

Abstract
Since their discovery in 2001, the T cell immunoglobulin mucin (TIM) family members have been shown to play important roles in the regulation of immune responses. The TIM family comprises of eight genes in the mouse, three of which are conserved in humans (TIM-1, TIM-3 and TIM-4). Initially, TIM-1 and TIM-3 were thought to be expressed solely on T cells. However, emerging data suggest a much broader expression pattern where their presence on APCs confers differing functions, including the ability to mediate phagocytosis. In contrast, TIM-4 is exclusively expressed on APCs. Together, the TIM molecules provide a functional repertoire for determining the fate of T cell activation and differentiation. To date, much of the knowledge about the TIM family members has been garnered from models of asthma, allergy and autoimmunity. More recently, data from experimental models of transplantation demonstrate that TIM family members are also key in alloimmunity. This review will serve to highlight the emerging data regarding this unique family of molecules, and to identify their potential in transplantation tolerance.
Keywords: transplant immunology, T cell immunoglobulin mucin (TIM) family, T cell activation, costimulatory molecules
INTRODUCTION
The T cell immunoglobulin mucin (TIM) family of genes encode type I glycoproteins, with a common immunoglobulin V-like domain, mucin-like domain, transmembrane domain, and a cytoplasmic region(1). TIM molecules are novel costimulatory molecules critical in T helper cell activation and differentiation. Recent evidence also points to a key function in regulating adaptive immunity through removal of apoptotic cells. The three human TIM genes are most similar to mouse TIM-1, TIM-3 and TIM-4. Their role in allergy, asthma, auto- and allo- immunity is increasingly appreciated.
TIM-1 OVERVIEW
TIM-1 was initially described as kidney injury molecule-1 (KIM-1), a cell surface protein highly expressed on proximal tubular epithelial cells following acute kidney injury (AKI) (2). KIM-1 expression on injured epithelial cells allows these cells to phagocytose apoptotic tubular cells through binding to phosphatidylserine (2). The soluble form of KIM-1 is now widely studied as a urinary biomarker for AKI.
TIM-1 is not expressed on naïve CD4+ T cells(3, 4), but becomes upregulated within hours of TCR stimulation(4) and is preferentially expressed on Th2 cells (3, 5). While its function on T cells has garnered the most attention, TIM-1 has also been found on cells of innate immunity such as mast cells(6), B cells(7), and invariant natural killer T cells(8). Initially, another member of the TIM family, TIM-4, was identified as the natural ligand for TIM-1(9). However, recent binding and co-crystallization studies have questioned this interaction(7) and a number of other potential ligands exist (Table 1).
Table 1.
| Expression | Signalling | Ligand | Innate immunity | Adaptive immunity | Role in disease states | |
|---|---|---|---|---|---|---|
| TIM-1 | Activated CD4+ T cells3,5 | Phosphorylation of intracellular tyrosine domain12,13. | Proposed ligands include: | Phosphotidylserine receptor16
|
Increased Th2 differentiation | Polymorphisms linked to increased risk of asthma4 and atopy1 |
| B cells7 | Activation of Akt and ERK/MAPK12,13. | Effects appear to vary with binding ligand and affinity | Increased expression in: | |||
| Mast cells6 | Increased transcription of IL-4 promoter12. | Upregulated on renal tubular epithelial cells in acute kidney injury and renal cell carcinoma2 (KIM-1) | ||||
| NK cells8 | ?TIM-1 ligation alone sufficient to induce T cell activation14,15 | |||||
|
| ||||||
| TIM-4 | Dendritic cells9 | Lacks intracellular tyrosine domain – Not known to mediate signalling | Proposed ligands include:
|
Phosphotidylserine receptor16 | Upregulation of Th2 responses18,19 | Upregulated in food allergy18,19 |
| Macrophages9 | Proliferation of activated T cells9,12 | Polymorphisms linked to asthma and atopy | ||||
|
| ||||||
| TIM-3 | Th1 cells1 | Phosphorylation of intracellular tyrosine domain by interleukin inducible T cell kinase22 | Galectin-921 | Limits macrophage expansion29. | Binding to Gal-9 leads to apoptosis of Th1 cells24. | Increased expression on exhausted T cells in chronic infection/malignancy16 |
| Th17 cells1 | ? Other unknown | Modulates CD80 and CTLA-4 expression on APCs28 | Blockade abrogates peripheral tolerance | Appears to be downregulated in multiple sclerosis16 | ||
| CD8+ T cells1 | Phosphotidylserine receptor16,32 | Asthma9 | ||||
| APCs1 | ||||||
Recognition of the role of TIM-1 in regulating Th1/Th2 cell differentiation stems from work in a Th-2 mediated experimental model of asthma(4). Polymorphisms within a single locus on murine chromosome 11 (now known to correspond to TIM-1) were demonstrated to confer protection against the development of airway hyperreactivity (AHR). In vitro, TIM-1 signaling, either by ectopic over-expression(10) or ligation with the anti-TIM-1 antibody 3B3(3) costimulates T cell proliferation and leads to enhanced IL-4 production. When administered during an immune response, 3B3, which binds to the IgV domain, augments T cell proliferation and increases the production of both Th1 and Th2 cytokines. In doing so, it abrogates the induction of high-dose tolerance(3). Other mAbs directed against the TIM-1 mucin domain also enhance Th2 cytokine production and exacerbate AHR(7). However, in direct contradistinction, two other mAbs (RMT1-10 and 4A2.2) generated against the same IgV domain as 3B3, reduce allergen-induced airway inflammation(7, 11). Similar paradoxical findings have been reported in a murine model of multiple sclerosis. Here, 3B3 mAb costimulates pathogenic Th1/Th17 responses, enhancing severity of experimental autoimmune encephalitis. In contrast, RMT1-10 appears to act antagonistically and decreases Th1/Th17 responses, thereby inhibiting development of EAE (11).
