

Abstract
Reactive oxygen species (ROS) are involved in numerous physiological and pathophysiological responses. Increasing evidence implicates ROS as signaling molecules involved in the propagation of cellular pathways. The NADPH oxidase (Nox) family of enzymes is a major source of ROS in the cell and has been related to the progression of many diseases and even in environmental toxicity. The complexity of this family’s effects on cellular processes stems from the fact that there are 7 members, each with unique tissue distribution, cellular localization and expression. Nox proteins also differ in activation mechanisms and the major ROS detected as their product. To add to this complexity, mounting evidence suggests that other cellular oxidases or their products may be involved in Nox regulation. The overall redox and metabolic status of the cell, specifically the mitochondria, also has implications on ROS signaling. Signaling of such molecules as electrophillic fatty acids has impact on many redox sensitive pathologies, and thus, as anti-inflammatory molecules, contributes to the complexity of ROS regulation. The following review is based on the proceedings of a recent international Oxidase Signaling Symposium at the University of Pittsburgh’s Vascular Medicine Institute and Department of Pharmacology and Chemical Biology, and encompasses further interaction and discussion among the presenters.
Keywords: ROS, reactive oxygen species, hydrogen peroxide, superoxide, NADPH oxidase, Nox, mitochondria, lipooxygenase, myleoperoxidase, xanthine oxidase, heme oxygenase 1, arsenic, hypertension, fibrosis, electrophilic fatty acid
1. Introduction
In 1933, a “burst of extra respiration” was described in phagocytosing leukocytes [1]. This discovery identified what came to be known as the phagocytic oxidative or respiratory burst and triggered the beginning of reactive oxygen species (ROS) research. In 1961 hydrogen peroxide (H2O2) was identified as the product of this burst [2] and in 1973 it was demonstrated that the H2O2 is derived from the monovalent reduction of oxygen (O2) to form superoxide (O2•−) [3].
Since those discoveries, H2O2 and O2•− have been implicated in many physiological and pathophysiological processes including signal transduction, cell proliferation, gene expression, angiogenesis, sepsis, diabetes, heart disease, neurodegenerative diseases, pulmonary diseases, and cancer [4, 5] [6–10]. H2O2 and O2•− were also described as causative factors in aging as early as 1956 [11], initially as agents of oxidative damage. Over the last decade, H2O2 and O2•−, often encompassed by the more generic term ROS, have emerged as mediators of intracellular signaling [12–14]. The concomitant identifications of reactive nitrogen species (RNS) and the interaction between ROS and RNS, specifically nitric oxide (•NO) and O2•−, uncovered a new level of complexity in how these biological processes and pathological pathways are modulated. Furthermore, evidence is mounting on cross-talk and feed-forward interactions among the different cellular oxidases. The current short review derives from a recent international Oxidase Meeting at the University of Pittsburgh’s Vascular Medicine Institute and Department of Pharmacology and Chemical Biology, focused on understanding the enzymology, structure-function, and signalling properties of oxidases in health and disease. This review highlights the mechanisms by which NADPH oxidase (Nox)-derived ROS interact with products of other oxidases and peroxidases, and covers new cutting-edge research tools and approaches in the study of ROS.
2. Reactive Oxygen Species
ROS are oxidants derived from the metabolism of oxygen which encompass a broad range of biochemical interactions and chemical properties. They include O2•−, H2O2, hydroxyl radical (•OH), peroxyl radical (ROO•), alkoxyl radical (RO•), ozone (O3) and singlet oxygen (1O2). O2•− is the precursor of most ROS in the cell and is dismuted to H2O2, either through the action of a family of enzymes, the superoxide dismutases (SOD), or spontaneously. Major cellular sources of ROS are mitochondria, cytochrome P450 family proteins, xanthine oxidoreductase, uncoupled nitric oxide synthase (NOS), peroxisomes, cyclooxygenases, lipoxygenases and the Nox family of enzymes [4, 15–22]. Of these, only the Nox enzymes possess pathways for the regulated generation of H2O2 and O2•−. Recently, it has been suggested that mitochondria may be able to generate regulated levels of O2•− at different sites in the respiratory chain but this remains controversial [23].
ROS can induce oxidative damage through lipid peroxidation of cellular membranes, DNA strand breaks, and protein oxidation [10, 24–26]. However, under normal physiological conditions, an overall reducing environment exists in cells and a number of protective mechanisms including the amino acid cysteine, glutathione (GSH), thioredoxin (Trx), SOD and catalase, among others, scavenge ROS and maintain oxidative damage at a minimum [8, 24]. The current paradigm describes a more elaborate role of regulated levels of ROS, specifically from the NADPH oxidases and mitochondria, as modulators of intracellular signaling pathways including those of angiotensin II (Ang II) [27, 28], platelet-derived growth factor (PDGF) [29–31], tumor necrosis factor α (TNF-α) [32–34], thrombin [35, 36], interferon-γ [37], insulin [38], [39], and bacterial lipopolysaccharide (LPS) [40, 41], to name a few. Indeed, there is mounting evidence of redox sensitivity of a number of the major cell signaling molecules, especially phosphatidylinositol-3 kinase (PI3K), c-Jun N-terminal kinase (JNK), mitogen activated protein kinases (MAPKs), their upstream modulators such as apoptosis signal-regulating kinase-1 (Ask-1), phosphatases such as protein tyrosine phosphatase 1B (PTP1B) and Src homology 2-containing inositol phosphatase 2 (SHP-2), transcription factors such as nuclear factor κB (NF-κB) and activator protein 1 (AP-1) [10, 24, 31, 42] [8, 9, 43–49], and more recently the cellular hemoglobin neuroglobin [50]. The specificity of ROS action on different signaling pathways is expected to depend on (a) the nature of the ROS produced, its amount and localization; (b) the redox status in the immediate proximity of the pathway; (c) the pKa of reactive thiols and/or nucleophilic moieties in signaling molecules; and (d) the overall redox status of the cell as determined by the balance of redox molecules including glutathione/glutathione disulfide (GSH/GSSG), NADPH/NADP, and NADH/NAD [10].
ROS are best known for their ability to effect cellular signaling via modifications of thiols on cysteine residues, or thioethers of methionines, on signaling proteins. 21,000 – 42,000 of all the unique cysteine residues in the human genome can be readily oxidized [42]. Under conditions of normal cellular pH, cysteine residues surrounded in the tertiary structure by basic amino acids have their thiol (-SH) group de-protonated to thiolate ion (-S) [10]. Thiolate is highly susceptible to oxidation resulting in formation of mixed disulfide bonds (-SS-), on the same or neighboring molecules, sulfenic acid or sulfenate ion (-SOH or – SO−), sulfinic acid or sulfinate ion (-SO2H or SO2−), or sulfonic acid or sulfonate ion (-SO3H or SO3−). These modifications lead to structural changes in the target molecule, which can result in either activation or inactivation [31, 42]. For example, PTP1B is inactivated by oxidation at cysteine 215 and such inactivation is true of other phosphatases [51] [52, 53], whereas Ask-1 appears to be activated by oxidation at cysteine 250 among others, and this ROS mediated activation is also true for other kinases of the MAPK kinase pathways [54–56]. The degree to which these cysteine residues are oxidized determines the reversibility or irreversibility of the modification, with the sulfonic acid being least likely to be reversibly reduced. Moreover, •NO and peroxynitrite (ONOO-) can result in modified cysteine-nitrosothiol and nitrothiol. Sulfenic acid and nitroso-thiol modifications of proteins undergo further oxidation leading to other oxidation products and contributing to mixed disulfide formation and thus, protein adducts. The stability or instability of these modifications depends greatly on thioredoxin reductase, glutaredoxin and protein disulfide reductase activities, proximity, and the disulfide accessibility within the target molecule, all of which determine the steady state level of the modified thiol, and thereby, its signaling potential.
3. Oxidases and Peroxidases
Of the cellular sources of ROS mentioned above, Nox enzymes are unique in that either H2O2 or O2•− are detected as their primary and regulated products. In combination with their diverse localization in the cell and juxtapositions to specific signaling pathways, tight regulation of the activity of Nox enzymes appears to provide precise modulation of ROS levels to control ROS-dependent signaling events. In addition, cells have developed elaborate mechanisms by which ROS and metabolites of different oxidases and peroxidases can interact with and regulate each other, thus introducing another level of complexity to the signaling of ROS in health and disease. Following is an overview of Nox enzymes and some of the cellular oxidases and peroxidases involved in these processes.
a. NADPH oxidases
NADPH oxidase activity was first described in liver microsomal isolates and bacteria in 1963 [57, 58]. Around the same time, human chronic granulomatous disease (CGD), a rare condition first described in 1957 characterized by susceptibility to recurrent infection due to failure of phagocytic microbicidal activity, was linked to defective respiratory burst of what was then referred to as an “NADH” oxidase [59–62]. The substrate of the oxidase was later revealed to be NADPH rather than NADH. In the 1980s and early 1990s, subunits of the phagocyte NADPH oxidase were characterized and a model for phagocytic NADPH oxidase was developed [16, 63–67]. The membrane component of the phagocyte NADPH oxidase that serves as the electron transferase is flavocytochrome b558, a heterodimer composed of gp91phox, currently referred to as Nox2, and p22phox, another membrane-spanning subunit. Based on sequence homology with gp91phox, other members of the NADPH oxidase (Nox) family were identified and their expression characterized in many tissues, including vascular smooth muscle and endothelial cells, and fibroblasts. Moreover, their role in numerous pathological and physiological conditions including heart disease, atherosclerosis, rheumatoid arthritis, sepsis, asthma, cystic fibrosis, diabetes, immune diseases, neurodegenerative diseases and cancer, was demonstrated [68–71] [4, 15, 16, 20, 72–77].
Structure and Function
The Nox family is defined by seven distinct membrane-integrated catalytic subunits that form the basis of the enzyme, Nox1 to 5 and Duox1 and Duox2, which give the individual oxidases their names. Nox1 – 4 are associated with p22phox, which appears to be involved in stabilization of these Nox proteins and is essential for their proper membrane targeting and activity. Nox5, Duox1 and Duox2 do not require p22phox for their activity. The cytosolic regulatory subunits that are responsible for activation of the Nox1- through 3-based enzymes, but not Nox4 or 5, are p47phox (also called Nox organizer 2, or NoxO2) or its homolog NoxO1, p67phox (also called Nox activator 2, or NoxA2) or its homolog NoxA1, and p40phox. p47phox and p67phox are the canonical regulatory subunits involved in Nox2 activation, whereas NoxO1 and NoxA1 are implicated as the major cytosolic subunits involved in the activity of Nox1. Nox3 seems to be activated by association with NoxO1 alone (human Nox3), NoxO1 and NoxA1 (murine Nox3), p47phox and p67phox, or p67phox alone, but this remains controversial. The small Rho family GTP-binding proteins Rac2 and Rac1 are essential for the catalytic activity of Nox2 and Nox1, respectively, and evidence suggests a need for Rac1 in the Nox3 system as well. Rac1 has also been shown to be important for Nox2 activation in some cells, including endothelial cells. In addition, p40phox and the GTPase Rap1A contribute to activation of the complex. Upon initiation of Nox2 oxidase activation, for example, p47phox, p67phox and p40phox translocate en bloc to the membrane, with transfer regulated by conformational changes in p47phox secondary to its phosphorylation. Concomitant with this translocation, Rac2 redistributes from cytoplasm to the membrane and completes the assembly process. In a similar process, Nox1 (and Nox3) likewise rely on NoxO1 and NoxA1 in complex activation. The activities of Duox1 and 2 depend on their association with the two membrane-bound proteins DuoxA1 and DuoxA2, which are important for trafficking of Duoxes from the ER to the Golgi and their final localization to vesicular structures or to the plasma membrane (figure 1) [4, 70, 78–103].
Figure 1. Current models of the active Nox complexes.

Each of the 7 members of the Nox family requires a different set of conditions and protein associations. This figure shows the cytosolic subunits that are beleived to assemble onto each Nox isoform in the active oxidase state.
All Nox family members contain NADPH- and flavin adenine dinucleotide (FAD)-binding domains in their C-terminal cytosolic tails as well as predicted six conserved transmembrane domains containing two highly conserved heme-binding histidines on transmembrane domains III and V bound to two asymmetrical heme groups [4, 104]. FAD is constitutively bound to the enzyme while NADPH serves as its substrate (figure 2). Nox enzymes catalyze the transport of electrons across biological membranes from NADPH to O2 generating O2•− [105]. In addition, Nox5, which has 5 isoforms (α, β, γ, δ and ε), displays an intracellular N-terminus that contains a calmodulin-like domain with four Ca2+-binding EF-hand motifs in all its isoforms with the exception of NOX5ε[85, 96, 106]. Also, Duox1 and 2 possess a seventh transmembrane domain, an extracellular N-terminus with a peroxidase-like domain and two Ca2+-binding EF-hand domains similar to Nox5, rendering these isozymes, as well as Nox5, highly regulated by changes in intracellular Ca2+ levels (figure 3). As with other Nox proteins, Nox5 is also regulated by PKC as well as c-Abl [107].
