

Abstract
S-Palmitoylation, the reversible post-translational acylation of specific cysteine residues with the fatty acid palmitate, promotes the membrane tethering and subcellular localization of proteins in several biological pathways. Although inhibiting palmitoylation holds promise as a means for manipulating protein targeting, advances in the field have been hampered by limited understanding of palmitoylation enzymology and consensus motifs. In order to define the complement of S-acylated proteins in the macrophage, we treated RAW 264.7 macrophage membranes with hydroxylamine to cleave acyl thioesters, followed by biotinylation of newly exposed sulfhydryls and streptavidin-agarose affinity chromatography. Among proteins identified by LC-MS/MS, S-acylation status was established by spectral counting to assess enrichment under hydroxylamine versus mock treatment conditions. Of 1183 proteins identified in four independent experiments, 80 proteins were significant for S-acylation at false discovery rate = 0.05, and 101 significant at false discovery rate = 0.10. Candidate S-acylproteins were identified from several functional categories, including membrane trafficking, signaling, transporters, and receptors. Among these were 29 proteins previously biochemically confirmed as palmitoylated, 45 previously reported as putative S-acylproteins in proteomic screens, 24 not previously associated with palmitoylation, and three presumed false-positives. Nearly half of the candidates were previously identified by us in macrophage detergent-resistant membranes, suggesting that palmitoylation promotes lipid raft-localization of proteins in the macrophage. Among the candidate novel S-acylproteins was phospholipid scramblase 3 (Plscr3), a protein that regulates apoptosis through remodeling the mitochondrial membrane. Palmitoylation of Plscr3 was confirmed through 3H-palmitate labeling. Moreover, site-directed mutagenesis of a cluster of five cysteines (Cys159–161-163–164-166) abolished palmitoylation, caused Plscr3 mislocalization from mitochondrion to nucleus, and reduced macrophage apoptosis in response to etoposide, together suggesting a role for palmitoylation at this site for mitochondrial targeting and pro-apoptotic function of Plscr3. Taken together, we propose that manipulation of protein palmitoylation carries great potential for intervention in macrophage biology via reprogramming of protein localization.
S-Palmitoylation is the post-translational modification of specific cysteine residues in proteins with the 16-carbon fatty acid palmitate, a process that promotes tethering of proteins to cellular membranes and thereby contributes to specifying protein subcellular localization. Unlike analogous lipid post-translational modifications with myristate (i.e. myristoylation) and isoprenoids (i.e. prenylation), palmitoylation is enzymatically reversible, being driven by palmitoyl acyl transferases (PATs)1 of the Asp-His-His-Cys (DHHC) family and reversed by palmitoyl-protein thioesterases (PPTs) (1, 2). Indeed, great interest has emerged in the potential for dynamic changes in palmitoylation to regulate cell biology. For example, β–adrenergic receptor activation accelerates depalmitoylation of receptor-associated Gαs, shifting Gαs to the cytoplasm (3), and glutamate receptor activation accelerates depalmitoylation of the postsynaptic scaffolding protein PSD-95, causing receptor endocytosis (4). On a broader scale, the fundamental significance of palmitoylation as an organizing principle in cell biology is suggested by the breadth of functional categories represented by confirmed palmitoylproteins (PPs), ranging from enzymes, to signaling proteins, to ion channels and receptors (1). As protein palmitoylation has been detected in several compartments of the cell, including the plasma membrane and mitochondria (1, 5), it has been speculated that different pools of PATs within the cell may possibly tag distinct populations of proteins for specific destinations within the cell.
Despite burgeoning interest, advances in the field of protein palmitoylation have unfortunately been hampered by the current limited understanding of the responsible enzymology and consensus motifs. Recently, however, a substantial advance was offered by the group of Roth and colleagues, who greatly expanded the number of known PPs in the yeast Saccharomyces cerevisiae through application of a novel affinity proteomics approach utilizing acyl-biotinyl exchange (ABE) chemistry (6). In this approach, which was recently also applied to neurons (7) and in a modified form to prostate cancer cells (8), PPs and other S-acylproteins are captured by cysteine-acyl thioester cleavage with hydroxylamine (HA), followed by biotinylation of newly exposed sulfhydryls and avidin-Sepharose chromatography. Specificity of S-acylprotein capture is achieved through MS quantitation of candidates under HA cleavage as compared with mock cleavage (buffer) conditions. Additional proteomic approaches employing click chemistry (9–11) or RNAi knockdown of PATs (12) have also recently been described. As protein expression, presumably including that of PATs and PPTs, varies widely among cell types, insights gained into palmitoylation in specific cells may well have limited generalizability to other cell types.
Herein, we report the first ABE proteomic analysis of the macrophage. Although very little is known about the role of protein palmitoylation in these immune cells, it is known that membrane trafficking by confirmed PPs, such as syntaxin 7, does play a pivotal role in hallmark functions of the macrophage, including phagocytosis (13). Moreover, it has recently been reported that palmitoylation can target select proteins to lipid raft microdomains in macrophages (14), as has been proposed for other cells. Given this and our recent finding that 3 PATs (DHHC13, DHHC20, and DHHC5) are expressed in macrophage rafts (15), we speculated that palmitoylation might indeed play an important role in protein localization within the macrophage.
Using a “Significance Analysis of Microarrays (SAM)”-type t-statistic and bootstrap methodology to assess S-acylation among ABE-enriched proteins, we have identified 80 candidate S-acylproteins at a false discovery rate (FDR) = 0.05, and 101 candidate S-acylproteins at FDR = 0.10 in RAW 264.7 macrophages. Of these 101 candidates, 29 are proteins for which palmitoylation has been previously biochemically confirmed, 45 are proteins identified as putative PPs in prior proteomic screens, and 24 represent candidate novel PPs. Of interest, nearly one-half of the candidates were detected by us in RAW 264.7 rafts (15), strongly suggesting that palmitoylation does promote raft localization in the macrophage. Moreover, among the PP candidates was phospholipid scramblase 3 (Plscr3), a protein that regulates mitochondrial function through remodeling of the mitochondrial outer membrane (16). Herein, we confirm palmitoylation of Plscr3 through 3H-palmitate labeling, identify the region of Plscr3 involved in palmitoylation, and demonstrate the requirement for palmitoylation at this locus for Plscr3 targeting to the mitochondrion, the first such direct demonstration, to our knowledge, of targeting of a protein to the mitochondrion by palmitoylation. Taken together, we propose that proteomic analysis may offer important new insights on a global scale into subcellular targeting of proteins in the macrophage, and we speculate that manipulation of protein palmitoylation may represent a fruitful opportunity for intervention in macrophage biology.
EXPERIMENTAL PROCEDURES
Reagents
Sequencing grade porcine trypsin was purchased from Promega Corp. (Madison, WI). Solvents for liquid chromatography were purchased as follows: HPLC-grade acetonitrile (Fisher Scientific, Fairlawn, NJ), formic acid (Sigma-Aldrich Corp., St. Louis, MO), and MassPREP Yeast Alcohol Dehydrogenase (Waters Corporation, Milford, MA). HEK293 and RAW 264.7 cells were from ATCC (Manassas, VA). Hydroxylamine, β-mercaptoethanol, and trichloroacetic acid were from Sigma. N-ethylmaleimide (NEM), biotin-HPDP (N-(6-(Biotinamido)hexyl)-3′-(2′-pyridyldithio)-propionamide, and streptavidin-agarose were from Pierce (Rockford, IL). Triton-X-100 and ethylenediaminetetraacetic acid (EDTA) were ultrapure grade from US Biochemical (Cleveland, OH).
