

Abstract
In this study we investigated the strengths and modes of selection associated with nucleosome positioning in the human lineage through the comparison of interspecies and intraspecies rates of divergence. We identify significant evidence for both positive and negative selection linked to human nucleosome positioning for the first time, implicating a widespread and important role for DNA sequence in the location of well-positioned nucleosomes. Selection appears to be acting on particular base substitutions to maintain optimum GC compositions in core and linker regions, with, e.g., unexpectedly elevated rates of C→T substitutions during recent human evolution at linker regions 60–90 bp from the nucleosome dyad but significant depletion of the same substitutions within nucleosome core regions. These patterns are strikingly consistent with the known relationships between genomic sequence composition and nucleosome assembly. By stratifying nucleosomes according to the GC content of their genomic neighborhood, we also show that the strength and direction of selection detected is dictated by local GC content. Intriguingly these signatures of selection are not restricted to nucleosomes in close proximity to exons, suggesting the correct positioning of nucleosomes is not only important in and around coding regions. This analysis provides strong evidence that the genomic sequences associated with nucleosomes are not evolving neutrally, and suggests that underlying DNA sequence is an important factor in nucleosome positioning. Recent signatures of selection linked to genomic features as ubiquitous as the nucleosome have important implications for human genome evolution and disease.
The fundamental level of chromatin compaction in the nucleus is the nucleosome, consisting of ∼147 bp of DNA wrapped around a histone octamer, with adjacent nucleosomes separated by variable length DNA linker sequences generally falling in the range of 20–80 bp. Due to the inherent inaccessibility of DNA compacted onto nucleosomes, the effect of nucleosomes and their positioning on transcription has been studied extensively, and it has been shown that there is a complex interplay between transcription factors, nucleosomes, and chromatin remodeling enzymes that together regulate the expression of genes (Cairns 2009). In addition to gene expression, nucleosome positioning has been shown to be associated with other key cellular processes, including mRNA splicing, DNA replication, and DNA repair (Berbenetz et al. 2010; Duan and Smerdon 2010; Tilgner and Guigó 2010). Consequently, determining the mechanisms involved in controlling the positioning of nucleosomes and their variants is fundamentally important not only to understanding a critical component of many biological processes but also to understanding the regulation of an epigenetic level associated with several diseases (Portela and Esteller 2010).
Although a number of chromatin remodeling enzymes have been identified, what controls the positioning of nucleosomes along DNA is still poorly understood. However, it has been proposed that the underlying DNA sequence itself may, to an extent, control nucleosome locations (Segal et al. 2006). To begin to test this hypothesis, two recent studies compared in vitro yeast nucleosome maps to those derived in vivo to begin to characterize any intrinsic affinity nucleosomes have for certain stretches of DNA (Kaplan et al. 2009; Zhang et al. 2009). In spite of the experimental similarities between these studies, they differed markedly in their estimates of the extent to which DNA plays a role in positioning nucleosomes (Kaplan et al. 2010; Zhang et al. 2010) and the extent to which DNA controls the location of nucleosomes remains unclear.
Certain links between nucleosomes and their underlying base composition are well known. The relationship between dinucleotide frequencies and the relative position from the nucleosome dyad (the mid-point of the nucleosome core) is well established, and a clear 10-bp periodicity in dinucleotides has been observed in a number of eukaryotes (Reynolds et al. 2010). A recent study has shown that there is also a marked, asymmetrical periodicity in mononucleotide patterns observed in both yeast and human genomes when DNA sequences are aligned at nucleosome dyads, and it was these mononucleotide patterns, rather than di- or trinucleotide frequencies, that were most informative in the prediction of nucleosome positions (Reynolds et al. 2010).
Periodicities in nucleotide frequencies have generally been thought to be a result of the requirement of DNA to curve around the histone octamer and the differing abilities of base pairs and DNA sequences to bend (Segal and Widom 2009). If this is the case, it is possible that base compositional biases are a result of selection for or against sequences that differ in their affinities for the nucleosome core, and there may therefore be detectable signatures of selection at the DNA level linked to nucleosome positioning. Examination of the correlation between sequence divergence and nucleosome positioning in coding regions has so far shown that substitution rates are lower in linker regions than at DNA wrapped around core histones, and this has been attributed to purifying selection at linker regions for DNA sequences that occlude nucleosome occupancy rather than selection for DNA curvature (Warnecke et al. 2008). There are, however, alternative explanations for lower divergence rates in linker regions, most notably mutational bias or positive selection in nucleosome core regions (Semple and Taylor 2009). There is substantial evidence that nucleosomes impede the access of DNA repair enzymes to underlying DNA sequences (Méndez-Acuña et al. 2010), and the base composition in linker regions is different from regions underlying nucleosomes (Reynolds et al. 2010), suggestive of different mutational loads. Consequently, any search for potential signatures of selection in and around nucleosomes must account for the differing rates of mutation and repair observed in these regions.
In this study we investigated whether signatures of selection associated with the positioning of nucleosomes could be observed in underlying DNA. If DNA plays a fundamentally important role in regulating nucleosome locations, there may be evidence that the DNA in and around nucleosomes is not evolving neutrally, and any observed deviations from selective neutrality should correlate with the associated chromatin structure.
Results
Complex patterns of divergence around nucleosomes
Two of the most comprehensive human nucleosome maps currently available are those produced by Schones et al. (2008) and Barski et al. (2007). Whereas the study of Barski et al. (2007) focused on the positioning of nucleosomes carrying at least one of 20 types of methylated histone, the study of Schones et al. (2008) examined nucleosome positioning irrespective of such modifications. Both data sets were generated in CD4+ T cells that play a central role in adaptive immunity and depend upon remodeling of chromatin structure for important aspects of their differentiation and function (Wilson et al. 2009), but the methodologies of the two studies differ extensively. In order to investigate signatures of selection in the human lineage, we first investigated human–chimpanzee sequence divergence patterns in and around the nucleosomes defined in these data sets.
As shown in Figure 1, human lineage-specific divergence rates were in general lower at nucleosomes in the Barski et al. (2007) data set, likely as a result of this data set being restricted to nucleosomes carrying modifications preferentially enriched at conserved regions in and around genes. Nucleosomes carrying two particular modifications (H2A.Z and H3K4me3) have recently been found to be associated with lower rates of genomic sequence variation compared with unmodified nucleosomes (Tolstorukov et al. 2011). The current data suggest that this phenomenon extends to nucleosomes carrying a broader range of histone modifications. As shown in other studies, in a range of eukaryotes (Warnecke et al. 2008; Washietl et al. 2008; Sasaki et al. 2009; Ying et al. 2010) the overall levels of divergence were observed to be lower in linker regions than at the cores of nucleosomes in both data sets (Fig. 1). Not only are strong peaks in divergence observed at the nucleosome dyads, the substitution rates in the linker regions immediately beside the nucleosomes are lower than the mean rates of divergence in flanking regions (defined as the sequences ±250–500 bp from the dyads). These low rates of divergence at linker DNA suggest that purifying selection may be occurring at these regions as previously proposed in other studies (Warnecke et al. 2008). However, other explanations are also possible, involving different combinations of mutation rate biases and variation in the mode and strength of selection present. Also, increases in the substitution rates of certain base changes could potentially mask decreases in others, which can further complicate analysis. To minimize this problem, we looked at each class of base change independently by determining the human–chimpanzee–orangutan ancestral base at each position and comparing it to the base observed in the human reference genome sequence. This allowed us to identify the relative contribution of each base change to the overall divergence patterns observed in Figure 1, and ensured that all patterns detected were specific to the human lineage since divergence from the chimpanzee.