Several intracellular signaling pathways may be activated following TIM-1 ligation. Cross-linking of TIM-1 with ‘agonistic’ 3B3 leads to phosphorylation of tyrosine residues in the TIM-1 cytoplasmic domain and downstream activation of Akt and ERK/MAPK(12, 13) Activation of TIM-1 also elicits a rise in intracellular calcium(14) and its interaction with TIM-4 triggers phosphorylation of the linker of activated T cells (LAT) (12), one of the most important T cell adapter proteins. Each of these events augments NFAT/AP-1 signaling, transcription factors critical for Th2 development. TIM-1 ligation may also direct T cell activation without concomitant TCR engagement. 3B3 can activate T cells through CD3 capping by increasing tyrosine phosphorylation of LAT(12), ZAP70 and ItK (15). However, this signaling mechanism has not been seen with the weaker affinity IgV mAb RMT1-10, or with mAbs directed against the mucin domain (3, 5, 12). Finally, TIM-1 promotes T cell viability through activation of PI3K/AKT pathway and induction of anti-apoptotic genes bcl-xL and bcl-2 (12, 14).
TIM-4 OVERVIEW
The role of TIM-4 in immune activation is less clearly defined. It is not expressed on T cells but is primarily found on APCs, including dendritic cells(9), macrophages(9) and peritoneal B-1 cells(16). TIM-4 was initially identified as the ligand for TIM-1(9) but it is now unclear whether direct interaction occurs(7) (Table 1). TIM-4 does act as a phosphatidylserine (PS) receptor, capable of binding and engulfing apoptotic bodies(17), but may also have other unidentified ligands.
Current evidence suggests TIM-4 interaction with its putative ligand promotes Th2 responses. TIM-4 upregulation on intestinal mucosal(18) and on bone marrow-derived(19) DCs (following exposure to Staphylococcus enterotoxin B or cholera toxin and peanut extract respectively) promotes Th2 polarization and intestinal allergy. Administration of TIM-4 antibody during either exposure restored oral tolerance. In vitro studies utilizing TIM-4 Ig fusion proteins have demonstrated conflicting results. Whereas high doses increase T cell proliferation, low doses appear inhibitory(9). TIM-4 Ig has also been shown to both activate (through enhanced ERK phosphorylation)(12) and inhibit(20) naïve T cells, whereas treatment of preactivated T cells enhances activation(20). In these studies, T cells were stimulated with anti-CD3 and anti-CD28 in the absence of APCs, rendering it difficult to translate these observations to an in vivo model. This is particularly true when trying to dissect the impact of TIM-4 as a PS receptor, since the absence of APCs eliminates the process of apoptotic body engulfment altogether. Overall, the data regarding the effect of TIM-4 binding to naïve T cells are conflicting and incomplete; much remains to be clarified regarding its activity in vivo. Unlike the other TIM family members, the cytoplasmic tail of TIM-4 lacks putative signaling motifs and therefore is unlikely to mediate direct inward signaling.
TIM-3 OVERVIEW
TIM-3 was initially identified on Th1 differentiated cells but is now known to be present on cells of both the innate (DCs, macrophages and mast cells) and adaptive (Th1, Th17, CD8+ T cells) immune system(1). Its ligand, galectin-9, is an S-type lectin(21) expressed on T cells, B cells, mast cells and on a range of non-immune cells. Finally, as with the other TIM family members, TIM-3 is known to be a phosphatidylserine receptor.
TIM-3 has diverse effects, involving both innate and adaptive immune responses. Upon ligation with galectin-9, TIM-3 intracellular tail is phosphorylated by the interleukin inducible T cell kinase (ITK)(22). Differing signaling pathways may exist in response to TIM-3 ligation in T cells versus DCs(23), but ERK phosphorylation appears common to both cell types.
Galectin-9:TIM-3 acts as a negative T cell costimulatory pathway. In an elegant negative feedback loop, IFNγ upregulates galectin-9, which in turn binds to TIM-3 to terminate Th1 responses by mediating calcium-calpain-caspase-dependent apoptosis (24)(21). Similarly, soluble galectin-9 administration decreases Th17 differentiation(25) while blockade of TIM-3 increases IL-17 production (26, 27).
The role of TIM-3 in innate immunity is conflicting. TIM-3 signaling on APCs reduces inflammation in models of viral inflammatory heart disease by downregulating CD80, CTLA-4 and TLR4 expression(28). Its blockade during induction of EAE leads to macrophage expansion and activation resulting in a more severe clinical phenotype(29). Furthermore, interruption of TIM-3:galectin-9 during liver ischemia-reperfusion injury increases neutrophil infiltration, hepatic apoptosis and proinflammatory cytokines release (30). In contrast, ex vivo studies show TIM-3:galectin-9 signaling acts synergistically with TLR stimuli to increase pro-inflammatory TNF-α secretion from DCs(23).
ROLE OF TIM PROTEINS AS PHOSPHATIDYLSERINE RECEPTORS
Each TIM family member also functions as a phosphatidylserine (PS) receptor. As such, they are capable of binding PS on dying cells thereby mediating engulfment of apoptotic bodies. Phagocytosis by professional APCs is central in maintaining tissue homeostasis and self-tolerance.