Acyl Biotinyl Exchange (ABE) Treatment of Macrophage Membrane Preparations
RAW 264.7 macrophage membranes were processed via HA or Tris (control) treatment followed by ABE chemistry and avidin-Sepharose chromatograpy, as previously described (17). Briefly, 5 × 107 macrophages were Dounce homogenized in ice cold lysis buffer (LB; 150 mm NaCl, 50 mm TrisHCl, 5 mm EDTA; pH 7.4) with 10 mm NEM plus protease inhibitors, and membranes collected by centrifugation (200,000 × g, 30 min, 4 °C). Membranes were resuspended in 3 ml of LB containing 10 mm NEM and solubilized by addition of Triton-X-100 to a final concentration of 1.7% using gentle rotation (4 °C, 1 h). Insoluble debris was removed by centrifugation and solubilized protein was precipitated (chloroform-methanol). Protein was resolubilized in 4% SDS buffer (4% SDS, 50 mm Tris, 5 mm EDTA; pH 7.4) with 10 mm NEM and incubated (10 min, 37 °C). Each tube was diluted with 3 volumes of LB with 1 mm NEM, protease inhibitors, and 0.2% Triton-X-100 for overnight incubation (4 °C). Excess NEM was removed by three sequential chloroform-methanol extractions after which each sample was dissolved in 4% SDS buffer and split into two equal portions: one for neutral pH HA treatment (HA+); the other for neutral pH Tris buffer treatment (HA−). The HA+ portion was diluted with 4 vol of HA+ buffer (0.7 m hydroxylamine pH 7.4, 1 mm HPDP-biotin crosslinker, 0.2% Triton X-100, protease inhibitors), and the HA− portion with 4 volumes of HA− buffer (50 mm Tris pH 7.4, 1 mm HPDP-biotin crosslinker 0.2% Triton X-100, protease inhibitors) and rotated (1 h, RT), with subsequent chloroform-methanol precipitation. Each portion was dissolved in 4% SDS buffer followed by addition of 4 volumes of low HPDP-biotin binding buffer (150 mm NaCl, 50 mm Tris pH 7.4, 5 mm EDTA, 0.2 mm HPDP-biotin, 0.2% Triton X-100) and another 1 h incubation with rotation and three sequential chloroform-methanol extractions to remove excess crosslinker. Protein pellets were solubilized in 2% SDS buffer (2% SDS, 50 mm Tris, 5 mm EDTA, pH 7.4) at 37 °C for 10–15 min and diluted 20-fold with LB containing 0.2% Triton-X-100 to a final concentration of 0.1% SDS. Samples were rotated (30 min, RT) and centrifuged (15,000 × g, 1 min) to remove particulates. Streptavidin-agarose was added at 15 μl beads/ml lysate and samples rotated (90 min, RT) to capture biotinylated proteins. Unbound proteins were removed by six sequential washes with LB containing 0.1% SDS and 0.2% Triton-X-100. Proteins were released by 150 μl LB containing 0.1% SDS, 0.2% Triton-X-100, 1% BME (15 min, 37 °C). Eluted proteins were precipitated (TCA), and redissolved in Laemmli buffer.
Protein Resolution and Digestion
ABE eluates from HA+ and HA− treatment conditions were resolved by 10% SDS-PAGE, and then stained with Sypro Ruby. Each gel lane was cut into 24 equal slices and digested in a 96-well tray using a Progest robotic digester (Genomic Solutions Inc., Ann Arbor, MI), as previously reported (15). Supernatants were pooled, lyophilized, and stored at −80 °C until analysis. Prior to MS, the lyophilized peptide samples were resuspended in 40 μl of 0.1% formic acid. Each resuspended peptide mixture in a 96-well tray was spiked with 2 μl of 400 fmol/μl of MassPREP Yeast Alcohol Dehydrogenase as an external standard to monitor the consistency of the individual LC-MS/MS runs.
NanoLC-MS/MS Analysis and Database Analysis
All nanoLC-MS/MS experiments were performed on an Agilent 1100 nanoLC system on-line with an Agilent XCT Ultra ion trap mass spectrometer with the chip cube interface (Agilent Technologies). Peptides were separated by reverse-phase LC with a trapping column or analytical column nano-flow setup (HPLC-Chip cube; Agilent Technologies) and analyzed in both the full scan MS and in the collision-induced dissociation tandem mass spectrometry (MS/MS) modes. Briefly, 20 μl of the peptide digest was loaded onto an Agilent C18 chip (75 μm x 43 mm) followed by a 15 min wash with 5% (v/v) acetonitrile/0.1% (v/v) formic acid. Peptides were eluted with a linear gradient of increasing concentration of acetonitrile (in 0.1% (v/v) formic acid) as follows: 0–45 min, 5–50% (v/v) acetonitrile, 45–50 min, acetonitrile increased to 95% (v/v), 50–60 min acetonitrile maintained at 95%. The mass spectrometer was operated in the positive ion, standard enhanced mode (allows for scan range of 50–2200 m/z at 5500 m/z/s) and included settings of a mass range from 200 to 2200 m/z, an ionization potential of 2.1 kV, an ion change control smart target (number of ions in the trap prior to scan out) of 100,000 or 200 ms of accumulation, and a 1.0-V fragmentation amplitude. MS/MS data were acquired using a data-dependent acquisition format with the six most abundant ions from each MS scan further interrogated by MS/MS. The automated switching for MS/MS required a threshold of 5000 counts. For all experiments, the isolation width was set to 4 m/z. Analyses by LC-MS/MS of HA+ and HA− gel lanes were alternated over time rather than run in groups in order to minimize the chance for technically introduced artifact.
Peak lists were generated from the data obtained from each nano liquid chromatography (LC)-electrospray ionization-tandem MS (MS/MS) analysis using the Data Extractor feature of Spectrum Mill MS Proteomics Workbench (version A.03.03.084 SR4, Agilent Technologies). The Data Extractor settings included limiting the data search to deconvoluted ions observed between 300 and 5000 Da and a retention time between 10 min and 60 min. MS scans with the same precursor mass (± 1.5 m/z) and retention time within 30 s were merged. Moreover, of the remaining MS/MS spectra, only spectra that contained sequence tag information greater than 2 residues were submitted for database searching. The resulting extracted data were then searched against the mouse NCBInr database (04/05/2008; 131,087 entries) using the MS/MS Search function in the Spectrum Mill MS Proteomics Workbench. Search settings included a trypsin specificity with up to one missed cleavage allowed, a precursor ion mass tolerance of 2 Da, a product ion mass tolerance of 1.0 Da, no fixed modifications, variable methionine oxidation and NEM modification of cysteines, and a minimum matched spectral intensity of 70%. Proteins identified with two unique peptides and a distinct summed MS/MS search score greater than 25 were autovalidated. When using these criteria, estimated FDR from searching a reversed database is <1%. Proteins with MS/MS search scores that were less than 25 but greater than 17 were manually validated. These validated proteins were then tabulated. In cases in which protein nomenclature associated with an NCBI protein database accession was obscure, protein identity was clarified by sequence alignment (http://blast.ncbi.nlm.nih.gov/Blast.cgi).
Site-directed Mutagenesis of Plscr3
A pCMV6 expression plasmid for murine Plscr3 was obtained from Origene (Rockville, MD). Cys→Ala mutations were introduced using the QuickChange Site-Directed Mutagenesis kit (Stratagene, La Jolla, CA). Briefly, the reaction mix was comprised of PAGE-purified mutant primers at 125 ng in a 50 μl PCR reaction with Phusion High Fidelity polymerase enzyme (New England BioLabs, Ipswich, MA), 10 mm dNTPs, and 25 ng of plasmid template. PCR was performed at 98 °C for 1 min, followed by 16–20 cycles of 98 °C for 15 s, 55 °C for 1 min, and 68 °C for 12 min, followed by a 4 °C hold. Dpn1 digestion took place after addition of NEB reaction buffer 4 (5.5 μl), and 0.55 μl Dpn1 (NEB, 20,000 U/ml) to each PCR reaction whereupon parental plasmids in samples were digested at 37 °C for 1 h. A 2 μl volume of this digestion was transformed into supercompetent XL1 cells (Stratagene, La Jolla, CA), followed by selection-plating and overnight incubation. Colonies were picked from 50 μg/ml kanamycin plates, DNA was prepared by miniprep, and sequenced by standard procedures.
Cell Culture and Transfection
RAW 264.7 macrophages and HEK293 cells were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% heat-inactivated fetal bovine serum, 2 mm l-glutamine, 100 μg/ml streptomycin, and 100 U/ml penicillin under a humidified 5% CO2 atmosphere at 37 °C. Transient transfection of plasmids was done using Fugene 6 (Roche Diagnostic, Indianopolis, IN) at a 3:1 ratio of reagent:plasmid in serum-free Optimem (Invitrogen-Invitrogen, Carlsbad, CA). After 25 min, reagent-DNA complexes were added to cells growing in complete media. Assays were performed 48 h post-transfection.
3H-Palmitate Labeling, Immunoprecipitation, and Immunoblotting
HEK293 cells transfected for 48 h with Plscr3 expression plasmids were incubated in DMEM containing 10% dialyzed, heat-inactivated fetal calf serum for 1 h prior to addition of 0.1mCi/ml 3H-palmitic acid (specific activity 50 Ci/mmole, Perkin Elmer, Waltham, MA) and incubation for 4 h. Media was removed and cells were washed with phosphate-buffered saline (PBS) prior to lysis (1% Nonidet P-40, 50 mm Tris HCl pH 8, 1 mm EDTA, 0.1% SDS, 0.5% sodium deoxycholate, 300 mm NaCl, protease inhibitors [Roche #1836153], 5 mm sodium fluoride, 1 mm sodium-o-vanadate and 1 mm PMSF). Lysates were sonicated on ice and debris pelleted by centrifugation at 4 °C. Lysates were made isotonic just before overnight immunoprecipitation with goat anti-Plscr3. Lysates were resolved by 10% SDS-PAGE, transferred to nitrocellulose (Bio-Rad), and then probed with goat anti-Plscr3 (Santa Cruz Biotechnology; Santa Cruz, CA). Membranes were then washed in Tween Tris Buffered Saline, and exposed for 60 min to a 1:5000 dilution of species-specific, HRP-conjugated secondary antibody (GE Healthcare) in 5% milk/Tween Tris Buffered Saline. After further Tween Tris Buffered Saline washes, signal was detected with exposure to ECL™ Western blot Detection Reagents (GE Healthcare), followed by application to film (GE Healthcare). Alternatively, 3H signal of the immunoprecipitate was detected by a ∼4 week autoradiogram exposure (−80 °C).
