

Abstract
Exposure of particulate air pollution is linked to increased incidences of cardiovascular diseases. Ambient ultra fine particles (UFP) from diesel vehicle engines have been shown to be pro-atherogenic in apoE knockout mice and may constitute a major cardiovascular risk in humans. We posited that circulating nano-sized particles from traffic pollution sources induced vascular oxidative stress via JNK activation in endothelial cells. Diesel UFP were collected from a 1998 Kenworth truck. Intra-cellular superoxide assay revealed that these UFP dose-dependently induced superoxide (O2·-) production in human aortic endothelial cells (HAEC). Flow cytometry (FACS) showed that UFP increased MitoSOX Red intensity specific for mitochondrial superoxide. Protein carbonyl content is increased by UFP as an indication of vascular oxidative stress. UFP also up-regulated hemeoxygenase-1 (HO-1) and tissue factor (TF) mRNA expression, and pre-treatment with antioxidant, N-acetyl cysteine (NAC), significantly decreased their expression. Furthermore, UFP transiently activated JNK in HAEC. Treatment with JNK inhibitor SP600125 and silencing of both JNK1 and JNK2 with siRNA inhibited UFP stimulated O2·- production and mRNA expression of HO-1 and TF. Our findings suggest that JNK activation play an important role in UFP-induced oxidative stress and stress response gene expression.
Keywords: Ultra fine particles (UFP), Human aortic endothelial cells, Oxidative stress, Superoxide, JNK, Diesel
Introduction
Atmospheric particulate matter (PM), a prominent and persistent air pollutant, is associated with increased incidences of cardiovascular and respiratory diseases [1] [2, 3]. Particles from diesel vehicle engines constitute a large composition of ambient urban ultra fine particles (UFP, defined as those smaller than 0.1 μm in diameter), a fraction of ambient PM [4]. They are comprised of carbonaceous cores, with aromatic hydrocarbons, quinones, and heterocyclic organic compounds condensed around them [5]. Araujo et al reported that chronic exposure of ApoE knock-out mice to these UFP accelerated the development of arteriosclerosis [6]. The mechanisms whereby exposure to UFP predisposes individuals to cardiopulmonary illness are emerging health and environmental interests.
Vascular oxidative stress is intimately related to cardiovascular diseases [7, 8]. Chronic exposure to UFP resulted in a decrease in the anti-inflammatory capacity of plasma high-density lipoprotein and an increase in oxidative stress in the arterial circulation of ApoE knockout mice [6]. Both atmospheric particulate matter (PM) and urban ultra fine particles (UFP) have been shown to induce oxidative stress in epithelial cells and macrophages [9-11]. UFP are associated with air pollution-induced asthma [12]. UFP were shown to modulate various gene expression, including tissue factor (TF) and hemooxygenase-1 (HO-1) in human pulmonary artery endothelial cells [13] and human microvascular endothelial cells[14]. Inhaled nano-sized particles in air pollutant can transmigrate across human pulmonary epithelium into systemic arterial circulation [15-17]. In this context, we propose that UFP from mobile sources of air pollution induce oxidative stress in vascular endothelial cells with relevance to endothelial cell dysfunction.
JNK is a major kinase of the mitogen-activated protein kinase (MAPK) family and is responsive to stress stimuli. JNK mediates signaling pathways in vascular endothelial cells [18]. JNK expression and activation were up-regulated in the atherosclerotic lesions [19]. JNK inhibitor, SP600125, reduced superoxide production and restored NO release in coronary arteries [20]. JNK2 knockout mice developed a low level of foam cells relevant to the initiation of atherosclerosis [21]. Pourazar et al demonstrated diesel exhaust (DE) significantly increased levels of nuclear phosphorylated JNK along with phosphorylated p38 kinase and NFκB in human airway epithelium[22]. Most recently, Kleinman et al reported that active JNK in central nerve system (CNS) was significantly increased in animals receiving ambient UFP suggesting a role of JNK in the effect of UFP in vivo [23]. In this study, we tested whether air pollutant nanoparticles from diesel vehicle engines induced vascular endothelial oxidative stress via JNK activation. We demonstrated that both JNK inhibitor and knock-down JNK decreased UFP-induced superoxide production and stress response gene expression in vascular endothelial cells.
Materials and Methods
Materials and Reagents
Endothelial cell culture media and reagents were obtained from Cell Application Inc. and Invitrogen Inc. FBS was obtained from Hyclone Inc. JNK inhibitor SP600125 and N-acetyl cysteine were purchased from Calbiochem. Protease inhibitor (PI) and phosphotase inhibitor cocktail were purchased from Sigma Inc. Anti-tubulin antibody was purchased from Upstate Biotech. Antibodies against phosphor-JNK, total JNK and HRP-conjugated secondary antibodies were obtained from Cell Signaling Inc. Scrambled control siRNA, JNK1 siRNA and JNK2 siRNA were obtained from Qiagen Inc
Collection and Preparation of Ultra Fine Particles (UFP)
The ultra fine particles used in the present study were collected from a 1998 Kenworth truck (11L diesel engine and a gross vehicle weight of about 80,000 lbs) at the California Air Resource Board (CARB) heavy duty diesel emission testing laboratory (HDETL) in downtown Los Angeles [24] [25]. A high volume sampler [26] operating at 450 lpm was employed to collect the PM mass on Teflon coated glass fiber filters (20 × 25 cm) (Pallflex Fiberfilm T60A20-8x10, Pall Corp., East Hills, NY). A portion of the filters was then analyzed by Shimadzu TOC-5000A liquid analyzer [27] for water soluble organic carbon (WSOC) and by ion chromatography (IC) technique for the chemical species. Another portion of these high volume samples was analyzed by gas chromatography-mass spectrometry (GC/MS) for organic compounds[28].
The remaining portion of the filters was used to prepare the suspension of PM for the cell exposure tests. The filters were first soaked in 10 ml of ultra-pure water (Milli-Q deionized water, resistively 18.2 megaohm; total organic compounds <10 ppb; particle-free; bacteria <1 colony forming unit/ml) for 30 minutes in endotoxin-free glass vial, followed by sonication for 30 minutes. The extracted filters were pre- and post-weighed after drying to determine the PM suspension mass concentration.
Cell Culture
Human aortic endothelial cells (HAEC) were purchased from Cell Application and cultured with endothelial cell growth media from Cell Application. The cells were used between passages 5 and 11. For treatment with UFP, the final media condition was M199/0.1%FBS. For treatment with inhibitors, cells were pretreated with inhibitor for 1 hour before UFP treatment.