T cells are not known to be capable of phagocytosis; therefore it appears more likely that TIM-1 on T cells mediates costimulation. However, TIM-1 expressed on proximal tubular epithelial cells after renal injury coordinates engulfment of apoptotic cells within the tubular lumen. This function appears central in recovery of renal function after AKI (2).
Over-expression of TIM-4 on APCs reduces the number of antigen-specific T cells remaining after immunization, a property attributed to increased phosphatidylserine receptor activity(31). Similarly, peritoneal macrophages and B-1 cells from TIM4-/- mice do not efficiently engulf apoptotic bodies(16). Surprisingly, phagocytic activity of splenic DCs from TIM-1-/- mice were unaltered, whereas others have shown CD8+ DC-phagocytic activity to be inhibited by TIM-3 blockade (32). In studies of both TIM-4 and TIM-3, blockade of PS receptor function led to dysregulated lymphocyte activation and signs of systemic autoimmunity.
MANIPULATION OF TIM SIGNALING IN TRANSPLANTATION
TIM-1 and TIM-3 have been studied in experimental transplant models and there are emerging data regarding their expression in human transplant recipients. The role of TIM-4 in transplantation has not yet been elucidated.
Murine models of alloimmunity have demonstrated a central role for TIM-1 in the balance between regulatory and effector T cells. As observed in models of asthma and autoimmunity, although anti-TIM-1 mAbs 3B3 and RMT 1-10 both bind the IgV domain, they demonstrate opposite effects. In vivo administration of ‘antagonistic’ RMT1-10 significantly prolongs fully mismatched cardiac allograft survival, and in combination with rapamycin, leads to tolerance(33). This is dependent on the presence of regulatory T cells and is associated with inhibition of alloreactive Th1 responses. Furthermore, in a Tbet deficient model resistant to tolerance induction with CTLA4 Ig and anti-CD154, RMT1-10 restored tolerance and specifically inhibited IL-17-producing CD8+ T cells, a subset thought to mediate resistance to tolerance induction(34). In an islet transplant model, TIM-1 signaling via the ‘agonistic’ antibody 3B3, increased expansion and survival of effector memory T cells, while inhibiting the conversion of Tregs. Furthermore, in vitro, 3B3 signaling ‘deprogramed’ Tregs into IL-17 producing cells. Overall, peripheral tolerance was hampered and commitment of alloreactive T cells to Treg versus Th1/Th17 phenotype was reciprocally altered(35).
To date, the mechanistic differences seen with activating versus inhibitory TIM-1 Abs remain incompletely explained, although several important differences exist. These agents have very similar specificities to the IgV domain but strikingly different binding affinities: 3B3 binds TIM-1 with 17 times greater affinity than RMT 1-10(11). This property likely accounts for differences in signals activated downstream of TCR activation. The observed differential effects of these antibodies may also relate to their ability to block interaction of TIM-1 with phosphotidylserine. 3B3 prevents TIM-1-mediated phagocytosis, whereas RMT1-10 demonstrates little or no capacity to do so(36). Finally, the role of TIM-1 signaling in non-T cells may also contribute to these differential effects. Most recently, TIM-1 expression has been found to identify IL-10-expressing regulatory B cells, and its ligation with RMT-10 promotes islet allograft tolerance (D. Rothstein, personal communication).
Taken together, these findings suggest that the effects of TIM-1 blockade in models of transplantation are dependent upon the binding characteristics of the antibody in question and its resultant downstream signaling. Furthermore, differing data from models of asthma, EAE and transplantation all indicate the particular susceptibility of the model in question plays an important role in the immune responses observed upon manipulation of TIM-1 signaling.
The pro-tolerogenic effects of TIM-1 blockade using RMT 1-10 suggest important potential in manipulation of this pathway for control of alloimmune responses but the conflicting data using 3B3 indicates great complexity within this system. Careful studies to more clearly evaluate the impact of TIM-1 signaling and blockade will be required before any potential therapeutic agent can be taken to clinical studies.
Recent studies of TIM-3 in allograft rejection have demonstrated its ability to broadly modulate alloimmune responses. Blockade of TIM-3:galectin-9 signaling using an anti-TIM-3 antibody (RMT 3-23), leads to accelerated rejection of cardiac allografts(27). This is characterised by increased donor-specific alloantibody production, increased Th1 and Th17 polarization, and suppression of adaptive Treg induction. These immunoregulatory functions of TIM-3 were dependent upon the presence of CD4+ T cells(27).
TIM-3:galectin-9 interactions also play an important role in Treg function. Galectin-9 expression on T cells is limited solely to Tregs(37, 38). TIM-3 blockade abrogates induced peripheral tolerance and causes significant attenuation of suppressive activity of natural Tregs leading to increased auto- and alloimmune responses including autoantibody production(37-40). Furthermore, the generation of allospecific regulatory T cells during tolerance induction has been demonstrated to be dependent upon intact TIM-3:galectin-9 pathway(38).
Administration of exogenous stable galectin-9 has been used both as a tool to investigate the role of the TIM-3:galectin-9 pathway in vivo but also as an agent to manipulate alloimmune responses. In a transplantation model, administration of exogenous stable galectin-9 in both murine skin and cardiac transplant recipients led to a prolonged allograft survival(41, 42). This was characterised by diminished Th1 and Th17 cytokine production and promotion of regulatory T cells(25, 42). These studies used models of both acute and chronic allograft rejection, but all show consistent evidence of Th1 and Th17 suppression by TIM-3:galectin-9 interaction. These data suggest that targeting this pathway pharmacologically holds significant promise in the management of detrimental alloresponses(41-43).