Subcellular Fractionation
A mitochondrial isolation procedure for cultured cells (Pierce, Rockford, IL) was followed with minor modifications. Briefly, 5 × 106 cells were solubilized in 800 μl of mitochondria isolation Reagent A and homogenized on ice. Cell lysate in Reagent A was added to an equal vol of Reagent C. Samples were centrifuged (700 × g, 10 min, 4 °C). The pellet was reserved for nuclear isolation. Supernatants were centrifuged (12,000 × g, 15 min, 4 °C) to pellet mitochondria. The post-mitochondrial supernatant was the cytosolic fraction. Mitochondria were washed once in Reagent C. Nuclei were isolated (18) by solubilizing cell membranes from the 700 × g pellet with 500 μl of low salt lysis buffer (20 mm Hepes, pH 7.4, 5 mm NaCl, 5 mm MgCl2, 0.5% Nonidet P-40, 0.1% sodium deoxycholate, protease inhibitors) for 10 min on ice, followed by centrifugation (1000 × g, 7 min, 4 °C). Nuclei were then washed once with low salt lysis buffer.
Establishment of Stable Expression RAW 264.7 Lines
RAW264.7 cells were transfected with empty vector or pCMV6-Plscr3 or pCMV6–5A-Mutant-Plscr3 constructs using Lipofectamine™ 2000 according to the manufacturer's instructions. Two days after transfection, selection medium containing 400 μg/ml G418 (Invitrogen) was added to cells plated at 10% confluence. Confluence and selection pressure were maintained by re-plating every 72 h thereafter. Complete cell death of untransfected controls was observed by 10 days. Stable clones were characterized by Western blotting and maintained in regular growth medium containing 400 μg/ml G418.
Immunofluorescence and Microscopy
HEK293 cells were grown on 0.1% gelatin- or poly-d-lysine-coated glass coverslips and transiently transfected with either wt or 5A-mutant-Plscr3 plasmids using Lipofectamine™ 2000 per manufacturer's instructions. At 48 h post-transfection, MitoTracker Red was added (100 nm) in Phenol Red-free complete DMEM. After 10 min at 37 °C, cells were rinsed with PBS, fixed with paraformaldehyde (4%, v/v) for 20 min, rinsed twice with PBS, and permeabilized with 0.2% (w/v) Triton X-100 in PBS (5 min, RT). After three PBS rinses, cells were placed in blocking solution (5% donkey serum, 2% bovine serum albumin, 0.05% Tween-20) at room temperature (1 h). Anti-Plscr3 was added (1 μg/ml) and incubation continued for an additional hr. After three PBS washes, Alexa Fluor 488-conjugated donkey anti-goat secondary antibody (Molecular Probes, Eugene, OR) was added (5 μg/ml) and incubated (45 min, RT). The cells were then PBS rinsed thrice, rinsed with water, and mounted on slides using Prolong Gold (Invitrogen) containing DAPI. Alexa Flour 488 was visualized (excitation, 488 nm), and MitoTracker Red visualized (excitation, 560 nm), using a Zeiss inverted laser-scanning confocal microscope (LSM-710) with 63×/1.4 NA oil objective. For colocalization, the dual color confocal images were scanned in two sequentially recorded channels that were calibrated with single label controls to eliminate bleed-through signal. Spectral recording with linear unmixing was used for finetuning of the two channel set up. Zen 2010 software was used to analyze the weighted colocalization coefficient.
Apoptosis Induction and Measurement by Flow Cytometry
Apoptosis in empty vector, wt Plscr3, and 5A- mutant Plscr3 stable RAW264.7 cells was determined after exposure to etoposide (Sigma) for 8 or 24 h. Cells were seeded at 0.75 million/well in a 12-well tissue culture plate 24 h prior to etoposide exposure in 0.1% DMSO. After exposure, media was removed and cells were incubated in 1 ml of Phenol Red-free DMEM (1% fetal calf serum) containing 1 μg/ml of the apoptosis probe, FAM-FLIVO (ImmunoChemistry Technologies, Bloomington, MN) per manufacturer's instructions. After 1.5 h, cells were lifted, centrifuged (5 min, 4 °C), resuspended in binding buffer, and treated with 7-aminoactinomycin D (BD Pharmingen; San Diego, CA) to probe membrane integrity. Cell staining was quantified on a BD LSR II cytometer (BD Biosciences) and analyzed using FACSDiVa software (version 6.1.3, Becton Dickinson).
Statistical Analysis
Analysis of S-acylation was limited to proteins with: (1) mean HA+ spectral count >mean HA− spectral count; and (2) ≥1 spectral count in ≥2 of 4 independent HA+ experiments (to avoid analysis of proteins detected in just a single HA+ experiment). Two hundred and ninety-seven proteins fulfilled these two criteria. A value of 0.2 was arbitrarily assigned to spectral count values of zero, in order to avoid division by zero, as previously reported (6). As the spectral count data had a skewed distribution, spectral count values for the 297 candidates were log-transformed. For each of the S-acylprotein candidates, a one-sided test was performed where the alternative hypothesis was that the mean of spectral counts under HA+ conditions was larger than the mean under HA− conditions. As the data were not normally distributed, a bootstrap methodology was used for testing each hypothesis, applying a “SAM” correction to the standard error. The software package ORIOGEN (19, 20) was used to implement the methodology. S-acylation status among the 297 candidates is reported at thresholds of FDR = 0.05 and FDR = 0.10. Principal component analysis was performed and displayed using Partek Genomics Suite 6.5 (Partek Inc. St. Louis, MO).
RESULTS
Pulldown of the Macrophage S-acylproteome
In an effort to profile the global S-acylproteome of the macrophage, we used ABE chemistry to capture S-acylproteins for proteomic identification, as recently described (17). Briefly, RAW 264.7 macrophage membranes were isolated, treated with the thiol-reactive reagent NEM to cloak free -SH groups, and then treated in parallel with either neutral pH HA (hereafter, “HA+”) to cleave cysteine-acyl thioester linkages, or Tris buffer (hereafter, “HA−“) to serve as a negative control (mock cleavage). The preparations were then treated with HPDP-biotin to tag the chemically de-acylated cysteine residues, followed by biotin tag capture by stepatividin-agarose chromatography.
Sypro staining of SDS-PAGE-resolved macrophage membrane preparations confirmed that HA+ treatment, as compared with HA− treatment, led to enrichment of several distinct protein bands, as expected (Fig. 1A). ABE procedures were completed on HA+ versus HA− treated membrane preparations from four independent macrophage cultures. For each of these experiments, HA+ and HA− gel lanes were excised and the proteins identified and quantified by LC-MS/MS employing spectral counting, as previously reported (6, 7, 17). Normalization of the raw spectral count values in individual LC-MS/MS runs to exogeneously spiked yeast alcohol dehydrogenase standard in order to address possible inter-run technical variability had no significant effect on the results (data not shown). A total of 1183 unique proteins were thus identified. The protein identities, raw spectral count values, and other related data are shown individually for the 4 experiments under HA+ and HA− conditions in supplemental Tables S1–S8, and are aligned in supplemental Table S9 for purpose of protein tracking across experiments.
Fig. 1.

Enrichment of candidate S-acylproteins in the macrophage through acyl- biotinyl exchange. A, RAW 264.7 membranes were subjected to acyl-biotin exchange under hydroxylamine (HA+) or mock (HA−) treatment conditions. Proteins precipitated by avidin-Sepharose were then resolved by 10% SDS-PAGE and detected by Sypro ruby stain. B, Mean spectral counts under HA+ and HA− treatment conditions was plotted for n = 297 proteins evaluated in the statistical analysis. Proteins identified as candidate S-acylproteins at FDR = 0.05 (see Table I) are shown in figure as open dots. Inset shows greater detail.
Previous ABE proteomic analyses have defined high confidence S-acylproteins with the use of arbitrary thresholds for the ratio of spectral counts under HA+ versus HA− conditions (e.g. HA+/HA− ratio>5.5) (6), to indicate enrichment by HA, or by using previously confirmed PPs within the analysis as benchmarks to define levels of confidence for new PP candidates (7). In an attempt to apply a rigorous new statistical approach to S-acylprotein identification, we first applied a priori criteria to the list of 1183 proteins to enrich for S-acylproteins. First, proteins that were detected in none or only one of the HA+ experiments (n = 586) were removed from further consideration (supplemental Table S9, yellow). Next, proteins with mean HA+ spectral counts ≤ mean HA− spectral counts (n = 300) were removed from consideration, as these proteins displayed no enrichment by the ABE procedure (supplemental Table S9, green). The mean spectral count values under HA+ and HA− treatment conditions for the remaining 297 S-acylprotein candidates (supplemental Table S9, white) are plotted in Fig. 1B.