Figure 1.

Human lineage-specific intra- and interspecies divergence rates around nucleosome dyads. Rates of intraspecies divergence are plotted on the secondary, right-hand y-axis, interspecies divergence on the primary, left-hand axis. Solid trend lines correspond to a sliding window size of 25 bp around each position. Nucleosome positioning data were derived independently from the Schones et al. (2008) and Barski et al. (2007) data sets.
Analysis of each possible base change independently clearly demonstrated (Fig. 2 [Schones et al. 2008 data set]; Supplemental Fig. 1 [Barski et al. 2007 data set]) that a number of different patterns of divergence contribute to the overall pattern (Fig. 1). Strong peaks of T→C, T→G, A→C, and A→G changes were observed at and around the nucleosome dyad with matching low rates of C→T, G→T, C→A, and G→A changes observed in the same regions. There consequently appears to be a strong preference for changes from AT to GC base pairs in regions underlying dyads.
Figure 2.

Interspecies rates of divergence around nucleosome dyads in the human lineage. Colored solid lines correspond to 25-bp sliding averages. Dotted vertical lines represent the estimated dyad position. Transversions are plotted on the secondary y-axis due to their substantially lower rates. Nucleosome positioning data were derived from the Schones et al. (2008) data set.
The overall pattern observed in Figure 1 is therefore in fact a composite of many different divergence traces, dominated by the more common transition changes. Although Figure 1 is generally suggestive of high substitution rates in nucleosome core regions, Figure 2 highlights that there are in fact both high and low rates of different base changes at these positions.
By using geographically diverse SNP data compiled from various recently published, high coverage, human whole-genome sequencing studies (Levy et al. 2007; Bentley et al. 2008; Wang et al. 2008; Wheeler et al. 2008; Ahn et al. 2009; Kim et al. 2009; Drmanac et al. 2010; Schuster et al. 2010; Tong et al. 2010), we also investigated the rates of intraspecies divergence relative to the same two data sets of nucleosomes. Analysis of this intraspecies polymorphism data showed that the broad variation in total polymorphism density in and around nucleosomes is similar to those patterns observed in the overall pattern of interspecies divergence; rates are highest at the nucleosome core and lowest toward the linker regions (Fig. 1). However, whereas the lowest rates of intraspecies divergence occur precisely at the edges of the predicted nucleosomes, i.e., ±70 bp from the dyad, the lowest rates of interspecies divergence occur further into the expected linker regions at around ±125 bp from the dyad (corresponding to the approximate mean mid-point of linker regions, i.e., half a nucleosome [∼75 bp] + 50 bp of linker). As with the treatment of interspecies divergence, we also measured the rate of each possible base change and calculated the number of observed intraspecies changes (polymorphisms) at each position relative to the nucleosome dyad. The ancestral base at each position was determined by comparison to the chimpanzee genome.
As can be seen in Figure 3 (Schones et al. 2008 data set; for equivalent Barski et al. 2007 data set graph, see Supplemental Fig. 2), the patterns of variation within the recent human lineage, like those of interspecies divergence, also fluctuate widely in and around nucleosomes. Most notably, the rates of T/C→A and G/A→T are depressed in the region of the nucleosome core. These patterns are consistent with observations that different classes of higher-order chromatin structure appear to suffer different mutational spectra (Prendergast et al. 2007), but here the differences seen are at the level of the nucleosome, the fundamental building block of higher-order structures.
Figure 3.

Intraspecies rates of divergence around nucleosome dyads in the human lineage. Colored solid lines correspond to 25-bp sliding averages. Dotted vertical lines represent the estimated dyad position. Transversions are plotted on the secondary y-axis due to their substantially lower rates. Nucleosome positioning data were derived from the Schones et al. (2008) data set.
Despite similarities, comparisons of inter- and intraspecies divergence rates were found to reveal surprising contrasts for certain classes of substitutions. For example, although there are strong peaks in the rate of interspecies T-to-G and A-to-C changes to either side of the nucleosome dyad, there are no matching peaks at the same positions in the corresponding intraspecies substitution rates.
Recent selection linked to human nucleosome positioning
Observed differences in rates of interspecies and intraspecies divergence, such as those seen in Figures 2 and 3, are potentially indicative of selection. Positive selection is expected to lead to an excess of interspecies divergence over intraspecies divergence; negative selection, the reverse. Such comparison of rates of fixed interspecies divergence and intraspecies polymorphisms has been formalized as the widely used McDonald-Kreitman (MK) test for selection and its variants (McDonald and Kreitman 1991). However since differences in substitution rates can be attributed to a number of other factors, such as altered rates of mutation and repair in a region (Semple and Taylor 2009), such tests need to account for these potentially confounding factors. Typically, in studies of protein-coding sequence, this has been achieved by comparing the divergence observed at sites of interest (e.g., nonsynonymous sites) to an estimate approximating the rate of neutral divergence and reflecting the background mutation rate (e.g., synonymous site divergence rates) (Hurst 2002). Therefore four rates of divergence are generally used in MK-type tests; not only the rates of inter- and intraspecies divergence at the sites of interest but also the inter- and intraspecies divergence rates at selectively “neutral” sites. However, there is increasing evidence that neutral proxies such as synonymous sites are actually evolving non-neutrally (Chamary and Hurst 2005; Prendergast et al. 2007), and given the ubiquitous presence of nucleosomes across the genome, there is no available estimate of neutral change that is not associated with either nucleosomes or linker regions. In this analysis, we therefore used the average rates of inter- and intraspecies divergence observed at −500 to −250 and +250 to +500 bp from the dyads of our sets of well-positioned nucleosomes as an indication of the average rates of divergence observed at flanking DNA sequences: those sequences not aligned according to nucleosome dyads (and likely to disproportionately contain nucleosomes less well positioned and under less control by DNA sequence). By taking matched flanking regions for each nucleosome, we expect to control for any local compositional, demographic, and substitution rate variation occurring in the region. Although these flanking regions will also be associated with nucleosome and linker regions to some extent, these will not be regularly arranged (due to variation in nucleosome density and linker length around the genome), and they therefore provide an estimate of the average rates of divergence at these regions. Since it is unlikely that all flanking regions are selectively neutral, our comparisons are potentially overly conservative, but they do allow a comparison of divergence rates in and around the positioned nucleosomes in the Schones et al. (2008) and Barski et al. (2007) data sets to an estimate of mean divergence in the same regions. If all regions relative to the nucleosome dyad are evolving neutrally, or under the same selective pressure, we would expect to see no significant deviations in the rates of Sx→y (the ratio of interspecies to intraspecies base changes corrected for flanking sequence rates; see Methods) across the 1-kb regions examined. However, positive selection will lead to elevated rates of Sx→y due to an excess of interspecies divergence relative to intraspecies polymorphism, and negative selection decreased rates (due to a relative excess of intraspecies polymorphism over interspecies substitutions).