Superoxide Assay
Intro-cellular superoxide production was measured using nitroblue tetrazolium (NBT) assay as previously described[29]. Mitochondrial O2·- production was measured with flow cytometry (FACS) after staining with mitochondrial superoxide specific dye MitoSOX Red(Invitrogen). HAEC cells were plated in 6-well plates and grown to proper density. The cells were then starved with Serum free defined media (Cell Application Inc) for at least 4 hours. The cells were washed with M199/0.1% FBS and incubated with 5uM of MitoSOX Red for 10 minutes. Cells were then treated with or without UFP in M199/0.1%FBS for 1 hour. Cells were then trypsinized and suspended in 1ml of PBS/2% paraformaldehyde for FACS analysis. Measurements were performed using FACS Caliber system (BD Biosciences) at the USC Center for Stem Cell and Regenerative Medicine Core. MitoSOX Red was excited at 396 nm [30] and the data were collected at 585/42 nm (FL2) channel. Quantifications were performed from mean intensity of MitoSOX fluorescence from triplicates.
Protein Carbonyl Assay
Protein oxidation of HAEC was assessed by measuring protein carbonyl levels. HAEC were plated in 100mm dishes and grown to confluence. The cells were then treated with or without UFP for 6 hours in M199/0.1%FBS. The cells were then washed with PBS and scrapped into PBS/0.5% Tween-20. After 30 min incubation in ice and spin at 14000rpm at 4 °C for 3 minutes, supernatant were collected as lysate. The total protein was assessed using DC protein assay (Bio-Rad). Lysates with equal amount of total protein were used to measure protein carbonyl levels following the instruction of assay kit from Cayman Chemicals [31]. The concentrations of protein carbonyl were calculated with the formula from the manufacturer and expressed as nmol/ml.
Quantitative RT-PCR (qRT-PCR)
mRNA expression of interested genes were measured with quantitative RT-PCR essentially as described previously[29]. The expression of target genes was calculated as fold of control and normalized to GAPDH. Primers used to measure HO-1, TF and JNK2 expression were as following: HO-1: forward: GGCAGAGAATGCTGAGTTCATGAGGA, reverse: ATAGATGTGGTACAGGGAGGCCATCA TF: forward: TTTGGAGTGGGAACCCAAACCCGTCA. reverse: ACCCGTGCCAAGTACGTCTGCTTCACAT, JNK2: forward: TCACATGGAGCTGGATCA TGAAAGAATGTC, reverse:TTCACAACAATGTTGCTAGGCTTCAAATCT.
siRNA Transfection
siRNA Transfection was done with Lipofectamine RNAiMax (Invitrogen). HAEC were plated in 6 well plates or 96-well plates without antibiotics the day before transfection. The cells were transfected with siRNA following manufacturer’s instruction. Transfection media were changed to normal growth media after 4-6 hours of transfection. Cells were used for confirmation of gene knockdown or function assay 48 hours after transfection.
Western Blot
After treatment, cells were lysed in RIPA buffer (50 mM Tris-HCl, pH 8.0, 150mM NaCl, 1% Triton X-100, 0.5% sodium deoxycholate supplemented with protease inhibitor (PI) cocktail) and phosphotase inhibitor cocktail at 4 °C for 30 min. After centrifugation for 5 min at 4 °C, supernatants were collected as whole cell lysates. Protein concentrations were determined. Equal protein amount of lysates were run on a 4-20% gradient SDS-PAGE gel. The proteins were then transferred to PVDF membrane and blotted with indicated primary and secondary antibodies. Signals were developed with Supersignal Western Pico (Pierce) and recorded with FluorChem FC2 (Alpha Inotech Inc). Densitometry scan of western blot were done with the software come with the FlorChem FC2 machine.
Statistical analysis
All experiments were done three or more times. Data were expressed as mean ± standard deviation (SD). For comparisons between two groups, student t-test was used for significance analysis.
Results
Ultra fine particles (UFP) increased endothelial superoxide production and protein oxidation
UFP from diesel engines were collected to examine their effects on HAEC. The inorganic and organic compound content of these particles is discussed in [32] and summarized in Supplemental table 1. UFP induced a dose-dependent response in intracellular superoxide (O2·-) production in HAEC as measured by NBT assay. In a representative experiment shown in Fig. 1A, treatment of HAEC with UFP at 12.5μg/ml resulted in a 4.2-fold increase in O2·- production as compared to vehicle control, and treatment with UFP at 50μg/ml resulted in a 13.8-fold increase in O2·- production (OD700nm: control=0.051±0.015, UFP at 12.5 μg/ml = 0.213±0.0164, p<0.0005; 25 μg/ml UFP=0.44±0.03, p<0.001; UFP at 50μg/ml=0.707 ±0.0738, p<0.005, n=3 in all cases).
Figure 1.
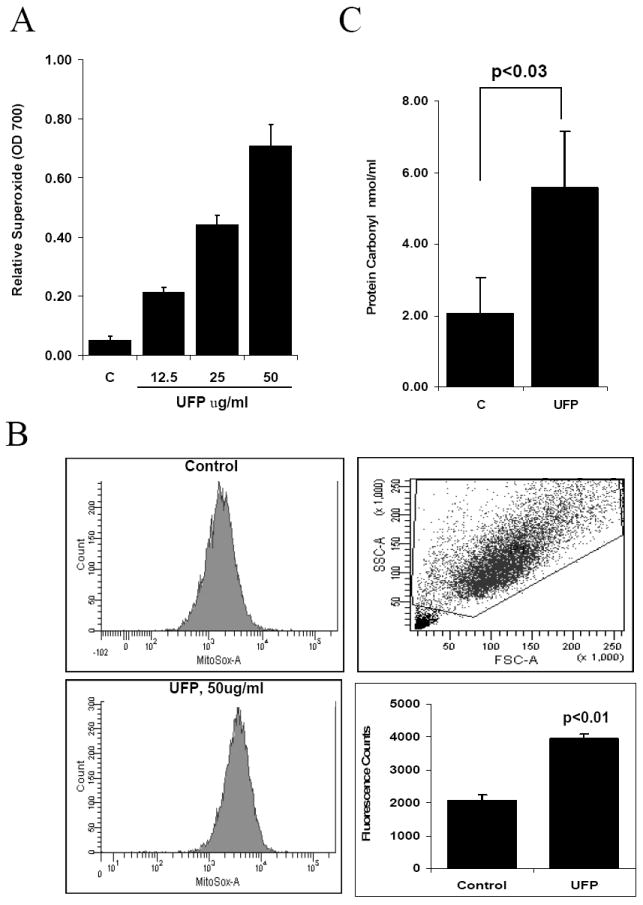
UFP Induces oxidative stress in HAEC. (A) UFP stimulated intracellular superoxide production. HAEC were treated with different concentration of UFP for 1 hour in the presence of NBT. Superoxide production was measured as absorbance at 700nm. (B) UFP stimulated mitochondrial superoxide production. HAEC were pretreated with mitochondrial superoxide specific dye MitoSOX Red for 10 minutes followed by treatment with 50ug/ml of UFP for 1 hour. The intensity of fluorescence was measured by FACS as quantification of mitochondrial superoxide. (C) UFP increased protein carbonyl contents. HAEC were treated with 50ug/ml of UFP for 6 hours. Cells were lysed and protein carbonyl content in equal amount of protein lysate was measured as described in methods.