TIM MOLECULES AS MARKERS OF ALLOGRAFT INJURY
KIM-1 (TIM-1) has been widely studied as a biomarker of acute kidney injury and its potential applications in the management of renal transplant recipients are an active area of investigation.
Several studies have examined the utility of KIM-1 in the prediction of delayed graft function (DGF). Intragraft KIM-1 correlates with donor GFR and creatinine at the time of organ procurement(44). Similarly, in the setting of donation after brain death, urinary KIM-1 is increased and a correlation has been reported between KIM-1 on day of transplant and creatinine at one-year follow up(45). However, in several larger studies, urinary or intragraft KIM-1 was not predictive of either DGF or GFR at three months or one year post transplantation(44, 46).
KIM-1 shows greater promise as a biomarker during follow up of transplant recipients. In biopsy studies, KIM-1 is an extremely sensitive marker of tubular cell injury(47). 92% of transplant biopsy specimens showing acute cellular rejection were KIM-1 positive and there was a significant correlation between KIM-1 positivity and increase in serum creatinine. Intriguingly, 28% of protocol biopsies without detectable tubular injury on histologic examination also stained positive for KIM-1, indicating its sensitivity in detecting subtle allograft injury(47). Furthermore, two studies have related elevated urinary KIM-1 with increased risk of progressive allograft dysfunction(48, 49) The highest tertile of urinary KIM-1 had a hazard ratio 5.1 (95% CI 1.5-17.8) for graft loss at four-year follow up(48).
TIM-3 is under investigation as a biomarker of Th1 activation and rejection in transplant recipients. TIM-3 mRNA levels are significantly higher within rejecting allografts and a strong correlation has been found between intragraft TIM-3 and IFN-γ levels. Interestingly, although TIM-3 levels were elevated in all cases of rejection, episodes that were refractory to treatment showed lower levels of TIM-3, suggesting a link between lack of negative costimulatory signaling and poorer allograft outcomes(50).
In a study of 115 patients, quantitative analysis of urinary and peripheral blood TIM-3 mRNA proved extremely accurate in the differentiation of DGF with acute tubular necrosis versus DGF with acute rejection(51). Furthermore, patients with acute rejection show much higher urine TIM-3 mRNA levels than those with other causes of allograft dysfunction or non-rejecting controls(51, 52). While larger studies will be needed to confirm these findings, these data suggest potential utility of TIM-3 mRNA measurement as a non-invasive tool in the diagnosis of allograft dysfunction.
CONCLUSIONS
Widely distributed across cells of the innate and adaptive immune system, as well as in non-myeloid cell lineages, TIM family members are emerging as central modulators of numerous facets of the immune response. Their costimulatory role in determining T cell activation and influence on T helper cell differentiation make them attractive targets for manipulation of unwanted effector T cell responses.
Much remains to be understood regarding the pleiotropic role of TIM molecules in immunity. Their role in alloimmunity, particularly in more complex in vivo models, needs to be more fully characterised. Future studies are required to comprehensively characterize TIM receptor/ligand network, to understand the effects of immune activation on the expression and functions of TIM family members and to elucidate the role of TIM signaling in innate immunity.
Modulating TIM pathways through the use of blocking antibodies or soluble fusion proteins may be complicated by the fact that TIM-ligand interactions appear to be cell and tissue-specific, with potentially divergent roles. Likewise, the observation that TIM antibodies with differing binding affinities or binding epitopes leads to differential effects across T cell subsets represents a considerable practical challenge to exploitation of these pathways. Furthermore, the recognition of the role of these molecules as PS receptors, capable of modulating innate immunity adds an additional layer of complexity. Recent studies indicating that blockade of the PS receptor function of TIM-3 and TIM-4 leads to autoimmunity is of concern. These are important considerations in the development of therapeutic agents targeting complex functions of TIM molecules in clinical practice.
Despite these challenges, these exciting molecules hold great potential to further our understanding of alloimmunity. Thus far, murine models of transplantation have shown that interruption of TIM-1 signalling and promotion of TIM-3:galectin-9 signalling all lead to improved allograft outcomes. Although the data on the function of TIM-4 in transplantation is lacking at this point, studies of food allergy suggest that this pathway may play a role in antigen specific tolerance. Thus, along with other TIM family members, TIM-4 may potentially regulate alloimmune responses. Another interesting question is whether and how TIM signaling in non-T cells influences adaptive immune responses to alloantigens. Addressing these issues will create a new avenue of investigation in both experimental and clinical transplantation.
Figure 1. Proposed model of TIM-1 signaling in activated T cells.
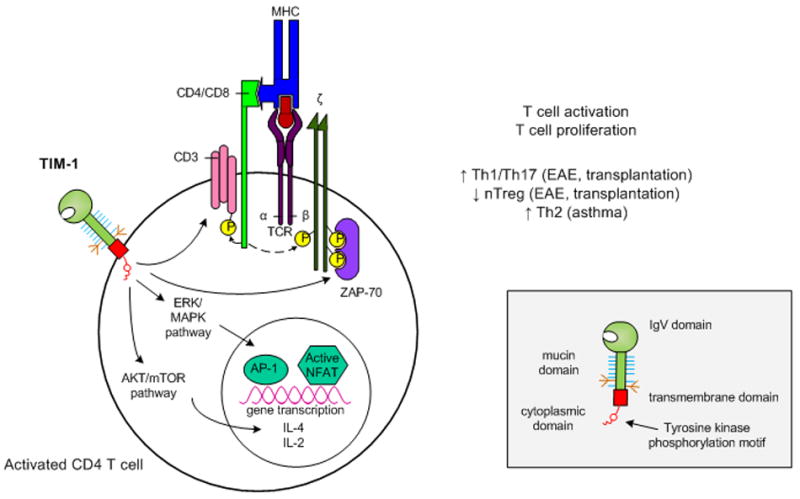
Upon ligation, tyrosine kinase residues in the TIM-1 cytoplasmic domain become phosphorylated. This leads to activation of Akt and ERK/MAPK which results in enhanced IL-2 and IL-4 transcription. In addition, TIM-1 ligation itself may be sufficient to initiate T cell activation through CD3 capping and increased phosphorylation of adaptor proteins associated with the TCR complex. Overall, TIM-1 signaling leads to enhanced T cell activation and proliferation, but the nature of the resulting T helper cell differentiation appears to be dependent on the susceptibility of the model system studied.