Figure 2. Schematic representation of common Nox features.

The “Nox” portion of each of the Nox family members contains a similar “Nox” component. Shown here are the key features in common for all Nox family members. The letter designations A – E signify the cytosolic/extracellular loop domains. Shown also are the heme binding sites (Fe) and the FAD and NADPH binding domains.
Figure 3. Outline of the structure of Nox family members.

Shown are the structural elements of the different members of the Nox family. The transmembrane domains are numbered I to VII. Shown are the calcium binding motifs (EF hands) and the peroxidase homology domain of Duox1/2.
In the active enzyme, two electrons are transferred from NADPH to FAD reducing it to FADH2 catalyzed by unique binding sites on the inner membrane C-terminal tail [108]. An electron is then transferred to the inner heme, which transfers it to the outer heme and then to a bound O2 molecule on the external membrane, generating O2•− [92]. The second electron is transferred in the same way. Different members of the Nox family, however, vary in the major ROS detected from their activity. For Nox1 – 3 and Nox5 O2•− is readily detected, whereas mainly H2O2 is detected from Duox1, Duox2 or Nox4 [109–112]. This difference might be connected to differences in Nox4/Duox folding, leading to distinct formation and membrane insertion of the heterodimer with p22phox or DuoxA1/2, or to interaction with yet unknown proteins. Thus, rapid dismutation of O2•− to H2O2 as a result of these differences may preclude from detection of significant amounts of O2•− by currently available methods.
Expression
Cellular distribution and subcellular localization of different Nox isoforms play an important role in how individual Nox proteins regulate signaling. Nox2, the first catalytic subunit to be identified, is, in human cells, a heavily glycosylated protein that migrates as a broad band of 60 – 91 kDa on Western blots. It is noteworthy that in murine cells, Nox2 is less glycosylated and migrates as a 55 – 60 kDa protein on Western blots. Nox2 is expressed on membranes of neutrophils, macrophages, endothelial cells, some vascular smooth muscle cells, fibroblasts, skeletal muscle cells, cardiomyocytes and hepatocytes [4, 72, 80, 104, 113–117]. In myeloid cells, localization to the plasma membrane and/or phagosomal membrane is clear. While some evidence suggests that the same may be true for other tissue cells with respect to plasma membrane localization of this isoform, this remains controversial and other evidence suggests intracellular, perinuclear distribution in some cells [118]. Nox1, initially identified in colonic epithelial cells, is a major isoform in large artery SMC [119, 120], and is also expressed in endothelial cells, osteoclasts and reproductive organs [70, 121, 122], where it is localized intracellularly in the proximity of the endoplasmic reticulum (ER) but also on the plasma membrane associated with caveolae [123–125]. Nox3, discovered in 2000 [126], appears to be more limited in distribution, being highly expressed in the inner ear and in low levels in skull bone, brain and some fetal tissues [90]. Nox4, originally described in the kidney as Renox, has a more ubiquitous expression profile, including smooth muscle cells, fibroblasts, hematopoietic stem cells, osteoclasts, neurons, and endothelial cells [113, 123, 127–133]. Within the cell Nox4 has been detected near focal adhesions [123], at the endoplasmic reticulum [129], in the nucleus [134], and in the mitochondria [135, 136], depending on the cell type. Nox5, discovered in 2001, is found in bone marrow, lymph nodes, spleen, reproductive tissues, stomach and pancreas, in addition to some fetal tissues and exists in a truncated form (Nox5-S) in vascular smooth muscle and endothelial cells [85, 96, 137–139]. Duox1 and 2 are highly expressed in the thyroid and are also found in airway epithelia and the prostate. Duox2 is also found in the digestive system. Within thyroid and epithelial cells, these two enzymes appear to localize to the apical membrane, but there is also evidence for an intracellular localization in these cells [93, 94, 140, 141]. It should be noted that species differences do exist in the expression profiles of Nox proteins. Specifically, Nox5 is not expressed in rodents and Duox are not expressed in mouse airway epithelial cells. Table 1 summarizes the cellular distribution and subcellular localization of Nox family members.
Table 1.
Tissues Distribution and Subcellular Localization of Nox Proteins
| Nox Isoform | Cellular Distribution | Subcellular Localization | References |
|---|---|---|---|
| Nox1 | Colon epithelium, vascular smooth muscle cells, endothelial cells, osteoclasts, reproductive organs | Intracellular membranes close to ER, endosomes, signalosome, caveolae | [119, 120], [70, 121, 122], [123, 124], [125] |
| Nox2 | neutrophils, macrophages, endothelial cells, vascular smooth muscle cells, fibroblasts, skeletal muscle cells, cardiomyocytes and hepatocyte | Cell membrane, phagosome, perinuclear | [4, 72, 80, 104,113 – 118] |
| Nox3 | Inner ear (vestibular system, cochlea), skull, brain, fetal tissues | Plasma membrane | [82, 90, 317, 318] |
| Nox4 | Kidney, vascular smooth muscle cells, fibroblasts, hematopoietic stem cells, osteoclasts, neurons, endothelial cells | Focal adhesions, ER, nucleus, mitochondria | [113,123,127 – 133], [134] |
| Nox5 | vascular smooth muscle, endothelial cells, bone marrow, lymph nodes, spleen, reproductive tissues, stomach, pancreas, fetal tissues | Plasma membrane, ER | [85, 96, 107, 137 – 139, 319 – 321] |
| Duox1/2 | Thyroid, airway epithelia, prostate, digestive system (Duox2) | Apical membrane | [93, 94,140] |
b. Peroxidases
Ubiquitously expressed throughout both plant and animal kingdoms, peroxidases consume H2O2 and catalyze the one- or two-electron oxidation of a remarkably diverse array of substrates. Recently, the heme-containing oxidoreductases have been classified based on their evolutionary relationships into the peroxidase-cyclooxygenase superfamily [142], which includes those enzymes previously designated as the “animal peroxidases”, namely myeloperoxidase (MPO), eosinophil peroxidase (EPO), lactoperoxidase (LPO), and thyroid peroxidase (TPO). Post-translational modifications of biological substrates by the action of peroxidases in general, and by mammalian peroxidases in particular, support a wide variety of physiologic functions, ranging from TPO-dependent thyroid hormone synthesis to the generation of hypobromous acid by EPO from stimulated eosinophils. With the exception of TPO, the members of this protein superfamily figure prominently in innate immunity, with each mediating functions specially tailored to their tissue location.
c. Xanthine Oxidoreductase
Xanthine oxidoreductase (XOR) catalyzes the terminal two steps of purine degradation (hypoxanthine → xanthine → uric acid) in humans. XOR is transcribed as a single gene product, xanthine dehydrogenase (XDH) where substrate-derived electrons at the molybdenum cofactor (Mo-cofactor) are transferred via two Fe/S centers to a FAD-cofactor where NAD+ is reduced to NADH. During inflammatory conditions, post-translational modification by oxidation of critical cysteine residues or limited proteolysis converts XDH to xanthine oxidase (XO) [143, 144]. The key difference distinguishing XO from XDH is the structural conformation and electrostatic microenvironment surrounding the FAD-cofactor resulting in lower affinity for NAD+ and enhancement of affinity for O2 [145]. Substrate-derived electrons at the Mo-cofactor of XO reduce O2 at the FAD-cofactor both divalently, forming H2O2 and univalently, generating O2•−. However, conversion to XO is not requisite for O2•− and H2O2 production, as XDH displays partial oxidase activity under conditions in which NAD+ levels are diminished, such as the ischemic/hypoxic microenvironment encountered in vascular inflammation [146]. This same inflammatory milieu leads to enhanced XO levels and thus increased XO-derived ROS formation resulting in activation of redox-dependent cell signaling reactions and alterations in vascular function. Evidence of this role for XO is exemplified by numerous studies in which XO inhibition attenuates symptoms of vascular disease including congestive heart failure, sickle cell anemia and diabetes [147–150].
d. Heme Oxygenase
Heme oxygenase (HO) catalyzes the degradation of Heme b (Fe-protoporphyrin-IX) to form tetrapyrrole biliverdin-IXα. Biliverdin-IXα is subsequently converted to bilirubin-IXα by a NADPH-dependent reductase [151]. HO enzymatic activity releases the heme core iron in the ferrous form Fe(II) and carbon monoxide (CO), a small gaseous molecule that is relatively stable in biological systems [151, 152]. Expression of HO-1, an inducible HO isoform, is usually undetectable under basal conditions, with the exception of spleen [153], the site of erythrocyte hemoglobin turnover, but responds to rapid transcriptional activation in response to biological stress, like LPS, NO, PDGF, Ang II, hypoxia and ROS. Besides its role in heme degradation, HO-1 also functions as an inducible stress protein with tissue and cell protective effects in numerous models of cellular stress and organ pathology [154]. These protective effects have been largely attributed to the production of CO, biliverdin and bilirubin.
e. Mitochondria
The mitochondrion is the site of oxidative phosphorylation, a process by which electrons are transferred through the mitochondrial respiratory chain, leading to the reduction of each protein complex, until the electrons are ultimately accepted by oxygen, yielding water (figure 4). This process is coupled to the production of ATP at complex V to generate 50–95% of cellular energy (depending on cell type). The majority of electrons entering the electron transport chain ultimately are accepted by oxygen to form water in a process that does not yield free radicals. However, it is estimated that under basal conditions approximately 1–3% of electrons at respiratory complexes distal to cytochrome c oxidase (complex IV) do not make it through the chain and instead can catalyze the univalent reduction of O2 to O2•−. Initially, this was considered to be a “leak” of electrons from the respiratory chain since it was induced by pathological conditions such as hyperoxia. Recently, it has been suggested that under physiological conditions this can be a regulated process involved in cell signaling and cellular responses to stress. Indeed, hypoxia, nitric oxide, and the PI3-kinase pathway all contribute to regulation of mitochondrial ROS [155–158]. The major points of O2•− production from the respiratory chain are complex I (NADH dehydrogenase), and complex III (ubiquinone-cytochrome c reductase). O2•− formation occurs at multiple flavin and heme sites of single electron reactions that exist within these multi-subunit complexes [17, 159, 160]. Notably, the majority of O2•− generated by the electron chain is rapidly dismuted to H2O2 by manganese SOD (MnSOD) localized within the mitochondrial matrix. O2•− generation at complexes I and III is regulated by the rate of respiration, with slower respiration leading to higher rates of ROS generation due to increased reduction of the chain. Interestingly, generation of ROS at different segments of the respiratory chain results in differential oxidative modification of the mitochondrial proteome [161]. Additionally, physiological regulators of respiration, such as •NO or CO, also modulate mitochondrial ROS generation [158]. This type of modulation has been implicated in the adaptive signaling observed in response to hypoxia [162]. In contrast, dysregulation of mitochondrial ROS generation has also been shown to contribute to the progression of pathologies such as ischemia/reperfusion injury and sepsis [163].
Figure 4. The mitochondrial electron transport chain.

This simplified diagram of the electron transport chain on the inner mitochondrial membrane shows the direction of electron (e−) flow along the chain (black arrows) and the direction of flow (red arrows) of hydrogen ion (H+) across the mitochondrial membrane. Dotted arrows show ROS production as a result of the electron leak. UQ refers to ubiquinone; Cyt C refers to cytochrome c.
4. Functional Distinctions Among Nox Isoforms: Heterodimerization and the “Nox” Domain
As mentioned above, the catalytic subunits Nox1 – 4 associate with p22phox and exist as integral membrane proteins. Heterodimer association is essential for Nox maturation, protein stabilization and function of these isoforms. Similarly, while p22phox does not associate with Duox proteins, heterodimerization with DuoxA1 and DuoxA2 is required for processing, subcellular localization and catalytic activity of Duox1 and Duox2, respectively [141, 164]. Not only do Nox isoforms differ in the presence of- and partners for- heterodimer formation, their dependence for activity on the different cytosolic subunits varies widely among members of this family [103]. The activities of Nox1 – 3/p22phox dimers require stimulus-mediated association with p47phox, p67phox, p40phox, NoxO1 and/or NoxA1, Rac activity and multiple phosphorylation events to form an active enzyme. In contrast to this spatial modulation, the Nox4/p22phox complex appears to be constitutively active and transcriptionally, rather than spatially, regulated. Some evidence, however, suggests also a possible regulation by polymerase [DNA-directed] delta-interacting protein 2 (poldip2) [165], but poldip2 associates with p22phox rather than with Nox4. Along the same lines, Laurindo and coworkers provide evidence for a possible regulatory association of Nox1, Nox2, Nox4 and p22phox with protein disulfide isomerase, a redox chaperone and member of the thioredoxin family [166–168]. Nox5 and Duox enzymes are activated by agonist-induced increases in intracellular calcium in concert with various phosphorylations [169]. Finally, H2O2 is detected as the primary product from Duox1, Duox2 and Nox4 rather than O2•− as is the case with the other isoforms. All these factors will have important implications with respect to cellular signaling responses initiated by different Nox isoforms.