As can be seen in Figure 4 (equivalent Barski et al. 2007 data set graph shown in Supplemental Fig. 3), a number of positions relative to the nucleosomal dyad showed a relative excess or depletion in the rate of interspecies divergence compared with intraspecies rates of change. For example, significantly elevated rates of interspecies C→T changes can be observed at around ±60–90 bp from the dyad, indicative of positive selection in the linker regions between nucleosomes. However, the area immediately around the dyad appears to be depleted for these changes, suggestive of negative selection for these substitutions in this region. Consequently, there is evidence for both elevated and depleted rates of interspecies divergence in close proximity for the same base changes. This is a striking result in view of the fact that A- and T-rich sequences are known to disfavor nucleosome assembly, and supports the view that such compositional preferences can be critical in nucleosome positioning and function (Henikoff 2008). It also suggests that broad patterns of nucleotide composition across the human genome have been influenced by complex, and sometimes opposing, forces of selection within the past few million years. Although others have postulated that selection may have acted upon human nucleosome positioning via sequence composition (Tolstorukov et al. 2011), to our knowledge the present study provides the first evidence for this. Examination of the selection on each possible base change at each position suggests that, in general, selection has acted to maintain higher GC compositions in and around nucleosome dyads and lower GC compositions at linker regions.
Figure 4.

Rates of selection in and around nucleosome dyads. Ratios of background corrected inter- and intraspecies divergence rates plotted against position from nucleosomal dyad (Sx→y scores). Dotted horizontal lines correspond to an uncorrected P-value of 0.004 (corrected P-value of 0.05). Nucleosome positioning data were derived from the Schones et al. (2008) data set.
MK test–inspired analyses have been shown to be potentially skewed by the presence of slightly deleterious mutations. To overcome this, previous studies have removed low frequency polymorphisms when using a MK-based test, as deleterious variants disproportionately segregate at low frequencies. We consequently repeated our analysis having removed SNPs with a minor allele frequency of <15% (Charlesworth and Eyre-Walker 2008; He et al. 2011). However, this had little effect on the broad patterns seen in our analysis, suggesting they are not being driven by the presence of an excess of low-frequency, deleterious variants (Supplemental Fig. 4). Similar significant deviations from the expected (flanking) interspecies-to-intraspecies divergence ratio are still observed.
As a result of the particular histone modifications examined in the Barski et al. (2007) ChIP-seq data, a disproportionate number of the nucleosomes in this data set are associated with exons and transcription start sites (TSSs), features that have also been associated with what have been termed “barrier” positions of low nucleosome occupancy (located at transcriptional start sites and the 3′ end of open reading frames) (Mavrich et al. 2008). We therefore investigated whether the signatures of selection seen in this data set were exclusively associated with nucleosomes near genes and barrier positions by restricting the analysis to only those (296,858) nucleosomes at least 500 bp from the nearest exon (around 69% of the Barski et al. 2007 data set). As can be seen in Supplemental Figure 5, nucleosomes not associated with exons show similar patterns of selection, suggesting that these signatures of selection are not restricted to nucleosomes in close proximity to coding regions and known nucleosome barriers.
Selection maintains optimal GC content for nucleosome positioning
These results (Fig. 4) suggest selection in the human lineage has acted to favor particular, complementary compositional biases at nucleosome cores and linker regions; high GC content at nucleosome cores and high AT composition at linker regions. If these patterns of selection were really linked to increasing the correct positioning of nucleosomes, it would be expected that selection along the human lineage has acted to increase the affinity of nucleosomes for their current positions in the human genome. Examination of mononucleotide and 5mer frequencies associated with current nucleosome positions in the human genome highlighted that nucleosomes do indeed preferentially assemble on DNA sequences of particular base compositions. As can be seen in Supplemental Figure 6, 5mers composed exclusively of AT base pairs are depleted at the nucleosome core. and the 32 5mers composed exclusively of A and T base pairs were observed to be the 32 most depleted sequences observed at the dyad (with respect to their levels in flanking sequences). This is in agreement with the known low nucleosome occupancies associated with AT-rich regions in other eukaryotes where 5mers composed exclusively of AT base pairs were observed to have the lowest occupancies (Kaplan et al. 2009). Consequently. the apparent observed positive selection for CG-to-AT substitutions and the negative selection against T→C and A→G changes at linker regions, over recent human evolution, are consistent with previous in vitro studies and the compositional biases observed in the human genome today. However, despite the selection for AT-to-CG changes at the dyad, this region is not enriched for 5mers composed exclusively of G and C bases. The 5mers most enriched around the dyad contain a mix of both CG and AT base pairs, with the most enriched sequence over its flanking levels being ACGTG in the Barski et al. (2007) data set (16th out of 1024 in the Schones et al. 2008 data set) and TGCCG in the Schones et al. (2008) data set (136th in the Barski et al. 2007 data set). Those 100 5mers most enriched at the dyad on average contained 1.8 and 1.4 A or T bases in the Barski et al. (2007) and Schones et al. (2008) data sets, respectively (compared to a genome-wide average of 2.5). Consequently, although some of the strongest signals of selection observed in Figure 4 are for TA-to-CG base changes at the nucleosome core and selection against G-to-A and C-to-T changes, nucleosomes only appear to favor regions of slight GC bias.
In order to begin to reconcile these observations, we directly examined how local GC composition affects the observed divergence rates. If selection is maintaining an optimum GC content at nucleosome cores and linkers, it would be expected that the strengths of selection would depend on the local GC content, with nucleosomes in regions of GC most distant from the optimum levels coming under the strongest selection. Examination of the relationship between underlying mononucleotide frequencies and the flanking (−500 to −250 and +250 to +500 bp from the dyads) GC content of nucleosomes highlighted that the elevated rates of G and C base pairs at the nucleosome core are indeed most noticeable where the flanking GC rate is low. At a flanking GC percentage of 60%–70%, the mononucleotide frequencies show the least difference between flanking and nucleosome core regions (Supplemental Figs. 7, 8), suggesting that this is the optimum equilibrium between AT and GC base pairs, recapitulating the apparent optimum of 64%–72% GC content (1.8 A or T bases) observed at the nucleosome cores in the 5mer analysis.