Inset: Domain structure of a TIM family member.
TIM members are composed of an IgV domain, mucin domain, transmembrane domain and cytoplasmic domain. Additionally, TIM-1 and TIM-3 possess tyrosine kinase phosphorylation motifs within their cytoplasmic domains.
Figure 2. Depiction of the costimulatory and phagocytic role ofTIM-4.
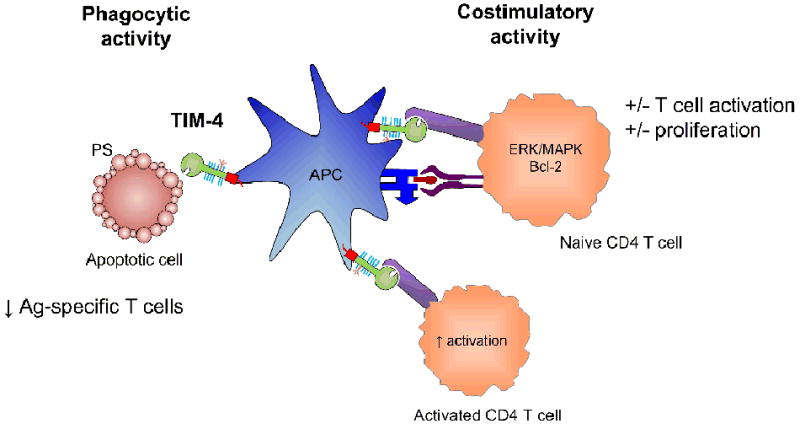
TIM-4 is expressed on antigen presenting cells. It is capable of phagocytosing apoptotic cells through the recognition of phosphatidylserine (PS) on the surface of these cells. Additionally, it has been shown to interact with unknown ligand(s) on naïve and activated T cells, and function as a costimulatory molecule. However, this role is less well defined, with reports of both activation and inhibition of T cell responses.
Figure 3. Mechanisms by which TIM-3 may modulate immune function.
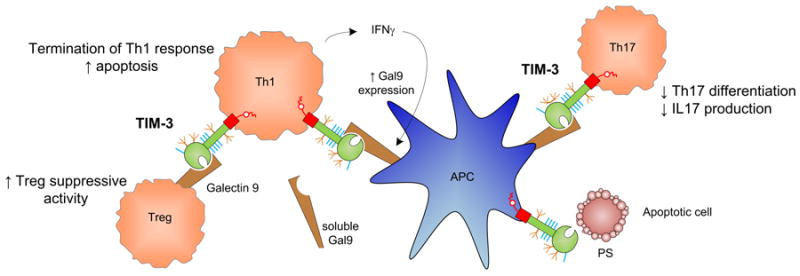
Interaction of TIM-3 on Th1 cells with its ligand galectin-9 (Gal-9) provides a negative costimulatory signal. Upon immune activation, Th1 cells produce IFNγ lead to upregulation of Gal-9 on APCs. Gal-9:TIM-3 signaling then promotes apoptosis and termination of Th1 responses. Their interaction on Th17 cells also inhibits Th17 differentiation and IL17 production. Furthermore, TIM-3:Gal-9 ligation increases the frequency of adaptive Tregs and may enhance Treg suppressive activity. Finally, TIM-3 has also been shown to be a phosphatidylserine receptor and is capable of recognizing apoptotic cells.
Acknowledgments
We are grateful to Ms. Melanie Yeung for technical assistance in preparing the figures. This work was supported by NIH grant R01AI070820.
Footnotes
DISCLOSURE: The authors of this manuscript have no conflicts of interest to disclose as described by the American Journal of Transplantation.