None of the Nox protein family members have been crystallized, but the putative structure of the “Nox” domain contains a cytosolic N-terminus (for Nox1 – 5 but not Duox 1 – 2), followed by six transmembrane α-helices (seven in the case of Duox 1 – 2) that are separated by three extracellular (A, C and E loops) and two intracellular loops (B and D loops). The cytosolic C-terminal region harbors the essential binding sites for FAD and NADPH, and may facilitate electron transfer via the heme coordination center embedded in the 3rd and 5th transmembrane domains by directly interacting with the B loop (figure 2) [170]. In the absence of a three-dimensional “Nox” structure, progress in understanding structural determinants of Nox function has relied on homology modeling, chimeric and mutagenesis studies, and analysis of naturally occurring Nox2 mutants identified in chronic granulomatous disease [171, 172]. These studies revealed that polybasic regions of the B loop are essential and unique for the catalytic activity of Nox2 and Nox4 [112, 170, 173]. Although replaceable among Nox family members, the D loop may control the electron transfer from FAD to O2 in Nox2 [112, 174]. Detailed understanding of the mechanism by which the active conformation of “Nox” is created is incomplete. The constitutive activity of the Nox4-p22phox complex in absence of any regulatory subunits, however, may provide important insights. For example, in depth mutagenesis studies have identified structural elements that are important either for Nox4 catalytic activity or for permitting assembly of the Nox4-p22phox complex. Cell-free assays and transfected cell models reveal that the dehydrogenase domain located in the Nox4 C terminus, as well as the D loop, are linked to an intrinsically activated state and increased ROS production [175, 176]. Furthermore, despite not confirming higher ROS output upon expression of Nox4-Nox2 chimeras, two independent studies in which the complete C terminal region of Nox4 was replaced with the analogous Nox2 domain succeeded in transforming the constitutively active Nox4 into an inducible, phorbol ester-stimulated Nox that requires the presence of regulatory subunits [112, 175]. Studying the differences in these regions between different Nox isoforms may contribute to understanding the conformational changes that occur in the “Nox” domain leading to activation and how this activation differs from one isoform to the other.
As mentioned above, to date, Nox4-p22phox heterodimerization is the only known prerequisite for Nox4 catalytic activity [109]. The determinants for the formation of the Nox4-p22phox complex differ from those for the association of Nox1 – 3 with p22phox. Truncation of the p22phox C terminus or a mutation in the last of the presumed four p22phox transmembrane domains (p22phox Y121H) does not affect Nox4-p22phox heterodimerization, Nox4 maturation or ROS generation, whereas Nox1 – 3 function is reduced under these conditions [82, 177–179]. In addition, the Nox4 D loop is critically involved in creating a functional complex with p22phox [112]. Although at this point speculative, formation of a unique quaternary structure might be key to constitutive Nox4 activity.
Similarly, as only H2O2 is detected in the extracellular milieu when Duox enzymes are stimulated, distinctive features in their heterodimer assembly with DuoxA subunits may drive this phenomenon. Indeed, DuoxA isoforms, and potentially also p22phox and other proteins associating to these subunits, are not only crucial for Duox (or Nox) maturation and formation of a catalytically functional complex, but also seem to play a role in intracellular targeting of the assembled oxidase [141]. Thus, a combination of structural and sequential motifs, together with the geometry of the heterodimer and the utilization of regulatory proteins, provide for individual functional dynamics and localization of Nox/Duox family members.
5. How Nox Isoforms Signal in Cells/Tissues: Compartmentalization
Most cells express multiple Nox isoforms, and in many cells, each Nox is linked to distinct cellular functions. This immediately raises the question of how enzymes that make identical products (O2•− and H2O2) can mediate different responses. The answer is most likely multifactorial, related to how their expression and activity is regulated, their subcellular localization, and the precise ratio of ROS species produced. For example, in vascular cells, as discussed above, Nox1 is found in caveolae [123] and signalosomes [125], is activated by growth-promoting stimuli such as Ang II, thrombin, prostaglandin F2α and PDGF [76, 180–182], and produces more detectable intracellular O2•− than H2O2 [183]. In endothelial cells, Nox2 is similarly localized and growth factor-sensitive [184, 185] but is also found in the perinuclear region [118]. Of interest, in these cells, Nox-derived O2•− is produced both extracellularly and intracellularly [186, 187]. In contrast, Nox4 is regulated by many factors including shear stress, ER stress, mitochondrial dysfunction, hypoxia, growth factors, Ang II and transforming growth factor (TGF)-β [132, 188–191] [192–194], and H2O2 is mainly detected as its product [109].
As a consequence of these differences in subcellular localization, each Nox is expected to activate specific, context-dependent signaling pathways in individual cell types via localized production of ROS. Interestingly, however, neither O2•− nor H2O2 are particularly chemically reactive. This allows for the possibility of their signal being effectively transduced by other regulatory proteins in the immediate vicinity of the compartment in which they are produced. For example, these proteins could be iron-containing proteins which activate H2O2 or a particularly reactive protein thiol. In vascular smooth muscle cells stimulated to grow, Nox1 activates tyrosine kinase pathways downstream of Ras that include Src, the EGF receptor kinase, Akt and variably, the MAPKs [27, 195–198]. In endothelial cells, similar growth-related, anti-apoptotic pathways are regulated by Nox2 rather than by Nox1 [199]. Although not all the direct targets of Nox-derived ROS are known, some of this stimulatory effect is due to inactivation of lipid or tyrosine phosphatases [200]. Another example of compartmentalized signaling occurs when cells are exposed to pro-inflammatory cytokines, such as TNF-α or IL-1β, or microbial agonists such as LPS. In such situations, endosomal Nox1 in smooth muscle cells or Nox2 in endothelial and cardiac cells stimulates NF-κB, leading to transcription of genes encoding pro-inflammatory proteins [125, 201, 202]. Moreover, the role of Nox proteins in signaling can vary according to the phenotype of the cells. Nox4 is involved in maintaining smooth muscle differentiation [203], but is also involved in the proliferation response to urokinase plasminogen activator [204]. In endothelial cells, endoplasmic reticular Nox4-mediated activation of protein tyrosine phosphatase-1B terminates epidermal growth factor-induced signaling [205], but Nox4 regulates Erk1/2 to promote proliferation in response to serum [199]. Nox4 has also been linked to multiple signaling pathways such as AP-1, Jak/Stat, E2F, pigment epithelium-derived factor (PEDF), PKCα and p38 MAPK [192–194, 206]. Finally, Nox proteins can cooperate to modulate a given physiological response. Redox signaling in migrating smooth muscle cells is comprised of Nox1-mediated actin polymerization in lamellipodia by virtue of its activation of the cofilin phosphatase slingshot-1L phosphatase [207] and of Nox4-regulated focal adhesion turnover [165]. Thus, recognition of the cellular context is essential to an appreciation of Nox-mediated signal transduction; one must consider the cell type, the functional endpoint, the complement of Nox proteins expressed in the cell, and the tissue environment.
6. Hypertension and Nox1, Nox4 and Nox5
a. Oxidative Stress and Human Hypertension
Compelling findings from experimental and animal studies suggest a causative role for oxidative stress in the pathogenesis of hypertension and a critical role for ROS in cardiovascular remodeling and hypertension-associated target organ damage [208, 209]. Almost all experimental models of hypertension display some form of oxidative excess. Sources of ROS in experimental hypertension include Nox1, Nox2 and Nox4, xanthine oxidase, uncoupled NOS and mitochondria [5, 210]. Mice deficient in ROS-generating enzymes have lower blood pressure (BP) and Ang II dependent hypertension compared with wild-type counterparts [211, 212]. Moreover in experimental hypertension, antioxidants and Nox inhibitors attenuate vascular remodeling and the development of hypertension [213, 214].
Despite extensive data supporting a role for oxidative stress in experimental hypertension, it is unclear whether oxidative stress causes hypertension in humans, and only a few small clinical studies demonstrate a blood pressure-lowering effect of anti-oxidants, with most large anti-oxidant clinical trials failing to demonstrate any cardiovascular benefit or reduction in blood pressure [215–218]. On the other hand, NADPH oxidase polymorphisms and decreased antioxidant capacity are associated with hypertension and cardiovascular disease [219, 220]. Normotensive subjects with a family history of hypertension have greater plasma H2O2 levels than blood pressure – matched normotensive subjects without a family history of hypertension [216]. While most of these studies show correlations between high BP and markers of oxidative stress, a causal relationship is yet to be established. Nevertheless, evidence does suggest that ROS do play a critical role in the molecular mechanisms associated with cardiovascular and renal injury in hypertension, and that hypertension itself can trigger to oxidative stress [221].
A number of reasons for the inconsistency of clinical findings and inconclusivity of large trial data have been proposed [222, 223]. Importantly, traditional antioxidant therapies may have failed due to slow reactivity and/or bioavailability. Theoretically, agents that reduce ROS formation in a Nox-specific manner should be more efficacious than non-specific, inefficient antioxidant vitamin scavengers, as used in most clinical trials [224]. Indeed, specific Nox inhibitors as therapeutic agents are currently under development for potential clinical use. Inhibiting Nox enzymes by targeting their substrate, NADPH [225], may be another theoretically interesting strategy that would involve the use inhibitors of glucose-6-phosphate dehydrogenase, a major source of NADPH. However, since affecting the hexose monophosphate shunt may have widespread off-target deleterious effects, this approach would pose significant challenges.
b. Nox4 and Nox5 in the Vascular System
Although there has been enormous progress in the biochemical, chemical and structural characterization of Nox isoforms, the physiological role of individual Nox proteins still remains largely unknown and the exact function in pathological processes awaits clarification. One of the key areas where the biology of the Nox enzymes has been developed is the cardiovascular system where Nox1, 2, 4 and 5 are the isoforms expressed. Whereas Nox1 and 2 have been well studied in the context of vascular biology, less is known about vascular Nox4 and Nox5.
Nox4 shares only 39% homology with Nox2 and 35% homology with Nox1 [109, 192], and as mentioned above, it is constitutively active. Its localization to the endoplasmic reticulum (ER) suggests it may be involved in ER stress [167, 205] and its association with stress fibers and focal adhesions signifies its role in cell differentiation and migration [165, 203]. The biological roles of vascular Nox4 in health and disease are unclear and data are conflicting. On the one hand, Nox4 may be pro-angiogenic, anti-apoptotic and important in VSMC differentiation; and, on the other hand, it is involved in cell senescence, apoptosis, VSMC migration, and VSMC hypertrophy [226–228]. The conflicting results from these studies have not been resolved by in vivo evidence. To date, very few studies reported on Nox4−/− mice. In one study, Nox4 was shown to be important in stroke, since Nox4-deficient mice were protected against oxidative stress and neurodegeneration post acute ischemic stroke [229]. In contrast, another study on Nox4−/− mice and transgenic mice with cardiomyocyte-targeted overexpression of Nox4 revealed a protective role of Nox4 in animals subjected to chronic pressure overload [230].
Nox5, the newest member of the Nox family [96], has close homology to Nox1 and Nox2. Since it is not expressed in rodents, the study of Nox5 is challenging as mouse or rat models cannot be easily used to examine its role in vascular disease. Thus, the functional significance of vascular Nox5 is unknown, although it has been implicated in endothelial cell proliferation and angiogenesis, in PDGF-induced proliferation of VSMCs and in oxidative damage in atherosclerosis [231, 232]. Vascular Nox5 is activated by thrombin, PDGF, Ang II, ET-1 and ionomycin through PKC and cAMP response element-binding protein (CREB) [233, 234]. Thus, although our knowledge of many of the features of Nox4 and Nox5 has increased recently, there is still a paucity of information regarding the role of these oxidases in vascular biology.