To formally test how selection was affected by local GC content, we examined modes and strengths of selection as before, but stratified the nucleosomes by the flanking GC content (−500 to −250 and +250 to +500 bp from the dyads). As can be seen in Figure 5, the signatures of selection (as measured by an excess or depletion in interspecies divergence rates) in the nucleosome core are strongest where the local GC content is low, with stronger selection appearing to have acted to overcome the local AT bias. However, a different picture emerges in linker regions, consistent with their biophysical preferences for relatively AT-rich sequences. Signatures of what is predominantly purifying selection at these regions are strongest in GC-rich neighborhoods, as the elevated rates of GC leads to strong selection against A-to-G and T-to-C changes in particular that would lead to an elevation in the GC content of already GC-rich regions, in areas where GC base pairs are disfavored. Consequently, strengths and directions of selection depend on the GC content of the genomic neighborhood nucleosomes are found in. It is known that large scale, multi-megabase fluctuations in GC content occur in the human genome, corresponding to variation in higher-order chromatin structures, such as replication timing domains (Hiratani et al. 2010) and lamin-associated domains (Peric-Hupkes et al. 2010). This suggests that a stretch of genomic DNA may be subject to conflicting compositional pressures from different levels of structural organization, leading to varying patterns of selection on the local positioning of nucleosomes between different genomic neighborhoods.
Figure 5.

Deviation of interspecies divergence rates from flanking rates in and around nucleosomes and at different flanking GC compositions. The percentage of enrichment (or depletion) of flanking corrected interspecies rates of changes with respect to corresponding observed rates of intraspecies change. Significantly elevated or depleted levels are indicated by * (uncorrected P-value of 0.05) and ** (uncorrected P-value of 0.00046, corrected P-value of 0.05). Nucleosome positioning data were derived from the Schones et al. (2008) data set.
Previous work has successfully used in vitro evolution to derive novel sequences that position nucleosomes more stably than those occurring in nature, and so it has been assumed that eukaryotic genome sequences have evolved to accommodate lower affinity “metastable” interactions with nucleosomes (Henikoff 2008). The current data are also consistent with this assumption and provide the first clear evidence for a complex interplay of selective forces in the human genome acting to produce the often delicately poised landscape of nucleosome associated DNA.
Extent of selection associated with nucleosome positioning
We next estimated the proportion of interspecies fixed differences in the human genome that is likely to be a consequence of selection. We compared the rates of substitution in and around nucleosomes to mean flanking sequence rates in the same regions (again estimated using the regions of DNA ±250–500 bp from our nucleosome midpoints; for more details, see Methods). The rates of A-to-C base changes (one of the substitutions observed to be strongly favored by selection) were shown to be on average ∼14.1% higher in the nucleosome core (dyad ±75 bp) in the Schones et al. (2008) data set (10.6% in Barski et al. 2007 data set), compared with the corresponding rates of intraspecies change at the same positions, suggesting there has been a 14.1% increase in the number of A-to-C changes in these regions as a result of positive selection (Fig. 5; Barski et al. 2007 data set shown in Supplemental Fig. 9). At nucleosomes where the GC percentage of the flanking sequence was <45% and where selection for A to C changes at the nucleosome core was observed to be the strongest, this figure was 16.3% (17.4% in the Barski et al. 2007 data set). Under the assumption that each nucleosome is associated with, on average, 60 bp of linker DNA, examination of ±105 bp of the nucleosome dyads (core region ±30 bp) illustrated that the proportion of the total data set–wide A-to-C changes likely to be a result of positive selection linked to nucleosome positioning is ∼12.6% in AT-rich regions and ∼10.5% in the data set as a whole (12.6% and 7.6% in the Barski et al. 2007 data set, respectively). Consequently, there appears to have been a substantial increase in the number of A-to-C changes in the human lineage as a result of positive selection for nucleosome occupancy. It should be noted that these figures are based on the assumption that these large nucleosome data sets are representative of the genome at large, though the techniques used to define these data are undoubtedly biased toward strongly positioned nucleosomes. It is possible (if not likely) that this minority subset of nucleosomes is under greater control of their positioning and is therefore associated with unusual levels of selection. However, our results comparing the observed patterns of selection at coding and noncoding regions and between nucleosomes carrying different modifications (Figs. 6, 7) suggest that the observed signatures of selection are broadly similar and a general feature of the well-positioned nucleosomes in these data sets.
Figure 6.

Histone modification-specific mononucleotide biases (1). The ratio of a variety of histone modification–specific nucleotide frequencies versus the nucleotide frequencies observed in the total pool of nucleosomes (restricted to nucleosomes with a flanking GC percentage between 30% and 40%).
Figure 7.

Histone modification–specific mononucleotide biases (2). The ratio of a variety of histone modification–specific nucleotide frequencies versus the nucleotide frequencies observed in the total pool of nucleosomes (restricted to nucleosomes with a flanking GC percentage between 30% and 40%).
It is also worth noting that the estimates of background (flanking region) substitution rates used here are unavoidably derived from a population of sequences at least partially occupied by nucleosomes themselves and therefore also putatively subject to any of the signatures of selection detected here. Similarly, the intraspecies rates of change are likely to have been affected, to an extent, by any significant selection linked to nucleosome positioning. These caveats suggest that our results are potentially conservative. However, even taken at face value, these data suggest that a substantial number of the fixed A-to-C base changes between human and chimpanzee are attributable to selection associated with nucleosome positioning, and that a not-insubstantial proportion of the human genome has been subject to recent selection linked to nucleosome positioning. The nucleosome data sets studied here encompass >160 Mb of genomic sequence, or over 5% of the human genome, exceeding the span of the protein-coding component of the genome for example.
Nucleosome modifications and compositional bias
Although the base compositional biases and patterns of selection at nucleosome dyads in the Schones et al. (2008) and Barski et al. (2007) data sets are broadly similar, differences can be observed between the two data sets (e.g., Fig. 5 vs. Supplemental Fig. 9). Histone modifications are known to affect the accessibility and functional role of the chromatin at a locus (Bell et al. 2010). It is therefore possible that these differences appear because the Barski et al. (2007) data set is restricted to nucleosomes carrying one of 20 different histone modifications, which may have distinct compositional biases and be under unusual modes and strengths of selection. Examination of the mononucleotide patterns underlying different modifications in the Barski et al. (2007) data set showed that for nucleosomes in similar GC environments, the broad patterns of nucleotide frequencies are similar. However, closer examination of the frequencies observed for nucleosomes carrying specific modifications versus those observed in the total pool of nucleosomes highlights that there are subtle differences in the biases for certain base pairs at given positions from the dyad in this data set. Although these biases are generally relatively small, different histone modifications clearly show distinct patterns of base composition in the underlying DNA even after controlling for local GC bias (Figs. 6, 7). We therefore tested whether modification-specific signatures of selection could also be detected. This was achieved by comparing the values of Sx→y observed at those nucleosomes carrying a modification of interest to all other nucleosomes in the data set. However, the vast majority of positions showed no significant difference in the Sx→y scores between nucleosomes carrying a modification and those that did not, and no broad patterns for or against particular substitutions were observed (data not shown). This potentially suggests that the differences in nucleotide patterns observed between modifications is a result of local mutational biases rather than selection; however, given the relatively small biases for certain base pairs observed in Figures 6 and 7 in conjunction with the relatively short evolutionary distance examined in this analysis, it is possible this analysis lacks the required power to detect what may be relatively subtle differences between nucleosomes carrying different modifications.