Mitochondrial metabolic homeostasis is relevant to endothelial function [8]. Mitochondrial O2·- production was assessed using MitoSOX Red fluorescence specific for mitochondrial O2·-. FACS analysis revealed that UFP at 50μg/ml significantly increased mitochondrial O2·- production (Fig. 1B, control =2067±175, UFP=3953±137, n= 3, p<0.01).
Production of O2·- is implicated in post-translational modification of protein and lipid as well as DNA damage [33]. Protein carbonyl assay showed that treatment with UFP at 50μg/ml for 6 hours increased protein carbonyl content to 5.58nmol/ml as compared to 2.04nmol/ml in control (n=3, p<0.03)(Fig. 1C), indicating that UFP induced vascular oxidative stress.
UFP up-regulated stress response gene expression
The dynamic changes in intracellular redox status are accompanied by UFP-mediated HO-1 and TF expression. UFP up-regulated HO-1 and TF mRNA expression in a dose-dependent manner in HAEC treated with UFP (Gene expression was expressed as fold of control, p-values vs control are all less than 0.02 or lower, n=3 in all cases) (Fig. 2). This finding is consistent with the dose-dependent increase of O2·- production in Fig. 1A. Treatment with N-Acetyl cysteine (NAC), an O2·- scavenger, attenuated UFP-induced HO-1 and TF mRNA expression by 51% (n=3, p<0.02) and 57% (n=3, p<0.01) respectively, implicating the role of UFP-induced O2·- production in HO-1 and TF stimulation (Fig. 3).
Figure 2.
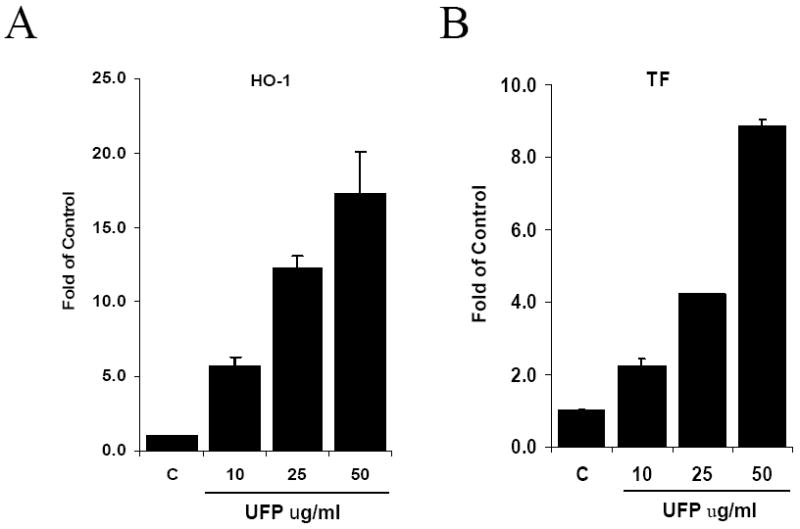
UFP stimulated the expression of HO-1 and TF. HAEC were treated for 4 hours with indicated concentration of UFP. The expression of HO-1 (A) and TF (B) mRNA was measured by qRT- PCR.
Figure 3.
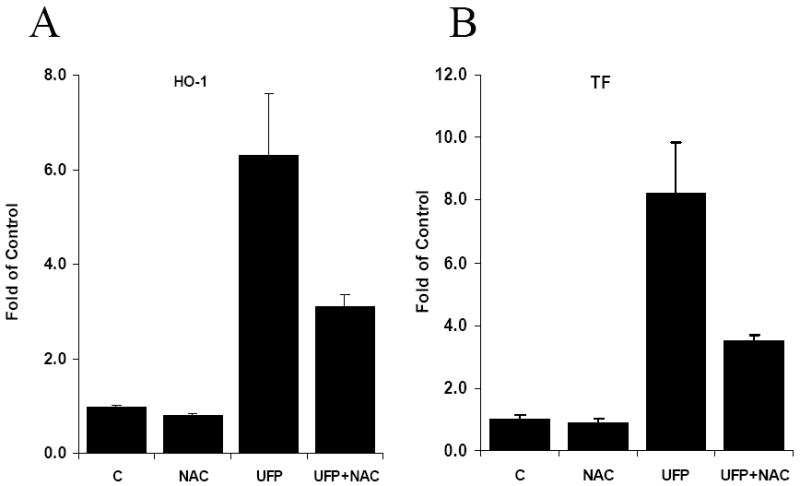
Antioxidant, N-Acetyl-Cysteine (NAC), inhibited UFP stimulated gene expression. HAEC were pretreated with 5mM of NAC for 1 hour in M199/25mM Hepes(pH7.4)/0.1%FBS followed by co-treatment of NAC with or without 50ug/ml of UFP. The expression of HO-1 (A) and TF (B) mRNA was measured by qRT- PCR.
UFP activated JNK
Western blot analysis demonstrated that UFP phosphorylated JNK 5 minutes after treatment, and phospho-JNK intensity peaked at 15 minutes in comparison with total JNK (Fig. 4). Two bands were observed in both phosphorylated and total-JNK, consistent with the presence of JNK1 and JNK2. The intensity of phospho-JNK decreased and was comparable to that of the background at 1 hour. Thus, our finding suggests that UFP induced a transient JNK activation.
Figure 4.