7. Lung Fibrosis and NADPH Oxidase
Fibrosis of the lung is typically the result of an ongoing reparative response to injury of the alveolar epithelium and/or capillary endothelium due to injurious agents that are airborne or bloodborne, respectively. Many acute injuries, such as seen in adult respiratory distress syndrome (ARDS), resolve without significant fibrosis, whereas, chronic inorganic/organic dust exposures can result in fibrosis, as seen in asbestosis and hypersensitivity pneumonitis [235]. A particularly enigmatic form of lung fibrosis is idiopathic pulmonary fibrosis (IPF), a disease of the elderly that has not been linked to any single etiological agent. IPF carries a high mortality rate with median survival rates less than 3 years [236], and there are currently no effective therapies that have been shown to influence survival.
Oxidative stress has been implicated in the pathogenesis of IPF [237]. Although precise sources of ROS/RNS and mechanisms remain unclear, enzymatic sources of ROS that have been implicated in lung fibrosis are Nox2 and Nox4. The evidence for Nox2 in lung fibrosis comes primarily from animal models that show protection from lung injury induced by bleomycin [238] or carbon nanotubes [239] in mice with a genetic deficiency of Nox2. Studies in human subjects with IPF are limited; however, some studies have demonstrated enhanced generation of ROS from alveolar inflammatory cells, primarily macrophages, in promoting injury to alveolar epithelial cells [240, 241].
A role for Nox4 in lung fibrosis has been defined more recently. The mRNA expression of Nox4 was found to be induced by the pro-fibrotic cytokine, transforming growth factor-β1 (TGF-β1), while other Nox/Duox isoforms were unaffected [242]. Nox4 mediates myofibroblast differentiation, contractility, and extracellular matrix production in lung fibroblasts [242], and is associated with fibroblastic foci within alveolar-interstitial and vascular remodeling in human IPF lung tissues [242]. RNAi-mediated knockdown of Nox4 protects against development of lung fibrosis in the two different murine models of lung injury [242]. Another more recent study by Amara et al. [243] demonstrates that lung fibroblasts express Nox4 in situ in the fibrotic IPF lung and that Nox4 is upregulated in IPF lung tissue-derived fibroblasts. There is also emerging evidence that Nox4 may be involved in the vascular remodeling associated with IPF [244–246]. These studies suggest that targeting of Nox4 may be an effective therapeutic strategy for IPF and potentially for hypoxia-induced vascular remodeling that leads to pulmonary hypertension [247].
8. Endothelial NADPH Oxidase in Arsenic Signaling: An Example of Environmental Vascular Pathogenesis
Among other environmental and nutritional insults, arsenic in the drinking water is a prototypical environmental hazard. It is a major worldwide public health concern, since exposure increases risks of cardiovascular and metabolic-related diseases, as well as a number of cancers. Arsenic directly affects the vasculature by enhancing angiogenesis and promoting vessel occlusion and atherogenesis [248]. However, the mechanisms that initiate arsenic signaling for pathogenic changes in the vasculature remain unclear. There is consensus from a number of reports that arsenic stimulates ROS production in vascular cells and that arsenic stimulation of Nox enzymes promotes both signaling events and toxicities in vitro [248–250]. Toxicity or vascular dysfunction may arise from generation of both H2O2 and peroxynitrite as arsenic stimulates both oxidant and reactive nitrogen formation [250–252]. Whereas cell culture-based studies provided a wealth of information regarding potential mechanisms for arsenic action through Nox, proof of functional consequence of this activation in arsenic-induced pathogenesis in vivo and identification of the mechanism through which arsenic activates the oxidase are lacking.
To address functional consequence in vivo, the role of Nox-derived arsenic stimulation of liver sinusoidal endothelial cell (LSEC) capillarization was investigated in genetic mouse models [253]. LSEC are critical for bulk removal of circulating lipids, lipoproteins, and modified proteins by the liver. Loss of these functions in capillarization, in LSEC differentiation, in aging and in oxidative injury may promote metabolic imbalance and age-related atherogenesis. Mice exposed to low levels (10–250 ppb) of arsenic (as arsenite) in drinking water have reduced LSEC fenestrations, as well as decreased protein and lipids scavenging by the liver. Capillarization and increased sinusoidal protein S-nitrosation are absent in arsenic-exposed p47phox null mice, indicating that Nox2-based oxidase activity is required for both events [253]. Ex vivo studies with primary LSEC isolates confirm the role of Nox2 in arsenic-stimulated O2•− generation, as this stimulation is inhibited by the Nox2 inhibitor, Nox2ds-tat peptide [253]. Furthermore, ex vivo studies with LSEC isolated from the p47phox null mice demonstrate that H2O2 is the primary oxidant signaling for defenestration [253]. Although these studies were the first in vivo confirmation that Nox2 mediates arsenic pathogenesis in the vasculature and that Nox2-derived oxidants promote capillarization, they did not identify the mechanism for arsenic stimulation of the enzyme complex. In vitro and in vivo studies indicate that arsenic causes Rac1 membrane mobilization [249, 254], and inhibition of Rac1 GTPase activity prevents arsenic-stimulated ex vivo LSEC capillarization [253]. In human microvascular lung endothelial cells (HMVEC) or LSEC treated with Pertussis toxin, arsenic fails to activate Rac1 GTPase activity or promote capillarization, respectively [255]. These results implicate a Gαi-protein coupled receptor in the activation pathway. A specific antagonist to the sphingosine-1-phosphate type 1 receptor (S1PR1) prevents arsenic-stimulated capillarization and O2•− generation in LSEC, as well as arsenic-stimulated increases in Rac1-GTPase activity, angiogenic gene expression, and in vitro tube formation in HMVEC. The primary role of S1PR1 in the actions of arsenite was confirmed with siRNA knockdown of the receptor in HMVECs [255]. These data support a mechanism in which arsenic co-opts signaling through S1P1 receptors to stimulate Rac1 and Nox2 activities required for the pathogenic ROS generation that underlies enhanced angiogenesis, vascular remodeling and endothelial dysfunction (e.g. loss of LSEC scavenging and increased peroxynitrite formation). These pathogenic changes may contribute to the etiology of both hepatic and systemic vascular diseases that result from environmental exposure to arsenic (figure 5).
Figure 5. Arsenic-stimulated capillarization of fenestrated liver sinusoidal microvessels.
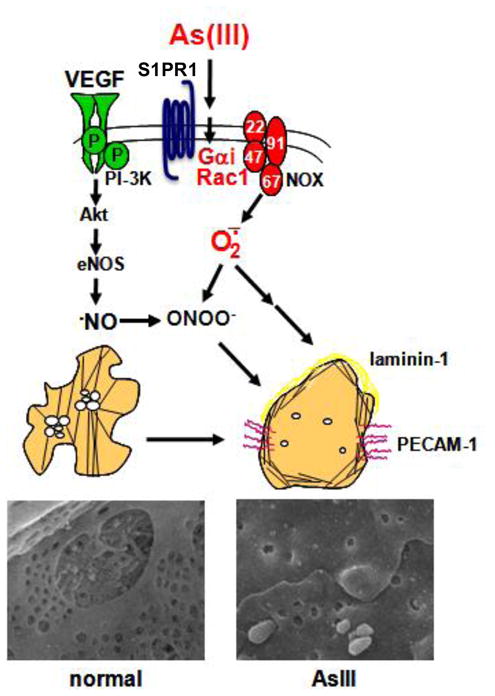
A model for the proposed mechanism of arsenic (As) action via Nox2 to induce phenotypic changes. SEM images are of liver sinusoids from mice drinking normal water or water containing 100 ppb arsenite for two weeks (modified from [322]).
9. Examples of Oxidase/Peroxidase Interaction
a. Airway Epithelia: NADPH Oxidase and the Action of Peroxidases in Host Defense
Polymorphonuclear neutrophils (PMN) constitute the dominant circulating cell in humans and, as phagocytic cells, contribute to the immediate innate immune response to infection. Once ingested by PMN, an invading microbe is compartmentalized in a phagosome, where it is exposed simultaneously to oxidants generated de novo by the NADPH oxidase [103] and to proteins stored in granules that fuse with the nascent phagosome [256]. The combined actions of the granule proteins and the varied oxidants synergize to create an environment toxic to many microbes and culminate, when successful, in the death and degradation of ingested prey (reviewed in [257]). PMN contain relatively large amounts of MPO, ~ 5% of the cell by weight [258] and predicted to reach millimolar concentrations in the restricted space of the phagosome, where the volume is estimated to be ~ 1.2 femtoliters [259]. MPO is structurally and functionally unique in the peroxidase-cyclooxygenase superfamily in having three, rather than two, covalent bonds between the heme group and the protein backbone and, consequently, in catalyzing the two-electron oxidation of Cl− at physiologic pH to generate HOCl− (reviewed in [260]). The chloride concentration in human PMN phagosomes is 73.3 ± 12.2 mM [261], with chloride delivered, in part, from the cytoplasm via the cystic fibrosis transmembrane conductance regulator (CFTR) [262]. Interestingly, CFTR is present in the secretory vesicles but not the plasma membrane of unstimulated human neutrophils [263]; during phagocytosis, secretory vesicles fuse with the nascent phagosome and thereby deliver CFTR to the membrane of the nascent phagosome. Patients with cystic fibrosis lack CFTR and their neutrophils exhibit depressed phagosomal chloride concentrations, defective chlorination of ingested targets, and depressed bacterial killing [263]. The HOCl produced by stimulated normal PMN represents as much as 72–90% of the oxygen consumed by the phagocyte NADPH oxidase [259, 264], demonstrating the extent to which phagocyte-generated oxidants supply HOCl to the antimicrobial machinery in the phagosomes. Given that HOCl targets vulnerable substrates regardless of their origin, there is extensive chlorination of host as well as microbial proteins present in the phagosome [265]. Overall, chlorination of bacteria directly correlates with the loss of microbial viability [265, 266], although identification of the critical targets that are modified remains the subject of study.
The principles of the peroxidase paradigm, exemplified by events in PMN phagosomes, apply to features of host defense in the airway. Airway epithelia constitutively produce H2O2 through the activity of Duox [267–269]. Goblet cells along the airway secrete LPO, which constitutes ~1% of the soluble protein in the airway lumen, thereby providing the peroxidase necessary to reduce the Duox-derived oxidants [270–272]. Airway epithelial cells also transport thiocyanate into the airway lumen, where OSCN− is generated and mediates bacteriostatic and bactericidal activities [268, 273]. Just as the combined activities of the phagocyte NADPH oxidase and MPO generate HOCl in phagosomes, Duox-derived H2O2 and LPO produce HOSCN in the human airway. The LPO-H2O2-OSCN system, earlier identified as the basis for the antimicrobial activity of saliva [274], oxidizes exposed and susceptible sulfhydryls to sulfenic acid and sulfenyl thiocyanite products, thereby compromising bacterial metabolism and viability [275].
Although the MPO- and LPO-mediated systems parallel each other in principle, significant differences exist. HOCl is a potent oxidant; for example, the k2 for HOCl reaction with free cysteines is 3.0 × 107 M−1 s−1 vs. 2.9 M−1 s−1 for H2O2 at pH 7.4 [276–278]. In contrast, HOSCN is a relatively weak oxidant, reacts primarily with sulfhydryls, and promotes modifications that are reversible in the absence of continuous SCN− or in the presence of reductants [275]. As a consequence of its nonselective reactivity, HOCl can mediate irreversible damage to targets, including both those on host and microbial proteins. HOSCN, on the other hand, because of its lower reactivity and more limited spectrum of susceptible substrates, can mediate more selective and reversible changes. In some cellular systems, the ED50 of HOSCN for host tissue is ~ 10 times higher than that for microbes, although it should not be assumed that the oxidant is uniformly nontoxic to tissues. HOSCN can oxidize intracellular thiols, thereby inhibiting thiol-dependent enzymes and compromising key metabolic activities and cell viability [[279, 280] and reviewed in [281]]. With that caveat in mind, there are data suggesting that SCN may blunt MPO-dependent cytotoxicity towards host cells and thereby serve as a protective agent against the oxidant damage in the airway of patients with excess inflammation, such as seen in patients with cystic fibrosis [282]. Several mechanisms may provide relative protection to host tissue in an inflamed airway where both the LPO- and MPO-dependent systems coexist. As LPO consumes H2O2, there will be less available to MPO for HOCl production. Low concentrations of SCN− compete effectively with Cl− for reaction with MPO, and concentrations above 100 μM decrease HOCl production and MPO-mediated cytotoxicity [282]. Lastly, SCN− directly and rapidly reduces OCl− in solution [283], thereby consuming the potentially damaging oxidant. Taken together, these reactions may provide a means by which innocent bystander damage in the airway due to HOCl produced by activated PMN recruited into the airway can be attenuated.
b. Vascular Xanthine Oxidoreductase: A Molecule in Need of Restraint or Nitrite?