Discussion
Previous studies have shown that rates of divergence differ in and around nucleosomes in various species, with divergence rates observed to be higher in nucleosome core regions than in linker DNA (Warnecke et al. 2008; Washietl et al. 2008; Sasaki et al. 2009). This has so far been attributed to negative selection in DNA sequences flanking nucleosomes (Warnecke et al. 2008), though there are potentially other explanations for these fluctuations in divergence rates. In this study we have shown that the patterns of interspecies divergence associated with nucleosomes are unexpectedly complex. Although the more common transitions, and in particular T→C and A→G changes, show elevated rates in the nucleosome core, rates of other changes, e.g., G→T, show substantially lower levels at DNA wrapped around the histones. By comparing rates of interspecies and intraspecies divergence, we have shown these differing rates of base change are not likely to be the result of altered mutation rates in and around nucleosomes but rather are a consequence of differing patterns of selection.
It is important to note that signatures of apparent selection are not always a result of the accumulation of adaptive changes. Recent studies have shown that what often appears to be positive selection is actually a result of the biased conversion of AT to GC base pairs by a process termed biased gene conversion (BGC) (Galtier and Duret 2007). BGC leads to the accumulation of AT-to-GC base changes via the biased repair of A:C and G:T mismatches through meiotic recombination. Therefore, in theory, BGC could underlie the subset of AT-to-GC changes observed at nucleosome cores in this study. It is, however, difficult to reconcile the current knowledge of BGC with the observations in this study. One of the fundamental observations underlying BGC is that it has been most prevalent in GC-rich, nucleosome-poor regions of the human genome, which are known to experience higher recombination rates (Duret and Galtier 2009). However, the highest rates of AT-to-CG fixation (importantly per A or T ancestral site) observed in this study were at nucleosomes in AT-rich regions, with little selection observed in GC-rich regions, the opposite of the pattern that would be expected under BGC. Critically, nucleosome cores have also been shown to occlude meiotic recombination (Getun et al. 2010); therefore to explain the results in this study, BGC would have to be highest specifically at the only regions where it is known to be occluded. Although it is of course possible that BGC may underlie a small number of base changes observed in our analysis, BGC does not appear to be a plausible explanation for the main trends observed here.
It has been shown by a number of approaches that AT-rich regions disfavor nucleosome assembly (Iyer and Struhl 1995; Kaplan et al. 2009), and in yeast, AT-rich, nucleosome occluding regions are thought to be maintained by a combination of selection against A/T-depleting substitutions and selection for A/T-gaining substitutions (Kenigsberg et al. 2010). We have shown that not only are similar substitution patterns observed in the linker regions of human nucleosomes but also an optimum GC content also appears to be maintained by selection at the core regions of human nucleosomes. Importantly, the strength of selection favoring this optimum appears to depend on the local GC content of the nucleosome, with substitution rates highest where the local GC content is furthest from the optimum. Consequently, the most parsimonious explanation for the results in this study is that differing modes and strengths of selection have acted to maintain favorable base compositions in both linker and core regions of human nucleosomes.
These data suggest that changes at the DNA level can affect nucleosome occupancy in a region and, ultimately, an organism's fitness. Although we have shown in this study that single base changes between 5mer sequences can dramatically affect their affinity for the nucleosome core, it may, at first, be difficult to see how single base changes could have a sufficiently large effect on nucleosome positioning for fitness to be affected. However, recent studies in yeast have shown that not only can nucleosome occupancy be predicted from DNA sequence but also the same models based on sequence alone can be used to predict changes in nucleosome occupancy between yeast species (Tirosh et al. 2010). Similarly, changes in gene expression have been linked to DNA sequence changes that directly alter the DNA-encoded nucleosome organization of yeast promoters (Field et al. 2009). Consequently, at least in yeast, single base changes can lead to predictable changes in nucleosome occupancy and alterations in gene expression. It is unlikely that all such changes would have no effect on fitness and be selectively neutral.
Although the direct effect of DNA sequence changes on nucleosome positioning in humans has been less well investigated, links between nucleosome positioning and single base changes have also been observed. For example, a polymorphism associated with asthma, type 1 diabetes, primary biliary cirrhosis, and Crohn's disease has been associated with allele-specific changes in nucleosome positioning (Verlaan et al. 2009). The discovery of widespread signatures of selection at DNA in and around nucleosomes therefore has substantial implications for the study of diseases and traits. It is possible that changes affecting nucleosome occupancy may be involved in a variety of diseases and help explain some of the variants emerging from genome-wide association studies, unlinked to genes and other known functional genomic regions (Manolio et al. 2009).
This novel link between divergence patterns and nucleosomes also brings into question how much of the genome is in fact evolving neutrally. Positive selection in the human lineage has previously been thought to be restricted to a relatively small proportion of genes and some noncoding regions (Kelley and Swanson 2008). Nucleosomes are a ubiquitous feature of DNA packaging, and signatures of positive selection linked to features that are so widespread appear to be unprecedented. We have estimated that there is an excess of up to 10.5% of certain base substitutions in the human lineage, as a result of selection linked to nucleosome positioning in the data sets examined. Although such estimates are derived from a group of relatively well-positioned nucleosomes potentially under unusual levels of selection, a substantial fraction of the human genome is implicated. Even if such signatures of selection are restricted to the DNA at the well-positioned nucleosomes examined in this study, they cover a greater proportion of DNA than protein coding genes (800,000 nucleosomes will cover ∼5%). The previously held belief of coding synonymous sites evolving neutrally has already been shown to be inappropriate (Chamary and Hurst 2005; Prendergast et al. 2007), and these results suggest that the positioning of nucleosomes is likely to impact the divergence of other traditionally “neutrally” evolving regions, such as intronic and intergenic DNA.
It is formally possible that the broad signatures of selection seen in this analysis are not directly linked to nucleosomes but are instead related to some category of functional sequence that to some extent co-occurs. However, this seems implausible given that these signatures are largely the same when only those nucleosomes distinct from exons and TSSs are examined. We see the same patterns in both data sets examined and when subdividing the nucleosome data set by their locations or histone modifications, illustrating that these results are not attributable to a small proportion of the nucleosomes examined but are a more general feature of the data sets.
Perhaps surprisingly, these signatures of selection are apparent over a relatively short evolutionary time: the past ∼5 Myr since human–chimpanzee divergence. Nucleosome positioning is critical to transcriptional activity, and patterns of transcription have been shown to differ extensively between humans and chimpanzees (Gilad et al. 2006). Divergence of expression patterns between yeast species has previously been associated with changes in nucleosome occupancy (Field et al. 2009). It is possible the novel lineage-specific signatures of selection observed here are associated with recent chromatin remodeling and nucleosome repositioning in primate evolution, contributing to the expression differences seen between species.