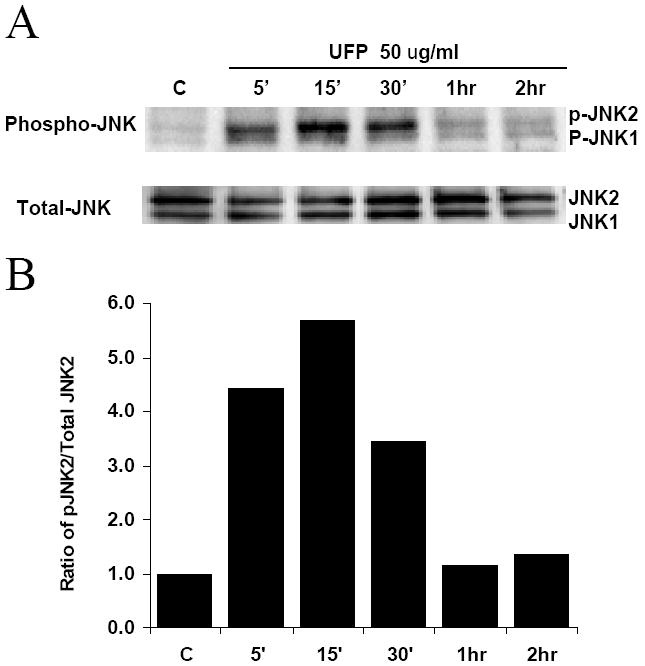
Transient activation of JNK by UFP. (A) HAEC were treated with 50 μg/ml of UFP for indicated time. Cells were lysed in RIPA buffer with protease and phosphatase inhibitors. 50μg of protein was loaded for SDS-PAGE in each lane. Activated JNK and total JNK were detected with anti-phospho-JNK and anti-total JNK antibodies. (B) Densitometry scan of (A). The relative activated JNK2 was expressed as ratio of phospho-JNK2 (p-JNK) to total JNK2.
UFP stimulated superoxide production and stress response gene expression via JNK activation
We tested the role of JNK in UFP-mediated O2·- production and gene expression by treatment with JNK inhibitor SP600125 and knock-down of JNK1 and JNK2. SP600125 at 10μM inhibited UFP-induced O2·- production by 41% ( Fig 5A)(n=3, p<0.003). SP600125 also inhibited UFP-induced HO-1 mRNA expression by 61.5% and TF mRNA expression by 62.9% (Fig 5B) (n=3, p<0.003).
Figure 5.
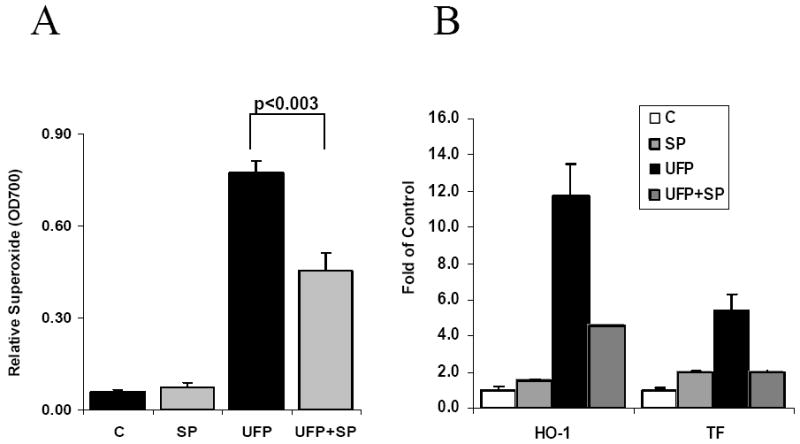
JNK inhibitor SP600125 (SP) inhibited UFP stimulated superoxide production and gene expression. (A) HAEC in 96 wells were pretreated with 10uM of SP600125 for 1 hours followed by co-treatment of 25ug/ml of UFP and SP600125 for 1 hour in the presence of NBT. Superoxide production was measured as O.D. 700nm. (B) HAEC were pre-treated with 10uM of SP600125 for 1 hour followed by co-treatment of inhibitor and 50ug/ml of UFP for another 4 hours. HO-1 and TF expression were measured by qRT- PCR.
To confirm the findings in JNK inhibitor study, we further silenced JNK1 and JNK2 expression. siJNK significantly reduced JNK mRNA and protein expression by 70% as compared to scrambled siRNA (Fig. 6A and 6B). We demonstrated that siJNK1 and siJNK2 reduced UFP-induced O2·- production by 40% in comparison with scrambled siJNK (Fig. 6C)(n=3, p<0.02). SiJNK1 and siJNK2 further reduced UFP-up-regulated mRNA expression of HO-1 by 52% (n=3, p<0.04) and TF by 60% (n=3, p<0.02) in comparison with scrambled siRNA (Fig. 6D). Taken together, our findings supported a role of JNK in UFP-induced oxidative stress.
Figure 6.
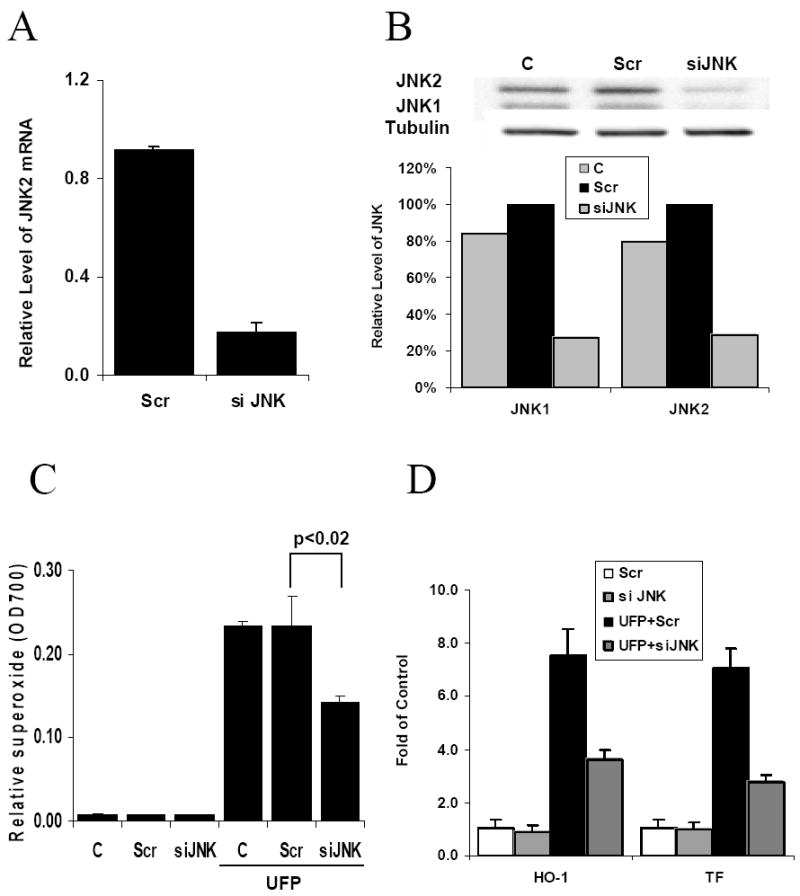
UFP stimulated superoxide production and gene expression is JNK-dependent. HAEC were transfected with scrambled control siRNA (Scr) or JNK1/JNK2 siRNA (siJNK). 48 hours after transfection, the cells were used for the following analysis. (A) Cells were lysed for measuring JNK2 mRNA expression by qRT-PCR. (B) Cell lysate was obtained to measure JNK1 and JNK2 protein expression by western blotting with anti-total JNK antibody. The relative levels of JNK1 and JNK2 were normalized to tubulin from densitometry scan data. (C) Cells in 96 well were used to measure superoxide production stimulated with or without 50ug/ml of UFP by NBT assay. (D) The expression of HO-1 and TF mRNA in cells treated with or without 50ug/ml of UFP was measured by qRT- PCR.