Xanthine oxidase (XO) is a critical source of reactive oxygen species contributing to vascular inflammation. However, reports of vascular production of ROS frequently assume XO as the O2•−-producing form of xanthine dehydrogenase (XDH) and, if mentioned, H2O2 is merely a secondary byproduct of spontaneous or enzymatic dismutation of O2•−. A crucial concept often overlooked is that XO-mediated O2•− production is directly proportional to O2 concentration and pH. 100% O2•− production is achieved only at conditions of 100% O2 saturation and pH 10 [284]. Under physiological conditions (21% O2 and pH 7.4), however, XO-catalyzed O2 reduction results in 72% of the product being H2O2 and only 28% being O2•−. Moreover, under hypoxic conditions (1% O2 and pH 7.4), XO-generated O2•− is decreased to 10% and H2O2 formation is increased to 90% [284, 285]. This O2-dependent effect on the relative proportions of H2O2 and O2•− generated by XO is enhanced ~30% when XO is immobilized on endothelial glycosaminoglycans (GAGs) resulting in further diminution of XO-derived O2•− formation [285]. Therefore, under inflammatory conditions where both vascular O2 and pH are significantly decreased while XO is released into the circulation and bound to endothelial GAGs, XO production of O2•− is greatly diminished, resulting in almost exclusive production of H2O2. The impact of these findings is exemplified in myograph studies where perfusion with 95% O2 is standard procedure. In this severely hyperoxic setting, O2•− formation is favored producing results and conclusions that overshadow the role of XO-derived H2O2. This is critical as H2O2 differs greatly from O2•− with regard to mobility and reactivity. For example, the negative charge on O2•− limits its ability to traverse lipid bilayers as well as react with DNA whereas can H2O2 freely diffuse through membranes where it can participate in a myriad of both autocrine and paracrine cellular signaling events [286]. In toto, this new perspective of XOR biochemistry demands a more critical evaluation of XO-derived ROS with attention focused on H2O2 under clinically-relevant O2 conditions.
Contrary to its deleterious effects as a source of ROS, emerging evidence has revealed a nitrite reductase activity of XO and, as such, identified XO as a source of •NO under hypoxic/inflammatory conditions. For example, under strictly anoxic conditions, using xanthine or NADH as reducing substrate, XO catalyzes the single electron reduction of NO2− to •NO at the molybdenum cofactor [287–289]. This evidence suggests a potentially beneficial role of XO as an •NO donor. However, the following major issues must be critically evaluated before an XOR-dependent mechanism for •NO generation from NO2− can be determined to be significant in vivo:
1) substrate concentration-reports of XOR-mediated •NO formation utilize excessive concentrations (1–50 mM) of NO2-, while tissue levels of NO2− rarely rise above 300 nM [290],
2) O2 concentration-all reports to date demonstrate XOR-driven •NO formation under anoxic conditions, while these conditions rarely exist for any appreciable time in arteries and arterioles,
3) allopurinol-identification of XOR-catalyzed •NO formation in tissues has been accomplished solely by inhibition with allopurinol without verification with alternative XOR-specific inhibitors, such as febuxostat. If XOR-mediated •NO production is functionally significant and beneficial, then inhibition of this process should produce undesirable outcomes. This is in contrast to numerous studies reporting positive effects of XOR inhibition with allopurinol under similar conditions. Thus, while it appears that XOR can mediate the reduction of NO2− to •NO in vitro, the in vivo significance of this finding is uncertain at present. Therefore, studies to delineate the relative impact of XOR-dependent generation of ROS vs. the potential beneficial effects of XOR-catalyzed •NO formation are crucial, and the interplay between these potential products and Nox activity would present a very interesting area for investigation (figure 6).
Figure 6. Scheme of xanthine oxidase-derived reactive intermediates.

Xanthine oxidase (XO) may produce ROS or •NO thereby contributing to either tissue dysfunction or protection, or both.
c. Regulation of Vascular Nox by Heme Oxygenase and Carbon Monoxide
A major product of heme oxygenase is CO, which acts as a heme iron ligand and forms complexes with hemoproteins and metalloenzymes. At low concentrations, CO has profound effects on intracellular signaling processes, including anti-inflammatory, antiproliferative, antiapoptotic and anticoagulative effects [154]. Indeed, studies have shown that CO may inhibit NADPH oxidase activity in lung smooth muscle cells [291], neutrophils [292] and macrophages [293]. Recently it was demonstrated that induction of HO-1 expression in rat aortic smooth muscle cells inhibits PDGF-induced migration [294], a process integral to the development of atherosclerosis and restenosis [295]. CO, but neither biliverdin nor bilirubin, inhibits Nox1-dependent PDGF-stimulated migration in RASMC by directly interacting with the enzyme. Inhibition of Nox1 activity by CO correlates with decreased phosphorylation of Akt, and the MAPKs p38, Erk1/2, and JNK1. Taken together, these studies reveal a novel mechanism by which increased HO-1 expression and activity, and HO-1-derived CO may mediate their beneficial effects in arterial inflammation and injury [294].
10. New Concepts in Studying ROS in Cardiovascular and Lung Disease: Mitochondrial reserve capacity, bioenergetics and therapeutic implications
Mitochondria are not only a source of ROS, but also represent a major target for ROS within the cell. For example, mitochondrial DNA is known to be damaged prior to nuclear DNA by cellular oxidative stress [296]. Additionally, multiple studies associate oxidative stress with changes in bioenergetics, and this mechanism is thought to underlie the pathogenesis of a number of diseases, including ischemia/reperfusion injury and cardiac failure [297, 298]. The importance of understanding the relationship between oxidative stress and mitochondrial bioenergetics is further highlighted by studies suggesting that specific isoforms of Nox may be localized within or in close proximity to the mitochondrion [135, 136]. Traditional studies of the effects of oxidative stress on mitochondrial function have focused on examining modifications of individual proteins within the mitochondria. However, with the advent of new technology, it is now possible to study bioenergetics as a whole in intact cells. Emerging from this new line of study is the concept that mitochondrial bioenergetic reserve capacity is important in regulating cellular response to oxidative stress [299]. In basal physiological conditions, cells respire at a rate that is approximately 20–50% of their maximal capacity. Reserve capacity of mitochondria is defined as the difference between the maximal and basal rates of respiration. Emerging data demonstrate that the amount of mitochondrial reserve capacity determines the response of a given cell to oxidative insults, such as the electrophilic lipid 4-hydroxynonenol or hydrogen peroxide [300]. Interestingly, oxidative and nitrosative stresses deplete the mitochondrial reserve capacity of the cell, and thereby decrease its ability to respond to stress [299]. The mitochondrion is now emerging as a center in which ROS-signaling can be integrated with cellular metabolism and the program leading to cell differentiation or death through apoptotic mechanisms. A recent concept that has emerged is that specific oxidation of the phospholipid cardiolipin by cytochrome c exposed to hydrogen peroxide can modulate the apoptotic process [301, 302]. Clearly, the basal bioenergetic status and extent of the mitochondrial reserve capacity are likely determinants of whether cells initiate a ROS-dependent proliferation or cell death signaling. Interestingly, aging and chronic inflammatory conditions generate ROS/RNS that deplete the reserve capacity which has been proposed as an underlying contributory factor to the bioenergetic defects associated with cardiovascular and neurodegenerative diseases.
11. Nitrated Fatty Acids: New Anti-Inflammatory Signaling Mediators
Electrophilic fatty acids mediate post-translational protein modifications (PTPMs) and serve as potent anti-inflammatory cell signaling mediators. Two classes of electrophilic fatty acids, nitro- and keto-derived fatty acids, have recently been identified. Electrophilic fatty acid oxidation products (EFOX) are α, β-unsaturated keto derivatives derived from omega-3 fatty acids and generated by a cyclooxygenase-2 (COX-2)-catalyzed mechanism in activated macrophages [303]. Electrophilic nitro-fatty acid (NO2-FA) derivatives are abundant bioactive oxides of nitrogen in blood and tissues. These electrophilic NO2-FAs are formed by oxidative inflammatory reactions that involve redox reactions of •NO and nitrite [304]. More recent data reveal that these electrophilic fatty acids can also be generated under mild acidic conditions in the presence of nitrite and unsaturated fatty acids (in preparation). Both classes of electrophilic fatty acids react with nucleophilic targets and thereby alter protein structure, function and cellular distribution through PTPMs.
The PTPMs induced by electrophilic fatty acids modulate the expression of mRNA and proteins under the regulation of Nrf2/Keap1 [305], NF-κB [306] and PPARγ [307]. Both EFOX and NO2-FA derivatives activate phase II gene transcription through the Nrf2/Keap1 pathway. These electrophilic derivatives target highly reactive cysteines on Keap1, thereby liberating Nrf2 and resulting in nuclear translocation, binding to antioxidant response elements (ARE) and activation of phase II genes such as HO-1 [308–311]. Additionally, EFOX and NO2-FAs inhibit LPS-induced NF-κB activation through similar electrophilic interactions. The dimeric interaction between p50 and p65 subunits of NF-κB promotes the progression of inflammation. The electrophilic fatty acid derivatives inhibit NF-κB activity by alkylating the p65 subunit. This electrophilic alkylation prevents NF-κB binding to promoter regions of inflammatory genes and thus exerts potent anti-inflammatory effects [303, 306]. Both EFOX and NO2-FA derivatives bind to a highly conserved cysteine residue in the PPARγ ligand-binding domain and thereby transactivate PPAR-responsive genes [303, 312]. These electrophilic fatty acid signaling mediators regulate metabolism and inflammation through PPAR activation [304, 311]. The clinically-relevant manifestations of the anti-inflammatory actions of these electrophilic nitro- and keto-derived fatty acid derivatives have recently been described using several murine models of disease. The functional outcomes of these electrophilic fatty acids have been shown to reduce atherosclerosis in an apoE−/− model [313], inhibit neointima formation in a model of restenosis [314], mediate protective effects in a murine model of focal cardiac ischemia and reperfusion injury [315] and normalize blood glucose levels in ob/ob mice in a model of type 2 diabetes [316]. These findings are summarized in figure 7.
Figure 7. Electrophilic fatty acids are potent anti-inflammatory cell signaling mediators.
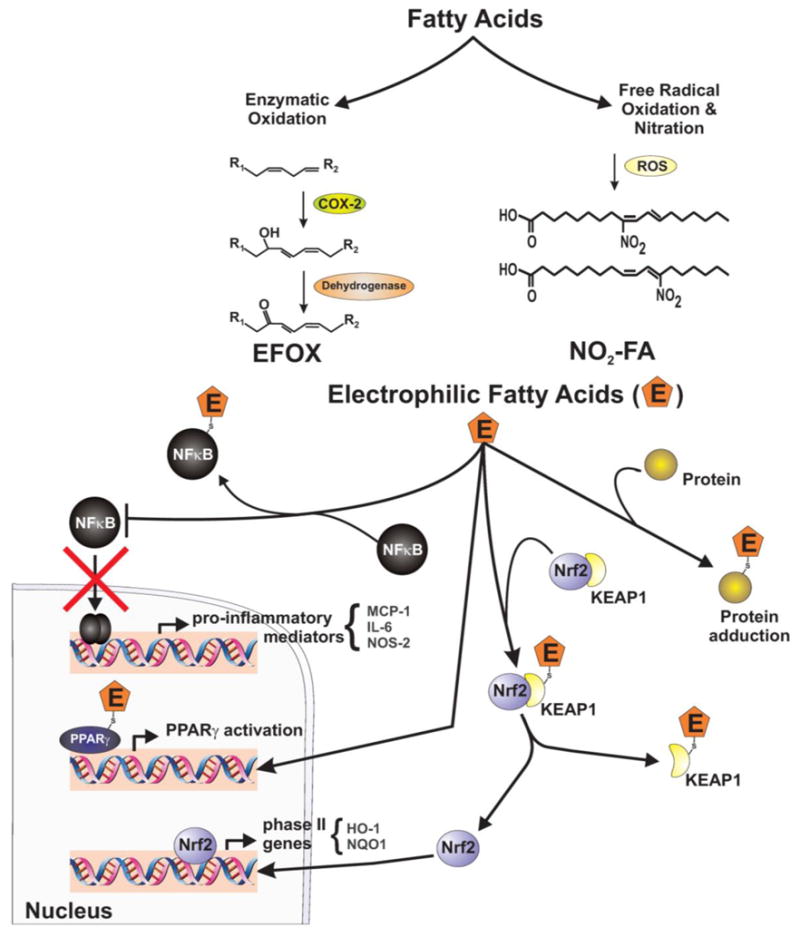
They are generated from fatty acids through enzymatic and non-enzymatic pathways, and modulate transcription factors, regulating inflammation and metabolism. Electrophilic fatty acids 1) adduct to specific cysteine residues on NFκB, resulting in inhibition of pro-inflammatory cytokine expression; 2) bind to PPARγ and transactivate downstream responsive genes; and 3) adduct to highly reactive cysteines on KEAP1 releasing Nrf2, causing its nuclear translocation, binding to ARE and activation of phase II genes.