The observation of distinct nucleotide patterns underlying different histone modifications is suggestive of DNA playing a role in controlling the positioning of specific histone modifications. However, the distribution of nucleosome modifications has been shown to differ between cell types, and therefore, further examination is required to investigate whether the biases observed in this study are specific to the CD4+ cells examined in the Schones et al. (2008) and Barski et al. (2007) data sets. Even if histone modifications generally show a bias toward certain underlying base compositions a number of other factors, such as histone acetylases and methyl transferases, are known to govern the distribution of histone modifications.
Finally, although the patterns of selection described in this study appear to be linked to the maintenance of optimum GC content, this explanation cannot underlie all the patterns of substitutions observed. For example, despite the apparent selection against A and T bases at the dyad, no significant selection against C→A, and its complement G→T, changes is observed. Therefore there appears to be much to learn about the factors driving sequence evolution in and around nucleosomes in the human genome.
Methods
Interspecies divergence rates
Nucleosome dyad positions were estimated from the Barski et al. (2007) data set of positioned nucleosomes derived using NPS (Zhang et al. 2008) by taking the midpoint of called nucleosomes as in the method of Reynolds et al. (2010). Dyad positions derived from the Schones et al. (2008) data set as used by Reynolds et al. (2010) were kindly provided by Sheila Reynolds and William Noble. In total, the Barski et al. (2007) and Schones et al. (2008) data sets contained the predicted positions of 432,541 and 817,774 autosomal nucleosomes, respectively. Human–chimpanzee–orangutan multiple sequence alignments were generated for each nucleosome, ±500 bp of the midpoint, using the pairwise alignments available at the UCSC Genome Browser (http://genome.ucsc.edu/cgi-bin/hgGateway; Homo sapiens build hg18, Pan troglodytes build 2, Pongo pygmaeus abelii build 2). To determine rates of human lineage-specific base changes at each position relative to the predicted dyad, the ancestral base at each position was determined to be the base shared by at least two of the three primates, with human-specific changes being where the human base did not match this ancestral base at the corresponding position. At the positions where ancestral bases could not be determined (e.g., due to gaps in the alignment or multiple differences between species), the corresponding position was excluded from further analyses.
Intraspecies divergence rates
SNP data from 10 geographical diverse fully sequenced human genomes were compiled into a MySQL database and used in the estimates of intraspecies divergence rates (both NA07022 and NA19240 were used from Drmanac et al. 2010, but only the Bantu genome from Schuster et al. 2010 was used due to the inclusion of extra exome data in the calling of the Khosian variants and potential resulting biases in SNP calling) (Levy et al. 2007; Bentley et al. 2008; Wang et al. 2008; Wheeler et al. 2008; Ahn et al. 2009; Kim et al. 2009; Drmanac et al. 2010; Schuster et al. 2010; Tong et al. 2010). The ancestral base at any position was assumed to be any observed allele (including that observed in the reference genome) that matched the corresponding base in the chimp genome. Sites where the ancestral base could not be determined were excluded. As with interspecies substitutions, human lineage-specific changes were deemed to be those changes observed in the human SNP data set not matching the ancestral base.
Rates of selection and significance
The rate of base changes at each position from the dyad of nucleosomes was measured by dividing the observed number of base changes by the total number of matching ancestral bases at each position (Equations 1 and 2).
Equations 1 and 2 calculate the rates of base change at given positions from the nucleosome dyad. x and y correspond to the bases before and after the specific change, respectively, x being the ancestral base and y being the base observed in the human lineage. interDiffx→y and intraDiffx→y are the total number of relevant interspecies and intraspecies changes observed at the position of interest relative to the dyad. ancestralBasex is the number of corresponding ancestral bases observed at the same position.
Due to the abnormal rates of divergence seen on the sex chromosomes (The Chimpanzee Sequencing and Analysis Consortium 2005), only nucleosomes from autosomes were used when investigating both inter- and intraspecies divergence rates.
Note that at shorter evolutionary distances, observed substitutions are expected to more closely reflect underlying rates of mutation; however, as evolutionary distance increases, the effect of selection will become more apparent. Consequently, where changes are selected against the ratio of fixed interspecies differences relative to intraspecies, changes will be lower relative to other areas of the genome. Where positive selection is occurring, there will be a high rate of accumulation of changes at a given position (in this case relative to nucleosome dyads), and the ratio of interspecies rates of change to intraspecies rates of change will be elevated relative to other positions.
Positions relative to the nucleosome dyad where interspecies rates of change showed unusual deviations from intraspecies rates were identified by first correcting each value of dInterx→y and dIntrax→y for flanking rates of divergence. Flanking rates of change were estimated by averaging over those 500 positions at ±250–500 bp from the nucleosome dyads. The rate of interspecies base changes observed across all nucleosomes at each position was then divided by the corresponding rate of intraspecies change to provide an indication of selection (Equation 3).
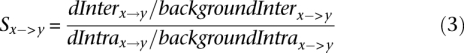 |
The ratio of background corrected inter- and intraspecies divergence rates was calculated for each base change and each position from the nucleosome dyad. BackgroundInter and backgroundIntra are the estimated background (flanking) rates of x→y changes.
Values of Sx→y > 1 indicate an excess of interspecies change and positive selection relative to flanking rates, and values <1 indicate an excess of intraspecies changes and negative selection. To assess whether a particular region relative to the nucleosome dyad exhibited significant evidence of selection, we ran a 25-bp sliding window with a 1-bp offset across our 1001 values of Sx→y (nucleosome midpoint ±500 bp). Windows significantly larger or smaller than expected by chance were determined by randomly permuting the positions of each value of Sx→y and rerunning the 25-bp sliding window analysis. This was repeated 10,000 times for each base change, and observed windows greater or smaller than 99.8% of all permuted windows across all positions were deemed significant (corresponding to a two tailed P-value of 0.004; although the permutation approach corrected for the number of windows tested, this cutoff corresponds to a P-value of 0.05 with a further Bonferroni correction for the 12 base changes tested). The proportion of elevated or depleted rates of interspecies divergence at different flanking GC compositions (Fig. 5; Supplemental Fig. 9) was assessed using a chi-square test. Raw counts of inter- and intraspecies divergence rates for both data sets are provided in Supplemental Table 1.
SNPs with a minor allele frequency >15% were identified by comparison to the frequencies observed at the same polymorphisms in the 1000 Genomes Project data set (which contains the majority of human, common SNPs) (The 1000 Genomes Project Consortium 2010). SNPs not detected in the 1000 Genomes Project, and consequently likely to be rare, were ignored.
To determine whether certain positions relative to nucleosome dyads were putatively under unusual levels of selection for subsets of nucleosomes with particular modifications, values of Sx→y calculated using only the subset of interest were compared to values of Sx→y obtained using all other positioned nucleosomes in the data set. Only modifications with at least 100,000 positioned nucleosomes in the data set were examined (H2AZ, H2BK5me1, H3K27me1, H3K36me3, H3K9me1, H3K4me1, H3K4me2, H3K4me3, and H4K20me1). Significance was assessed by randomly sampling the same number of nucleosomes from the total data set and again comparing the resulting values of Sx→y to the remaining set. This was repeated 100 times for each histone modification and base change, providing a distribution of Sx→y ratios. This distribution was used to calculate standard (z) scores and corresponding P-values. These P-values were converted to q values using the R qvalue package for FDR calculations (Storey and Tibshirani 2003).