As the spectral count values for these candidates had a skewed distribution, the spectral count values were log-transformed. S-acylation status of the 297 candidates was next tested using a SAM-type methodology (19, 20). Using this approach, we determined that 80 proteins were significant for S-acylation at FDR = 0.05, and an additional 21, significant at FDR = 0.10 but not FDR = 0.05. The 80 proteins that were significant for S-acylation at FDR = 0.05 are indicated by open circles in Fig. 1B, showing that they indeed appear to represent a population distinguished by its relative enrichment under HA+ treatment conditions.
Principal component analysis (PCA) is a qualitative assessment tool widely used in systems biology that allows one to visualize complex, higher-dimensional data by projecting it into lower (e.g. 3-)dimensional space, in the process revealing hidden structure (e.g. clustering) among the data by reducing complexity and filtering out noise. This unsupervised analysis plots complex data as a scatter plot in three-dimensional space by constructing artificial variables (“principal components”) that best explain the variance of the data. PCA plots can be useful to qualitatively confirm the similarity/distinctness of protein expression under different experimental conditions, as well as whether an experimental procedure for selecting proteins indeed yields an informative protein set (21, 22). A PCA plot based on all 297 proteins that were entered into our statistical procedure for S-acylprotein identification indicated separate clustering of proteins under HA+ and HA− conditions (Fig. 2A), thus confirming that the ABE procedure had delineated discrete protein populations (on the basis of HA treatment). As shown, this plot accounts for 77.5% of the variability in the data. Moreover, a PCA plot based on just the 80 S-acylprotein candidates that were found to be significant at FDR = 0.05 revealed yet further delineation of the HA+ and HA− clusters (Fig. 2B), in the process accounting for almost all (93.6%) of the variability in the data. Taken together, the PCA therefore qualitatively supports the success of our joint ABE biochemical-statistical procedure for identifying HA-enriched proteins, indicating that the 80 S-acylprotein candidates identified clearly discriminate between the HA+ and HA− condition.
Fig. 2.

Principal component analysis of candidate S-acylproteins. PCA plots of the data were performed using (A) the full set of 297 proteins that were entered into the statistical analysis of S-acylation, or (B) just the 80 S-acylprotein candidates that were significant at FDR = 0.05 (Table I). These unsupervised plots, accounting for 77.5% (A) and 93.6% (B) of the variability in the data, respectively, reveal discrete clustering of proteins on the basis of HA treatment. This serves to confirm the selectivity of the ABE-statistical procedure, verifying that the 80 S-acylprotein candidates identified clearly discriminate between the HA+ and HA− conditions.
The 101 FDR = 0.10 S-acylation candidates are listed by functional category in Table I, along with whether they have been previously biochemically confirmed to be PPs, or, alternatively, remain unconfirmed but have been identified in previous published palmitoylproteomic screens of mammalian cells. The associated supporting data, including p values for S-acylation, HA+/HA− ratios, and citations to prior confirmation of palmitoylation, are listed in supplemental Table S10.
Table I. Candidate S-acylproteins by functional category. All significant for S-acylation at FDR = 0.05, except for those with an asterix (*), which were significant at FDR = 0.10 but not at FDR = 0.05. Status column indicates whether candidates are already biochemically confirmed as palmitoylproteins in the literature in human (H), mouse (M), rat (R), or bovine (B) species, or alternatively, if they are unconfirmed but previously identified in proteomic screens by: (7–11). Proteins without an entry in Status column represent candidate novel S-acylproteins.
| Membrane Trafficking | Receptors | Miscellaneous | ||||||
| Acc. # | Gene | Status | Acc. # | Gene | Status | Acc. # | Gene | Status |
| 6678553 | Vamp3 | (1,2,4) | 729081 | Cd36 | H | 148676911 | Mtdh | (1–4) |
| 148707350 | Vamp4 | (3) | 6680878 | Srb2* | (2,3) | 13385560 | b | (2) |
| 56541060 | Vamp5 | (1) | 74225999 | M6pr | B | 148687610 | Bri3bp | |
| 33468929 | Vamp7 | (2,3) | 85540471 | Cd44 | H | 120587021 | Pigt | |
| 12844640 | Stx6 | R | 266394 | Itga4* | 29788753 | Pigk | ||
| 20139979 | Stx7 | H | 148670103 | Igf2r* | (1–3,5) | 13507626 | Ifitm2 | |
| 9055356 | Stx8 | H | 29789020 | Cd74 | M | 12841777 | Ifitm3 | (4) |
| 14715019 | Stx12 | R | 6680351 | Irgm1* | (4) | |||
| 62471433 | Plscr1 | H | Enzyme/metabolic | 33859722 | Tmx1 | (4) | ||
| 12963735 | Plscr3 | (2,3) | Acc. # | Gene | Status | 20070422 | Praf2 | (1,3) |
| 74149227 | Scamp1 | R | 148702068 | Adam17 | (1,2) | 74191638 | Rpl3* | (2,3,4) |
| 12331398 | Scamp2 | (1–3) | 148694017 | Adpgk | 14149647 | Rpl9 | (2) | |
| 74179717 | Scamp3 | (3,4,5) | 9789991 | Hsd17b12 | (3) | 26336098 | Rp2h | |
| 148707755 | Rab7b* | (3) | 31559956 | Daglb | (1,2) | 148686436 | Arl15 | (1–3) |
| 148696058 | Snap23 | H | 34328288 | Aldh3b1 | (2) | 148683525 | Golim4* | (2) |
| 7304929 | Trappc3 | H | 148708180 | Ugt1a7c | (4) | 31980808 | Eif3g | |
| 50510831 | Ergic1 | 27370240 | Lnpep | (1,2,5) | 109809743 | Golga7 | H | |
| 34849462 | Ergic3 | (1–3) | 26352009 | Agpat1 | (1–3) | 12963539 | Ethe1 | |
| 28372380 | Steap3 | 13385658 | Mrpl10 | 38142456 | Fam108b | (1–3,5) | ||
| 8134596 | Npc1* | (2,5) | 26343873 | Cybb | 2493276 | Bak1 | ||
| 74152765 | Cyba | 62740082 | Tecr* | |||||
| Signaling | 13938651 | Efr3a | (2) | |||||
| Acc. # | Gene | Status | Membrane/structural | 74149009 | c | |||
| 21703986 | Pi4k2a | R | Acc. # | Gene | Status | 10141011 | Ly9 | |
| 9910604 | Lat2 | 6679809 | Flot1* | M | 12856757 | Ccny | (1,2) | |
| 148672975 | Fyn* | H | 7710018 | Stom | H | 84095197 | Tmem55b | (1–3) |
| 161760636 | Lyn* | H | 62526118 | Ckap4 | H | 50510847 | Myof | (2) |
| 41054806 | Gnai2 | R | 94536791 | Flot2 | R | 82881899 | Rpl27-ps1* | (3) |
| 13386338 | Rap2b | H | 148681238 | Pppde1 | ||||
| 149265925 | Cdc42se2* | H | Channels/transporters | 74150483 | Rpn2 | (3) | ||
| 148690841 | R-ras | H | Acc. # | Gene | Status | 149273155 | d | |
| 131884 | N-ras | H | 148691502 | Slc35b2* | (1,2) | 29747827 | Spcs2* | |
| 149250541 | Rgs19 | H | 114326474 | Slc1a5 | (1,2,4,5) | 74211556 | Nfu1* | |
| 12851417 | Rala | (2,3) | 123229245 | Slc44a1 | (2,3) | 116517301 | Ptbp1* | (1,4,5) |
| 15743554 | Spred1 | (3) | ||||||
| 81903514 | a* | (2,3) | False-positive | Chaperones | ||||
| 6677905 | Glg1* | (3,5) | Acc. # | Gene | Acc. # | Gene | Status | |
| 84662745 | Gnaq | H | 31542559 | Dlat* | (3,5) | 6671664 | Canx | M |
| 74204211 | Ube2l3 | (3,5) | 37359796 | Mlec | (4) | |||
| 9837286 | Ubc | (3) | 88909653 | Sil1 | ||||
| 15126683 | Dnajc5 | R | ||||||
a Gene = 2400001E08Rik; protein = RhoA activator C11orf59 homolog.
b 2900010M23Rik.
c 2510039O18Rik.
d LOC100041852. Dlat and Ube2l3 are presumed false-positive (non-acylated), thioester-utilizing proteins (see text); Ubc is a presumed false-positive as reported by (7).