12. Summary and Future Directions
An emerging principle in cellular responses to various stimuli is the cross-talk between what initially were thought to be distinct signaling pathways. Oxidases are no exception. The different sources, localization and temporal patterns of oxidases’ actions determine the final outcome in both physiological and pathophysiological conditions. Indeed, oxidases, especially Nox proteins, are involved in a plethora of pathophysiological conditions. We have highlighted only a few of these conditions here, showing that different Nox isoforms signal differently, and that they play an important role in hypertension, lung fibrosis and arsenic poisoning. Importantly, the roles of Nox proteins in different tissues are influenced by the activity of other oxidases and peroxidases, such as myeloperoxidase, heme oxygenase and xanthine oxidase. New discoveries on mitochondrial bioenergetic profiles and reserve capacity help in enriching our understanding of the role of mitochondria in regulation of cellular redox status, possibly providing the opportunity for the study of more elaborate regulation mechanisms between mitochondria and Nox proteins. Furthermore, the emergence of electrophilic fatty acids as anti-inflammatory agents with therapeutic implications and the advent of new approaches to regulating oxidases will further our knowledge of their actions and interactions, and more importantly, will pave the way for the development of novel therapeutic strategies. Technological advances in measurement of oxidase activity have contributed and will continue to contribute to this goal.
Acknowledgments
Dr. Pagano receives support from NIH R01HL079207 and is an Established Investigator of the American Heart Association. Dr. Gladwin receives research support from NIH grants R01HL098032, RO1HL096973, RC1DK085852. All Vascular Medicine Institute investigators receive support from the Institute for Transfusion Medicine and the Hemophilia Center of Western Pennsylvania.
14. List of abbreviations
- ROS
Reactive oxygen species
- Nox
NADPH oxidase
- O2•−
Superoxide
- H2O2
Hydrogen peroxide
- RNS
Reactive nitrogen species
- •NO
Nitric oxide
- •OH
Hydroxyl radical
- ROO•
Peroxyl radical
- RO•
Alkoxyl radical
- O3
Ozone
- 1O2
Singlet oxygen
- SOD
Superoxide dismutase
- NOS
Nitric oxide synthase
- GSH
Glutathione
- Trx
Thioredoxin
- AngII
Angiotensin II
- PDGF
Platelet-derived growth factor
- TNF-α
Tumor necrosis factor-α
- LPS
Lipopolysaccharide
- PI3K
Phosphatidylinositol-3 kinase
- JNK
c-Jun N-terminal kinase
- MAPK
Mitogen activated protein kinase
- Ask-1
Apoptosis signal-regulating kinase-1
- PTP1B
Protein tyrosine phosphatase 1B
- SHP-2
Src homology 2-containing inositol phosphatase 2
- NF-κB
Nuclear factor κB
- AP-1
Activator protein 1
- CGD
Chronic granulomatous disease
- FAD
Flavine adenine dinuculeotide
- MPO
Myeloperoxidase
- EPO
Eosinophil peroxidase
- LPO
Lactoperoxidase
- TPO
Thyroid peroxidase
- XOR
Xanthine oxidoreductase
- XDH
Xanthine dehydrogenase
- XO
Xanthine oxidase
- HO-1
Heme oxygenase 1
- CO
Carbon monoxide
- poldip2
Polymerase [DNA-directed] delta-interacting protein 2
- TGF-β/1
Transforming growth factor-β/1
- PEDF
Pigment epithelium-derived factor
- CREB
cAMP response element-binding protein
- ARDS
Adult respiratory distress syndrome
- IPF
Idiopathic pulmonary fibrosis
- S1PR1
Sphingosine-1-phosphate type 1 receptor
- EFOX
Electrophilic fatty acid oxidation products
- COX-2
Cyclooxygenase-2
- NO2-FA
Nitro-fatty acid
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Baldridge CW, Gerard RW. THE EXTRA RESPIRATION OF PHAGOCYTOSIS. AJP - Legacy. 1932;103:235–236. [Google Scholar]
- 2.Iyer GYN, Islam MF, Quastel H. Biochemical Aspects of Phagocytosis. Nature. 1961:535–541. [Google Scholar]
- 3.Babior BM, Kipnes RS, Curnutte JT. Biological defense mechanisms. The production by leukocytes of superoxide, a potential bactericidal agent. J Clin Invest. 1973;52:741–744. doi: 10.1172/JCI107236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bedard K, Krause KH. The NOX family of ROS–generating NADPH oxidases: physiology and pathophysiology. Physiol Rev. 2007;87:245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- 5.Lassegue B, Griendling KK. NADPH oxidases: functions and pathologies in the vasculature. Arterioscler Thromb Vasc Biol. 30:653–661. doi: 10.1161/ATVBAHA.108.181610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Trachootham D, Alexandre J, Huang P. Targeting cancer cells by ROS-mediated mechanisms: a radical therapeutic approach? Nat Rev Drug Discov. 2009;8:579–591. doi: 10.1038/nrd2803. [DOI] [PubMed] [Google Scholar]
- 7.Cabello CM, Bair WB, 3rd, Wondrak GT. Experimental therapeutics: targeting the redox Achilles heel of cancer. Curr Opin Investig Drugs. 2007;8:1022–1037. [PubMed] [Google Scholar]
- 8.Droge W. Free radicals in the physiological control of cell function. Physiol Rev. 2002;82:47–95. doi: 10.1152/physrev.00018.2001. [DOI] [PubMed] [Google Scholar]
- 9.Thannickal VJ, Fanburg BL. Reactive oxygen species in cell signalling. Am J Physiol Lung Cell Mol Physiol. 2000;279:L1005–L1028. doi: 10.1152/ajplung.2000.279.6.L1005. [DOI] [PubMed] [Google Scholar]
- 10.Liu H, Colavitti R, Rovira II, Finkel T. Redox-dependent transcriptional regulation. Circ Res. 2005;97:967–974. doi: 10.1161/01.RES.0000188210.72062.10. [DOI] [PubMed] [Google Scholar]
- 11.Harman D. Aging: a theory based on free radical and radiation chemistry. J Gerontol. 1956;11:298–300. doi: 10.1093/geronj/11.3.298. [DOI] [PubMed] [Google Scholar]
- 12.Muller FL, Lustgarten MS, Jang Y, Richardson A, Van RH. Trends in oxidative aging theories. Free Radic Biol Med. 2007;43:477–503. doi: 10.1016/j.freeradbiomed.2007.03.034. [DOI] [PubMed] [Google Scholar]
- 13.Finkel T. Oxygen radicals and signaling. Curr Opin Cell Biol. 1998;10:248–253. doi: 10.1016/s0955-0674(98)80147-6. [DOI] [PubMed] [Google Scholar]
- 14.D’Autreaux B, Toledano MB. ROS as signalling molecules: mechanisms that generate specificity in ROS homeostasis. Nat Rev Mol Cell Biol. 2007;8:813–824. doi: 10.1038/nrm2256. [DOI] [PubMed] [Google Scholar]
- 15.Lassegue B, Clempus RE. Vascular NAD(P)H oxidases: specific features, expression, and regulation. Am J Physiol Regul Integr Comp Physiol. 2003;285:R277–R297. doi: 10.1152/ajpregu.00758.2002. [DOI] [PubMed] [Google Scholar]
- 16.Quinn MT, Gauss KA. Structure and regulation of the neutrophil respiratory burst oxidase: comparison with nonphagocyte oxidases. J Leukoc Biol. 2004;76:760–781. doi: 10.1189/jlb.0404216. [DOI] [PubMed] [Google Scholar]
- 17.Turrens JF. Mitochondrial formation of reactive oxygen species. J Physiol. 2003;552:335–344. doi: 10.1113/jphysiol.2003.049478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Turrens JF. Superoxide production by the mitochondrial respiratory chain. Biosci Rep. 1997;17:3–8. doi: 10.1023/a:1027374931887. [DOI] [PubMed] [Google Scholar]
- 19.White CR, Darley-Usmar V, Berrington WR, McAdams M, Gore JZ, Thompson JA, Parks DA, Tarpey MM, Freeman BA. Circulating plasma xanthine oxidase contributes to vascular dysfunction in hypercholesterolemic rabbits. Proceedings of the National Academy of Sciences. 1996;93:8745–8749. doi: 10.1073/pnas.93.16.8745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Geiszt M, Leto TL. The Nox family of NAD(P)H oxidases: host defense and beyond. J Biol Chem. 2004;279:51715–51718. doi: 10.1074/jbc.R400024200. [DOI] [PubMed] [Google Scholar]
- 21.Leto TL, Geiszt M. Role of Nox family NADPH oxidases in host defense. Antioxid Redox Signal. 2006;8:1549–1561. doi: 10.1089/ars.2006.8.1549. [DOI] [PubMed] [Google Scholar]
- 22.Takeya R, Sumimoto H. Regulation of novel superoxide-producing NAD(P)H oxidases. Antioxid Redox Signal. 2006;8:1523–1532. doi: 10.1089/ars.2006.8.1523. [DOI] [PubMed] [Google Scholar]
- 23.Murphy E, Bers D, Rizzuto R. Mitochondria: from basic biology to cardiovascular disease. J Mol Cell Cardiol. 2009;46:765–766. doi: 10.1016/j.yjmcc.2009.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Magder S. Reactive oxygen species: toxic molecules or spark of life? Crit Care. 2006;10:208. doi: 10.1186/cc3992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Salvemini D, Cuzzocrea S. Oxidative stress in septic shock and disseminated intravascular coagulation. Free Radic Biol Med. 2002;33:1173–1185. doi: 10.1016/s0891-5849(02)00961-9. [DOI] [PubMed] [Google Scholar]
- 26.Halliwell B. The role of oxygen radicals in human disease, with particular reference to the vascular system. Haemostasis. 1993;23(Suppl 1):118–126. doi: 10.1159/000216921. [DOI] [PubMed] [Google Scholar]
- 27.Ushio-Fukai M, Alexander RW, Akers M, Yin Q, Fujio Y, Walsh K, Griendling KK. Reactive oxygen species mediate the activation of Akt/protein kinase B by angiotensin II in vascular smooth muscle cells. J Biol Chem. 1999;274:22699–22704. doi: 10.1074/jbc.274.32.22699. [DOI] [PubMed] [Google Scholar]
- 28.Zafari AM, Ushio-Fukai M, Akers M, Yin Q, Shah A, Harrison DG, Taylor WR, Griendling KK. Role of NADH/NADPH oxidase-derived H 2 O 2 in angiotensin II-induced vascular hypertrophy. Hypertension. 1998;32:488–495. doi: 10.1161/01.hyp.32.3.488. [DOI] [PubMed] [Google Scholar]
- 29.Sundaresan M, Yu ZX, Ferrans VJ, Irani K, Finkel T. Requirement for generation of H 2 O 2 for platelet-derived growth factor signal transduction. Science. 1995;270:296–299. doi: 10.1126/science.270.5234.296. [DOI] [PubMed] [Google Scholar]
- 30.Lange S, Heger J, Euler G, Wartenberg M, Piper HM, Sauer H. Platelet-derived growth factor BB stimulates vasculogenesis of embryonic stem cell-derived endothelial cells by calcium-mediated generation of reactive oxygen species. Cardiovasc Res. 2009;81:159–168. doi: 10.1093/cvr/cvn258. [DOI] [PubMed] [Google Scholar]
