreys AJ, Barber R, Bois P, Buard J, Dubrova YE, Grant G, Hollies CR, May CA, Neumann R, Panayi M, Ritchie AE, Shone AC, Signer E, Stead JD, Tamaki K. Human minisatellites, repeat DNA instability and meiotic recombination. Electrophoresis. 1999;20:1665–1675. doi: 10.1002/(SICI)1522-2683(19990101)20:8<1665::AID-ELPS1665>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- Jordan P, Snyder LA, Saunders NJ. Diversity in coding tandem repeats in related Neisseria spp. BMC Microbiol. 2003;3:23. doi: 10.1186/1471-2180-3-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemp DJ, Coppel RL, Anders RF. Repetitive proteins and genes of malaria. Annu Rev Microbiol. 1987;41:181–208. doi: 10.1146/annurev.mi.41.100187.001145. [DOI] [PubMed] [Google Scholar]
- Kenneson A, Zhang F, Hagedorn CH, Warren ST. Reduced FMRP and increased FMR1 transcription is proportionally associated with CGG repeat number in intermediate-length and premutation carriers. Hum Mol Genet. 2001;10:1449–1454. doi: 10.1093/hmg/10.14.1449. [DOI] [PubMed] [Google Scholar]
- Kidgell C, Volkman SK, Daily J, Borevitz JO, Plouffe D, Zhou Y, Johnson JR, Le Roch K, Sarr O, Ndir O, Mboup S, Batalov S, Wirth DF, Winzeler EA. A systematic map of genetic variation in Plasmodium falciparum. PLoS Pathog. 2006;2:e57. doi: 10.1371/journal.ppat.0020057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K, Weiss LM. Toxoplasma gondii: the model apicomplexan. Int J Parasitol. 2004;34:423–432. doi: 10.1016/j.ijpara.2003.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kochan J, Perkins M, Ravetch JV. A tandemly repeated sequence determines the binding domain for an erythrocyte receptor binding protein of P falciparum. Cell. 1986;44:689–696. doi: 10.1016/0092-8674(86)90834-2. [DOI] [PubMed] [Google Scholar]
- Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- Levdansky E, Romano J, Shadkchan Y, Sharon H, Verstrepen KJ, Fink GR, Osherov N. Coding tandem repeats generate diversity in Aspergillus fumigatus genes. Eukaryot Cell. 2007;6:1380–1391. doi: 10.1128/EC.00229-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levinson G, Gutman GA. Slipped-strand mispairing: a major mechanism for DNA sequence evolution. Mol Biol Evol. 1987;4:203–221. doi: 10.1093/oxfordjournals.molbev.a040442. [DOI] [PubMed] [Google Scholar]
- Metzgar D, Bytof J, Wills C. Selection against frameshift mutations limits microsatellite expansion in coding DNA. Genome Res. 2000;10:72–80. [PMC free article] [PubMed] [Google Scholar]
- Morgante M, Hanafey M, Powell W. Microsatellites are preferentially associated with nonrepetitive DNA in plant genomes. Nat Genet. 2002;30:194–200. doi: 10.1038/ng822. [DOI] [PubMed] [Google Scholar]
- Mu J, Awadalla P, Duan J, McGee KM, Keebler J, Seydel K, McVean GA, Su XZ. Genome-wide variation and identification of vaccine targets in the Plasmodium falciparum genome. Nat Genet. 2007;39:126–130. doi: 10.1038/ng1924. [DOI] [PubMed] [Google Scholar]
- Nair S, Nash D, Sudimack D, Jaidee A, Barends M, Uhlemann AC, Krishna S, Nosten F, Anderson TJ. Recurrent gene amplification and soft selective sweeps during evolution of multidrug resistance in malaria parasites. Mol Biol Evol. 2007;24:562–573. doi: 10.1093/molbev/msl185. [DOI] [PubMed] [Google Scholar]
- O’Donnell RA, Freitas-Junior LH, Preiser PR, Williamson DH, Duraisingh M, McElwain TF, Scherf A, Cowman AF, Crabb BS. A genetic screen for improved plasmid segregation reveals a role for Rep20 in the interaction of Plasmodium falciparum chromosomes. EMBO J. 2002;21:1231–1239. doi: 10.1093/emboj/21.5.1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Dushlaine CT, Edwards RJ, Park SD, Shields DC. Tandem repeat copy-number variation in protein-coding regions of human genes. Genome Biol. 2005;6:R69. doi: 10.1186/gb-2005-6-8-r69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oquendo P, Goman M, Mackay M, Langsley G, Walliker D, Scaife J. Characterisation of a repetitive DNA sequence from the malaria parasite, Plasmodium falciparum. Mol Biochem Parasitol. 1986;18:89–101. doi: 10.1016/0166-6851(86)90053-8. [DOI] [PubMed] [Google Scholar]