An example of an abundant protein that was not enriched by the ABE procedure but was likely retained through nonspecific binding to streptavidin-agarose is M2-type pyruvate kinase (acc# 1405933). This protein had similar spectral counts across all HA+ and HA− samples (supplemental Table S9). An MS/MS spectrum of ion m/z 714.4 is shown in Fig. 3A. Precursor mass and extensive b- and y-ion series identify the sequence as NTGIICTIGPASR from pyruvate kinase, with the cysteine modified by NEM, consistent with it being nonacylated. An extracted ion chromatogram (EIC) for m/z 714.4 from the region of the gel containing pyruvate kinase from the first HA+ sample set is shown in Fig. 3B as a dotted line; the EIC for m/z 714.4 from the corresponding gel slice from the matched HA− sample is shown as a solid line. These EICs demonstrate qualitatively that there are similar amounts of this NEM-cysteine-containing peptide from pyruvate kinase under HA+ and HA− treatments, consistent with its lack of enrichment by ABE.
Fig. 3.

Comparative MS analysis of proteins enriched and not enriched by acyl-biotinyl exchange procedure. A, MS/MS of ion m/z 714.4, corresponding to a tryptic peptide of M2-pyruvate kinase, a protein not enriched by acyl-biotinyl exchange procedure (see text). Analysis is consistent with NEM modification of the cysteine residue. B, Extracted ion chromatogram (EIC) of ion m/z 714.4 from HA+ -treated condition (dashed line) and HA− -treated condition (solid line). C, MS/MS of ion m/z 861.6, corresponding to tryptic peptide 12 of stomatin, a known palmitoylprotein that was enriched by acyl-biotinyl exchange (see text). Analysis indicates that the cysteine residue is unmodified. D, EIC of ion m/z 885.5, corresponding to peptide in C with cysteine oxidized to cysteic acid, from HA+ -treated condition (dashed line) and HA− -treated condition (solid lines).
Conversely, an example of a protein that was enriched by ABE is the confirmed PP stomatin (acc# 7710018) (23), which displayed a mean HA+/HA− spectral count ratio of 10.6 across the four experiments (Table I, supplemental Table S9). An MS/MS spectrum of a doubly charged ion m/z 861.6 is shown in Fig. 3C, where the precursor mass and extensive b- and y-ion series identify the peptide as GPGLFFILPCTDSLIK from stomatin, in which the cysteine is unmodified. A more abundant and consistently observed form of this doubly charged peptide is at m/z 885.5, likely corresponding to oxidation of cysteine to cysteic acid (supplemental Fig. S1). Oxidation of this cysteine most likely occurred during electrophoresis, as a variety of artifactual modifications, including oxidation, are a common occurrence during electrophoresis for several amino acids including cysteine (24–28). Indeed, enrichment of this peptide by ABE provides indirect support for its postprocedure artifactual oxidation, as cysteic acid would not be expected to be affinity-enriched by the biotin-reagent. This peptide was also observed with the cysteine residue modified by BME (supplemental Fig. S2), consistent with its modification during elution.
An EIC for m/z 885.5 from the region of the gel containing stomatin from the first HA+ experiment is shown in Fig. 3D (dotted line). The EICs for m/z 885.5 from the corresponding gel slice in the matched HA− treatment, as well as the six gel slices above and below this corresponding slice (to account for the possibility of different migration or errors in gel cutting) are overlaid as solid lines in Fig. 3D. Similar results were obtained for the HA+ and HA− treatments in the three other experiments (supplemental Fig. S3). These EICs demonstrate qualitatively that although this free cysteine-containing peptide from stomatin is essentially absent in the HA− treatments, it is relatively abundant in the HA+ treatments. Although not definitive proof of acylation, the absence of NEM on this cysteine (as well as the HA+ enrichment of this cysteinyl peptide) is consistent with its acylation. Indeed, this specific residue in stomatin was previously reported to be palmitoylated (23).
Identification of Established and Putative Novel Palmitoylproteins
Several functional categories are represented among the high- and intermediate-confidence S-acylation candidates that were identified in our analysis (Table I). Notably, among the 101 candidates identified at FDR = 0.10 are 29 previously confirmed PPs (25 of these at FDR = 0.05), 45 proteins not previously confirmed but proposed as PPs from prior proteomic screens by other investigators (7–11) (33 of these at FDR = 0.05), and 24 proteins representing candidate novel PPs. Consistent with prior reports, we found a predominance of S-acylation candidates in the categories of membrane trafficking and cell signaling. Among the membrane trafficking S-acylproteins identified were multiple soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) proteins that have been previously reported to be palmitoylated in other cell types, including vesicle-associated membrane proteins (VAMPs) 3, 4, 5, and 7; syntaxins 6, 7, 8, and 12, and SNAP-23. Pathway analysis (Ingenuity Systems) confirmed marked representation of proteins involved in protein transport (13 proteins, p = 4.87E-10). Among the cell signaling proteins identified were kinases (Fyn, Lyn), GTPases (Rap2b, R-Ras, N-Ras, Gnaq, and Gnai2), adaptors (linker for activated T cells 2; Lat2), and integrins (Itga4). Whereas Lat1 is well-described as a palmitoylated transmembrane adaptor protein in T cells (29), its more recently described homologue in non-T cells, Lat2/NTAL, has not been previously reported as a PP to our knowledge. Whereas several integrins have been previously confirmed as being palmitoylated (e.g. α3, α6, and β4), we identify integrin α4 (VLA-4) as a putative S-acylprotein in the macrophage.
It has previously been reported that the ABE enrichment method for PPs has the potential for identification of proteins that accept groups other than fatty acids in labile thioester linkage (6, 7). Indeed, Dlat (E2 component of pyruvate dehydrogenase complex), identified by us as a candidate S-acylprotein (Table I), has been acknowledged as a probable “false-positive ” (nonacylated) protein in prior ABE proteomic investigations as it accepts acetyl moieties in thioester linkage to its lipoic acid prosthetic group (7, 30). Component X of the pyruvate dehydrogenase complex (Pdhx) has been similarly acknowledged as a probable false-positive (7); although enriched by our ABE procedure (HA+/HA− spectral count ratio = 5.5 [Table S9]), it was not significant at FDR = 0.10 in our analysis. Ube2l3 (ubiquitin-conjugating enzyme; accepts ubiquitin in thioester linkage), and polyubiqutin C (Ubc), both identified by us as S-acylprotein candidates (Table I) have been previously acknowledged as probable false-positives (7). Conversely, three previously confirmed PPs were enriched by the ABE procedure—transferrin receptor (HA+/HA− = 3.29); phosphatidylinositol 4-kinase type 2 beta (HA+/HA− = 6.75); and scavenger receptor class B member 1 (HA+/HA− = 5.5)—but did not meet significance at FDR = 0.10 (supplemental Table S9), thus representing false-negatives within the parameters of our analysis.
Palmitoylation is thought to promote protein localization to lipid rafts (1); indeed for some proteins, such as Rap2b, a small GTPase found in our analysis (Table I), palmitoylation is not required for generic membrane localization, but is required for specific localization of proteins to rafts (31). It is estimated that in MDCK cells about one-half of proteins in detergent-resistant membranes (a cell fraction approximating lipid rafts (15)) may be palmitoylated (32). In order to investigate the degree of representation of raft proteins in our analysis, we looked for expression of the 98 FDR = 0.10 S-acylprotein candidates (excluding the three false-positives) within the RAW 264.7 detergent-resistant membrane proteome recently reported by our group (15). Interestingly, 47 (48%) of the S-acylproteins identified in the present analysis were detected in macrophage detergent-resistant membranes (supplemental Table S11), and thus may be raft proteins. Conversely, the remaining 51 S-acylprotein candidates were not detected in raft preparations (supplemental Table S12). Because of limitations of MS sensitivity and other technical variables, our not having detected a protein in raft preparations cannot be taken as conclusive evidence that the protein does not localize to rafts wholly or in part. Although caution is therefore warranted in drawing inferences about the features of S-acylproteins localizing or not to rafts, we note that all membrane or structural and channel or transporter proteins as well as almost all SNARE proteins (i.e. syntaxins, VAMPs) of the membrane trafficking category (Table I) were detected in rafts. By contrast, only some of the S-acylprotein candidates of the signaling classification (Table I) were detected in rafts.
Confirmation of Plscr3 as a Palmitoylprotein
Among the high-confidence putative novel S-acylproteins identified in our analysis was Plscr3 (Table I). The Plscr proteins play a critical role in regulating the asymmetry of membranes through interleaflet phospholipid translocation, and in this capacity have been implicated in several critical cellular events, ranging from cell activation to apoptosis. Four phospholipid scramblases have been identified in mice and humans (i.e. Plscr1- Plscr4) (33). Of these, hPlscr1, the best-studied, has been biochemically confirmed to undergo multiple palmitoylation at a locus of 5 cysteines (184CCCPCC189) (34); of note, we also identify mPlscr1 in the present analysis of RAW 264.7 murine macrophages as a candidate PP (Table I). Murine and human Plscr proteins have nuclear localization signals (35), and it has been shown that prevention of Plscr1 palmitoylation through cysteine-to-alanine mutagenesis at 184CCCPCC189 causes mislocalization of the protein from the plasma membrane to the nucleus (34). By contrast, Plscr3, not previously biochemically confirmed as a PP to our knowledge, is reported to localize to the outer mitochondrial membrane, at which site it is thought to regulate mitochondrial function and apoptosis (16).