- 31.Kang SW. Two axes in platelet-derived growth factor signaling: tyrosine phosphorylation and reactive oxygen species. Cell Mol Life Sci. 2007;64:533–541. doi: 10.1007/s00018-007-6437-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hennet T, Richter C, Peterhans E. Tumour necrosis factor-alpha induces superoxide anion generation in mitochondria of L929 cells. Biochem J. 1993;289(Pt 2):587–592. doi: 10.1042/bj2890587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Meier B, Radeke HH, Selle S, Younes M, Sies H, Resch K, Habermehl GG. Human fibroblasts release reactive oxygen species in response to interleukin-1 or tumour necrosis factor-alpha. Biochem J. 1989;263:539–545. doi: 10.1042/bj2630539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.De Keulenaer GW, Alexander RW, Ushio-Fukai M, Ishizaka N, Griendling KK. Tumour necrosis factor alpha activates a p22phox-based NADH oxidase in vascular smooth muscle. Biochem J. 1998;329(Pt 3):653–657. doi: 10.1042/bj3290653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Holland JA, Meyer JW, Chang MM, O’Donnell RW, Johnson DK, Ziegler LM. Thrombin stimulated reactive oxygen species production in cultured human endothelial cells. Endothelium. 1998;6:113–121. doi: 10.3109/10623329809072198. [DOI] [PubMed] [Google Scholar]
- 36.Patterson C, Ruef J, Madamanchi NR, Barry-Lane P, Hu Z, Horaist C, Ballinger CA, Brasier AR, Bode C, Runge MS. Stimulation of a vascular smooth muscle cell NAD(P)H oxidase by thrombin. Evidence that p47 phox may participate in forming this oxidase in vitro and in vivo. J Biol Chem. 1999;274:19814–19822. doi: 10.1074/jbc.274.28.19814. [DOI] [PubMed] [Google Scholar]
- 37.Pawate S, Shen Q, Fan F, Bhat NR. Redox regulation of glial inflammatory response to lipopolysaccharide and interferongamma. J Neurosci Res. 2004;77:540–551. doi: 10.1002/jnr.20180. [DOI] [PubMed] [Google Scholar]
- 38.May JM, de Haen C. Insulin-stimulated intracellular hydrogen peroxide production in rat epididymal fat cells. J Biol Chem. 1979;254:2214–2220. [PubMed] [Google Scholar]
- 39.Ceolotto G, Bevilacqua M, Papparella I, Baritono E, Franco L, Corvaja C, Mazzoni M, Semplicini A, Avogaro A. Insulin generates free radicals by an NAD(P)H, phosphatidylinositol 3’-kinase-dependent mechanism in human skin fibroblasts ex vivo. Diabetes. 2004;53:1344–1351. doi: 10.2337/diabetes.53.5.1344. [DOI] [PubMed] [Google Scholar]
- 40.Park HS, Chun JN, Jung HY, Choi C, Bae YS. Role of NADPH oxidase 4 in lipopolysaccharide-induced proinflammatory responses by human aortic endothelial cells. Cardiovasc Res. 2006;72:447–455. doi: 10.1016/j.cardiores.2006.09.012. [DOI] [PubMed] [Google Scholar]
- 41.Al Ghouleh I, Magder S. Nicotinamide adenine dinucleotide phosphate (reduced form) oxidase is important for LPS-induced endothelial cell activation. Shock. 2008;29:553–559. doi: 10.1097/SHK.0b013e318157ebc8. [DOI] [PubMed] [Google Scholar]
- 42.Jones DP. Radical-free biology of oxidative stress. Am J Physiol Cell Physiol. 2008;295:C849–C868. doi: 10.1152/ajpcell.00283.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gloire G, Legrand-Poels S, Piette J. NF-kappaB activation by reactive oxygen species: fifteen years later. Biochem Pharmacol. 2006;72:1493–1505. doi: 10.1016/j.bcp.2006.04.011. [DOI] [PubMed] [Google Scholar]
- 44.Kabe Y, Ando K, Hirao S, Yoshida M, Handa H. Redox regulation of NF-kappaB activation: distinct redox regulation between the cytoplasm and the nucleus. Antioxid Redox Signal. 2005;7:395–403. doi: 10.1089/ars.2005.7.395. [DOI] [PubMed] [Google Scholar]
- 45.Janssen-Heininger YMW, Poynter ME, Baeuerle PA. Recent advances torwards understanding redox mechanisms in the activation of nuclear factor [kappa]b. Free Radical Biology and Medicine. 2000;28:1317–1327. doi: 10.1016/s0891-5849(00)00218-5. [DOI] [PubMed] [Google Scholar]
- 46.Janssen-Heininger YM, Macara I, Mossman BT. Cooperativity between oxidants and tumor necrosis factor in the activation of nuclear factor (NF)-kappaB: requirement of Ras/mitogen-activated protein kinases in the activation of NF-kappaB by oxidants. Am J Respir Cell Mol Biol. 1999;20:942–952. doi: 10.1165/ajrcmb.20.5.3452. [DOI] [PubMed] [Google Scholar]
- 47.Tabet F, Schiffrin EL, Callera GE, He Y, Yao G, Ostman A, Kappert K, Tonks NK, Touyz RM. Redox-sensitive signaling by angiotensin II involves oxidative inactivation and blunted phosphorylation of protein tyrosine phosphatase SHP-2 in vascular smooth muscle cells from SHR. Circ Res. 2008;103:149–158. doi: 10.1161/CIRCRESAHA.108.178608. [DOI] [PubMed] [Google Scholar]
- 48.Hsieh HL, Wang HH, Wu CY, Yang CM. Reactive Oxygen Species-Dependent c-Fos/Activator Protein 1 Induction Upregulates Heme Oxygenase-1 Expression by Bradykinin in Brain Astrocytes. Antioxid Redox Signal. 2010;13:1829–1844. doi: 10.1089/ars.2009.2957. [DOI] [PubMed] [Google Scholar]
- 49.Brown DI, Griendling KK. Nox proteins in signal transduction. Free Radic Biol Med. 2009;47:1239–1253. doi: 10.1016/j.freeradbiomed.2009.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tiso M, Tejero J, Basu S, Azarov I, Wang X, Simplaceanu V, Frizzell S, Jayaraman T, Geary L, Shapiro C, Ho C, Shiva S, Kim-Shapiro DB, Gladwin MT. Human neuroglobin functions as a redox regulated nitrite reductase. J Biol Chem. doi: 10.1074/jbc.M110.159541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Barrett WC, DeGnore JP, Keng YF, Zhang ZY, Yim MB, Chock PB. Roles of superoxide radical anion in signal transduction mediated by reversible regulation of protein-tyrosine phosphatase 1B. J Biol Chem. 1999;274:34543–34546. doi: 10.1074/jbc.274.49.34543. [DOI] [PubMed] [Google Scholar]
- 52.Lee SR, Kwon KS, Kim SR, Rhee SG. Reversible inactivation of protein-tyrosine phosphatase 1B in A431 cells stimulated with epidermal growth factor. J Biol Chem. 1998;273:15366–15372. doi: 10.1074/jbc.273.25.15366. [DOI] [PubMed] [Google Scholar]
- 53.Denu JM, Tanner KG. Specific and reversible inactivation of protein tyrosine phosphatases by hydrogen peroxide: evidence for a sulfenic acid intermediate and implications for redox regulation. Biochemistry. 1998;37:5633–5642. doi: 10.1021/bi973035t. [DOI] [PubMed] [Google Scholar]
- 54.Kurata S. Selective activation of p38 MAPK cascade and mitotic arrest caused by low level oxidative stress. J Biol Chem. 2000;275:23413–23416. doi: 10.1074/jbc.C000308200. [DOI] [PubMed] [Google Scholar]
- 55.Torres M, Forman HJ. Redox signaling and the MAP kinase pathways. Biofactors. 2003;17:287–296. doi: 10.1002/biof.5520170128. [DOI] [PubMed] [Google Scholar]
- 56.Nadeau PJ, Charette SJ, Landry J. REDOX reaction at ASK1-Cys250 is essential for activation of JNK and induction of apoptosis. Mol Biol Cell. 2009;20:3628–3637. doi: 10.1091/mbc.E09-03-0211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nishibayashi H, Omura T, Sato R. A flavoprotein oxidizing NADPH isolated from liver microsomes. Biochim Biophys Acta. 1963;67:520–522. doi: 10.1016/0006-3002(63)91861-4. [DOI] [PubMed] [Google Scholar]
- 58.Nishibayashi-Yamashita H, Sato R. Vitamin K3-dependent NADPH oxidase of liver microsomes. Purification, properties, and identity with microsomal NADPH-cytochrome c reductase. J Biochem. 1970;67:199–210. doi: 10.1093/oxfordjournals.jbchem.a129243. [DOI] [PubMed] [Google Scholar]
- 59.Quie PG, White JG, Holmes B, Good RA. In vitro bactericidal capacity of human polymorphonuclear leukocytes: diminished activity in chronic granulomatous disease of childhood. J Clin Invest. 1967;46:668–679. doi: 10.1172/JCI105568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Berendes H, Bridges RA, Good RA. A fatal granulomatosus of childhood: the clinical study of a new syndrome. Minn Med. 1957;40:309–312. [PubMed] [Google Scholar]
- 61.Baehner RL, Nathan DG. Leukocyte oxidase: defective activity in chronic granulomatous disease. Science. 1967;155:835–836. doi: 10.1126/science.155.3764.835. [DOI] [PubMed] [Google Scholar]
- 62.Holmes B, Page AR, Good RA. Studies of the metabolic activity of leukocytes from patients with a genetic abnormality of phagocytic function. J Clin Invest. 1967;46:1422–1432. doi: 10.1172/JCI105634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Royer-Pokora B, Kunkel LM, Monaco AP, Goff SC, Newburger PE, Baehner RL, Cole FS, Curnutte JT, Orkin SH. Cloning the gene for an inherited human disorder--chronic granulomatous disease--on the basis of its chromosomal location. Nature. 1986;322:32–38. doi: 10.1038/322032a0. [DOI] [PubMed] [Google Scholar]
- 64.Teahan C, Rowe P, Parker P, Totty N, Segal AW. The X-linked chronic granulomatous disease gene codes for the beta-chain of cytochrome b-245. Nature. 1987;327:720–721. doi: 10.1038/327720a0. [DOI] [PubMed] [Google Scholar]
- 65.Dinauer MC, Orkin SH, Brown R, Jesaitis AJ, Parkos CA. The glycoprotein encoded by the X-linked chronic granulomatous disease locus is a component of the neutrophil cytochrome b complex. Nature. 1987;327:717–720. doi: 10.1038/327717a0. [DOI] [PubMed] [Google Scholar]
- 66.Segal AW. Absence of both cytochrome b -245 subunits from neutrophils in X-linked chronic granulomatous disease. Nature. 1987;326:88–91. doi: 10.1038/326088a0. [DOI] [PubMed] [Google Scholar]
- 67.Babior BM. NADPH oxidase: an update. Blood. 1999;93:1464–1476. [PubMed] [Google Scholar]
- 68.Hultqvist M, Holmdahl R. Ncf1 (p47phox) polymorphism determines oxidative burst and the severity of arthritis in rats and mice. Cell Immunol. 2005;233:97–101. doi: 10.1016/j.cellimm.2005.04.008. [DOI] [PubMed] [Google Scholar]
- 69.Gelderman KA, Hultqvist M, Olsson LM, Bauer K, Pizzolla A, Olofsson P, Holmdahl R. Rheumatoid Arthritis: The Role of Reactive Oxygen Species in Disease Development and Therapeutic Strategies. Antioxid Redox Signal. 2007 doi: 10.1089/ars.2007.1569. [DOI] [PubMed] [Google Scholar]
- 70.Griendling KK, Sorescu D, Ushio-Fukai M. NAD(P)H oxidase: role in cardiovascular biology and disease. Circ Res. 2000;86:494–501. doi: 10.1161/01.res.86.5.494. [DOI] [PubMed] [Google Scholar]
- 71.Taniyama Y, Griendling KK. Reactive oxygen species in the vasculature. Molecular and cellular mechanisms. Hypertension. 2003;42:1075–1081. doi: 10.1161/01.HYP.0000100443.09293.4F. [DOI] [PubMed] [Google Scholar]
- 72.Jones SA, O’Donnell VB, Wood JD, Broughton JP, Hughes EJ, Jones OTG. Expression of phagocyte NADPH oxidase components in human endothelial cells. Am J Physiol. 1996;271:H1626–H1634. doi: 10.1152/ajpheart.1996.271.4.H1626. [DOI] [PubMed] [Google Scholar]
- 73.Li JM, Shah AM. Endothelial cell superoxide generation: regulation and relevance for cardiovascular pathophysiology. Am J Physiol Regul Integr Comp Physiol. 2004;287:R1014–1030. doi: 10.1152/ajpregu.00124.2004. [DOI] [PubMed] [Google Scholar]
- 74.Ray R, Shah AM. NADPH oxidase and endothelial cell function. Clin Sci (Lond) 2005;109:217–226. doi: 10.1042/CS20050067. [DOI] [PubMed] [Google Scholar]