- Pagani F, Buratti E, Stuani C, Romano M, Zuccato E, Niksic M, Giglio L, Faraguna D, Baralle FE. Splicing factors induce cystic fibrosis transmembrane regulator exon 9 skipping through a nonevolutionary conserved intronic element. J Biol Chem. 2000;275:21041–21047. doi: 10.1074/jbc.M910165199. [DOI] [PubMed] [Google Scholar]
- Pain A, Bohme U, Berry AE, Mungall K, Finn RD, Jackson AP, Mourier T, Mistry J, Pasini EM, Aslett MA, Balasubrammaniam S, Borgwardt K, Brooks K, Carret C, Carver TJ, Cherevach I, Chillingworth T, Clark TG, Galinski MR, Hall N, Harper D, Harris D, Hauser H, Ivens A, Janssen CS, Keane T, Larke N, Lapp S, Marti M, Moule S, Meyer IM, Ormond D, Peters N, Sanders M, Sanders S, Sargeant TJ, Simmonds M, Smith F, Squares R, Thurston S, Tivey AR, Walker D, White B, Zuiderwijk E, Churcher C, Quail MA, Cowman AF, Turner CM, Rajandream MA, Kocken CH, Thomas AW, Newbold CI, Barrell BG, Berriman M. The genome of the simian and human malaria parasite Plasmodium knowlesi. Nature. 2008;455:799–803. doi: 10.1038/nature07306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pizzi E, Frontali C. Low-complexity regions in Plasmodium falciparum proteins. Genome Res. 2001;11:218–229. doi: 10.1101/gr.152201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rando OJ, Verstrepen KJ. Timescales of genetic and epigenetic inheritance. Cell. 2007;128:655–668. doi: 10.1016/j.cell.2007.01.023. [DOI] [PubMed] [Google Scholar]
- Rathod PK, McErlean T, Lee PC. Variations in frequencies of drug resistance in Plasmodium falciparum. Proc Natl Acad Sci USA. 1997;94:9389–9393. doi: 10.1073/pnas.94.17.9389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribacke U, Mok BW, Wirta V, Normark J, Lundeberg J, Kironde F, Egwang TG, Nilsson P, Wahlgren M. Genome wide gene amplifications and deletions in Plasmodium falciparum. Mol Biochem Parasitol. 2007;155:33–44. doi: 10.1016/j.molbiopara.2007.05.005. [DOI] [PubMed] [Google Scholar]
- Rice P, Longden I, Bleasby A. EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet. 2000;16:276–277. doi: 10.1016/s0168-9525(00)02024-2. [DOI] [PubMed] [Google Scholar]
- Rich SM, Ayala FJ. Population structure and recent evolution of Plasmodium falciparum. Proc Natl Acad Sci USA. 2000;97:6994–7001. doi: 10.1073/pnas.97.13.6994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rich SM, Leendertz FH, Xu G, LeBreton M, Djoko CF, Aminake MN, Takang EE, Diffo JL, Pike BL, Rosenthal BM, Formenty P, Boesch C, Ayala FJ, Wolfe ND. The origin of malignant malaria. Proc Natl Acad Sci USA. 2009;106:14902–14907. doi: 10.1073/pnas.0907740106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richard GF, Dujon B. Molecular evolution of minisatellites in hemiascomycetous yeasts. Mol Biol Evol. 2006;23:189–202. doi: 10.1093/molbev/msj022. [DOI] [PubMed] [Google Scholar]
- Schlotterer C. The evolution of molecular markers—just a matter of fashion? Nat Rev Genet. 2004;5:63–69. doi: 10.1038/nrg1249. [DOI] [PubMed] [Google Scholar]
- Schofield L. On the function of repetitive domains in protein antigens of Plasmodium and other eukaryotic parasites. Parasitol Today. 1991;7:99–105. doi: 10.1016/0169-4758(91)90166-l. [DOI] [PubMed] [Google Scholar]
- Siwach P, Pophaly SD, Ganesh S. Genomic and evolutionary insights into genes encoding proteins with single amino acid repeats. Mol Biol Evol. 2006;23:1357–1369. doi: 10.1093/molbev/msk022. [DOI] [PubMed] [Google Scholar]
- Snell RG, MacMillan JC, Cheadle JP, Fenton I, Lazarou LP, Davies P, MacDonald ME, Gusella JF, Harper PS, Shaw DJ. Relationship between trinucleotide repeat expansion and phenotypic variation in Huntington’s disease. Nat Genet. 1993;4:393–397. doi: 10.1038/ng0893-393. [DOI] [PubMed] [Google Scholar]
- Stahl HD, Crewther PE, Anders RF, Brown GV, Coppel RL, Bianco AE, Mitchell GF, Kemp DJ. Interspersed blocks of repetitive and charged amino acids in a dominant immunogen of Plasmodium falciparum. Proc Natl Acad Sci USA. 1985;82:543–547. doi: 10.1073/pnas.82.2.543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su X, Wellems TE. Toward a high-resolution Plasmodium falciparum linkage map: polymorphic markers from hundreds of simple sequence repeats. Genomics. 1996;33:430–444. doi: 10.1006/geno.1996.0218. [DOI] [PubMed] [Google Scholar]