First, as sequence alignment revealed homology of Plscr3 at the palmitoylation locus of Plscr1 (159CGCSCCPC166 in Plscr3) (35), we questioned whether this homologous 5-cysteine cluster was also a locus of palmitoylation in Plscr3. Second, by analogy to Plscr1, we questioned whether preventing palmitoylation of Plscr3 through mutagenesis of this cysteine cluster would cause its mislocalization. In order to confirm that Plscr3 is indeed a PP, we expressed wild-type (wt) Plscr3 in HEK293 cells, cultured the cells in the presence of 3H-palmitate, and then immunoprecipitated Plscr3 (34). Autoradiography of the immunoprecipitate indeed revealed 3H-palmitate signal, confirming Plscr3 palmitoylation (Fig. 4). A weak, but detectable palmitate signal was also noted upon immunoprecipitation of the native Plscr3 protein in cells transfected with empty vector. By contrast, compared with overexpressed wt protein, alanine mutagenesis of all five cysteines in 159CGCSCCPC166 (hereafter, “5A ” mutant) led to loss of the palmitoylation signal, suggesting that one or more of these cysteine residues is the site of palmitoylation. More selective mutagenesis of just the two N-terminal cysteines of the cluster, Cys159 and Cys161 (2A mutant) led to partial loss of 3H-palmitate signal, whereas mutagenesis of the three C-terminal cysteines of the cluster, Cys163, Cys164, and Cys166 (3A) led to minimal change in the palmitoylation signal (Fig. 4). Taken together, this suggests that palmitoylation occurs within the 159CGCSCCPC166 region of Plscr3, likely at >1 cysteine residue, as is reported to be the case for the homologous five-cysteine cluster in Plscr1 (34).
Fig. 4.
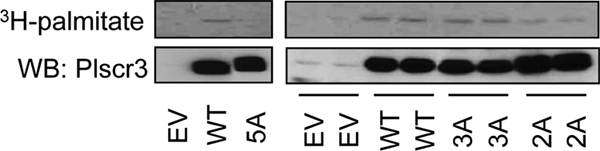
Identification of palmitoylated residues in Plscr3. HEK293 cells were transiently transfected with empty vector (EV), wild type (WT) Plscr3, or with cysteine-to-alanine mutant Plscr3 in which all 5 cysteines (5A), the 3 C-terminal cysteines (3A), or the 2 N-terminal cysteines (2A) of the 159CGCSCCPC166 sequence were mutagenized. Transfected cells were exposed to 0.1mCi/ml 3H-palmitate (4h), Plscr3 was immunoprecipitated, and precipitates were then probed for Plscr3 by immunoblot and imaged by autoradiography. Samples in right figure represent biological duplicates. Data are representative of three independent experiments.
Palmitoylation Regulates Plscr3 Subcellular Localization
In order to test the effect of palmitoylation upon subcellular targeting of Plscr3 to the mitochondrion, we expressed wt and Cys/Ala mutant Plscr3 in HEK293 cells and then performed subcellular fractionation. Immunoblotting confirmed that overexpressed wt Plscr3 was found in the mitochondrial fraction, and, to a much lesser extent, in the nucleus (Fig. 5, supplemental Fig. S4). By contrast, the non-palmitoylated 5A mutant was shifted from mitochondrion to the nucleus. This suggests that palmitoylation does play a pivotal role in targeting of Plscr3 to the mitochondrion, and that, in the absence of this lipid modification, the nuclear localization sequence is sufficient to target the protein to the nucleus. The 3A and 2A Plscr3 mutants also had increased protein levels in the nucleus compared with wt, albeit much more modest reductions in protein level in the mitochondria compared with the 5A mutant. Plscr1 has been shown upon nuclear localization to act as a transcription factor, inducing the target gene inositol 1,4,5-triphosphate receptor type 1 (IP3R1) (36). As Plscr3, like Plscr1, has a DNA-binding motif (35), this raises the interesting possibility that it too may act as palmitoylation-sensitive transcription factor, potentially subserving distinct cellular functions at its different subcellular sites. In order to confirm and further characterize the subcellular localization of Plscr3, we performed anti-Plscr3 immunocytochemistry upon HEK293 cells expressing either wt or 5A mutant Plscr3. Although not exclusive to the mitochondrion, wt Plscr3 did display mitochondrial localization (Fig. 5B), and this was significantly reduced for 5A mutant Plscr3, as judged by the weighted coefficient of intensity-independent colocalization with MitoTracker Red-stained mitochondria (Fig. 5C). Compared with wt, 5A mutant Plscr3 generally displayed a less punctuate appearance, and across a population of screened cells was found to adopt increased localization to the nucleus as well as to extra-nuclear sites (Fig. 5B).
Fig. 5.

Palmitoylation of Plscr3 regulates its subcellular localization. A, HEK293 cells were transiently transfected with empty vector (EV), WT Plscr3, or with 5A, 3A, or 2A cysteine-to-alanine Plscr3 mutants as described in Fig. 4. Cells were then fractionated into mitochondrial and nuclear fractions, and both whole cell lysate (WCL) and fractions immunoblotted (IB) for Plscr3. Data are representative of three independent experiments. B, HEK293 cells stably transfected with EV, wt Plscr3, or 5A mutant Plscr3 were imaged (63×/1.4 NA oil objective) after immunostaining with anti-Plscr3 antibody (primary) and Alexa Fluor 488-conjugated donkey anti-goat secondary antibody, MitoTracker Red to detect mitochondria, and DAPI to detect nuclei. Two examples of 5A Plscr3-transfected fields are shown to illustrate extranuclear (middle panels) and nuclear (bottom panels) staining patterns. C, Mitochondrial localization of WT and 5A mutant Plscr3 in transfected HEK293 cells was quantified by determining the weighted colocalization coefficient to reveal intensity-independent colocalization of Plscr3 with Mitotracker Red-stained mitochondria. Data are mean ± S.E. (n = 9 fields [1–4 cells/fields] per condition; *, p < 0.0001).
Plscr3 is reported to promote apoptosis through translocation of cardiolipin from the inner to the outer mitochondrial membrane, an event that leads to tBid-induced cytochrome C release into the cytosol (16). We thus predicted that mislocalization of Plscr3 away from the mitochondrion through preventing palmitoylation might reduce this pro-apoptotic function of the protein. In order to test this, we generated RAW 264.7 macrophages stably expressing wt Plscr3, 5A mutant Plscr3, or EV, treated them with the pro-apoptotic agent etoposide (or vehicle), and then performed flow cytometric quantitation of apoptosis using a fluorescent caspase activity probe (37). As shown in Fig. 6, 5A mutant Plscr3-expressing macrophages indeed had lesser apoptosis than either EV or wt Plscr3-expressing counterparts at both 8 and 24 h post-treatment with etoposide.
Fig. 6.

Palmitoylation-deficient mutant Plscr3 confers reduced apoptotic function. A, RAW 264.7 macrophages stably expressing EV, wt Plscr3, or 5A mutant Plscr3 were treated with vehicle (DMSO) or different concentrations of etoposide (Etop) for 24 h. Cells were then treated with a cell-permeant caspase activity probe (FAM-FLIVO™, Immunochemistry) and 7-aminoactinomycin D (AAD) to probe membrane integrity. Apoptosis was quantified by flow cytometry as the sum of FLIVO+ 7-AAD− (lower right quadrants in B) and FLIVO+ 7-AAD+ (upper right quadrants in B) cells by flow cytometry. Data are mean ± S.E., and are representative of two independent experiments performed in duplicate. B, Representative flow cytometry plots for data in A. C, Cells as in A were treated with vehicle or 8 nm etoposide for 8 h, and then apoptosis was quantified as in A. Data are mean ± S.E., and are representative of two independent experiments performed in triplicate.
DISCUSSION
We report the first S-acylproteomic analysis of the macrophage, a cell type in which protein palmitoylation has been little studied to date. In our analysis, we identified 29 proteins that have previously been confirmed biochemically to be PPs, and an additional 45 that have not been confirmed to date, but were identified as putatively acylated in proteomic screens conducted in either neuronal cells (7), Jurkat T cells (9, 10), prostate cancer cells (8), or a dendritic cell line (11). For one such putative PP, Plscr3, a protein that plays a critical role in remodeling of the mitochondrial membrane during apoptosis, we confirmed its palmitoylation though 3H-palmitate labeling, and used site-directed mutagenesis to identify the probable locus of palmitoylation as well as the necessity for palmitoylation in targeting of this protein to the mitochondrion and in macrophage apoptosis. In addition to these 74 proteins for which palmitoylation has previously been either confirmed or proposed, we report an additional 24 proteins as novel S-acylprotein candidates.