Acknowledgments
We thank Sheila Reynolds (Institute for Systems Biology, Seattle, WA) and William Noble (University of Washington, Seattle, WA) for providing their Schones et al. (2008) data set–derived nucleosome positions. We also thank Martin Taylor, Wendy Bickmore, Nick Gilbert, and Jim Allan for their helpful comments and discussions regarding this project. We thank the UK Medical Research Council for financial support and the three anonymous reviewers for their very helpful comments.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.122275.111.
References
- The 1000 Genomes Project Consortium 2010. A map of human genome variation from population-scale sequencing. Nature 467: 1061–1073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn SM, Kim TH, Sunghoon L, Kim D, Ghang H, Kim DS, Kim BC, Kim SY, Kim WY, Kim C, et al. 2009. The first Korean genome sequence and analysis: full genome sequencing for a socio-ethnic group. Genome Res 19: 1622–1629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barski A, Cuddapah S, Cui K, Roh T-Y, Schones DE, Wang Z, Wei G, Chepelev I, Zhao K 2007. High-resolution profiling of histone methylations in the human genome. Cell 129: 823–837 [DOI] [PubMed] [Google Scholar]
- Bell O, Schwaiger M, Oakeley EJ, Lienert F, Beisel C, Stadler MB, Schubeler D 2010. Accessibility of the Drosophila genome discriminates PcG repression, H4K16 acetylation and replication timing. Nat Struct Mol Biol 17: 894–900 [DOI] [PubMed] [Google Scholar]
- Bentley DR, Balasubramanian S, Swerdlow HP, Smith GP, Milton J, Brown CG, Hall KP, Evers DJ, Barnes CL, Bignell HR, et al. 2008. Accurate whole human genome sequencing using reversible terminator chemistry. Nature 456: 53–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berbenetz NM, Nislow C, Brown GW 2010. Diversity of eukaryotic DNA replication origins revealed by genome-wide analysis of chromatin structure. PLoS Genet 6: e1001092 doi: 10.1371/journal.pgen.1001092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cairns BR 2009. The logic of chromatin architecture and remodelling at promoters. Nature 461: 193–198 [DOI] [PubMed] [Google Scholar]
- Chamary JV, Hurst LD 2005. Evidence for selection on synonymous mutations affecting stability of mRNA secondary structure in mammals. Genome Biol 6: R75 doi: 10.1186/gb-2005-6-9-r75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth J, Eyre-Walker A 2008. The McDonald-Kreitman test and slightly deleterious mutations. Mol Biol Evol 25: 1007–1015 [DOI] [PubMed] [Google Scholar]
- The Chimpanzee Sequencing and Analysis Consortium 2005. Initial sequence of the chimpanzee genome and comparison with the human genome. Nature 437: 69–87 [DOI] [PubMed] [Google Scholar]
- Drmanac R, Sparks AB, Callow MJ, Halpern AL, Burns NL, Kermani BG, Carnevali P, Nazarenko I, Nilsen GB, Yeung G, et al. 2010. Human genome sequencing using unchained base reads on self-assembling DNA nanoarrays. Science 327: 78–81 [DOI] [PubMed] [Google Scholar]
- Duan M-R, Smerdon MJ 2010. UV damage in DNA promotes nucleosome unwrapping. J Biol Chem 285: 26295–26303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duret L, Galtier N 2009. Biased gene conversion and the evolution of mammalian genomic landscapes. Annu Rev Genomics Hum Genet 10: 285–311 [DOI] [PubMed] [Google Scholar]
- Field Y, Fondufe-Mittendorf Y, Moore IK, Mieczkowski P, Kaplan N, Lubling Y, Lieb JD, Widom J, Segal E 2009. Gene expression divergence in yeast is coupled to evolution of DNA-encoded nucleosome organization. Nat Genet 41: 438–445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galtier N, Duret L 2007. Adaptation or biased gene conversion? Extending the null hypothesis of molecular evolution. Trends Genet 23: 273–277 [DOI] [PubMed] [Google Scholar]
- Getun IV, Wu ZK, Khalil AM, Bois PRJ 2010. Nucleosome occupancy landscape and dynamics at mouse recombination hotspots. EMBO Rep 11: 555–560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilad Y, Oshlack A, Smyth GK, Speed TP, White KP 2006. Expression profiling in primates reveals a rapid evolution of human transcription factors. Nature 440: 242–245 [DOI] [PubMed] [Google Scholar]
- He BZ, Holloway AK, Maerkl SJ, Kreitman M 2011. Does positive selection drive transcription factor binding site turnover? A test with Drosophila cis-regulatory modules. PLoS Genet 7: e1002053 doi: 10.1371/journal.pgen.1002053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henikoff S 2008. Nucleosome destabilization in the epigenetic regulation of gene expression. Nat Rev Genet 9: 15–26 [DOI] [PubMed] [Google Scholar]
- Hiratani I, Ryba T, Itoh M, Rathjen J, Kulik M, Papp B, Fussner E, Bazett-Jones DP, Plath K, Dalton S, et al. 2010. Genome-wide dynamics of replication timing revealed by in vitro models of mouse embryogenesis. Genome Res 20: 155–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurst LD 2002. The Ka/Ks ratio: diagnosing the form of sequence evolution. Trends Genet 18: 486–487 [DOI] [PubMed] [Google Scholar]
- Iyer V, Struhl K 1995. Poly(dA:dT), a ubiquitous promoter element that stimulates transcription via its intrinsic DNA structure. EMBO J 14: 2570–2579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan N, Moore IK, Fondufe-Mittendorf Y, Gossett AJ, Tillo D, Field Y, LeProust EM, Hughes TR, Lieb JD, Widom J, et al. 2009. The DNA-encoded nucleosome organization of a eukaryotic genome. Nature 458: 362–366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan N, Moore I, Fondufe-Mittendorf Y, Gossett AJ, Tillo D, Field Y, Hughes TR, Lieb JD, Widom J, Segal E 2010. Nucleosome sequence preferences influence in vivo nucleosome organization. Nat Struct Mol Biol 17: 918–920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley JL, Swanson WJ 2008. Positive selection in the human genome: From genome scans to biological significance. Annu Rev Genomics Hum Genet 9: 143–160 [DOI] [PubMed] [Google Scholar]
- Kenigsberg E, Bar A, Segal E, Tanay A 2010. Widespread compensatory evolution conserves DNA-encoded nucleosome organization in yeast. PLoS Comput Biol 6: e1001039 doi: 10.1371/journal.pcbi.1001039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JI, Ju YS, Park H, Kim S, Lee S, Yi JH, Mudge J, Miller NA, Hong D, Bell CJ, et al. 2009. A highly annotated whole-genome sequence of a Korean individual. Nature 460: 1011–1015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy S, Sutton G, Ng PC, Feuk L, Halpern AL, Walenz BP, Axelrod N, Huang J, Kirkness EF, Denisov G, et al. 2007. The diploid genome sequence of an individual human. PLoS Biol 5: e254 doi: 10.1371/journal.pbio.0050254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manolio TA, Collins FS, Cox NJ, Goldstein DB, Hindorff LA, Hunter DJ, McCarthy MI, Ramos EM, Cardon LR, Chakravarti A, et al. 2009. Finding the missing heritability of complex diseases. Nature 461: 747–753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mavrich TN, Ioshikhes IP, Venters BJ, Jiang C, Tomsho LP, Qi J, Schuster SC, Albert I, Pugh BF 2008. A barrier nucleosome model for statistical positioning of nucleosomes throughout the yeast genome. Genome Res 18: 1073–1083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald JH, Kreitman M 1991. Adaptive protein evolution at the Adh locus in Drosophila. Nature 351: 652–654 [DOI] [PubMed] [Google Scholar]
- Méndez-Acuña L, Di Tomaso MV, Palitti F, Martínez-López W 2010. Histone post-translational modifications in DNA damage response. Cytogenet Genome Res 128: 28–36 [DOI] [PubMed] [Google Scholar]
- Peric-Hupkes D, Meuleman W, Pagie L, Bruggeman SWM, Solovei I, Brugman W, Gräf S, Flicek P, Kerkhoven RM, van Lohuizen M, et al. 2010. Molecular maps of the reorganization of genome-nuclear lamina interactions during differentiation. Mol Cell 38: 603–613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portela A, Esteller M 2010. Epigenetic modifications and human disease. Nat Biotechnol 28: 1057–1068 [DOI] [PubMed] [Google Scholar]
- Prendergast JGD, Campbell H, Gilbert N, Dunlop MG, Bickmore WA, Semple CAM 2007. Chromatin structure and evolution in the human genome. BMC Evol Biol 7: 72 doi: 10.1186/1471-2148-7-72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds SM, Bilmes JA, Noble WS 2010. Learning a weighted sequence model of the nucleosome core and linker yields more accurate predictions in Saccharomyces cerevisiae and Homo sapiens. PLoS Comput Biol 6: e1000834 doi: 10.1371/journal.pcbi.1000834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki S, Mello CC, Shimada A, Nakatani Y, Hashimoto S-I, Ogawa M, Matsushima K, Gu SG, Kasahara M, Ahsan B, et al. 2009. Chromatin-associated periodicity in genetic variation downstream of transcriptional start sites. Science 323: 401–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schones DE, Cui K, Cuddapah S, Roh T-Y, Barski A, Wang Z, Wei G, Zhao K 2008. Dynamic regulation of nucleosome positioning in the human genome. Cell 132: 887–898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuster SC, Miller W, Ratan A, Tomsho LP, Giardine B, Kasson LR, Harris RS, Petersen DC, Zhao F, Qi J, et al. 2010. Complete Khoisan and Bantu genomes from southern Africa. Nature 463: 943–947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal E, Widom J 2009. What controls nucleosome positions? Trends Genet 25: 335–343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal E, Fondufe-Mittendorf Y, Chen L, Thastrom A, Field Y, Moore IK, Wang JPZ, Widom J 2006. A genomic code for nucleosome positioning. Nature 442: 772–778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semple CAM, Taylor MS 2009. Molecular biology: The structure of change. Science 323: 347–348 [DOI] [PubMed] [Google Scholar]
- Storey JD, Tibshirani R 2003. Statistical significance for genomewide studies. Proc Natl Acad Sci 100: 9440–9445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilgner H, Guigó R 2010. From chromatin to splicing: RNA-processing as a total artwork. Epigenetics 5: 180–184 [DOI] [PubMed] [Google Scholar]
- Tirosh I, Sigal N, Barkai N 2010. Divergence of nucleosome positioning between two closely related yeast species: genetic basis and functional consequences. Mol Syst Biol 6: 365 doi: 10.1038/msb.2010.20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolstorukov MY, Volfovsky N, Stephens RM, Park PJ 2011. Impact of chromatin structure on sequence variability in the human genome. Nat Struct Mol Biol 18: 510–515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong P, Prendergast JG, Lohan AJ, Farrington SM, Cronin S, Friel N, Bradley DG, Hardiman O, Evans A, Wilson JF, et al. 2010. Sequencing and analysis of an Irish human genome. Genome Biol 11: R91 doi: 10.1186/gb-2010-11-9-r91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verlaan DJ, Berlivet S, Hunninghake GM, Madore A-M, Larivière M, Moussette S, Grundberg E, Kwan T, Ouimet M, Ge B 2009. Allele-specific chromatin remodeling in the ZPBP2/GSDMB/ORMDL3 locus associated with the risk of asthma and autoimmune disease. Am J Hum Genet 85: 377–393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Wang W, Li R, Li Y, Tian G, Goodman L, Fan W, Zhang J, Li J, Zhang J, et al. 2008. The diploid genome sequence of an Asian individual. Nature 456: 60–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warnecke T, Batada NN, Hurst LD 2008. The impact of the nucleosome code on protein-coding sequence evolution in yeast. PLoS Genet 4: e1000250 doi: 10.1371/journal.pgen.1000250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Washietl S, Machné R, Goldman N 2008. Evolutionary footprints of nucleosome positions in yeast. Trends Genet 24: 583–587 [DOI] [PubMed] [Google Scholar]
- Wheeler DA, Srinivasan M, Egholm M, Shen Y, Chen L, McGuire A, He W, Chen YJ, Makhijani V, Roth GT, et al. 2008. The complete genome of an individual by massively parallel DNA sequencing. Nature 452: 872–876 [DOI] [PubMed] [Google Scholar]
- Wilson CB, Rowell E, Sekimata M 2009. Epigenetic control of T-helper-cell differentiation. Nat Rev Immunol 9: 91–105 [DOI] [PubMed] [Google Scholar]
- Ying H, Epps J, Williams R, Huttley G 2010. Evidence that localized variation in primate sequence divergence arises from an influence of nucleosome placement on DNA repair. Mol Biol Evol 27: 637–649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Shin H, Song J, Lei Y, Liu XS 2008. Identifying positioned nucleosomes with epigenetic marks in human from ChIP-Seq. BMC Genomics 9: 537 doi: 10.1186/1471-2164-9-537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Moqtaderi Z, Rattner BP, Euskirchen G, Snyder M, Kadonaga JT, Liu XS, Struhl K 2009. Intrinsic histone-DNA interactions are not the major determinant of nucleosome positions in vivo. Nat Struct Mol Biol 16: 847–852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Moqtaderi Z, Rattner BP, Euskirchen G, Snyder M, Kadonaga JT, Liu XS, Struhl K 2010. Evidence against a genomic code for nucleosome positioning. Reply to “Nucleosome sequence preferences influence in vivo nucleosome organization.” Nat Struct Mol Biol 17: 920–923 [DOI] [PMC free article] [PubMed] [Google Scholar]