- Su X, Ferdig MT, Huang Y, Huynh CQ, Liu A, You J, Wootton JC, Wellems TE. A genetic map and recombination parameters of the human malaria parasite Plasmodium falciparum. Science. 1999;286:1351–1353. doi: 10.1126/science.286.5443.1351. [DOI] [PubMed] [Google Scholar]
- Tan JC, Patel JJ, Tan A, Blain JC, Albert TJ, Lobo NF, Ferdig MT. Optimizing comparative genomic hybridization probes for genotyping and SNP detection in Plasmodium falciparum. Genomics. 2009;93:543–550. doi: 10.1016/j.ygeno.2009.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toth G, Gaspari Z, Jurka J. Microsatellites in different eukaryotic genomes: survey and analysis. Genome Res. 2000;10:967–981. doi: 10.1101/gr.10.7.967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Triglia T, Stahl HD, Crewther PE, Scanlon D, Brown GV, Anders RF, Kemp DJ. The complete sequence of the gene for the knob-associated histidine-rich protein from Plasmodium falciparum. EMBO J. 1987;6:1413–1419. doi: 10.1002/j.1460-2075.1987.tb02382.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trotta RF, Brown ML, Terrell JC, Geyer JA. Defective DNA repair as a potential mechanism for the rapid development of drug resistance in Plasmodium falciparum. Biochemistry. 2004;43:4885–4891. doi: 10.1021/bi0499258. [DOI] [PubMed] [Google Scholar]
- van Belkum A, Scherer S, van Alphen L, Verbrugh H. Short-sequence DNA repeats in prokaryotic genomes. Microbiol Mol Biol Rev. 1998;62:275–293. doi: 10.1128/mmbr.62.2.275-293.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verstrepen KJ, Jansen A, Lewitter F, Fink GR. Intragenic tandem repeats generate functional variability. Nat Genet. 2005;37:986–990. doi: 10.1038/ng1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinayak S, Mittra P, Sharma YD. Wide variation in microsatellite sequences within each Pfcrt mutant haplotype. Mol Biochem Parasitol. 2006;147:101–108. doi: 10.1016/j.molbiopara.2006.01.013. [DOI] [PubMed] [Google Scholar]
- Vinces MD, Legendre M, Caldara M, Hagihara M, Verstrepen KJ. Unstable tandem repeats in promoters confer transcriptional evolvability. Science. 2009;324:1213–1216. doi: 10.1126/science.1170097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkman SK, Lozovsky E, Barry AE, Bedford T, Bethke L, Myrick A, Day KP, Hartl DL, Wirth DF, Sawyer SA. Genomic heterogeneity in the density of noncoding single-nucleotide and microsatellite polymorphisms in Plasmodium falciparum. Gene. 2007a;387:1–6. doi: 10.1016/j.gene.2006.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkman SK, Sabeti PC, DeCaprio D, Neafsey DE, Schaffner SF, Milner DA, Jr, Daily JP, Sarr O, Ndiaye D, Ndir O, Mboup S, Duraisingh MT, Lukens A, Derr A, Stange-Thomann N, Waggoner S, Onofrio R, Ziaugra L, Mauceli E, Gnerre S, Jaffe DB, Zainoun J, Wiegand RC, Birren BW, Hartl DL, Galagan JE, Lander ES, Wirth DF. A genome-wide map of diversity in Plasmodium falciparum. Nat Genet. 2007b;39:113–119. doi: 10.1038/ng1930. [DOI] [PubMed] [Google Scholar]
- Wang Z, Weber JL, Zhong G, Tanksley SD. Survey of plant short tandem DNA repeats. Theor Appl Genet. 1994;88:1–6. doi: 10.1007/BF00222386. [DOI] [PubMed] [Google Scholar]
- Wellems TE, Panton LJ, Gluzman IY, do Rosario VE, Gwadz RW, Walker-Jonah A, Krogstad DJ. Chloroquine resistance not linked to mdr-like genes in a Plasmodium falciparum cross. Nature. 1990;345:253–255. doi: 10.1038/345253a0. [DOI] [PubMed] [Google Scholar]
- Whetstine JR, Witt TL, Matherly LH. The human reduced folate carrier gene is regulated by the AP2 and sp1 transcription factor families and a functional 61-base pair polymorphism. J Biol Chem. 2002;277:43873–43880. doi: 10.1074/jbc.M208296200. [DOI] [PubMed] [Google Scholar]
- WHO. World malaria report 2009. World Health Organization; Geneva: 2009. [Google Scholar]
- Wootton JC, Federhen S. Analysis of compositionally biased regions in sequence databases. Meth Enzymol. 1996;266:554–571. doi: 10.1016/s0076-6879(96)66035-2. [DOI] [PubMed] [Google Scholar]
- Ye J, McGinnis S, Madden TL. BLAST: improvements for better sequence analysis. Nucleic Acids Res. 2006;34:W6–W9. doi: 10.1093/nar/gkl164. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.