In a previous report, we detected expression of the PATs DHHC13, DHHC20, and DHHC5 in raft preparations from RAW 264.7 macrophages (15). It has been proposed that the specific subcellular localization of the different DHHC enzymes may account for the selective local targeting within the cell of their respective substrates. Moreover, it is thought that palmitoylation promotes localization of proteins to lipid rafts (32), a concept supported by our striking finding that 48% of the PP candidates identified in our analysis were previously detected by us in RAW 264.7 macrophage detergent-resistant membranes (supplemental Table S11). Given their raft-localization, we speculate that it is likely that DHHC5, DHHC13, and/or DHHC20 may play a role in localization of PPs to lipid rafts in the macrophage. DHHC6 and DHHC20 have previously been identified as putative PPs in proteomic screens (7, 9), suggesting their potential for autopalmitoylation. Although detected in the present analysis, we did not find them to be candidate PPs, as they showed minimal/inconsistent enrichment by the ABE procedure (supplemental Table S9). Conversely, DHHC13, not previously reported as a PP to our knowledge, did show enrichment by the ABE procedure (HA+/HA− ratio = 18 [Table S9]), but did not emerge from the analysis statistically significant at FDR = 0.10 for S-acylation.
Using the ABE proteomic approach, we identified 74 previously confirmed or proposed PPs. Nevertheless, limitations to the ABE approach should also be acknowledged. The detection of many proteins under HA- conditions, as found in prior reports (6–8), suggests that the initial NEM treatment step may provide incomplete cloaking of sulfhydryl groups. A modification to the ABE protocol that may reduce this background was recently reported by Yang and colleagues (8). Despite the use of HA enrichment to address the potentially incomplete chemistry of NEM, it is also likely that our analysis failed to recognize S-acylproteins in addition to the false-negatives we recognized. Conversely, it is also possible that the list of novel putative S-acylproteins candidates we provide includes false-positives beyond those of which we are aware.
An important insight derived from our analysis that may inform future S-acylproteomic investigations (as well as analogous affinity proteomics platforms) is that statistical analysis of replicate experiments may help to identify S-acylproteins in some situations where using an arbitrary HA+/HA− ratio cutpoint such as 5.5 fails. Indeed, in our analysis, there were 24 S-acylprotein candidates that had an HA+/HA− ratio <5.5 but were significant at FDR = 0.10 (supplemental Table S10); of these, 15 have been previously confirmed as PPs or reported as PP candidates in screens, and three are previously reported thioester-utilizing false-positives. For example, among the previously confirmed PPs in our FDR = 0.05 list (Table I) were Gnai2 (HA+/HA− ratio = 3.3), Fyn (HA+/HA− ratio = 4.2), and N-Ras (HA+/HA− ratio = 4.7). A statistical approach to S-acylprotein identification not only rewarding high HA+/HA− ratios but also evaluating interexperimental variability in spectral counts may account for these successful low ratio identifications.
Biological insights provided by our analysis should also be highlighted. In the present study, we confirmed that Plscr3, a protein that regulates apoptosis and mitochondrial function (16), is a PP, and determined that mutagenesis at a conserved cysteine cluster abrogated palmitoylation, mislocalizing the protein from the mitochondrion to the nucleus. To our knowledge, this is the first example of targeting of a protein to the mitochondrion by palmitoylation. In parallel, we found that palmitoylation-deficient Plscr3 conferred reduced apoptotic response to etoposide. Interestingly, EV and wt Plscr3-transfected macrophages displayed comparable apoptotic responses, whereas 5A Plscr3-transfected cells were relatively “protected ” from apoptosis. Although the full implications of these findings are not yet clear, this suggests that palmitoylation-deficient, mislocalized Plscr3 may exercise a dominant negative effect on pro-apoptotic function, perhaps reducing apoptosis through competition for, or mislocalization of, pro-apoptotic cofactors.
These findings also add Plscr3 to a list of several other proteins whose subcellular localization is known to shift from membrane to nucleus upon depalmitoylation. For example, blocking palmitoylation of Plscr1, R7BP, Wrch-1, and steroid receptors has been shown to shift them from the plasma membrane to the nucleus (34, 38–40). In the case of the membrane remodeling protein Plscr1, the protein assumes the function of a transcription factor upon localization to the nucleus (34). Taken together, this phenomenon of regulation of dual-localized proteins by palmitoylation suggests several interesting hypotheses: (1) that some membrane-associated proteins may have a “default ” localization to the nucleus; (2) that palmitoylation may couple very different functions occurring at different locations within the cell by acting as a switch to determine the “zip code ” of dual function proteins; and (3) that functional communication between different subcellular sites may be driven by protein palmitoylation and perhaps by the subcellular availability of acyl metabolites and activity of local PATs. As Plscr3, like Plscr1, has a DNA-binding motif (35), future studies will be needed to determine whether it too functions as a transcription factor under depalmitoylated conditions, and whether its family of target genes have any evident teleological connection to the function Plscr3 serves at its mitochondrial location.
In summary, we report the first analysis of the complement of S-acylated proteins in the macrophage, and use the example of Plscr3 to demonstrate the potential importance of protein acylation to the biology of this important immune cell. We speculate that it is likely that abnormalities of macrophage function are an unrecognized feature of at least some of the diseases that have been associated with aberrant function of DHHC acyltransferases, and, conversely, that manipulation of protein palmitoylation may offer a promising opportunity for intervention in macrophage biology.
Acknowledgments
We thank Dr. Leesa J. Deterding for helpful discussions, Jeff Tucker and Agnes Janoshazi of the NIEHS Fluorescence Microscopy and Imaging Center for assistance with cell imaging, and Dr. Carl Bortner and Maria Sifre of the NIEHS Flow Cytometry Center for assistance with flow cytometry.
Footnotes
* This research was supported by the Intramural Research Program of the National Institutes of Health, National Institute of Environmental Health Sciences Z01 ES102005 and ES050171.
 This article contains supplemental Figs. S1 to S4 and Tables S1 to S12.
This article contains supplemental Figs. S1 to S4 and Tables S1 to S12.
1 The abbreviations used are:
- ABE
- acyl biotinyl exchange
- Biotin-HPDP
- N-[6-(biotinamido)hexyl]-3′-(2′-pyridyldithio)propionamide
- DHHC
- Asp-His-His-Cys
- EIC
- extracted ion chromatogram
- FDR
- false discovery rate
- HA
- hydroxylamine
- NEM
- N-ethylmaleimide
- PAT
- palmitoyl acyltransferase
- Plscr
- phospholipid scramblase
- PP
- palmitoylprotein
- PPT
- protein palmitoylthiesterase
- SAM
- Significance Analysis of Microarrays.