Discussion
Numerous epidemiological studies support the notion that air pollution is closely associated with increased incidence of cardiovascular diseases [1-3], and a direct study in mouse demonstrated that ultra fine particles (UFP) promote atherosclerosis [6]. Our study demonstrated that UFP emitted from diesel engines induced vascular oxidative stress via JNK activation in HAEC. We showed that (1) UFP induced cytosolic and mitochondrial superoxide production; (2) UFP increased the expression of oxidative stress response genes; namely, HO-1 and coagulant gene TF; (3) UFP induced a transient JNK activation that peaked at 15 minutes; and (4) JNK activation played an important role in UFP-induced superoxide production and stress response gene expression. Thus, our finings implicated a link between UFP-induced vascular oxidative stress and endothelial dysfunction.
Various mechanisms have linked the adverse health effects of airborne particulate matter (PM) with inflammatory responses, endotoxin effects, pro-coagulant effect and oxidative stress. The generation of superoxide and subsequent oxidative stress has been demonstrated to be an important mechanism [34-36]. In the mice models, diesel extracted particle (DEP)-mediated superoxide production played a critical role in reactive airway disease such as asthma, and superoxide dismutase (SOD) or nitric oxide synthetase (NOS) inhibitor was reported to suppress this oxidative effect[37, 38]. Antioxidant NAC has also been shown to suppress the adjuvant effects of DEP in allergic response [39]. In ApoE knockout mice, UFP significantly increased lipid peroxidation and the expression of antioxidant gene such as catalase and SOD2[6]. Previous in vitro studies showed that PM stimulated oxidative stress in macrophages and epithelial cells [10, 40, 41]. Our study demonstrated that UFP from mobile pollution sources, such as diesel engines, induced oxidative stress in endothelial cells.
Cellular reactive oxygen species (ROS) arise from multiple sources. NADPH oxidase is believed to be the major source of superoxide generation in the vasculature [42, 43]. We observed that Apocynin, which affects the assembly of NADPH oxidase subunits, inhibited UFP stimulated gene expression (unpublished data). We showed that mitochondria also contributed to UFP-induced superoxide production as measured by mitochondria specific dye MitoSOX Red. While UFP-induced intracellular superoxide production was inhibited by JNK inhibitor (Fig.5A) and JNK siRNA(Fig. 6C), our preliminary study revealed that UFP-induced mitochondrial superoxide generation in HAEC was insensitive to JNK inhibitor SP600125 (data not shown). Thus, UFP can induce superoxide production via more than one mechanism.
In our study, UFP stimulated the expression of HO-1 and TF in a dose-dependent manner, consistent with other studies using different sources of airborne PM and type of cells [6, 13, 14, 44]. Our data also showed that antioxidant, NAC, inhibited the induction of HO-1 and TF expression by UFP. However, inflammatory cytokine expression and cytotoxicity as reported by others[13, 45] were not significantly induced by UFP at the dose and time frame of treatment used in our study (data not shown). According to the hierarchical oxidative stress hypothesis, different doses of air pollutant induced tiered cellular responses [46]. Low dose exposure of cells to air pollutant particles activated antioxidant responses including an increase in the expression of HO-1, whereas higher dose exposure led to pro-inflammatory responses including generation of cytokines such as IL-8 and TNF-α and cytotoxicity. Araujo et al. reported that UFP promoted atherosclerosis in mice in response to chronic UFP exposure for a total 75 hours over a 5 -week period[6]. The effects of repeated and long- term exposure of HAEC to UFP would be of interest to assess inflammatory response and cytotoxicity.
Accumulated evidence supports that oxidative stress plays a major role on airborne particle-associated diseases. However, the mechanism whereby PM induces oxidative stress remains undefined. In this study, we demonstrated that UFP-simulated superoxide production in endothelial cells is, in part, mediated by JNK activation. JNK is the major member in the MAPK superfamily that mediates stress responses. Both exogenous and endogenous ROS activate JNK via different pathways involving redox sensing proteins such as thioredoxin, glutaredoxin and glutathione transferase pi (GSTπ)[47]. Interestingly, JNK can also mediate the generation of ROS in cells treated with stress stimuli. For example, JNK activation by TNF leads to an increase in ROS formation which is required in TNFα-induced necrosis, and this effect is significantly reduced in JNK−/− fibroblasts [48]. In diallyl tridulfide (DATS) induced cell cycle arrest, ROS generation was shown to be a critical event, which is dependent on JNK signaling[49]. Analogous to other oxidant stimuli, our study suggest that UFP can induce oxidative stress via JNK activation. While our data showed that UFP increased the levels of total phospho-JNK in HAEC, DE was shown to increase nuclear phospho-JNK in human airway[22]. This difference could be due to the time points at which phospho-JNK was assessed. We demonstrated that UFP-stimulated HO-1 and TF expression was inhibited by JNK inhibitor, SP600125, and antioxidant, NAC. JNK siRNA further corroborated the role of JNK in oxidative stress. Recently, Hartz et al. reported that DEP-upregulated P-glycoprotein involved JNK in the brain capillaries [50] in which JNK activation seemed to be downstream from DEP-induced oxidative stress. Their findings suggested an autocrine or paracrine effect of DEP-induced TNF-α release. In our study, JNK was activated in less than 5 minutes, suggesting that it was unlikely to be a secondary effect. Both SP600125 and JNK siRNA reduced UFP-stimulated superoxide production, supporting the notion that JNK activation was upstream from UFP-induced oxidative stress in HAEC. It was also likely that oxidative stress induced by UFP further activated JNK to a positive feedback loop between ROS production and JNK activation [47, 48]. Recently, JNK2 knockout mice on the high cholesterol diet were reported to harbor a significant decrease in vascular oxidative stress as evidenced by a decrease in superoxide, peroxynitrite and lipid peroxidation levels as compared to wild type mice; thus, supporting JNK as a key mediator of oxidative stress [51].