References
- 1.Freeman GJ, Casasnovas JM, Umetsu DT, DeKruyff RH. TIM genes: a family of cell surface phosphatidylserine receptors that regulate innate and adaptive immunity. Immunol Rev. May;235(1):172–89. doi: 10.1111/j.0105-2896.2010.00903.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ichimura T, Asseldonk EJ, Humphreys BD, Gunaratnam L, Duffield JS, Bonventre JV. Kidney injury molecule-1 is a phosphatidylserine receptor that confers a phagocytic phenotype on epithelial cells. J Clin Invest. 2008 May;118(5):1657–68. doi: 10.1172/JCI34487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Umetsu SE, Lee WL, McIntire JJ, Downey L, Sanjanwala B, Akbari O, et al. TIM-1 induces T cell activation and inhibits the development of peripheral tolerance. Nat Immunol. 2005 May;6(5):447–54. doi: 10.1038/ni1186. [DOI] [PubMed] [Google Scholar]
- 4.McIntire JJ, Umetsu SE, Akbari O, Potter M, Kuchroo VK, Barsh GS, et al. Identification of Tapr (an airway hyperreactivity regulatory locus) and the linked Tim gene family. Nat Immunol. 2001 Dec;2(12):1109–16. doi: 10.1038/ni739. [DOI] [PubMed] [Google Scholar]
- 5.Meyers JH, Chakravarti S, Schlesinger D, Illes Z, Waldner H, Umetsu SE, et al. TIM-4 is the ligand for TIM-1, and the TIM-1-TIM-4 interaction regulates T cell proliferation. Nat Immunol. 2005 May;6(5):455–64. doi: 10.1038/ni1185. [DOI] [PubMed] [Google Scholar]
- 6.Nakae S, Iikura M, Suto H, Akiba H, Umetsu DT, Dekruyff RH, et al. TIM-1 and TIM-3 enhancement of Th2 cytokine production by mast cells. Blood. 2007 Oct 1;110(7):2565–8. doi: 10.1182/blood-2006-11-058800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sizing ID, Bailly V, McCoon P, Chang W, Rao S, Pablo L, et al. Epitope-dependent effect of anti-murine TIM-1 monoclonal antibodies on T cell activity and lung immune responses. J Immunol. 2007 Feb 15;178(4):2249–61. doi: 10.4049/jimmunol.178.4.2249. [DOI] [PubMed] [Google Scholar]
- 8.Khademi M, Illes Z, Gielen AW, Marta M, Takazawa N, Baecher-Allan C, et al. T Cell Ig- and mucin-domain-containing molecule-3 (TIM-3) and TIM-1 molecules are differentially expressed on human Th1 and Th2 cells and in cerebrospinal fluid-derived mononuclear cells in multiple sclerosis. J Immunol. 2004 Jun 1;172(11):7169–76. doi: 10.4049/jimmunol.172.11.7169. [DOI] [PubMed] [Google Scholar]
- 9.Meyers JH, Sabatos CA, Chakravarti S, Kuchroo VK. The TIM gene family regulates autoimmune and allergic diseases. Trends Mol Med. 2005 Aug;11(8):362–9. doi: 10.1016/j.molmed.2005.06.008. [DOI] [PubMed] [Google Scholar]
- 10.De Souza AJ, Oriss TB, O’Malley KJ, Ray A, Kane LP. T cell Ig and mucin 1 (TIM-1) is expressed on in vivo-activated T cells and provides a costimulatory signal for T cell activation. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(47):17113–8. doi: 10.1073/pnas.0508643102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xiao S, Najafian N, Reddy J, Albin M, Zhu C, Jensen E, et al. Differential engagement of Tim-1 during activation can positively or negatively costimulate T cell expansion and effector function. J Exp Med. 2007 Jul 9;204(7):1691–702. doi: 10.1084/jem.20062498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rodriguez-Manzanet R, Meyers JH, Balasubramanian S, Slavik J, Kassam N, Dardalhon V, et al. TIM-4 expressed on APCs induces T cell expansion and survival. Journal of Immunology. 2008;180(7):4706–13. doi: 10.4049/jimmunol.180.7.4706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.De Souza AJ, Oak JS, Jordanhazy R, DeKruyff RH, Fruman DA, Kane LP. T cell Ig and mucin domain-1-mediated T cell activation requires recruitment and activation of phosphoinositide 3-kinase. Journal of Immunology. 2008;180(10):6518–26. doi: 10.4049/jimmunol.180.10.6518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mariat C, Degauque N, Balasubramanian S, Kenny J, DeKruyff RH, Umetsu DT, et al. Tim-1 signaling substitutes for conventional signal 1 and requires costimulation to induce T cell proliferation. J Immunol. 2009 Feb 1;182(3):1379–85. doi: 10.4049/jimmunol.182.3.1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Binne LL, Scott ML, Rennert PD. Human TIM-1 associates with the TCR complex and up-regulates T cell activation signals. Journal of Immunology. 2007;178(7):4342–50. doi: 10.4049/jimmunol.178.7.4342. [DOI] [PubMed] [Google Scholar]
- 16.Rodriguez-Manzanet R, DeKruyff R, Kuchroo VK, Umetsu DT. The costimulatory role of TIM molecules. Immunol Rev. 2009 May;229(1):259–70. doi: 10.1111/j.1600-065X.2009.00772.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kobayashi N, Karisola P, Pena-Cruz V, Dorfman DM, Jinushi M, Umetsu SE, et al. TIM-1 and TIM-4 Glycoproteins Bind Phosphatidylserine and Mediate Uptake of Apoptotic Cells. Immunity. 2007;27(6):927–40. doi: 10.1016/j.immuni.2007.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang PC, Xing Z, Berin CM, Soderholm JD, Feng BS, Wu L, et al. TIM-4 expressed by mucosal dendritic cells plays a critical role in food antigen-specific Th2 differentiation and intestinal allergy. Gastroenterology. 2007 Nov;133(5):1522–33. doi: 10.1053/j.gastro.2007.08.006. [DOI] [PubMed] [Google Scholar]