REFERENCES
- 1. Iwanaga T., Tsutsumi R., Noritake J., Fukata Y., Fukata M. (2009) Dynamic protein palmitoylation in cellular signaling. Prog. Lipid Res. 48, 117–127 [DOI] [PubMed] [Google Scholar]
- 2. Resh M. D. (2006) Palmitoylation of ligands, receptors, and intracellular signaling molecules. Sci. STKE. 2006, re14. [DOI] [PubMed] [Google Scholar]
- 3. Wedegaertner P. B., Bourne H. R. (1994) Activation and depalmitoylation of Gs alpha. Cell 77, 1063–1070 [DOI] [PubMed] [Google Scholar]
- 4. El-Husseini Ael-D., Schnell E., Dakoji S., Sweeney N., Zhou Q., Prange O., Gauthier-Campbell C., Aguilera-Moreno A., Nicoll R. A., Bredt D. S. (2002) Synaptic strength regulated by palmitate cycling on PSD-95. Cell 108, 849–863 [DOI] [PubMed] [Google Scholar]
- 5. Kostiuk M. A., Keller B. O., Berthiaume L. G. (2009) Non-radioactive detection of palmitoylated mitochondrial proteins using an azido-palmitate analogue. Methods Enzymol. 457, 149–165 [DOI] [PubMed] [Google Scholar]
- 6. Roth A. F., Wan J., Bailey A. O., Sun B., Kuchar J. A., Green W. N., Phinney B. S., Yates J. R., 3rd, Davis N. G. (2006) Global analysis of protein palmitoylation in yeast. Cell 125, 1003–1013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kang R., Wan J., Arstikaitis P., Takahashi H., Huang K., Bailey A. O., Thompson J. X., Roth A. F., Drisdel R. C., Mastro R., Green W. N., Yates J. R., 3rd, Davis N. G., El-Husseini A. (2008) Neural palmitoyl-proteomics reveals dynamic synaptic palmitoylation. Nature 456, 904–909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yang W., Di Vizio D., Kirchner M., Steen H., Freeman M. R. (2010) Proteome-scale characterization of human s-acylated proteins in lipid raft-enriched and non-raft membranes. Mol. Cell Proteomics 9, 54–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Martin B. R., Cravatt B. F. (2009) Large-scale profiling of protein palmitoylation in mammalian cells. Nat. Methods 6, 135–138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wilson J. P., Raghavan A. S., Yang Y. Y., Charron G., Hang H. C. (2011) Proteomic analysis of fatty-acylated proteins in mammalian cells with chemical reporters reveals S-acylation of histone H3 variants. Mol. Cell. Proteomics 10, M110.001198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yount J. S., Moltedo B., Yang Y. Y., Charron G., Moran T. M., López C. B., Hang H. C. (2010) Palmitoylome profiling reveals S-palmitoylation-dependent antiviral activity of IFITM3. Nat. Chem. Biol. 6, 610–614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhang J., Planey S. L., Ceballos C., Stevens S. M., Jr., Keay S. K., Zacharias D. A. (2008) Identification of CKAP4/p63 as a major substrate of the palmitoyl acyltransferase DHHC2, a putative tumor suppressor, using a novel proteomics method. Mol. Cell. Proteomics 7, 1378–1388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Collins R. F., Schreiber A. D., Grinstein S., Trimble W. S. (2002) Syntaxins 13 and 7 function at distinct steps during phagocytosis. J. Immunol. 169, 3250–3256 [DOI] [PubMed] [Google Scholar]
- 14. Gonnord P., Delarasse C., Auger R., Benihoud K., Prigent M., Cuif M. H., Lamaze C., Kanellopoulos J. M. (2009) Palmitoylation of the P2X7 receptor, an ATP-gated channel, controls its expression and association with lipid rafts. Faseb J. 23, 795–805 [DOI] [PubMed] [Google Scholar]
- 15. Dhungana S., Merrick B. A., Tomer K. B., Fessler M. B. (2009) Quantitative proteomics analysis of macrophage rafts reveals compartmentalized activation of the proteasome and of proteasome-mediated ERK activation in response to lipopolysaccharide. Mol. Cell. Proteomics 8, 201–213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Liu J., Dai Q., Chen J., Durrant D., Freeman A., Liu T., Grossman D., Lee R. M. (2003) Phospholipid scramblase 3 controls mitochondrial structure, function, and apoptotic response. Mol. Cancer Res. 1, 892–902 [PubMed] [Google Scholar]
- 17. Wan J., Roth A. F., Bailey A. O., Davis N. G. (2007) Palmitoylated proteins: purification and identification. Nat. Protoc. 2, 1573–1584 [DOI] [PubMed] [Google Scholar]
- 18. Deryckere F., Gannon F. (1994) A one-hour minipreparation technique for extraction of DNA-binding proteins from animal tissues. BioTechniques 16, 405 [PubMed] [Google Scholar]
- 19. Peddada S., Harris S., Zajd J., Harvey E. (2005) ORIOGEN: order restricted inference for ordered gene expression data. Bioinformatics 21, 3933–3934 [DOI] [PubMed] [Google Scholar]
- 20. Peddada S. D., Lobenhofer E. K., Li L., Afshari C. A., Weinberg C. R., Umbach D. M. (2003) Gene selection and clustering for time-course and dose-response microarray experiments using order-restricted inference. Bioinformatics 19, 834–841 [DOI] [PubMed] [Google Scholar]
- 21. Gerling I. C., Singh S., Lenchik N. I., Marshall D. R., Wu J. (2006) New data analysis and mining approaches identify unique proteome and transcriptome markers of susceptibility to autoimmune diabetes. Mol. Cell. Proteomics 5, 293–305 [DOI] [PubMed] [Google Scholar]
- 22. Yohannes E., Chang J., Christ G. J., Davies K. P., Chance M. R. (2008) Proteomics analysis identifies molecular targets related to diabetes mellitus-associated bladder dysfunction. Mol. Cell. Proteomics 7, 1270–1285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Snyers L., Umlauf E., Prohaska R. (1999) Cysteine 29 is the major palmitoylation site on stomatin. FEBS Lett. 449, 101–104 [DOI] [PubMed] [Google Scholar]
- 24. Brewer J. M. (1967) Artifact produced in disc electrophoresis by ammonium persulfate. Science 156, 256–257 [DOI] [PubMed] [Google Scholar]
- 25. Froelich J. M., Reid G. E. (2008) The origin and control of ex vivo oxidative peptide modifications prior to mass spectrometry analysis. Proteomics 8, 1334–1345 [DOI] [PubMed] [Google Scholar]
- 26. Pelletier O. (1966) Determination of cystine as cysteic acid after low voltage paper electrophoresis. J. Agric. Food Chem. 14, 496–498 [Google Scholar]
- 27. Sun G., Anderson V. E. (2004) Prevention of artifactual protein oxidation generated during sodium dodecyl sulfate-gel electrophoresis. Electrophoresis 25, 959–965 [DOI] [PubMed] [Google Scholar]
- 28. Swiderek K. M., Davis M. T., Lee T. D. (1998) The identification of peptide modifications derived from gel-separated proteins using electrospray triple quadrupole and ion trap analyses. Electrophoresis 19, 989–997 [DOI] [PubMed] [Google Scholar]
- 29. Zhang W., Trible R. P., Samelson L. E. (1998) LAT palmitoylation: its essential role in membrane microdomain targeting and tyrosine phosphorylation during T cell activation. Immunity 9, 239–246 [DOI] [PubMed] [Google Scholar]
- 30. Mattevi A., Obmolova G., Kalk K. H., Teplyakov A., Hol W. G. (1993) Crystallographic analysis of substrate binding and catalysis in dihydrolipoyl transacetylase (E2p). Biochemistry 32, 3887–3901 [DOI] [PubMed] [Google Scholar]
- 31. Canobbio I., Trionfini P., Guidetti G. F., Balduini C., Torti M. (2008) Targeting of the small GTPase Rap2b, but not Rap1b, to lipid rafts is promoted by palmitoylation at Cys176 and Cys177 and is required for efficient protein activation in human platelets. Cell. Signal. 20, 1662–1670 [DOI] [PubMed] [Google Scholar]
- 32. Melkonian K. A., Ostermeyer A. G., Chen J. Z., Roth M. G., Brown D. A. (1999) Role of lipid modifications in targeting proteins to detergent-resistant membrane rafts. Many raft proteins are acylated, while few are prenylated. J. Biol. Chem. 274, 3910–3917 [DOI] [PubMed] [Google Scholar]
- 33. Wiedmer T., Zhou Q., Kwoh D. Y., Sims P. J. (2000) Identification of three new members of the phospholipid scramblase gene family. Biochim. Biophys. Acta 1467, 244–253 [DOI] [PubMed] [Google Scholar]
- 34. Wiedmer T., Zhao J., Nanjundan M., Sims P. J. (2003) Palmitoylation of phospholipid scramblase 1 controls its distribution between nucleus and plasma membrane. Biochemistry 42, 1227–1233 [DOI] [PubMed] [Google Scholar]
- 35. Sahu S. K., Gummadi S. N., Manoj N., Aradhyam G. K. (2007) Phospholipid scramblases: an overview. Arch. Biochem. Biophys. 462, 103–114 [DOI] [PubMed] [Google Scholar]
- 36. Zhou Q., Ben-Efraim I., Bigcas J. L., Junqueira D., Wiedmer T., Sims P. J. (2005) Phospholipid scramblase 1 binds to the promoter region of the inositol 1,4,5-triphosphate receptor type 1 gene to enhance its expression. J. Biol. Chem. 280, 35062–35068 [DOI] [PubMed] [Google Scholar]
- 37. Riol-Blanco L., Delgado-Martin C., Sánchez-Sánchez N., Alonso C. L., Gutierrez-López M. D., Del Hoyo G. M., Navarro J., Sánchez-Madrid F., Cabañas C., Sánchez-Mateos P., Rodríguez-Fernández J. L. (2009) Immunological synapse formation inhibits, via NF-kappaB and FOXO1, the apoptosis of dendritic cells. Nat. Immunol. 10, 753–760 [DOI] [PubMed] [Google Scholar]
- 38. Berzat A. C., Buss J. E., Chenette E. J., Weinbaum C. A., Shutes A., Der C. J., Minden A., Cox A. D. (2005) Transforming activity of the Rho family GTPase, Wrch-1, a Wnt-regulated Cdc42 homolog, is dependent on a novel carboxyl-terminal palmitoylation motif. J. Biol. Chem. 280, 33055–33065 [DOI] [PubMed] [Google Scholar]
- 39. Drenan R. M., Doupnik C. A., Boyle M. P., Muglia L. J., Huettner J. E., Linder M. E., Blumer K. J. (2005) Palmitoylation regulates plasma membrane-nuclear shuttling of R7BP, a novel membrane anchor for the RGS7 family. J. Cell Biol. 169, 623–633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Pedram A., Razandi M., Sainson R. C., Kim J. K., Hughes C. C., Levin E. R. (2007) A conserved mechanism for steroid receptor translocation to the plasma membrane. J. Biol. Chem. 282, 22278–22288 [DOI] [PubMed] [Google Scholar]