Considerable studies support the involvement of JNK in atherogenesis. Increased expression and activation of JNK was shown to present in atherosclerotic lesions [19, 52]. Recently, we also demonstrated prominent presence of phospho-JNK in both endothelial and smooth muscle cells of human coronary arteries in patients with ischemic cardiomyopathy (unpublished data). Studies with knockout mice under ApoE-/- background demonstrated the requirement of JNK2 in foam cell formation in atherosclerosis [21]. Given the intimate relation between the JNK-mediated vascular oxidative stress and atherosclerosis, it is reasonable to test our hypothesis that UFP-activated JNK and subsequent superoxide production induce endothelial dysfunction and the initiation of atherosclerosis in our future investigation. In summary, we demonstrated that UFP emitted from diesel engines induced JNK activation with implication to superoxide production and up-regulation of stress response genes (Fig 7). The results pave a foundation for further assessment of the mechanism of UFP-promoted atherosclerosis.
Figure 7.
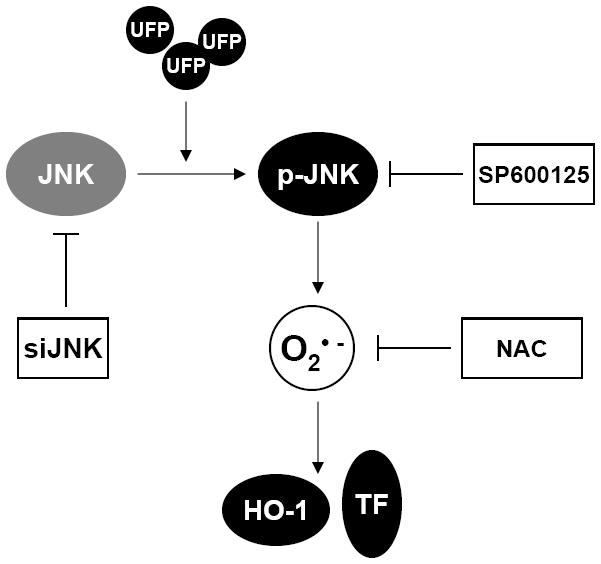
Mode of action of UFP stimulated effect in HAEC. UFP treatment activates JNK, which lead to increased intracellular superoxide production that eventually results in increased expression of stress response gene HO-1 and TF. Knockdown of JNK by siRNA or JNK inhibitor SP6000125 attenuate UFP stimulated superoxide production and gene expression. Scavenger of superoxide by Antioxidant NAC inhibits UF stimulated gene expression.
Acknowledgments
The authors are grateful to Dr. Michael Kahn for providing the flow cytometry facility in the Center of Stem Cell and Regenerative Medicine at USC. These studies were supported by AHA GIA 0655051Y (TKH), NIH HL068689 (TKH), and NIH HL083015 (TKH). Additional support was provided by the California Air Resources Board, the South Coast Air Quality Management District (Grant No. 05-308) and by the Southern California Particle Center (SCPC), funded by EPA under the STAR program through Grant RD-8324- 1301-0 to the University of Southern California.
List of Abbreviations
- UFP
ultra fine particles
- PM
particulate matters
- HAEC
human aortic endothelial cells
- HO-1
hemooxygenase-1
- TF
tissue factor
- NAC
N-acetyl cysteine
- NBT
nitroblue tetrazolium
- qRT-PCR
quantitative reverse transcription-polymerase chain reaction
- DE
diesel exhaust
- DEP
diesel extracted particle
- ROS
reactive oxygen species
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Brunekreef B, Holgate ST. Air pollution and health. Lancet. 2002;360:1233–1242. doi: 10.1016/S0140-6736(02)11274-8. [DOI] [PubMed] [Google Scholar]
- 2.Knutsen S, Shavlik D, Chen LH, Beeson WL, Ghamsary M, Petersen F. The association between ambient particulate air pollution levels and risk of cardiopulmonary and all-cause mortality during 22 years follow-up of a non-smoking cohort. Results from the AHSMOG study. Epidemiology. 2004;15:S45–S45. [Google Scholar]
- 3.Ostro B. The Association of Air-Pollution and Mortality - Examining the Case for Inference. Arch Environ Health. 1993;48:336–342. doi: 10.1080/00039896.1993.9936722. [DOI] [PubMed] [Google Scholar]
- 4.Kittelson DB. Engines and nanoparticles: A review. J Aerosol Sci. 1998;29:575–588. [Google Scholar]
- 5.Reynolds LJ, Murphy SA, Richards RJ. Toxicity of modified and nonmodified diesel exhaust particles on different lung alveolar epithelial cell cultures. In Vitro Mol Toxicol. 2000;13:173–179. [Google Scholar]
- 6.Araujo JA, Barajas B, Kleinman M, Wang X, Bennett BJ, Gong KW, Navab M, Harkema J, Sioutas C, Lusis AJ, Nel AE. Ambient particulate pollutants in the ultrafine range promote early atherosclerosis and systemic oxidative stress. Circ Res. 2008;102:589–596. doi: 10.1161/CIRCRESAHA.107.164970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Giordano FJ. Oxygen, oxidative stress, hypoxia, and heart failure. J Clin Invest. 2005;115:500–508. doi: 10.1172/JCI200524408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Griendling KK, FitzGerald GA. Oxidative stress and cardiovascular injury: Part II: animal and human studies. Circulation. 2003;108:2034–2040. doi: 10.1161/01.CIR.0000093661.90582.c4. [DOI] [PubMed] [Google Scholar]
- 9.Gonzalez-Flecha B. Oxidant mechanisms in response to ambient air particles. Mol Aspects Med. 2004;25:169–182. doi: 10.1016/j.mam.2004.02.017. [DOI] [PubMed] [Google Scholar]
- 10.Li N, Sioutas C, Cho A, Schmitz D, Misra C, Sempf J, Wang M, Oberley T, Froines J, Nel A. Ultrafine particulate pollutants induce oxidative stress and mitochondrial damage. Environ Health Perspect. 2003;111:455–460. doi: 10.1289/ehp.6000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Risom L, Moller P, Loft S. Oxidative stress-induced DNA damage by particulate air pollution. Mutat Res. 2005;592:119–137. doi: 10.1016/j.mrfmmm.2005.06.012. [DOI] [PubMed] [Google Scholar]
- 12.Li N, Hao M, Phalen RF, Hinds WC, Nel AE. Particulate air pollutants and asthma. A paradigm for the role of oxidative stress in PM-induced adverse health effects. Clin Immunol. 2003;109:250–265. doi: 10.1016/j.clim.2003.08.006. [DOI] [PubMed] [Google Scholar]