- 75.Meier B, Cross AR, Hancock JT, Kaup FJ, Jones OTG. Identification of a superoxide-generating NADPH oxidase system in human fibroblasts. Biochem J. 1991;275:241–245. doi: 10.1042/bj2750241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Griendling KK, Minieri CA, Ollerenshaw JD, Alexander RW. Angiotensin II stimulates NADH and NADPH oxidase activity in cultured vascular smooth muscle cells. Circ Res. 1994;74:1141–1148. doi: 10.1161/01.res.74.6.1141. [DOI] [PubMed] [Google Scholar]
- 77.Geiszt M. NADPH oxidases: new kids on the block. Cardiovasc Res. 2006;71:289–299. doi: 10.1016/j.cardiores.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 78.Kawahara T, Ritsick D, Cheng G, Lambeth JD. Point mutations in the proline-rich region of p22 phox are dominant inhibitors of Nox1- and Nox2-dependent reactive oxygen generation. J Biol Chem. 2005;280:31859–31869. doi: 10.1074/jbc.M501882200. [DOI] [PubMed] [Google Scholar]
- 79.Cave AC, Brewer AC, Narayanapanicker A, Ray R, Grieve DJ, Walker S, Shah AM. NADPH oxidases in cardiovascular health and disease. Antioxid Redox Signal. 2006;8:691–728. doi: 10.1089/ars.2006.8.691. [DOI] [PubMed] [Google Scholar]
- 80.Quinn MT, Ammons MC, Deleo FR. The expanding role of NADPH oxidases in health and disease: no longer just agents of death and destruction. Clin Sci (Lond) 2006;111:1–20. doi: 10.1042/CS20060059. [DOI] [PubMed] [Google Scholar]
- 81.Parkos CA, Allen RA, Cochrane CG, Jesaitis AJ. Purified cytochrome b from human granulocyte plasma membrane is comprised of two polypeptides with relative molecular weights of 91,000 and 22,000. J Clin Invest. 1987;80:732–742. doi: 10.1172/JCI113128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Nakano Y, Banfi B, Jesaitis AJ, Dinauer MC, Allen LA, Nauseef WM. Critical roles for p22phox in the structural maturation and subcellular targeting of Nox3. Biochem J. 2007;403:97–108. doi: 10.1042/BJ20060819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ambasta RK, Kumar P, Griendling KK, Schmidt HH, Busse R, Brandes RP. Direct interaction of the novel Nox proteins with p22phox is required for the formation of a functionally active NADPH oxidase. J Biol Chem. 2004;279:45935–45941. doi: 10.1074/jbc.M406486200. [DOI] [PubMed] [Google Scholar]
- 84.Parkos CA, Dinauer MC, Walker LE, Allen RA, Jesaitis AJ, Orkin SH. Primary structure and unique expression of the 22-kilodalton light chain of human neutrophil cytochrome b. Proceedings of the National Academy of Sciences. 1988;85:3319–3323. doi: 10.1073/pnas.85.10.3319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Cheng G, Cao Z, Xu X, van Meir EG, Lambeth JD. Homologs of gp91phox: cloning and tissue expression of Nox3, Nox4, and Nox5. Gene. 2001;269:131–140. doi: 10.1016/s0378-1119(01)00449-8. [DOI] [PubMed] [Google Scholar]
- 86.Bayraktutan U, Blayney L, Shah AM. Molecular characterization and localization of the NAD(P)H oxidase components gp91-phox and p22-phox in endothelial cells. Arterioscler Thromb Vasc Biol. 2000;20:1903–1911. doi: 10.1161/01.atv.20.8.1903. [DOI] [PubMed] [Google Scholar]
- 87.Hanna IR, Hilenski LL, Dikalova A, Taniyama Y, Dikalov S, Lyle A, Quinn MT, Lassegue B, Griendling KK. Functional association of nox1 with p22phox in vascular smooth muscle cells. Free Radic Biol Med. 2004;37:1542–1549. doi: 10.1016/j.freeradbiomed.2004.08.011. [DOI] [PubMed] [Google Scholar]
- 88.Ueno N, Takeya R, Miyano K, Kikuchi H, Sumimoto H. The NADPH oxidase Nox3 constitutively produces superoxide in a p22phox-dependent manner: its regulation by oxidase organizers and activators. J Biol Chem. 2005;280:23328–23339. doi: 10.1074/jbc.M414548200. [DOI] [PubMed] [Google Scholar]
- 89.Banfi B, Clark RA, Steger K, Krause KH. Two novel proteins activate superoxide generation by the NADPH oxidase NOX1. J Biol Chem. 2003;278:3510–3513. doi: 10.1074/jbc.C200613200. [DOI] [PubMed] [Google Scholar]
- 90.Banfi B, Malgrange B, Knisz J, Steger K, Dubois-Dauphin M, Krause KH. NOX3, a superoxide-generating NADPH oxidase of the inner ear. J Biol Chem. 2004;279:46065–46072. doi: 10.1074/jbc.M403046200. [DOI] [PubMed] [Google Scholar]
- 91.Kiss PJ, Knisz J, Zhang Y, Baltrusaitis J, Sigmund CD, Thalmann R, Smith RJH, Verpy E, Bnfi B. Inactivation of NADPH oxidase organizer 1 results in severe imbalance. Curr Biol. 2006;16:208–213. doi: 10.1016/j.cub.2005.12.025. [DOI] [PubMed] [Google Scholar]
- 92.Cross AR, Segal AW. The NADPH oxidase of professional phagocytes--prototype of the NOX electron transport chain systems. Biochim Biophys Acta. 2004;1657:1–22. doi: 10.1016/j.bbabio.2004.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.De Deken X, Wang D, Many MC, Costagliola S, Libert F, Vassart G, Dumont JE, Miot F. Cloning of two human thyroid cDNAs encoding new members of the NADPH oxidase family. J Biol Chem. 2000;275:23227–23233. doi: 10.1074/jbc.M000916200. [DOI] [PubMed] [Google Scholar]
- 94.De DX, Wang D, Dumont JE, Miot F. Characterization of ThOX proteins as components of the thyroid H(2)O(2)-generating system. Exp Cell Res. 2002;273:187–196. doi: 10.1006/excr.2001.5444. [DOI] [PubMed] [Google Scholar]
- 95.Wang D, De DX, Milenkovic M, Song Y, Pirson I, Dumont JE, Miot F. Identification of a novel partner of duox: EFP1, a thioredoxin-related protein. J Biol Chem. 2005;280:3096–3103. doi: 10.1074/jbc.M407709200. [DOI] [PubMed] [Google Scholar]
- 96.Banfi B, Molnar G, Maturana A, Steger K, Hegedus B, Demaurex N, Krause KH. A Ca(2+)-activated NADPH oxidase in testis, spleen, and lymph nodes. J Biol Chem. 2001;276:37594–37601. doi: 10.1074/jbc.M103034200. [DOI] [PubMed] [Google Scholar]
- 97.Grasberger H, Refetoff S. Identification of the maturation factor for dual oxidase. Evolution of an eukaryotic operon equivalent. J Biol Chem. 2006;281:18269–18272. doi: 10.1074/jbc.C600095200. [DOI] [PubMed] [Google Scholar]
- 98.Bokoch GM, Diebold BA. Current molecular models for NADPH oxidase regulation by Rac GTPase. Blood. 2002;100:2692–2696. doi: 10.1182/blood-2002-04-1149. [DOI] [PubMed] [Google Scholar]
- 99.Nisimoto Y, Freeman JL, Motalebi SA, Hirshberg M, Lambeth JD. Rac binding to p67(phox). Structural basis for interactions of the Rac1 effector region and insert region with components of the respiratory burst oxidase. J Biol Chem. 1997;272:18834–18841. doi: 10.1074/jbc.272.30.18834. [DOI] [PubMed] [Google Scholar]
- 100.Sumimoto H, Hata K, Mizuki K, Ito T, Kage Y, Sakaki Y, Fukumaki Y, Nakamura M, Takeshige K. Assembly and activation of the phagocyte NADPH oxidase. Specific interaction of the N-terminal Src homology 3 domain of p47phox with p22phox is required for activation of the NADPH oxidase. J Biol Chem. 1996;271:22152–22158. doi: 10.1074/jbc.271.36.22152. [DOI] [PubMed] [Google Scholar]
- 101.Sumimoto H. Structure, regulation and evolution of Nox-family NADPH oxidases that produce reactive oxygen species. FEBS J. 2008;275:3249–3277. doi: 10.1111/j.1742-4658.2008.06488.x. [DOI] [PubMed] [Google Scholar]
- 102.Cheng G, Ritsick D, Lambeth JD. Nox3 regulation by NOXO1, p47phox, and p67phox. J Biol Chem. 2004;279:34250–34255. doi: 10.1074/jbc.M400660200. [DOI] [PubMed] [Google Scholar]
- 103.Nauseef WM. Biological roles for the NOX family NADPH oxidases. J Biol Chem. 2008;283:16961–16965. doi: 10.1074/jbc.R700045200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Vignais PV. The superoxide-generating NADPH oxidase: structural aspects and activation mechanism. Cell Mol Life Sci. 2002;59:1428–1459. doi: 10.1007/s00018-002-8520-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Takeya R, Sumimoto H. Molecular mechanism for activation of superoxide-producing NADPH oxidases. Molecules and Cells. 2003;16:271–277. [PubMed] [Google Scholar]
- 106.Fulton DJ. Nox5 and the regulation of cellular function. Antioxid Redox Signal. 2009;11:2443–2452. doi: 10.1089/ars.2009.2587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.El Jamali A, Valente AJ, Lechleiter JD, Gamez MJ, Pearson DW, Nauseef WM, Clark RA. Novel redox-dependent regulation of NOX5 by the tyrosine kinase c-Abl. Free Radic Biol Med. 2008;44:868–881. doi: 10.1016/j.freeradbiomed.2007.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Nisimoto Y, Motalebi S, Han CH, Lambeth JD. The p67(phox) activation domain regulates electron flow from NADPH to flavin in flavocytochrome b(558) J Biol Chem. 1999;274:22999–23005. doi: 10.1074/jbc.274.33.22999. [DOI] [PubMed] [Google Scholar]
- 109.Martyn KD, Frederick LM, von Loehneysen K, Dinauer MC, Knaus UG. Functional analysis of Nox4 reveals unique characteristics compared to other NADPH oxidases. Cell Signal. 2006;18:69–82. doi: 10.1016/j.cellsig.2005.03.023. [DOI] [PubMed] [Google Scholar]
- 110.Geiszt M, Witta J, Baffi J, Lekstrom K, Leto TL. Dual oxidases represent novel hydrogen peroxide sources supporting mucosal surface host defense. FASEB J. 2003;17:1502–1504. doi: 10.1096/fj.02-1104fje. [DOI] [PubMed] [Google Scholar]
- 111.Ameziane-El-Hassani R, Morand S, Boucher JL, Frapart YM, Apostolou D, Agnandji D, Gnidehou S, Ohayon R, Noel-Hudson MS, Francon J, Lalaoui K, Virion A, Dupuy C. Dual oxidase-2 has an intrinsic Ca2+-dependent H2O2-generating activity. J Biol Chem. 2005;280:30046–30054. doi: 10.1074/jbc.M500516200. [DOI] [PubMed] [Google Scholar]
- 112.von Lohneysen K, Noack D, Wood MR, Friedman JS, Knaus UG. Structural insights into Nox4 and Nox2: motifs involved in function and cellular localization. Mol Cell Biol. 30:961–975. doi: 10.1128/MCB.01393-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Petry A, Djordjevic T, Weitnauer M, Kietzmann T, Hess J, Gorlach A. NOX2 and NOX4 mediate proliferative response in endothelial cells. Antioxid Redox Signal. 2006;8:1473–1484. doi: 10.1089/ars.2006.8.1473. [DOI] [PubMed] [Google Scholar]
- 114.Gorlach A, Brandes RP, Nguyen K, Amidi M, Dehghani F, Busse R. A gp91phox containing NADPH oxidase selectively expressed in endothelial cells is a major source of oxygen radical generation in the arterial wall. Circ Res. 2000;87:26–32. doi: 10.1161/01.res.87.1.26. [DOI] [PubMed] [Google Scholar]
- 115.Touyz RM, Chen X, Tabet F, Yao G, He G, Quinn MT, Pagano PJ, Schiffrin EL. Expression of a functionally active gp91phox-containing neutrophil-type NAD(P)H oxidase in smooth muscle cells from human resistance arteries. Regulation by angiotensin II. Circ Res. 2002;90:1205–1213. doi: 10.1161/01.res.0000020404.01971.2f. [DOI] [PubMed] [Google Scholar]
- 116.Javeshghani D, Hussain SNA, Scheidel J, