

Abstract
Alcohol is readily distributed throughout the body in the blood stream and crosses biological membranes, which affect virtually all biological processes inside the cell. Excessive alcohol consumption induces numerous pathological stress responses, part of which is endoplasmic reticulum (ER) stress response. ER stress, a condition under which unfolded/misfolded protein accumulates in the ER, contributes to alcoholic disorders of major organs such as liver, pancreas, heart, and brain. Potential mechanisms that trigger the alcoholic ER stress response are directly or indirectly related to alcohol metabolism, which includes toxic acetaldehyde and homocysteine, oxidative stress, perturbations of calcium or iron homeostasis, alterations of S-adenosylmethionine to S-adenosylhomocysteine ratio, and abnormal epigenetic modifications. Interruption of the ER stress triggers is anticipated to have therapeutic benefits for alcoholic disorders.
1. Introduction
Alcohol is the most socially accepted addictive drug. Alcohol abuse and dependence causes social problems such as domestic violence and loss of productivity in work place as well as traffic accident-related injuries and chronic organ disorders. Excessive alcohol use is the third leading cause of preventable death in the United States and is responsible for 3.8% of deaths worldwide [1–3]. Alcohol-related medical problems can be improved upon a good understanding of pathogenesis of alcohol-induced injuries. After its consumption, alcohol is readily distributed throughout the body in the blood stream and crosses biological membranes which affect virtually all organs and biological processes in the body. Most of the alcohol that enters the body is first oxidized to toxic acetaldehyde, which is catalyzed by the cytosolic alcohol dehydrogenase (ADH) (Figure 1). Acetaldehyde is then converted by acetaldehyde dehydrogenase (ALDH) to acetic acid, which occurs primarily in the liver [4]. Alcohol can also be oxidized to acetaldehyde by cytochrome P450IIE1 (CYP2E1) which generates hydrogen peroxide. Alcohol-related medical illness results directly or indirectly from the toxic alcohol metabolites in cells and tissues. Alcoholic injuries can be found in most organs including brain, gastrointestinal tract, immune system, kidney, lung, heart, pancreas, and most frequently liver (reviewed in [1, 5–13]). Alcohol-induced liver disease (ALD) is better characterized than in other organs. The progression of ALD includes a spectrum of liver diseases, ranging from steatosis, steatohepatitis, fibrosis, to cirrhosis and even cancer [1, 7, 13]. However, the underlying molecular mechanisms of ALD are not completely understood. Both primary factors and cofactors are involved in the pathogenesis of ALD. Primary factors include but are not limited to increased oxidative stress mainly from mitochondrial malfunction and CYP2E1, increased endotoxin production and TNF signaling, impaired innate and adaptive immunity, hypoxia, impaired methionine metabolism, and epigenetic modifications [7, 9, 10, 13–18]. Cofactors may include malnutrition or complications with diabetes, obesity, smoking, or HCV/HIV infections [1, 9, 10, 13]. Alcohol-induced perturbations of homeostasis in the endoplasmic reticulum (ER) have evolved as an important factor contributing to fatty liver disease, which has been reviewed by a few comprehensive reviews [19–22]. Evidence for the involvement of ER in the pathogenesis of alcoholic injury is now accumulating beyond the liver. The purpose of this review is to highlight phenomenological evidence for alcohol-induced ER stress in select organ disorders and to discuss potential molecular mechanisms causing alcoholic ER stress.
Figure 1.
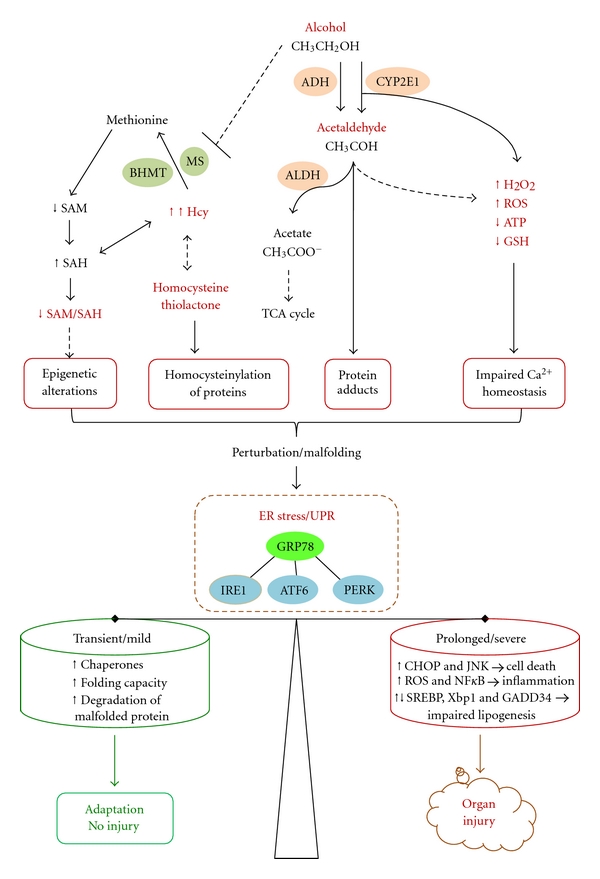
Mechanisms of alcohol-induced endoplasmic reticulum (ER) stress and organ injuries. ADH: alcohol dehydrogenase; ALDH: acetaldehyde dehydrogenase; CYP2E1: cytochrome P450 2E1; ROS: reactive oxidative stress; GSH: glutathione; BHMT: betaine-homocysteine methyltransferase; MS: methionine synthase; Hcy, homocysteine; SAM: S-adenosylmethionine, SAH: S-adenosylhomocysteine; TCA: tricarboxylic acid; UPR: unfolded protein response; GRP78: glucose-regulated protein 78; IRE1: inositol requiring enzyme; ATF6:activating transcription factor 6; PERK: protein kinase ds RNA-dependent-like ER kinase; CHOP: C/EBP-homologous protein; JNK, c-jun-N-terminal kinase; NFκB, nuclear factor κB; SREBP: sterol regulatory element binding protein; Xbp-1: X box binding protein 1; GADD34: growth arrest and DNA damage-inducible protein. See the context for details.
2. ER Stress and the Unfolded Protein Response (UPR)
The ER is an essential organelle for protein synthesis and modifications, for storing and releasing Ca2+, for the biosynthesis of lipids and sterols, and for detoxification of certain drugs. ER stress is a condition under which unfolded or malfolded proteins accumulate in the ER (reviewed in [18–21]). ER stress results from perturbations in ER homeostasis such as calcium depletion, inhibition of glycosylation, alterations of the redox state, or lipid overloading. ER stress triggers the unfolded protein response (UPR), which constitutes a series of ER-to-nucleus signaling mediated by three ER resident transmembrane sensor proteins, inositol requiring protein 1 (IRE1), ds-RNA-activated protein kinase (PKR) like ER kinase (PERK), and activating transcription factor 6 (ATF6) (Figure 1). The three sensors are activated upon dissociation from their inhibitory binding with the chaperone GRP78/BiP. IRE1, which has kinase and endoribonuclease activities, is activated by transautophosphorylation. The activated IRE1 processes the transcription factor X-box binding protein-1 (XBP1) mRNA via the unconventional splicing to form transcriptionally active spliced XBP1 (sXBP1). sXBP1 activates UPR target genes, including chaperones and ER-associated degradation (ERAD) pathway genes. The second sensor PERK phosphorylates the eukaryotic initiation factor 2α-subunit (eIF2α), leading to an inhibition of the initiation of translation and a global attenuation in protein translation. Phosphorylation of eIF2α selectively activates activating transcription factor 4 (ATF4), which regulates ER chaperone genes, ERAD pathway genes, amino acid metabolism genes, and the transcription factor C/EBP homologous protein (CHOP) [19–21]. The third sensor ATF6 is cleaved in the Golgi to form a transcriptionally active fragment that traffics to the nucleus to activate UPR target genes. In general the UPR results in reduced synthesis of nascent proteins, increased unloading of unfolded proteins, and increased capacity of folding, which lead to restoration of ER homeostasis.
However, prolonged or severe UPR provokes a complex network of interacting and parallel responses contributing to pathological consequences such as apoptosis, inflammation, and fat accumulation [19–24]. The ER stress-induced apoptosis is mediated by a few factors. CHOP regulates growth arrest and DNA damage-inducible protein (GADD34). GADD34 binds protein phosphatase-1 and enhances eIF2α dephosphorylation, leading to premature restoration of translation and enhanced ER stress. CHOP can also regulate expression of the TRAIL receptor DR5, pro- and antiapoptotic Bcl-2 family protein Bim, Bax and Bcl-2 modulating cell death [19–21]. Sustained activation of IRE1 recruits the adaptor protein TRAF2 and activates JNK and NF-κB, both of which mediate apoptosis [23]. In addition, alterations in ER calcium homeostasis, upregulation of ER oxidase 1 (ERO1) by CHOP, activation of caspase 12, and activation of GSK3β by tribbles 3 (TRB3) and AKT are other mechanisms underlying ER stress-induced inflammation and apoptosis [21, 23, 25]. Lipid accumulation is also a main pathological feature of prolonged ER stress, and each of the three ER sensor pathways has direct molecular effects on lipid synthesis. The IRE1α-XBP1 branch regulates C/EBPα and C/EBPβ that control directly the expression of genes involved in de novo fatty acid biosynthesis [26]. The ATF6 branch is involved in phospholipid biosynthesis as well as in fatty acid oxidation and lipoprotein secretion [27, 28]. The PERK-eIF2α branch influences expression of C/EBP family and PPARγ transcription factors via the eIF2α-specific phosphatase GADD34 and regulates SREBP1-related de novo lipid synthesis and accumulation [18–24, 29, 30].
3. ER Stress in Alcoholic Organ Injuries
3.1. Liver
Alcohol is mainly metabolized in the liver, and liver cells are rich in ER which assumes synthesis of a large amount of secretory and membrane proteins [19, 20, 29]. Partial role of ER in alcohol metabolism was initially realized decades ago as NADH from the hepatic oxidation of ethanol to acetaldehyde by ADH was found to support also microsomal ethanol oxidations [14, 15]. The inducible microsomal ethanol oxidizing system (MEOS) is associated with proliferation of the ER and a concomitant induction of cytochrome P4502E1 (CYP2E1) in rats and in humans. Free radical release as a consequence of CYP2E1 function in the ER and subsequent oxidative stress and lipid peroxidation generally contribute to ALD [14, 15]. However, alcohol-induced ER stress response was not recognized until recently. Molecular evidence for an impaired UPR was first found in the intragastric alcohol-fed mice using microarray gene expression profiling [18]. The alterations of selected ER stress markers were associated with severe steatosis, scattered apoptosis, and necroinflammatory foci. Moderate upregulation of expression of SREBP-1c and SREBP-2 and their responsive genes was detected by immuoblotting [18]. SREBP-1c knockout mice were protected against triglyceride accumulation [30–32]. Knocking out CHOP resulted in minimal alcohol-induced apoptosis in mouse liver [32–34]. In a setting of alcohol infusion and moderate obesity, there are synergistic effects of accentuated ER and mitochondrial stress, nitrosative stress mediated by M1 macrophage activation, and adiponectin resistance on hepatic necroinflammation and steatohepatitis [35]. In micropigs fed alcohol, liver steatosis and apoptosis were shown to be accompanied by increased mRNA levels of CYP2E1, GRP78 and SREBP-1c, and protein levels of CYP2E1, GRP78, activated SREBP and caspase 12 [36]. In addition, the ER stress response was correlated with elevated transcripts of lipogenic enzymes such as fatty acid synthase (FAS), acetyl-CoA carboxylase (ACC), and stearoyl-CoA desaturase (SCD). Further, alcohol-induced lipopolysaccharide (LPS) is linked to impaired UPR and advanced hepatic injury [37–39]. In cirrhotic rat livers, only eIF2α was activated in the basal state. After LPS challenge, full UPR as indicated by activation of IRE1α, ATF-6, and eIF2α was detected [37]. However, LPS-induced accumulation of NF-κB-dependent antiapoptotic proteins was not observed, suggesting that the UPR sensitized the cirrhotic livers to LPS/TNFα-mediated apoptosis. Alcohol-induced hepatic ER stress response not only occurs in rodents but also in livers of baboon and human patients [40, 41]. In baboon fed alcohol orally, upregulation of calpain 2, calpain p94, and ERD21 and downregulation of eIF2α were among the genes of altered expression that was revealed by using cDNA array analysis [41]. Gene expression profiling of cirrhotic liver samples from human alcoholics also revealed alterations of calpain and calreticulin that are indicative of ER malfunction.
3.2. Pancreas
The pancreas is one of the important digestive organs adversely affected by alcohol abuse. Pancreatitis is among the most common alcohol-related hospital diagnosis in USA [11]. The underlying mechanisms for alcohol-induced pancreatitis are not well understood. Similar to the liver, the pancreas has the capacity to metabolize alcohol via both the oxidative and nonoxidative pathways yielding toxic metabolites such as acetaldehyde and lipid esters. Fatty acid ethyl and cholesteryl esters are known to accumulate in the acinar cell after chronic alcohol consumption which decreases the stability of the membranes of zymogen granules and lysosomes [42, 43], which cause a premature activation of intracellular digestive enzyme and may predispose the gland to autodigestive inflammation and injury. In respect to the role of organelles in alcoholic pancreatic injury, the ER has been considered as the acinar cell has the highest rate of protein synthesis among all tissues in adult organism. In fact, perturbations of ER homeostasis are found in acute pancreatitis [44, 45], and all the three ER stress/UPR transducers (i.e., IRE1, ATF6, and PERK) and their downstream pathways are activated. However, chronic alcohol feeding alone causes minimal pancreatic tissue injury in animal models [45, 46]. Further studies demonstrate that alcohol feeding activates the UPR in pancreas with upregulation of the transcription factor XBP1 in the intragastric alcohol infusion model [47, 48]. This suggests that alcohol induces a physiologic adaptive UPR that may prevent pathophysiologic pancreatitis responses. Indeed, heterozygous deletion of the XBP1 gene prevents XBP1 upregulation and results in pathologic changes including extensive dilation of the ER with occasional dense luminal inclusions, hallmarks of ER stress, and significant accumulation of autophagic vacuoles in acinar cells [48]. Thus, impaired UPR in the pancreas can potentiate alcohol-induced toxicity and aggravate pancreatic damages.
3.3. Brain
Alcohol exposure during development has devastating effects on the loss of neurons in selected brain areas, which leads to profound damages to the central nervous system (CNS). Alcohol consumption during pregnancy causes fetal alcohol spectrum disorders (FASDs) [1, 49]. Microcephaly, abnormal cortical thickness, reduced cerebral white matter volume, ventriculomegaly, and cerebellar hypoplasia are the prominent CNS abnormalities in FASDs. Children with (FASD) have a variety of cognitive, behavioral, and neurological impairments [49]. What cause ethanol-induced neurodegeneration are not clear. Considering that ER stress plays a role in the pathogenesis of several popular neurological diseases such as Huntington's disease, brain ischemia, Alzheimer's disease, and Parkinson's disease [50–53], an involvement of ER stress in alcohol-induced neuron toxicity has been hypothesized [54]. Recent evidence from both in vitro and in vivo tests appears to support the assumption. Exposure of SH-SY5Y neuroblastoma cells or primary cerebellar granule neurons to ethanol alone had little effect on the expression of ER stress markers [54]; however, ethanol markedly increased the expression of GRP78, CHOP, ATF4, ATF6, and phosphorylated PERK and eIF2α in the presence of tunicamycin or thapsigargin, which was accompanied with increased cell death. Acute exposure of seven-day-old mice to ethanol by subcutaneous injection at a dose of 5 g/kg significantly increased ER stress response. Increase of ATF6, CHOP, GRP78, and mesencephalic astrocyte-derived neurotrophic factor as well as the phosphorylation of IRE1, eIF2α, PERK, and PKR were detected within 24 hours after the ethanol exposure. Further, the ethanol-induced increase in phosphorylated eIF2α, caspase-12 and CHOP was distributed in neurons of specific areas of the cerebral cortex, hippocampus, and thalamus. Since the age of the animals used in this experiment is equivalent to the third trimester of pregnancy in humans, the above evidence suggests that ethanol directly induce ER stress in the developing brain.
3.4. Heart
It is well documented that chronic heavy alcohol drinking is a risk factor for cardiovascular disorders including cardiac hypertrophy, myofibrillar disruption, reduced contractility, and decreased ejection fraction [55]. Alcohol may change the circulatory hemodynamics resulting in stress on the heart. The stressed heart demands more cardiac output which leads to compensative hypertrophic responses such as neurohormonal activation and increased growth factors and cytokines, resulting in enlarged cardiomyocytes and increased sarcomere assembly. ER stress may play a critical role in regulating protein synthesis in cardiac myocytes, and thereby produce cell enlargement and cardiac hypertrophy. Chronic alcohol consumption by FVB (Friend virus-B type) albino mice at 4% of diet for 12 weeks resulted in increased heart weight and heart-to-body weight ratio [56]. In the myocardium of the FVB mice chronically fed alcohol, GRP78, CHOP, and IRE1a protein expression levels were increased, indicative of the UPR. Class I alcohol dehydrogenase efficiently oxidizes alcohol resulting in increased production of acetaldehyde. Overexpressing alcohol dehydrogenase in the FVB mice during chronic ethanol treatment resulted in a greater UPR upregulation [56]. The finding indicates that acetaldehyde from alcohol metabolism may induce ER stress. Furthermore, overexpressing of the antioxidant protein metallothionein in FVB mice significantly reduced peak shortening and maximal shortening velocity of cardiac myocytes by LPS, which is often elevated in alcoholics [13–15, 39, 40]. In parallel, the transgenic FVB mice displayed decreased protein levels of GRP78, CHOP, PERK, and IRE1 whereas the wild type FVB displayed a significant increase in the protein levels of PERK, phospho-JNK, and phospho-p38 in the myocardium in response to LPS [56, 57].
4. Mechanisms of Alcohol-Induced ER Stress
4.1. Acetaldehyde Adducts and ER Stress
Alcohol-derived acetaldehyde is highly reactive [58–62]. At physiological temperature and pH, acetaldehyde reacts with nucleophilic groups in proteins, such as α-amino groups of internal lysine residues and the ε-amino group on the N-terminal amino acid of unblocked proteins forming unstable Schiff base acetaldehyde adducts. In addition, ethanol abuse may also lead to the formation of other types of protein adducts, such as malondialdehyde-acetaldehyde hybrids and α-hydroxyethyl protein-adducts. The acetaldehyde adducts initiate immunogenic reactions, cause conformational changes and inactivation of the adducted targets, or trigger aberrant protein degradation, which contribute to the development of alcoholic organ diseases (Figure 1). Malondialdehyde–acetaldehyde adduct is found to be the dominant epitope after malondialdehyde modification of proteins in atherosclerosis [63]. Antibodies to the aldehyde adducts have been detected in the serum of patients with atherosclerotic lesions and correlate with the progression of atherosclerosis. It is known that atherosclerosis develops as a result of protein unfolding and modification of protein and/or macromolecular complex function at the cellular level [63]. In supporting this, evidence for ER stress response was found in transgenic mice with cardiac overexpression of ADH that increased acetaldehyde exposure [56, 57]. The ADH transgene increased induction of IRE1, eIF-2α, GRP78, and CHOP and exacerbated chronic alcohol ingestion-induced myocardial dysfunction and hypertrophy. Further, in a mouse model of acute ethanol intoxication, inhibition of ADH causes downregulation of GRP78 mRNA levels [64]. This suggests a causal relationship between ethanol metabolism and ER stress response. Acetaldehyde adducts also affect ER Ca2+ handling in rat ventricular myocytes [65, 66], which may disturb ER calcium homeostasis playing a critical role in stress-mediated cellular injury [67]. In response to alcohol dosing in vivo, the actin in Type I and Type II fibre predominant muscles of rats was found to form stable covalent adducts with acetaldehyde [68]. Histochemical analysis showed that unreduced-acetaldehyde-protein adducts were located within the sarcolemmal (i.e., muscle membrane) and subsarcolemmal regions, which perturbed the membranes and increased protein and enzyme activity of sarcoplasmic-ER Ca2+-ATPase, resulting in muscle cell death and alcoholic myopathy. In addition, acetaldehyde adducts are found in the central nervous system which may be responsible for alcoholic ER stress response. In the brain of a heavy drinker who had died suddenly while drinking continuously, acetaldehyde adducts were immunologically identified [69]. In a mouse model administered with the Lieber-DeCarli liquid diet and alcohol, acetaldehyde adducts were readily detected in degenerated neurons in the cerebral cortex [70]. The neural region that alcoholic ER stress response occurred colocalized with the acetaldehyde adducts. In young mice, ethanol-induced increase in ER stress protein markers was found to be distributed in the immature neurons of specific areas of the cerebral cortex, hippocampus and thalamus [54]. Thus, while most organs of the body can be affected by alcohol-derived acetaldehyde, cardiac and skeletal muscle cells and neurons appear to be particularly susceptible to acetaldehyde adducts that cause ER stress and injury.
4.2. Homocysteine Toxicity and ER Stress
Homocysteine (Hcy) is a normal intermediate involved in the metabolism of the essential amino acid-methionine (Figure 1). Excessive Hcy is toxic to cells. An abnormally elevated level of Hcy in the blood, a medical condition termed hyperhomocysteinemia (HHcy), is an independent risk factor in cardiovascular, neurodegenerative diseases, diabetes, obesity, and hepatic steatosis [32, 71–73]. It is generally accepted that aminoacyl thioester homocysteine thiolactone (HTL) derived from Hcy editing during protein synthesis contributes to the most of Hcy toxicity [74, 75]. HTL undergoes not only nucleophilic, which can be facilitated in the presence of acetaldehyde, but also electrophilic reactions to form protein adducts or homocysteinylation of protein lysine side chains and/or other free amine groups [75]. These reactions cause malfolding of proteins and trigger ER stress response. Evidence linking HHcy to ER stress and alcoholic liver injury has well been established in cell and animal models [16, 18–20, 32]. The intragastric alcohol feeding exhibited a greater than 5-fold increase in mouse plasma Hcy [18, 34, 35]. Hcy is metabolized normally by remethylation to methionine which is catalyzed by methionine synthase (MS) using folate as a methyl donor and by betaine-homocysteine methyltransferase (BHMT) using betaine as a methyl donor. Chronic alcohol-induced disturbance of methionine metabolism appears to contribute to the alcoholic HHcy. Alcohol inhibits enzyme activity of MS in mice and rats and reduces mRNA expression of BHMT and MS in mice [16, 17, 34, 76–79]. Simultaneous betaine feeding in the intragastric alcohol-fed mice decreased alcoholic HHcy and abrogated ER stress response in parallel with decreased ALT and amelioration of alcohol-induced necroinflammation, apoptosis, and fatty liver [18]. In cultured HepG2 cells, BHMT overexpression inhibited Hcy-induced ER stress response, lipid accumulation, and cell death [77]. In primary mouse hepatocytes, suppression of BHMT by RNA interference potentiated Hcy-induced but not tunicamycin-induced ER stress response and cell injury [77]. Transgenic mice expressing human BHMT in organs peripheral to the liver are resistant to alcohol or a high methionine and low folate diet induced HHcy and fatty liver [78]. In intragastric alcohol-fed rats, BHMT is induced, which minimizes the effect of inhibited MS on Hcy levels and subsequent ER stress response and injury [79]. In a survey using 14 mouse strains, Ivan Rusyn has found that the alcoholic HHcy is correlated with alcohol-induced liver jury (personal communication, 2011). Therefore, the above several lines of evidence support Hcy toxicity as a pathogenic factor contributing to alcohol-induced disorders.
4.3. SAM/SAH Ratio, Epigenetic Alterations and ER Stress
There are two types of important epigenetic regulations of gene expression: DNA methylation of cytosines within CpG dinucleotides and histone modifications [80, 81]. Aberrant epigenetic changes are involved in the etiology of a growing number of disorders such as alcohol dependence. Both global hypomethylation of DNA in liver and hypermethylation of DNA from peripheral blood cells have been reported in animal models and in human subjects with alcohol dependence [82–86]. This is because DNA methylation in general depends on the methyl donor S-adenosylmethionine (SAM) and is inhibited by S-adenosylhomocysteine (SAH). Both SAM and SAH are involved in methionine metabolism [87, 88]. Inside the cell, SAM is demethylated to SAH, which is readily converted to Hcy which is remethylated to methionine. Plasma Hcy is not metabolized and represents the cumulative export of Hcy from liver and other tissues. Alcohol consumption decreases levels of SAM and increases levels of SAH and/or Hcy resulting in a decrease in SAM to SAH ratio (Figure 1) [76, 78, 87–92]. Thus, alcohol has a marked impact on the hepatic methylation capacity. Evidence demonstrating epigenetic effects on alcoholic ER stress is emerging [17, 82]. In 66 male alcoholic patients with alcohol dependence, chronically elevated Hcy levels are associated with increased DNA methylation in the promoter region of homocysteine-inducible ER protein (HERP) and decreased expression of HERP mRNA in the blood [93, 94]. The decrease in HERP levels is followed by a lethal ER stress, mitochondrial dysfunction, and cell death in neurons of the developing and adult brain [94]. Thus it is conceivable that alcoholic Hcy regulates HERP and causes ER stress and injury through an epigenetic mechanism. In respect to the epigenetic modifications of histone, it is reported that alcohol causes a dose- and time-dependent selective acetylation of histone H3-K9 in cultured hepatocytes [95, 96]. Intragastric administration of ethanol increases the levels of acetylated H3-K9 by 2-3 folds in the liver of mice after 12 h [97]. Further analysis indicates that the increased acetylation is tissue specific as it is noted in liver, lung, and spleen but not in tissues from other organs tested. Thus, while other stress pathways such as the MAPK signaling may be involved, the alcoholic epigenetic effects on the ER stress pathways can be more relevant. For instance, in both cystathionine beta synthase heterozygous (CBS+/−) and wild type (WT) mice fed ethanol diets by intragastric infusion for 4 weeks, steatohepatitis, reduction in liver SAM, elevation in liver SAH, and reduction in the SAM/SAH ratio were observed [17]. Hepatic ER stress markers including GRP78, ATF4, CHOP, caspase 12, and SREBP-1c were upregulated and negative correlated with the SAM/SAH ratio in response to alcohol. Further, trimethylated histone H3 lysine-9 (3meH3K9) protein levels in centrilobular regions revealed by immunohistochemistry were reduced in ethanol-fed mice. The levels of 3meH3K9 in the promoter regions of GRP78, SREBP-1c, and CHOP revealed specifically by a chromatin immunoprecipitation assay were decreased only in CBS+/− mice fed alcohol. Since CBS is involved in transsulfuration of Hcy, the findings imply that interactions of CBS ablation and alcohol feeding impair methionine metabolism, which leads to epigenetic modifications of ER stress signaling pathways. In addition, the key modulator of UPR, sXBP1 has recently been found to be a nonhistone protein target of acetylation mediated by p300 and deacetylation mediated by the NAD+-dependent class III deacetylase SIRT1 (sirtuin 1) [98, 99]. SIRT1 is demonstrated to be one of the major targets of alcohol action which influences TNF-α production in macrophages and alters glucose and lipid metabolism in the liver leading to hepatic steatosis and inflammation [100–102]. SIRT1 may also play a role in alcohol-induced ER stress response and injury through an epigenetic mechanism.
4.4. Oxidative Stress and Disruption of Ca2+ or Iron Homeostasis and ER Stress
In the ER, proteins undergo oxidative protein folding. PDI is a critical oxoreductase that catalyzes disulfide bond formation with consequent generation of reactive oxygen species (ROS) during the oxidative protein folding [19, 103]. ROS is normally under control due to cellular glutathione that sustains PDI ability to regenerate and form disulfide bridges repeatedly [103–105]. However, chronic ethanol consumption increases the production of a variety of ROS, including superoxide, H2O2, lipid peroxides, and peroxynitrite [1, 13–15]. Alcoholic ROS reduce glutathione level and increase oxidized glutathione, which breaks the redox status of the ER (Figure 1). This loss of redox homeostasis perturbs the oxidative folding and makes PDI ineffective in the catalytic redox cycles leading to more utilization of reduced glutathione. Depletion of glutathione generates excessive ROS which triggers ER stress. Antioxidant treatment, CHOP deletion, or translation attenuation has been shown to reduce oxidative stress and preserve ER function [19–23]. Ethanol rapidly caused oxidative stress in cultured neuronal cells and antioxidants blocked alcoholic potentiation of ER stress and cell death [54]. An association of ER stress response with increased oxidized glutathione was found in the pancreatic acinar cell of the ethanol-fed rats [47]. In HepG2 cells, acetaldehyde impaired mitochondrial glutathione transport and stimulated mitochondrial cholesterol content, the latter of which was preceded by increased levels of CHOP and SREBP1 [106]. Chronic exposure of animals to alcohol or overexpression of cytochrome CYP2E1 in hepatocytes increases the expression of superoxide dismutase (SOD) and activates nuclear factor erythroid 2-related factor 2 (Nrf2), which is an ER stress responsive factor [14, 107–109]. These lines of evidence suggest an intimate relationship between ER stress and ROS production. Furthermore, alcoholic oxidative stress plays a critical role in possible interplays between ER stress and mitochondrial stress, which can be mediated either by intracellular calcium or iron. Alcohol or Hcy induces alterations of lipid composition in the ER and affected ratio of phosphatidylcholine (PC) to phosphatidylethanolamine (PE) [20, 78]. Alterations of the PC/PE ratio disrupt ER calcium homeostasis causing ER stress [110]. Under ER stress, abnormal release of intracellular Ca2+ from the ER via inositol 1,4,5-triphosphate receptor (IP3R) channels leads to excessive mitochondrial Ca2+ uptake, which in turn promotes ROS production and apoptosis via multiple effects on the mitochondria [67, 111, 112]. Elevated serum iron indices (transferrin saturation, ferritin) and hepatic iron overloading are often observed in patients with alcoholic liver disease [113–117]. Excessive iron damages mitochondrial iron–sulfur clusters that generate defects in heme-containing cytochrome c and cytochrome oxidase leading to excess mitochondrial ROS [118]. Iron homeostasis is regulated by hepcidin, a circulatory antimicrobial peptide synthesized in hepatocytes [119]. Critically, ER stress response can regulate expression of hepcidin [19, 29, 120]. Thus a vicious cycle exists: alcoholic ROS and/or ER stress damage mitochondria through iron, which in return augments ROS and stresses the ER further, all of which probably act synergistically to cause severe alcoholic injury.
4.5. Synergistic ER Stress by Alcohol, Drugs, Viral Infection and Environments
Acute alcohol or chronic alcohol at moderate concentrations may not induce readily detectable ER stress response in some cell and animal models [29, 47]. This does not rule out the doomed potential of alcohol to induce ER stress. Indeed, ER stress can be synergistically induced by alcohol in the presence of environmental factors, genetic predispositions, drugs, or virus infection. First, it is recently noted that an accelerated development of pancreatitis in alcoholic patients who smoke may result from an additive or multiplicative effect that is mediated by ER stress response [47]. Second, in a mouse model with liver-specific deletion of Grp78, low-level oral alcohol feeding did not induce HHcy that is often seen in mice fed high doses of alcohol [29]. However, the low alcohol feeding activated SREBP1 and unconventional splicing of Xbp1 (sXbp1) and decreased Insig 1 and ATF6 and its downstream targets such as ERp57 and Derl3 in the liver GRP78 knockouts, leading to aggravated lipid accumulation in the liver. Thus, compared to the aforementioned Hcy-ER stress mechanism, Grp78 deletion represents a genetic predisposition that unmasks a distinct mechanism by which alcohol induces ER stress, one that normally is largely obscured by compensatory changes in normal animals or presumably in the majority of human population who have low-to-moderate drinking. Similarly, certain drugs potentiate alcoholic ER stress response. For instance, some HIV protease inhibitors (HIV PIs) used in anti-HIV therapeutics can cause adverse side effects such as dyslipidemia and liver injury [29, 121, 122]. Portion of HIV-infected patients often concomitantly consume or abuse alcohol leading to more severe liver injury. One of the underlying mechanisms is severe ER stress responses that are caused by both alcohol and the HIV drugs. It has been demonstrated that single gavage dosing for alcohol alone or ritonavir and lopinavir combined did not induce detectable liver injury in wild type [29]. However, the gavage treatment with alcohol plus the two HIV drugs caused significant increase in plasma ALT as well as activation of CHOP, ATF4, and sXbp1. Thus, alcohol exacerbates some HIV drug-induced ER stress and subsequent injury. Third, it is known that both alcoholic activation of the ER stress sensor-IRE1α and alcohol-induced accumulation of proinflammatory cytokines such as TNFα, IL-6, and MCP-1 activate JNK and/or NF-κB pathways that mediate tissue/organ injuries [9, 10, 23, 29, 37–39]. This pathway overlap may be a result of interactions between ER stress and inflammation. The likely scenario is that mild ER stress under moderate alcohol dosing has a negative impact on ER function, which makes cells more susceptible to inflammatory signals, which subsequently augments ER stress response and injury via the JNK pathway. Fourth, alcohol may sensitize virus-infected cells to ER stress and apoptosis. It is reported that hepatitis C (HCV) infection causes ER stress in cell and animal models as well as in patients with chronic HCV [123–125]. HCV directly induces steatosis and development of hepatocellular carcinoma (HCC), which is correlated with a state of oxidative stress in mice transgenic for the HCV core protein [126, 127]. There is clinical evidence indicating that alcohol metabolism increases HCV replication and modulates the host response to HCV [128, 129]. The HCV nonstructural protein 5A (NS5A) localizes to the ER and is part of the HCV replication complex that forms altered cytoplasmic membrane structures. The membrane structure triggers ER stress and the UPR, leading to a release of ER Ca2+ stores and subsequent oxidative stress [124]. In addition, interactions between HCV core and destabilization of the mitochondrial electron transport chain result in increased production of ROS [130, 131]. Since alcohol alone perturbs Ca2+ homeostasis and stimulates ROS generation, it is conceivable that ROS mediates the synergistic interactions between alcohol consumption and HCV infection.
5. Concluding Remarks
While a large number of different stress responses and pathological pathways have been implicated in ethanol-induced injury [1, 7, 13–15], the occurrence of ER stress in the major organs including liver, brain, pancreas, and heart firmly supports its contributing role to alcoholic disorders. Alcohol causes alterations in many specific steps involved in the ER stress and UPR. The potential causes for alcohol-induced ER stress are directly or indirectly related to alcohol metabolism, which include but may not be limited to toxic acetaldehyde and homocysteine modifying proteins, oxidative stress from impaired CYP2E1 function and perturbations of calcium or iron homeostasis, alterations of SAM to SAH ratio and subsequent biochemical or epigenetic modifications, and, most importantly, interactions between these factors. Each of the factors may contribute more or less to the induction of the ER stress depending on tissues/organs or experimental models, dosage and duration of alcohol exposure, and presence of other environmental factors. Current investigations and conclusions on alcoholic ER stress appear depending on positive identifications of selective molecular markers of ER stress response, conclusions from which can be misleading sometimes. For instance, the ER stress-induced UPR is dynamic. It can be protective when most of the ER markers are positively detected or detrimental when most markers are latent or disappearing. The timing and quantity of the protection cannot be defined currently. Thus, circumstantially negative observations of the ER stress markers may not necessary rule out an existence of alcoholic ER stress. Future research should be directed at developing sensitive markers, particularly epigenetic markers, for identifying the alcoholic ER stress, and at defining timing and dynamics of the alcoholic ER stress and injuries using both acute and chronic models. Another point is that the ER is a cytosolic network that communicates readily with other cellular loci such as mitochondria, lysosome, cytoplasm, and nucleus. Simultaneous appearance of alcoholic dysfunctions of the other loci such as ATP depletion, abnormal degradation of the inside materials, oxidative stress, and numerous other stress responses could overshadow the role of ER stress in alcoholic diseases. Thus, the role of alcoholic ER stress in organ disorders can be defined precisely by studying complex interplays among the organelles and loci in disease pathogenesis, which could provide better therapeutic strategies targeting the ER. Finally, with respect to the therapeutic interventions at alcoholic ER stress, possible approaches include lowering homocysteine and raising SAM by nutritional support with betaine or folate [16, 20, 32], improving protein folding by using chemical chaperone PBA (sodium 4-phenylbutyrate) and TUDCA [19, 20, 29], blocking eIF2α dephosphorylation by using salubrinal [132], and ameliorating ROS production from the oxidative protein folding by using antioxidants. However, results of clinical trials are not available. Each of the individual approaches alone may not be a simple or universal cure as alcohol-induced pathogenesis is very complex. It is anticipated that properly combined treatments with all the beneficial agents can be effective.
Acknowledgments
This work has been supported by NIH Grants R01AA018846, R01AA018612, and R01AA014428 and by the USC Research Center for Liver Disease (P30 DK48522) and the Southern California Research Center for ALPD and Cirrhosis (P50 AA11999). The author thanks Dr. N. Kaplowitz and the graduate students and fellows who contributed to the studies.
References
- 1.Gunzerath L, Hewitt BG, Li TK, Warren KR. Alcohol research: past, present, and future. Annals of the New York Academy of Sciences. 2011;1216(6648):1–23. doi: 10.1111/j.1749-6632.2010.05832.x. [DOI] [PubMed] [Google Scholar]
- 2.Heron MP, Hoyert DL, Murphy SL, Xu JQ, Kochanek KD, Tejada-Vera B. Deaths: final data for 2006. National Vital Statistics Reports. 2009;57(14):1–134. [PubMed] [Google Scholar]
- 3.Rehm J, Mathers C, Popova S, Thavorncharoensap M, Teerawattananon Y, Patra J. Global burden of disease and injury and economic cost attributable to alcohol use and alcohol-use disorders. The Lancet. 2009;373(9682):2223–2233. doi: 10.1016/S0140-6736(09)60746-7. [DOI] [PubMed] [Google Scholar]
- 4.Zakhari S. Overview: how is alcohol metabolized by the body? Alcohol Research and Health. 2006;29(4):245–254. [PMC free article] [PubMed] [Google Scholar]
- 5.Neiman J. Alcohol as a risk factor for brain damage: neurologic aspects. Alcoholism: Clinical and Experimental Research. 1998;22(7):346–351. doi: 10.1097/00000374-199807001-00011. [DOI] [PubMed] [Google Scholar]
- 6.Epstein M. Alcohol’s impact on kidney function. Alcohol Health and Research World. 1997;21(1):84–92. [PMC free article] [PubMed] [Google Scholar]
- 7.French SW. The mechanism of organ injury in alcoholics: implications for therapy. Alcohol and Alcoholism Supplement. 1991;1:57–63. [PubMed] [Google Scholar]
- 8.Bode C, Bode JC. Alcohol’s role in gastrointestinal tract disorders. Alcohol Health and Research World. 1997;21(1):76–83. [PMC free article] [PubMed] [Google Scholar]
- 9.Szabo G, Wands JR, Eken A, et al. Alcohol and hepatitis C virus-interactions in immune dysfunctions and liver damage. Alcoholism: Clinical and Experimental Research. 2010;34(10):1675–1686. doi: 10.1111/j.1530-0277.2010.01255.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gao B, Seki E, Brenner DA, et al. Innate immunity in alcoholic liver disease. American Journal of Physiology—Gastrointestinal and Liver Physiology. 2011;300(4):516–525. doi: 10.1152/ajpgi.00537.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yang AL, Vadhavkar S, Singh G, Omary MB. Epidemiology of alcohol-related liver and pancreatic disease in the United States. Archives of Internal Medicine. 2008;168(6):649–656. doi: 10.1001/archinte.168.6.649. [DOI] [PubMed] [Google Scholar]
- 12.George A, Figueredo VM. Alcohol and arrhythmias: a comprehensive review. Journal of Cardiovascular Medicine. 2010;11(4):221–228. doi: 10.2459/JCM.0b013e328334b42d. [DOI] [PubMed] [Google Scholar]
- 13.Frazier TH, Stocker AM, Kershner NA, Marsano LS, McClain CJ. Treatment of alcoholic liver disease. Therapeutic Advances in Gastroenterology. 2011;4(1):63–81. doi: 10.1177/1756283X10378925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cederbaum AI, Lu Y, Wu D. Role of oxidative stress in alcohol-induced liver injury. Archives of Toxicology. 2009;83(6):519–548. doi: 10.1007/s00204-009-0432-0. [DOI] [PubMed] [Google Scholar]
- 15.Thurman RG, Ji S, Lemasters JJ. Alcohol-induced liver injury. The role of oxygen. Recent Developments in Alcoholism. 1984;2:103–117. [PubMed] [Google Scholar]
- 16.Halsted CH. Nutrition and alcoholic liver disease. Seminars in Liver Disease. 2004;24(3):289–304. doi: 10.1055/s-2004-832941. [DOI] [PubMed] [Google Scholar]
- 17.Esfandiari F, Medici V, Wong DH, et al. Epigenetic regulation of hepatic endoplasmic reticulum stress pathways in the ethanol-fed cystathionine β synthase-deficient mouse. Hepatology. 2010;51(3):932–941. doi: 10.1002/hep.23382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ji C, Kaplowitz N. Betaine decreases hyperhomocysteinemia, endoplasmic reticulum stress, and liver injury in alcohol-fed mice. Gastroenterology. 2003;124(5):1488–1499. doi: 10.1016/s0016-5085(03)00276-2. [DOI] [PubMed] [Google Scholar]
- 19.Malhi H, Kaufman RJ. Endoplasmic reticulum stress in liver disease. Journal of Hepatology. 2011;54(4):795–809. doi: 10.1016/j.jhep.2010.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ji C. Dissection of endoplasmic reticulum stress signaling in alcoholic and non-alcoholic liver injury. Journal of Gastroenterology and Hepatology. 2008;23(1):S16–S24. doi: 10.1111/j.1440-1746.2007.05276.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ron D, Hubbard SR. How IRE1 Reacts to ER Stress. Cell. 2008;132(1):24–26. doi: 10.1016/j.cell.2007.12.017. [DOI] [PubMed] [Google Scholar]
- 22.Colgan SM, Hashimi AA, Austi RC. Endoplasmic reticulum stressand lipid dysregulation. Expert Reviews in Molecular Medicine. 2011;13:p. e4. doi: 10.1017/S1462399410001742. [DOI] [PubMed] [Google Scholar]
- 23.Kitamura M. Control of NF-κB and inflammation by the unfolded protein response. International Reviews of Immunology. 2011;30(1):4–15. doi: 10.3109/08830185.2010.522281. [DOI] [PubMed] [Google Scholar]
- 24.Lee AH, Scapa EF, Cohen DE, Glimcher LH. Regulation of hepatic lipogenesis by the transcription factor XBP1. Science. 2008;320(5882):1492–1496. doi: 10.1126/science.1158042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bommiasamy H, Back SH, Fagone P, et al. ATF6α induces XBP1-independent expansion of the endoplasmic reticulum. Journal of Cell Science. 2009;122(10):1626–1636. doi: 10.1242/jcs.045625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rutkowski DT, Wu J, Back SH, et al. UPR pathways combine to prevent hepatic steatosis caused by ER stress-mediated suppression of transcriptional master regulators. Developmental Cell. 2008;15(6):829–840. doi: 10.1016/j.devcel.2008.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Oyadomari S, Harding HP, Zhang Y, Oyadomari M, Ron D. Dephosphorylation of translation initiation factor 2α enhances glucose tolerance and attenuates hepatosteatosis in mice. Cell Metabolism. 2008;7(6):520–532. doi: 10.1016/j.cmet.2008.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bobrovnikova-Marjon E, Hatzivassiliou G, Grigoriadou C, et al. PERK-dependent regulation of lipogenesis during mouse mammary gland development and adipocyte differentiation. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(42):16314–16319. doi: 10.1073/pnas.0808517105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ji C, Kaplowitz N, Lau MY, Kao E, Petrovic LM, Lee AS. Liver-specific loss of glucose-regulated protein 78 perturbs the unfolded protein response andexacerbates a spectrum of liver diseases in mice. Hepatology. 2011;54(1):229–239. doi: 10.1002/hep.24368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ji C, Chan C, Kaplowitz N. Predominant role of sterol response element binding proteins (SREBP) lipogenic pathways in hepatic steatosis in the murine intragastric ethanol feeding model. Journal of Hepatology. 2006;45(5):717–724. doi: 10.1016/j.jhep.2006.05.009. [DOI] [PubMed] [Google Scholar]
- 31.Ji C, Kaplowitz N. ER stress: can the liver cope? Journal of Hepatology. 2006;45(2):321–333. doi: 10.1016/j.jhep.2006.06.004. [DOI] [PubMed] [Google Scholar]
- 32.Dara L, Ji C, Kaplowitz N. The contribution of endoplasmic reticulum stress to liver diseases. Hepatology. 2011;53(5):1752–1763. doi: 10.1002/hep.24279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ji C, Mehrian-Shai R, Chan C, Hsu YH, Kaplowitz N. Role of CHOP in hepatic apoptosis in the murine model of intragastric ethanol feeding. Alcoholism: Clinical and Experimental Research. 2005;29(8):1496–1503. doi: 10.1097/01.alc.0000174691.03751.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ji C, Deng Q, Kaplowitz N. Role of TNF-α in ethanol-induced hyperhomocysteinemia and murine alcoholic liver injury. Hepatology. 2004;40(2):442–451. doi: 10.1002/hep.20309. [DOI] [PubMed] [Google Scholar]
- 35.Xu J, Lai KK, Verlinsky A, et al. Synergistic steatohepatitis by moderate obesity and alcohol in mice despite increased adiponectin and p-AMPK. Journal of Hepatology. 2011;55(3):673–682. doi: 10.1016/j.jhep.2010.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Esfandiari F, Villanueva JA, Wong DH, French SW, Halsted CH. Chronic ethanol feeding and folate deficiency activate hepatic endoplasmic reticulum stress pathway in micropigs. American Journal of Physiology—Gastrointestinal and Liver Physiology. 2005;289(1):G54–G63. doi: 10.1152/ajpgi.00542.2004. [DOI] [PubMed] [Google Scholar]
- 37.Tazi KA, Bièche I, Paradis V, et al. In vivo altered unfolded protein response and apoptosis in livers from lipopolysaccharide-challenged cirrhotic rats. Journal of Hepatology. 2007;46(6):1075–1088. doi: 10.1016/j.jhep.2007.01.034. [DOI] [PubMed] [Google Scholar]
- 38.Järveläinen HA, Oinonen T, Lindros KO. Alcohol-induced expression of the CD14 endotoxin receptor protein in rat Kupffer cells. Alcoholism: Clinical and Experimental Research. 1997;21(8):1547–1551. [PubMed] [Google Scholar]
- 39.Su GL, Rahemtulla A, Thomas P, Klein RD, Wang SC, Nanji AA. CD14 and lipopolysaccharide binding protein expression in a rat model of alcoholic liver disease. American Journal of Pathology. 1998;152(3):841–849. [PMC free article] [PubMed] [Google Scholar]
- 40.He L, Marecki JC, Serrero G, Simmen FA, Ronis MJ, Badger TM. Dose-dependent effects of alcohol on insulin signaling: partial explanation for biphasic alcohol impact on human health. Molecular Endocrinology. 2007;21(10):2541–2550. doi: 10.1210/me.2007-0036. [DOI] [PubMed] [Google Scholar]
- 41.Seth D, Leo MA, McGuinness PH, et al. Gene expression profiling of alcoholic liver disease in the baboon (Papiohamadryas) and human liver. American Journal of Pathology. 2003;163(6):2303–2317. doi: 10.1016/S0002-9440(10)63587-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pandol SJ, Periskic S, Gukovsky I, et al. Ethanol diet increases the sensitivity of rats to pancreatitis induced by cholecystokinin octapeptide. Gastroenterology. 1999;117(3):706–716. doi: 10.1016/s0016-5085(99)70465-8. [DOI] [PubMed] [Google Scholar]
- 43.Gukovskaya AS, Mareninova OA, Odinokova IV, et al. Cell death in pancreatitis: effects of alcohol. Journal of Gastroenterology and Hepatology. 2006;21(3):S10–S13. doi: 10.1111/j.1440-1746.2006.04571.x. [DOI] [PubMed] [Google Scholar]
- 44.Kubisch CH, Sans MD, Arumugam T, Ernst SA, Williams JA, Logsdon CD. Early activation of endoplasmic reticulum stress is associated with arginine-induced acute pancrea-titis. American Journal of Physiology—Gastrointestinal and Liver Physiology. 2006;291(2):G238–G245. doi: 10.1152/ajpgi.00471.2005. [DOI] [PubMed] [Google Scholar]
- 45.Gukovsky I, Lugea A, Shahsahebi M, et al. A rat model reproducing key pathological responses of alcoholic chronic pancreatitis. American Journal of Physiology—Gastrointestinal and Liver Physiology. 2007;294(1):G68–G79. doi: 10.1152/ajpgi.00006.2007. [DOI] [PubMed] [Google Scholar]
- 46.Singh M, LaSure MM, Bockman DE. Pancreatic acinar cell function and morphology in rats chronically fed on ethanol diet. Gastroenterology. 1982;82(3):425–434. [PubMed] [Google Scholar]
- 47.Pandol SJ, Gorelick FS, Gerloff A, Lugea A. Alcohol abuse, endoplasmic reticulum stress and pancreatitis. Digestive Diseases. 2010;28(6):776–782. doi: 10.1159/000327212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lugea A, Tischler D, Nguyen J, et al. Adaptive unfolded protein response attenuates alcohol-induced pancreatic damage. Gastroenterology. 2011;140(3):987–997. doi: 10.1053/j.gastro.2010.11.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Riley EP, Infante MA, Warren KR. Fetal alcohol spectrum disorders: an overview. Neuropsychology Review. 2011;21(2):73–80. doi: 10.1007/s11065-011-9166-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tajiri S, Oyadomari S, Yano S, et al. Ischemia-induced neuronal cell death is mediated by the endoplasmic reticulum stress pathway involving CHOP. Cell Death and Differentiation. 2004;11(4):403–415. doi: 10.1038/sj.cdd.4401365. [DOI] [PubMed] [Google Scholar]
- 51.Katayama T, Imaizumi K, Manabe T, Hitomi J, Kudo T, Tohyama M. Induction of neuronal death by ER stress in Alzheimer’s disease. Journal of Chemical Neuroanatomy. 2004;28(1-2):67–78. doi: 10.1016/j.jchemneu.2003.12.004. [DOI] [PubMed] [Google Scholar]
- 52.Silva RM, Ries V, Oo TF, et al. CHOP/GADD153 is a mediator of apoptotic death in substantia nigra dopamine neurons in an in vivo neurotoxin model of parkinsonism. Journal of Neurochemistry. 2005;95(4):974–986. doi: 10.1111/j.1471-4159.2005.03428.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Scheper W, Hoozemans JJ. Endoplasmic reticulum protein quality control in neurodegenerative disease: the good, the bad and the therapy. Current Medicinal Chemistry. 2009;16(5):615–626. doi: 10.2174/092986709787458506. [DOI] [PubMed] [Google Scholar]
- 54.Ke Z, Wang X, Liu Y, et al. Ethanol induces endoplasmic reticulum stress in the developing brain. Alcoholism: Clinical and Experimental Research. 2011;35(9):1574–1583. doi: 10.1111/j.1530-0277.2011.01503.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Spies CD, Sander M, Stangl K, et al. Effects of alcohol on the heart. Current Opinion in Critical Care. 2001;7(5):337–343. doi: 10.1097/00075198-200110000-00004. [DOI] [PubMed] [Google Scholar]
- 56.Li SY, Ren J. Cardiac overexpression of alcohol dehydrogenase exacerbates chronic ethanol ingestion-induced myocardial dysfunction and hypertrophy: role of insulin signaling and ER stress. Journal of Molecular and Cellular Cardiology. 2008;44(6):992–1001. doi: 10.1016/j.yjmcc.2008.02.276. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 57.Li SY, Gilbert SA, Li Q, Ren J. Aldehyde dehydrogenase-2 (ALDH2) ameliorates chronic alcohol ingestion-induced myocardial insulin resistance and endoplasmic reticulum stress. Journal of Molecular and Cellular Cardiology. 2009;47(2):247–255. doi: 10.1016/j.yjmcc.2009.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Setshedi M, Wands JR, Monte SM. Acetaldehyde adducts in alcoholic liver disease. Oxidative Medicine and Cellular Longevity. 2010;3(3):178–185. doi: 10.4161/oxim.3.3.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Donohue TM, Tuma DJ, Sorrell MF. Binding of metabolically derived acetaldehyde to hepatic proteins in vitro. Laboratory Investigation. 1983;49(2):226–231. [PubMed] [Google Scholar]
- 60.Stevens VJ, Fanil WJ, Newman CB, Sims RV, Cerami A, Peterson CM. Acetaldehyde adduct with hemoglobin. Journal of Clinical Investigation. 1981;67(2):361–369. doi: 10.1172/JCI110043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hoerner M, Behrens UJ, Worner T, Lieber CS. Humoral immune response to acetaldehyde adducts in alcoholic patients. Research Communications in Chemical Pathology and Pharmacology. 1986;54(1):3–12. [PubMed] [Google Scholar]
- 62.Israel Y, Hurwitz E, Niemela O, Arnon R. Monoclonal and polyclonal antibodies against acetaldehyde-containing epitopes in acetaldehyde-protein adducts. Proceedings of the National Academy of Sciences of the United States of America. 1986;83(20):7923–7927. doi: 10.1073/pnas.83.20.7923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ursini F, Davies KJ, Maiorino M, Parasassi T, Sevanian A. Atherosclerosis: another protein misfolding disease? Trends in Molecular Medicine. 2002;8(8):370–374. doi: 10.1016/s1471-4914(02)02382-1. [DOI] [PubMed] [Google Scholar]
- 64.Nishitani Y, Matsumoto H. Ethanol rapidly causes activation of JNK associated with ER stress under inhibition of ADH. FEBS Letters. 2006;580(1):9–14. doi: 10.1016/j.febslet.2005.11.030. [DOI] [PubMed] [Google Scholar]
- 65.Mills KR, Ward K, Martin F, Peters TJ. Peripheral neuropathy and myopathy in chronic alcoholism. Alcohol and Alcoholism. 1986;21(4):357–362. [PubMed] [Google Scholar]
- 66.Sun YH, Li YQ, Feng SL, et al. Calcium-sensing receptor activation contributed to apoptosis stimulates TRPC6 channel in rat neonatal ventricular myocytes. Biochemical and Biophysical Research Communications. 2010;394(4):955–961. doi: 10.1016/j.bbrc.2010.03.096. [DOI] [PubMed] [Google Scholar]
- 67.Mekahli D, Bultynck G, Parys JB, de Smedt H, Missiaen L. Endoplasmic-reticulum calcium depletion and disease. Cold Spring Harbor Perspectives in Biology. 2011;3(6) doi: 10.1101/cshperspect.a004317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Worrall S, Niemela O, Parkkila S, Peters TJ, Preedy VR. Protein adducts in type I and type II fibre predominant muscles of the ethanol-fed rat: preferential localisation in the sarcolemmal and subsarcolemmal region. European Journal of Clinical Investigation. 2001;31(8):723–730. doi: 10.1046/j.1365-2362.2001.00848.x. [DOI] [PubMed] [Google Scholar]
- 69.Nakamura K, Iwahashi K, Furukawa A, et al. Acetaldehyde adducts in the brain of alcoholics. Archives of Toxicology. 2003;77(10):591–593. doi: 10.1007/s00204-003-0465-8. [DOI] [PubMed] [Google Scholar]
- 70.Nakamura K, Iwahashi K, Itoh M, et al. Immunohistochemical study on acetaldehyde adducts in alcohol-fed mice. Alcoholism: Clinical and Experimental Research. 2000;24(4):93–96. [PubMed] [Google Scholar]
- 71.Lentz SR. Mechanisms of homocysteine-induced atherothrombosis. Journal of Thrombosis and Haemostasis. 2005;3(8):1646–1654. doi: 10.1111/j.1538-7836.2005.01364.x. [DOI] [PubMed] [Google Scholar]
- 72.Seshadri S, Beiser A, Selhub J, et al. Plasma homocysteine as a risk factor for dementia and Alzheimer’s disease. New England Journal of Medicine. 2002;346(7):476–483. doi: 10.1056/NEJMoa011613. [DOI] [PubMed] [Google Scholar]
- 73.Ji C, Kaplowitz N. Hyperhomocysteinemia, endoplasmic reticulum stress, and alcoholic liver injury. World Journal of Gastroenterology. 2004;10(12):1699–1708. doi: 10.3748/wjg.v10.i12.1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Jakubowski H. Mechanism of the condensation of homocysteine thiolactone with aldehydes. Chemistry. 2006;12(31):8039–8043. doi: 10.1002/chem.200600785. [DOI] [PubMed] [Google Scholar]
- 75.Jakubowski H. Molecular basis of homocysteine toxicity in humans. Cellular and Molecular Life Sciences. 2004;61(4):470–487. doi: 10.1007/s00018-003-3204-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kenyon SH, Nicolaou A, Gibbons WA. The effect of ethanol and its metabolites upon methionine synthase activity in vitro. Alcohol. 1998;15(4):305–309. doi: 10.1016/s0741-8329(97)00134-1. [DOI] [PubMed] [Google Scholar]
- 77.Ji C, Shinohara M, Kuhlenkamp J, Chan C, Kaplowitz N. Mechanisms of protection by the betaine-homocysteine methyltransferase/betaine system in HepG2 cells and primary mouse hepatocytes. Hepatology. 2007;46(5):1586–1596. doi: 10.1002/hep.21854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ji C, Shinohara M, Vance D, et al. Effect of transgenic extrahepatic expression of betaine-homocysteine methyltransferase on alcohol or homocysteine-induced fatty liver. Alcoholism: Clinical and Experimental Research. 2008;32(6):1049–1058. doi: 10.1111/j.1530-0277.2008.00666.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Shinohara M, Ji C, Kaplowitz N. Differences in betaine-homocysteine methyltransferase expression, endoplasmic reticulum stress response, and liver injury between alcohol-fed mice and rats. Hepatology. 2010;51(3):796–805. doi: 10.1002/hep.23391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Brown R, Strathdee G. Epigenomics and epigenetic therapy of cancer. Trends in Molecular Medicine. 2002;8(4):S43–S48. doi: 10.1016/s1471-4914(02)02314-6. [DOI] [PubMed] [Google Scholar]
- 81.Fischle W, Wang Y, Allis CD. Histone and chromatin cross-talk. Current Opinion in Cell Biology. 2003;15(2):172–178. doi: 10.1016/s0955-0674(03)00013-9. [DOI] [PubMed] [Google Scholar]
- 82.Oliva J, Dedes J, Li J, French SW, Bardag-Gorce F. Epigenetics of proteasome inhibition in the liver of rats fed ethanol chronically. World Journal of Gastroenterology. 2009;15(6):705–712. doi: 10.3748/wjg.15.705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Garro AJ, Espina N, McBeth D, Wang SL, Wu-Wang CY. Effects of alcohol consumption on DNA methylation reactions and gene expression: implications for increased cancer risk. European Journal of Cancer Prevention. 1992;1(3):19–23. doi: 10.1097/00008469-199210003-00003. [DOI] [PubMed] [Google Scholar]
- 84.Garro AJ, McBeth DL, Lima V, Lieber CS. Ethanol consumption inhibits fetal DNA methylation in mice: implications for the fetal alcohol syndrome. Alcoholism: Clinical and Experimental Research. 1991;15(3):395–398. doi: 10.1111/j.1530-0277.1991.tb00536.x. [DOI] [PubMed] [Google Scholar]
- 85.Choi SW, Stickel F, Baik HW, Kim YI, Seitz HK, Mason JB. Chronic alcohol consumption induces genomic but not p53-specific DNA hypomethylation in rat colon. Journal of Nutrition. 1999;129(11):1945–1950. doi: 10.1093/jn/129.11.1945. [DOI] [PubMed] [Google Scholar]
- 86.Bleich S, Lenz B, Ziegenbein M, et al. Epigenetic DNA hypermethylation of the HERP gene promoter induces down-regulation of its mRNA expression in patients with alcohol dependence. Alcoholism: Clinical and Experimental Research. 2006;30(4):587–591. doi: 10.1111/j.1530-0277.2006.00068.x. [DOI] [PubMed] [Google Scholar]
- 87.Lu SC, Mato JM. Role of methionine adenosyltransferase and S-adenosylmethionine in alcohol-associated liver cancer. Alcohol. 2005;35(3):227–234. doi: 10.1016/j.alcohol.2005.03.011. [DOI] [PubMed] [Google Scholar]
- 88.Finkelstein JD. Methionine metabolism in mammals. Journal of Nutritional Biochemistry. 1990;1(5):228–237. doi: 10.1016/0955-2863(90)90070-2. [DOI] [PubMed] [Google Scholar]
- 89.Kharbanda KK, Rogers DD, II, Mailliard ME, et al. A comparison of the effects of betaine and S-adenosylmethionine on ethanol-induced changes in methionine metabolism and steatosis in rat hepatocytes. Journal of Nutrition. 2005;135(3):519–524. doi: 10.1093/jn/135.3.519. [DOI] [PubMed] [Google Scholar]
- 90.Barak AJ, Beckenhauer HC, Mailliard ME, Kharbanda KK, Tuma DJ. Betaine lowers elevated S-adenosylhomocysteine levels in hepatocytes from ethanol-fed rats. Journal of Nutrition. 2003;133(9):2845–2848. doi: 10.1093/jn/133.9.2845. [DOI] [PubMed] [Google Scholar]
- 91.Trimble KC, Molloy AM, Scott JM, Weir DG. The effect of ethanol on one-carbon metabolism: increased methionine catabolism and lipotrope methyl-group wastage. Hepatology. 1993;18(4):984–989. doi: 10.1002/hep.1840180433. [DOI] [PubMed] [Google Scholar]
- 92.Giovannucci E, Chen J, Smith-Warner SA, et al. Methylenetetrahydrofolate reductase, alcohol dehydrogenase, diet, and risk of colorectal adenomas. Cancer Epidemiology Biomarkers and Prevention. 2003;12(10):970–979. [PubMed] [Google Scholar]
- 93.Lenz B, Bleich S, Beutler S, et al. Homocysteine regulates expression of herp by DNA methylation involving the AARE and CREB binding sites. Experimental Cell Research. 2006;312(20):4049–4055. doi: 10.1016/j.yexcr.2006.09.004. [DOI] [PubMed] [Google Scholar]
- 94.Ma Y, Hendershot LM. Herp is dually regulated by both the endoplasmic reticulum stress-specific branch of the unfolded protein response and a branch that is shared with other cellular stress pathways. Journal of Biological Chemistry. 2004;279(14):13792–13799. doi: 10.1074/jbc.M313724200. [DOI] [PubMed] [Google Scholar]
- 95.Park PH, Miller R, Shukla SD. Acetylation of histone H3 at lysine 9 by ethanol in rat hepatocytes. Biochemical and Biophysical Research Communications. 2003;306(2):501–504. doi: 10.1016/s0006-291x(03)01040-4. [DOI] [PubMed] [Google Scholar]
- 96.Park PH, Lim RW, Shukla SD. Involvement of histone acetyltransferase (HAT) in ethanol-induced acetylation of histone H3 in hepatocytes: potential mechanism for gene expression. American Journal of Physiology—Gastrointestinal and Liver Physiology. 2005;289(6):G1124–G1136. doi: 10.1152/ajpgi.00091.2005. [DOI] [PubMed] [Google Scholar]
- 97.Kim JS, Shukla SD. Acute in vivo effect of ethanol (binge drinking) on histone H3 modifications in rat tissues. Alcohol and Alcoholism. 2006;41(2):126–132. doi: 10.1093/alcalc/agh248. [DOI] [PubMed] [Google Scholar]
- 98.Wang FM, Chen YJ, Ouyang HJ. Regulation of unfolded protein response modulator XBP1s by acetylation and deacetylation. Biochemical Journal. 2011;433(1):245–252. doi: 10.1042/BJ20101293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Li Y, Xu S, Giles A, et al. Hepatic overexpression of SIRT1 in mice attenuates endoplasmic reticulum stress and insulin resistance in the liver. FASEB Journal. 2011;25(5):1664–1679. doi: 10.1096/fj.10-173492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Lieber CS, Leo MA, Wang X, DeCarli LM. Effect of chronic alcohol consumption on Hepatic SIRT1 and PGC-1α in rats. Biochemical and Biophysical Research Communications. 2008;370(1):44–48. doi: 10.1016/j.bbrc.2008.03.005. [DOI] [PubMed] [Google Scholar]
- 101.Shen Z, Ajmo JM, Rogers CQ, et al. Role of SIRT1 in regulation of LPS- or two ethanol metabolites-induced TNF-α production in cultured macrophage cell lines. American Journal of Physiology—Gastrointestinal and Liver Physiology. 2009;296(5):G1047–G1053. doi: 10.1152/ajpgi.00016.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Purushotham A, Schug TT, Xu Q, Surapureddi S, Guo X, Li X. Hepatocyte-specific deletion of SIRT1 alters fatty acid metabolism and results in hepatic steatosis and inflammation. Cell Metabolism. 2009;9(4):327–338. doi: 10.1016/j.cmet.2009.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Shimizu Y, Hendershot LM. Oxidative folding: cellular strategies for dealing with the resultant equimolar production of reactive oxygen species. Antioxidants and Redox Signaling. 2009;11(9):2317–2331. doi: 10.1089/ars.2009.2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Malhotra JD, Kaufman RJ. Endoplasmic reticulum stress and oxidative stress: a vicious cycle or a double-edged sword? Antioxidants and Redox Signaling. 2007;9(12):2277–2293. doi: 10.1089/ars.2007.1782. [DOI] [PubMed] [Google Scholar]
- 105.Görlach A, Klappa P, Kietzmann T. The endoplasmic reticulum: folding, calcium homeostasis, signaling, and redox control. Antioxidants and Redox Signaling. 2006;8(9-10):1391–1418. doi: 10.1089/ars.2006.8.1391. [DOI] [PubMed] [Google Scholar]
- 106.Lluis JM, Colell A, García-Ruiz C, Kaplowitz N, Fernández-Checa JC. Acetaldehyde impairs mitochondrial glutathione transport in HepG2 cells through endoplasmic reticulum stress. Gastroenterology. 2003;124(3):708–724. doi: 10.1053/gast.2003.50089. [DOI] [PubMed] [Google Scholar]
- 107.Bae SH, Sung SH, Cho EJ, et al. Concerted action of sulfiredoxin and peroxiredoxin I protects against alcohol-induced oxidative injury in mouse liver. Hepatology. 2011;53(3):945–953. doi: 10.1002/hep.24104. [DOI] [PubMed] [Google Scholar]
- 108.Koch OR, Pani G, Borrello S, et al. Oxidative stress and antioxidant defenses in ethanol-induced cell injury. Molecular Aspects of Medicine. 2004;25(1-2):191–198. doi: 10.1016/j.mam.2004.02.019. [DOI] [PubMed] [Google Scholar]
- 109.Gong P, Cederbaum AI. Nrf2 is increased by CYP2E1 in rodent liver and HepG2 cells and protects against oxidative stress caused by CYP2E1. Hepatology. 2006;43(1):144–153. doi: 10.1002/hep.21004. [DOI] [PubMed] [Google Scholar]
- 110.Fu S, Yang L, Li P, et al. Aberrant lipid metabolism disrupts calcium homeostasis causing liver endoplasmic reticulum stress in obesity. Nature. 2011;473(7348):528–531. doi: 10.1038/nature09968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Deniaud A, El Dein OS, Maillier E, et al. Endoplasmic reticulum stress induces calcium-dependent permeabilitytransition, mitochondrial outer membranepermeabilization and apoptosis. Oncogene. 2008;27(3):285–299. doi: 10.1038/sj.onc.1210638. [DOI] [PubMed] [Google Scholar]
- 112.Peng TI, Jou MJ. Oxidative stress caused by mitochondrial calcium overload. Annals of the New York Academy of Sciences. 2010;1201:183–188. doi: 10.1111/j.1749-6632.2010.05634.x. [DOI] [PubMed] [Google Scholar]
- 113.Chapman RW, Morgan MY, Laulicht M, Hoffbrand AV, Sherlock S. Hepatic iron stores and markers of iron overload in alcoholics and patients with idiopathic hemochromatosis. Digestive Diseases and Sciences. 1982;27(10):909–916. doi: 10.1007/BF01316575. [DOI] [PubMed] [Google Scholar]
- 114.Potter BJ, Chapman RW, Nunes RM, Sorrentino D, Sherlock S. Transferrin metabolism in alcoholic liver disease. Hepatology. 1985;5(5):714–721. doi: 10.1002/hep.1840050503. [DOI] [PubMed] [Google Scholar]
- 115.Irving MG, Halliday JW, Powell LW. Association between alcoholism and increased hepatic iron stores. Alcoholism: Clinical and Experimental Research. 1988;12(1):7–13. doi: 10.1111/j.1530-0277.1988.tb00124.x. [DOI] [PubMed] [Google Scholar]
- 116.de Feo TM, Fargion S, Duca L, et al. Non-transferrin-bound iron in alcohol abusers. Alcoholism: Clinical and Experimental Research. 2001;25(10):1494–1499. doi: 10.1097/00000374-200110000-00013. [DOI] [PubMed] [Google Scholar]
- 117.Whitfield JB, Zhu G, Heath AC, Powell LW, Martin NG. Effects of alcohol consumption on indices of iron stores and of iron stores on alcohol intake markers. Alcoholism: Clinical and Experimental Research. 2001;25(7):1037–1045. [PubMed] [Google Scholar]
- 118.Napoli E, Taroni F, Cortopassi GA. Frataxin, iron-sulfur clusters, heme, ROS, and aging. Antioxidants and Redox Signaling. 2006;8(3-4):506–516. doi: 10.1089/ars.2006.8.506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Pigeon C, Ilyin G, Courselaud B, Leroyer P, Turlin B, Brissot P. A new mouse liver-specific gene, encoding a protein homologous to humanantimicrobial peptide hepcidin, is overexpressed during iron overload. Journal of Biological Chemistry. 2001;276(11):7811–7819. doi: 10.1074/jbc.M008923200. [DOI] [PubMed] [Google Scholar]
- 120.Vecchi C, Montosi G, Zhang K, et al. ER stress controls iron metabolism through induction of hepcidin. Science. 2009;325(5942):877–880. doi: 10.1126/science.1176639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Bertholet N, Cheng DM, Samet JH, Quinn E, Saitz R. Alcohol consumption patterns in HIV-infected adults with alcohol problems. Drug and Alcohol Dependence. 2010;112(1-2):160–163. doi: 10.1016/j.drugalcdep.2010.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Galvan FH, Bing EG, Fleishman JA, et al. The prevalence of alcohol consumption and heavy drinking among people with HIV in the United States: results from the HIV cost and services utilization study. Journal of Studies on Alcohol. 2002;63(2):179–186. doi: 10.15288/jsa.2002.63.179. [DOI] [PubMed] [Google Scholar]
- 123.Asselah T, Bièche I, Mansouri A, et al. In vivo hepatic endoplasmic reticulum stress in patients with chronic hepatitis C. Journal of Pathology. 2010;221(3):264–274. doi: 10.1002/path.2703. [DOI] [PubMed] [Google Scholar]
- 124.Joyce MA, Walters KA, Lamb SE, et al. HCV induces oxidative and ER stress, and sensitizes infected cells to apoptosis in SCID/Alb-uPA mice. PLoS Pathogens. v2009;5(2):p. e1000291. doi: 10.1371/journal.ppat.1000291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Tardif KD, Waris G, Siddiqui A. Hepatitis C virus, ER stress, and oxidative stress. Trends in Microbiology. 2005;13(4):159–163. doi: 10.1016/j.tim.2005.02.004. [DOI] [PubMed] [Google Scholar]
- 126.Lerat H, Kammoun HL, Hainault I, et al. Hepatitis C virus proteins induce lipogenesis and defective triglyceride secretion in transgenic mice. Journal of Biological Chemistry. 2009;284(48):33466–33474. doi: 10.1074/jbc.M109.019810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Lerat H, Honda M, Beard MR, et al. Steatosis and liver cancer in transgenic mice expressing the structural and nonstructural proteins of hepatitis C virus. Gastroenterology. 2002;122(2):352–365. doi: 10.1053/gast.2002.31001. [DOI] [PubMed] [Google Scholar]
- 128.Safdar K, Schiff ER. Alcohol and hepatitis C. Seminars in Liver Disease. 2004;24(3):305–315. doi: 10.1055/s-2004-832942. [DOI] [PubMed] [Google Scholar]
- 129.Pessione F, Degos F, Marcellin P, et al. Effect of alcohol consumption on serum hepatitis C virus RNA and histological lesions in chronic hepatitis C. Hepatology. 1998;27(6):1717–1722. doi: 10.1002/hep.510270635. [DOI] [PubMed] [Google Scholar]
- 130.Otani K, Korenaga M, Beard MR, et al. Hepatitis C virus core protein, cytochrome P450 2E1, and alcohol produce combined mitochondrial injury and cytotoxicity in hepatoma cells. Gastroenterology. 2005;128(1):96–107. doi: 10.1053/j.gastro.2004.10.045. [DOI] [PubMed] [Google Scholar]
- 131.Korenaga M, Okuda M, Otani K, Wang T, Li Y, Weinman SA. Mitochondrial dysfunction in hepatitis C. Journal of Clinical Gastroenterology. 2005;39(4):S162–S166. doi: 10.1097/01.mcg.0000155517.02468.46. [DOI] [PubMed] [Google Scholar]
- 132.Boyce M, Bryant KF, Jousse C, et al. A selective inhibitor of elF2α dephosphorylation protects cells from ER stress. Science. 2005;307(5711):935–939. doi: 10.1126/science.1101902. [DOI] [PubMed] [Google Scholar]