- 13.Karoly ED, Li Z, Dailey LA, Hyseni X, Huang YC. Up-regulation of tissue factor in human pulmonary artery endothelial cells after ultrafine particle exposure. Environ Health Perspect. 2007;115:535–540. doi: 10.1289/ehp.9556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gong KW, Zhao W, Li N, Barajas B, Kleinman M, Sioutas C, Horvath S, Lusis AJ, Nel A, Araujo JA. Air-pollutant chemicals and oxidized lipids exhibit genome-wide synergistic effects on endothelial cells. Genome Biol. 2007;8:R149. doi: 10.1186/gb-2007-8-7-r149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nemmar A, Hoylaerts MF, Hoet PH, Nemery B. Possible mechanisms of the cardiovascular effects of inhaled particles: systemic translocation and prothrombotic effects. Toxicol Lett. 2004;149:243–253. doi: 10.1016/j.toxlet.2003.12.061. [DOI] [PubMed] [Google Scholar]
- 16.Takenaka S, Karg E, Kreyling WG, Lentner B, Moller W, Behnke-Semmler M, Jennen L, Walch A, Michalke B, Schramel P, Heyder J, Schulz H. Distribution pattern of inhaled ultrafine gold particles in the rat lung. Inhal Toxicol. 2006;18:733–740. doi: 10.1080/08958370600748281. [DOI] [PubMed] [Google Scholar]
- 17.Takenaka S, Karg E, Kreyling WG, Lentner B, Schulz H, Ziesenis A, Schramel P, Heyder J. Fate and toxic effects of inhaled ultrafine cadmium oxide particles in the rat lung. Inhal Toxicol. 2004;16(Suppl 1):83–92. doi: 10.1080/08958370490443141. [DOI] [PubMed] [Google Scholar]
- 18.Surapisitchat J, Hoefen RJ, Pi X, Yoshizumi M, Yan C, Berk BC. Fluid shear stress inhibits TNF-alpha activation of JNK but not ERK1/2 or p38 in human umbilical vein endothelial cells: Inhibitory crosstalk among MAPK family members. Proc Natl Acad Sci U S A. 2001;98:6476–6481. doi: 10.1073/pnas.101134098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Metzler B, Hu Y, Dietrich H, Xu Q. Increased expression and activation of stress-activated protein kinases/c-Jun NH(2)-terminal protein kinases in atherosclerotic lesions coincide with p53. Am J Pathol. 2000;156:1875–1886. doi: 10.1016/S0002-9440(10)65061-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang C, Hein TW, Wang W, Ren Y, Shipley RD, Kuo L. Activation of JNK and xanthine oxidase by TNF-alpha impairs nitric oxide-mediated dilation of coronary arterioles. J Mol Cell Cardiol. 2006;40:247–257. doi: 10.1016/j.yjmcc.2005.11.010. [DOI] [PubMed] [Google Scholar]
- 21.Ricci R, Sumara G, Sumara I, Rozenberg I, Kurrer M, Akhmedov A, Hersberger M, Eriksson U, Eberli FR, Becher B, Boren J, Chen M, Cybulsky MI, Moore KJ, Freeman MW, Wagner EF, Matter CM, Luscher TF. Requirement of JNK2 for scavenger receptor A-mediated foam cell formation in atherogenesis. Science. 2004;306:1558–1561. doi: 10.1126/science.1101909. [DOI] [PubMed] [Google Scholar]
- 22.Pourazar J, Mudway IS, Samet JM, Helleday R, Blomberg A, Wilson SJ, Frew AJ, Kelly FJ, Sandstrom T. Diesel exhaust activates redox-sensitive transcription factors and kinases in human airways. Am J Physiol Lung Cell Mol Physiol. 2005;289:L724–730. doi: 10.1152/ajplung.00055.2005. [DOI] [PubMed] [Google Scholar]
- 23.Kleinman MT, Araujo JA, Nel A, Sioutas C, Campbell A, Cong PQ, Li H, Bondy SC. Inhaled ultrafine particulate matter affects CNS inflammatory processes and may act via MAP kinase signaling pathways. Toxicol Lett. 2008;178:127–130. doi: 10.1016/j.toxlet.2008.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Biswas S, Hu S, Verma V, Herner J, Robertson WJ, Ayala A, Sioutas C. Physical Properties of particulate matter (PM) from late model heavy duty diesel vehicles operating with advanced emission control technologies. Atomspheric Environment. 2008 [Google Scholar]
- 25.Holmen BA, Ayala A. Ultrafine PM emissions from natural gas, oxidation-catalyst diesel, and particle-trap diesel heavy-duty transit buses. Environ Sci Technol. 2002;36:5041–5050. doi: 10.1021/es015884g. [DOI] [PubMed] [Google Scholar]
- 26.Misra C, Kim S, Shen S, Sioutas C. A high flow rate, very low pressure drop impactor for inertial separation of ultrafine from accumulation mode particles. J Aerosol Sci. 2002;33:735–752. [Google Scholar]
- 27.Decesari S, Facchini MC, Matta E, Lettini F, Mircea M, Fuzzi S, Tagliavini E, Putaud JP. Chemical features and seasonal variation of fine aerosol water-soluble organic compounds in the Po Valley, Italy. Atmospheric Environment. 2001;35:3691–3699. [Google Scholar]
- 28.Schauer JJ, Fraser MP, Cass GR, Simoneit BR. Source reconciliation of atmospheric gas-phase and particle-phase pollutants during a severe photochemical smog episode. Environ Sci Technol. 2002;36:3806–3814. doi: 10.1021/es011458j. [DOI] [PubMed] [Google Scholar]
- 29.Li R, Chen W, Yanes R, Lee S, Berliner JA. OKL38 is an oxidative stress response gene stimulated by oxidized phospholipids. J Lipid Res. 2007;48:709–715. doi: 10.1194/jlr.M600501-JLR200. [DOI] [PubMed] [Google Scholar]
- 30.Robinson KM, Janes MS, Pehar M, Monette JS, Ross MF, Hagen TM, Murphy MP, Beckman JS. Selective fluorescent imaging of superoxide in vivo using ethidium-based probes. Proc Natl Acad Sci U S A. 2006;103:15038–15043. doi: 10.1073/pnas.0601945103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Levine RL, Williams JA, Stadtman ER, Shacter E. Carbonyl assays for determination of oxidatively modified proteins. Methods Enzymol. 1994;233:346–357. doi: 10.1016/s0076-6879(94)33040-9. [DOI] [PubMed] [Google Scholar]
- 32.Lough GC, Schauer JJ, Park JS, Shafer MM, Deminter JT, Weinstein JP. Emissions of metals associated with motor vehicle roadways. Environ Sci Technol. 2005;39:826–836. doi: 10.1021/es048715f. [DOI] [PubMed] [Google Scholar]