- 19.Feng BS, Chen X, He SH, Zheng PY, Foster J, Xing Z, et al. Disruption of T-cell immunoglobulin and mucin domain molecule (TIM)-1/TIM4 interaction as a therapeutic strategy in a dendritic cell-induced peanut allergy model. Journal of Allergy and Clinical Immunology. 2008;122(1):55–61. e7. doi: 10.1016/j.jaci.2008.04.036. [DOI] [PubMed] [Google Scholar]
- 20.Mizui M, Shikina T, Arase H, Suzuki K, Yasui T, Rennert PD, et al. Bimodal regulation of T cell-mediated immune responses by TIM-4. International Immunology. 2008;20(5):695–708. doi: 10.1093/intimm/dxn029. [DOI] [PubMed] [Google Scholar]
- 21.Zhu C, Anderson AC, Schubart A, Xiong H, Imitola J, Khoury SJ, et al. The Tim-3 ligand galectin-9 negatively regulates T helper type 1 immunity. Nat Immunol. 2005 Dec;6(12):1245–52. doi: 10.1038/ni1271. [DOI] [PubMed] [Google Scholar]
- 22.van de Weyer PS, Muehlfeit M, Klose C, Bonventre JV, Walz G, Kuehn EW. A highly conserved tyrosine of Tim-3 is phosphorylated upon stimulation by its ligand galectin-9. Biochem Biophys Res Commun. 2006 Dec 15;351(2):571–6. doi: 10.1016/j.bbrc.2006.10.079. [DOI] [PubMed] [Google Scholar]
- 23.Anderson AC, Anderson DE, Bregoli L, Hastings WD, Kassam N, Lei C, et al. Promotion of tissue inflammation by the immune receptor Tim-3 expressed on innate immune cells. Science. 2007 Nov 16;318(5853):1141–3. doi: 10.1126/science.1148536. [DOI] [PubMed] [Google Scholar]
- 24.Kashio Y, Nakamura K, Abedin MJ, Seki M, Nishi N, Yoshida N, et al. Galectin-9 induces apoptosis through the calcium-calpain-caspase-1 pathway. J Immunol. 2003 Apr 1;170(7):3631–6. doi: 10.4049/jimmunol.170.7.3631. [DOI] [PubMed] [Google Scholar]
- 25.Seki M, Oomizu S, Sakata KM, Sakata A, Arikawa T, Watanabe K, et al. Galectin-9 suppresses the generation of Th17, promotes the induction of regulatory T cells, and regulates experimental autoimmune arthritis. Clin Immunol. 2008 Apr;127(1):78–88. doi: 10.1016/j.clim.2008.01.006. [DOI] [PubMed] [Google Scholar]
- 26.Hastings WD, Anderson DE, Kassam N, Koguchi K, Greenfield EA, Kent SC, et al. TIM-3 is expressed on activated human CD4+ T cells and regulates Th1 and Th17 cytokines. Eur J Immunol. 2009 Sep;39(9):2492–501. doi: 10.1002/eji.200939274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Boenisch O, D’Addio F, Watanabe T, Elyaman W, Magee CN, Yeung MY, et al. TIM-3: a novel regulatory molecule of alloimmune activation. J Immunol. Nov 15;185(10):5806–19. doi: 10.4049/jimmunol.0903435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Frisancho-Kiss S, Nyland JF, Davis SE, Barrett MA, Gatewood SJ, Njoku DB, et al. Cutting edge: T cell Ig mucin-3 reduces inflammatory heart disease by increasing CTLA-4 during innate immunity. J Immunol. 2006 Jun 1;176(11):6411–5. doi: 10.4049/jimmunol.176.11.6411. [DOI] [PubMed] [Google Scholar]
- 29.Monney L, Sabatos CA, Gaglia JL, Ryu A, Waldner H, Chernova T, et al. Th1-specific cell surface protein Tim-3 regulates macrophage activation and severity of an autoimmune disease. Nature. 2002 Jan 31;415(6871):536–41. doi: 10.1038/415536a. [DOI] [PubMed] [Google Scholar]
- 30.Uchida Y, Ke B, Freitas MC, Yagita H, Akiba H, Busuttil RW, et al. T-Cell Immunoglobulin Mucin-3 Determines Severity of Liver Ischemia/Reperfusion Injury in Mice in a TLR4-Dependent Manner. Gastroenterology. Jul 13; doi: 10.1053/j.gastro.2010.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Albacker LA, Karisola P, Chang YJ, Umetsu SE, Zhou M, Akbari O, et al. TIM-4, a receptor for phosphatidylserine, controls adaptive immunity by regulating the removal of antigen-specific T cells. Journal of Immunology. 185(11):6839–49. doi: 10.4049/jimmunol.1001360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nakayama M, Akiba H, Takeda K, Kojima Y, Hashiguchi M, Azuma M, et al. Tim-3 mediates phagocytosis of apoptotic cells and cross-presentation. Blood. 2009 Apr 16;113(16):3821–30. doi: 10.1182/blood-2008-10-185884. [DOI] [PubMed] [Google Scholar]