- 33.Hwang ES, Kim GH. Biomarkers for oxidative stress status of DNA, lipids, and proteins in vitro and in vivo cancer research. Toxicology. 2007;229:1–10. doi: 10.1016/j.tox.2006.10.013. [DOI] [PubMed] [Google Scholar]
- 34.Li N, Xia T, Nel AE. The role of oxidative stress in ambient particulate matter-induced lung diseases and its implications in the toxicity of engineered nanoparticles. Free Radic Biol Med. 2008;44:1689–1699. doi: 10.1016/j.freeradbiomed.2008.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nel A, Xia T, Madler L, Li N. Toxic potential of materials at the nanolevel. Science. 2006;311:622–627. doi: 10.1126/science.1114397. [DOI] [PubMed] [Google Scholar]
- 36.Romieu I, Castro-Giner F, Kunzli N, Sunyer J. Air pollution, oxidative stress and dietary supplementation: a review. Eur Respir J. 2008;31:179–197. doi: 10.1183/09031936.00128106. [DOI] [PubMed] [Google Scholar]
- 37.Lim HB, Ichinose T, Miyabara Y, Takano H, Kumagai Y, Shimojyo N, Devalia JL, Sagai M. Involvement of superoxide and nitric oxide on airway inflammation and hyperresponsiveness induced by diesel exhaust particles in mice. Free Radic Biol Med. 1998;25:635–644. doi: 10.1016/s0891-5849(98)00073-2. [DOI] [PubMed] [Google Scholar]
- 38.Sagai M, Saito H, Ichinose T, Kodama M, Mori Y. Biological effects of diesel exhaust particles. I. In vitro production of superoxide and in vivo toxicity in mouse. Free Radic Biol Med. 1993;14:37–47. doi: 10.1016/0891-5849(93)90507-q. [DOI] [PubMed] [Google Scholar]
- 39.Whitekus MJ, Li N, Zhang M, Wang M, Horwitz MA, Nelson SK, Horwitz LD, Brechun N, Diaz-Sanchez D, Nel AE. Thiol antioxidants inhibit the adjuvant effects of aerosolized diesel exhaust particles in a murine model for ovalbumin sensitization. J Immunol. 2002;168:2560–2567. doi: 10.4049/jimmunol.168.5.2560. [DOI] [PubMed] [Google Scholar]
- 40.Li N, Wang M, Oberley TD, Sempf JM, Nel AE. Comparison of the pro-oxidative and proinflammatory effects of organic diesel exhaust particle chemicals in bronchial epithelial cells and macrophages. J Immunol. 2002;169:4531–4541. doi: 10.4049/jimmunol.169.8.4531. [DOI] [PubMed] [Google Scholar]
- 41.Marano F, Boland S, Bonvallot V, Baulig A, Baeza-Squiban A. Human airway epithelial cells in culture for studying the molecular mechanisms of the inflammatory response triggered by diesel exhaust particles. Cell Biol Toxicol. 2002;18:315–320. doi: 10.1023/a:1019548517877. [DOI] [PubMed] [Google Scholar]
- 42.Mohazzab KM, Kaminski PM, Wolin MS. NADH oxidoreductase is a major source of superoxide anion in bovine coronary artery endothelium. Am J Physiol. 1994;266:H2568–2572. doi: 10.1152/ajpheart.1994.266.6.H2568. [DOI] [PubMed] [Google Scholar]
- 43.Harrison D, Griendling KK, Landmesser U, Hornig B, Drexler H. Role of oxidative stress in atherosclerosis. Am J Cardiol. 2003;91:7A–11A. doi: 10.1016/s0002-9149(02)03144-2. [DOI] [PubMed] [Google Scholar]
- 44.Hirano S, Furuyama A, Koike E, Kobayashi T. Oxidative-stress potency of organic extracts of diesel exhaust and urban fine particles in rat heart microvessel endothelial cells. Toxicology. 2003;187:161–170. doi: 10.1016/s0300-483x(03)00053-2. [DOI] [PubMed] [Google Scholar]
- 45.Su DS, Serafino A, Muller JO, Jentoft RE, Schlogl R, Fiorito S. Cytotoxicity and inflammatory potential of soot particles of low-emission diesel engines. Environ Sci Technol. 2008;42:1761–1765. doi: 10.1021/es0716554. [DOI] [PubMed] [Google Scholar]
- 46.Xiao GG, Wang M, Li N, Loo JA, Nel AE. Use of proteomics to demonstrate a hierarchical oxidative stress response to diesel exhaust particle chemicals in a macrophage cell line. J Biol Chem. 2003;278:50781–50790. doi: 10.1074/jbc.M306423200. [DOI] [PubMed] [Google Scholar]
- 47.Shen HM, Liu ZG. JNK signaling pathway is a key modulator in cell death mediated by reactive oxygen and nitrogen species. Free Radic Biol Med. 2006;40:928–939. doi: 10.1016/j.freeradbiomed.2005.10.056. [DOI] [PubMed] [Google Scholar]
- 48.Ventura JJ, Cogswell P, Flavell RA, Baldwin AS, Jr, Davis RJ. JNK potentiates TNF-stimulated necrosis by increasing the production of cytotoxic reactive oxygen species. Genes Dev. 2004;18:2905–2915. doi: 10.1101/gad.1223004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Antosiewicz J, Herman-Antosiewicz A, Marynowski SW, Singh SV. c-Jun NH(2)-terminal kinase signaling axis regulates diallyl trisulfide-induced generation of reactive oxygen species and cell cycle arrest in human prostate cancer cells. Cancer Res. 2006;66:5379–5386. doi: 10.1158/0008-5472.CAN-06-0356. [DOI] [PubMed] [Google Scholar]
- 50.Hartz AM, Bauer B, Block ML, Hong JS, Miller DS. Diesel exhaust particles induce oxidative stress, proinflammatory signaling, and P-glycoprotein up-regulation at the blood-brain barrier. FASEB J. 2008;22:2723–2733. doi: 10.1096/fj.08-106997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Osto E, Matter CM, Kouroedov A, Malinski T, Bachschmid M, Camici GG, Kilic U, Stallmach T, Boren J, Iliceto S, Luscher TF, Cosentino F. c-Jun N-terminal kinase 2 deficiency protects against hypercholesterolemia-induced endothelial dysfunction and oxidative stress. Circulation. 2008;118:2073–2080. doi: 10.1161/CIRCULATIONAHA.108.765032. [DOI] [PubMed] [Google Scholar]
- 52.Nishio H, Matsui K, Tsuji H, Tamura A, Suzuki K. Immunohistochemical study of the phosphorylated and activated form of c-Jun NH2-terminal kinase in human aorta. Histochem J. 2001;33:167–171. doi: 10.1023/a:1017952310800. [DOI] [PubMed] [Google Scholar]