- 33.Ueno T, Habicht A, Clarkson MR, Albin MJ, Yamaura K, Boenisch O, et al. The emerging role of T cell Ig mucin 1 in alloimmune responses in an experimental mouse transplant model. J Clin Invest. 2008 Feb;118(2):742–51. doi: 10.1172/JCI32451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yuan X, Ansari MJ, D’Addio F, Paez-Cortez J, Schmitt I, Donnarumma M, et al. Targeting Tim-1 to overcome resistance to transplantation tolerance mediated by CD8 T17 cells. Proc Natl Acad Sci U S A. 2009 Jun 30;106(26):10734–9. doi: 10.1073/pnas.0812538106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Degauque N, Mariat C, Kenny J, Zhang D, Gao W, Vu MD, et al. Immunostimulatory Tim-1-specific antibody deprograms Tregs and prevents transplant tolerance in mice. J Clin Invest. 2008 Feb;118(2):735–41. doi: 10.1172/JCI32562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee HH, Meyer EH, Goya S, Pichavant M, Kim HY, Bu X, et al. Apoptotic cells activate NKT cells through T cell Ig-like mucin-like-1 resulting in airway hyperreactivity. Journal of Immunology. 185(9):5225–35. doi: 10.4049/jimmunol.1001116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang F, Wan L, Zhang C, Zheng X, Li J, Chen ZK. Tim-3-Galectin-9 pathway involves the suppression induced by CD4+CD25+ regulatory T cells. Immunobiology. 2009;214(5):342–9. doi: 10.1016/j.imbio.2008.10.007. [DOI] [PubMed] [Google Scholar]
- 38.Sanchez-Fueyo A, Tian J, Picarella D, Domenig C, Zheng XX, Sabatos CA, et al. Tim-3 inhibits T helper type 1-mediated auto- and alloimmune responses and promotes immunological tolerance. Nat Immunol. 2003 Nov;4(11):1093–101. doi: 10.1038/ni987. [DOI] [PubMed] [Google Scholar]
- 39.Muthukumarana PA, Zheng XX, Rosengard BR, Strom TB, Metcalfe SM. In primed allo-tolerance, TIM-3-Ig rapidly suppresses TGFbeta, but has no immediate effect on Foxp3. Transpl Int. 2008 Jun;21(6):593–7. doi: 10.1111/j.1432-2277.2008.00654.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sabatos CA, Chakravarti S, Cha E, Schubart A, Sanchez-Fueyo A, Zheng XX, et al. Interaction of Tim-3 and Tim-3 ligand regulates T helper type 1 responses and induction of peripheral tolerance. Nat Immunol. 2003 Nov;4(11):1102–10. doi: 10.1038/ni988. [DOI] [PubMed] [Google Scholar]
- 41.Wang F, He W, Yuan J, Wu K, Zhou H, Zhang W, et al. Activation of Tim-3-Galectin-9 pathway improves survival of fully allogeneic skin grafts. Transpl Immunol. 2008 Apr;19(1):12–9. doi: 10.1016/j.trim.2008.01.008. [DOI] [PubMed] [Google Scholar]
- 42.He W, Fang Z, Wang F, Wu K, Xu Y, Zhou H, et al. Galectin-9 significantly prolongs the survival of fully mismatched cardiac allografts in mice. Transplantation. 2009 Sep 27;88(6):782–90. doi: 10.1097/TP.0b013e3181b47f25. [DOI] [PubMed] [Google Scholar]
- 43.Boenisch O, D’Addio F, Watanabe T, Elyaman W, Magee CN, Yeung MY, et al. TIM-3: A Novel Regulatory Molecule of Alloimmune Activation. J Immunol. Oct 18; doi: 10.4049/jimmunol.0903435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schroppel B, Kruger B, Walsh L, Yeung M, Harris S, Garrison K, et al. Tubular expression of KIM-1 does not predict delayed function after transplantation. J Am Soc Nephrol. Mar;21(3):536–42. doi: 10.1681/ASN.2009040390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nijboer WN, Schuurs TA, Damman J, van Goor H, Vaidya VS, van der Heide JJ, et al. Kidney injury molecule-1 is an early noninvasive indicator for donor brain death-induced injury prior to kidney transplantation. Am J Transplant. 2009 Aug;9(8):1752–9. doi: 10.1111/j.1600-6143.2009.02713.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hall IE, Yarlagadda SG, Coca SG, Wang Z, Doshi M, Devarajan P, et al. IL-18 and urinary NGAL predict dialysis and graft recovery after kidney transplantation. J Am Soc Nephrol. Jan;21(1):189–97. doi: 10.1681/ASN.2009030264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang J, Gu Y, Xu C, Qu X. Increased T cell immunoglobulin mucin-3 and its ligand in acquired aplastic anemia. Eur J Haematol. 2008 Aug;81(2):130–9. doi: 10.1111/j.1600-0609.2008.01095.x. [DOI] [PubMed] [Google Scholar]
- 48.van Timmeren MM, Vaidya VS, van Ree RM, Oterdoom LH, de Vries AP, Gans RO, et al. High urinary excretion of kidney injury molecule-1 is an independent predictor of graft loss in renal transplant recipients. Transplantation. 2007 Dec 27;84(12):1625–30. doi: 10.1097/01.tp.0000295982.78039.ef. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Szeto CC, Kwan BC, Lai KB, Lai FM, Chow KM, Wang G, et al. Urinary expression of kidney injury markers in renal transplant recipients. Clin J Am Soc Nephrol. Dec;5(12):2329–37. doi: 10.2215/CJN.01910310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ponciano VC, Renesto PG, Nogueira E, Rangel EB, Cenedeze MA, Franco MF, et al. Tim-3 expression in human kidney allografts. Transpl Immunol. 2007 Apr;17(3):215–22. doi: 10.1016/j.trim.2006.11.003. [DOI] [PubMed] [Google Scholar]
- 51.Manfro RC, Aquino-Dias EC, Joelsons G, Nogare AL, Carpio VN, Goncalves LF. Noninvasive Tim-3 messenger RNA evaluation in renal transplant recipients with graft dysfunction. Transplantation. 2008 Dec 27;86(12):1869–74. doi: 10.1097/TP.0b013e3181914246. [DOI] [PubMed] [Google Scholar]
- 52.Renesto PG, Ponciano VC, Cenedeze MA, Saraiva Camara NO, Pacheco-Silva A. High expression of Tim-3 mRNA in urinary cells from kidney transplant recipients with acute rejection. Am J Transplant. 2007 Jun;7(6):1661–5. doi: 10.1111/j.1600-6143.2007.01795.x. [DOI] [PubMed] [Google Scholar]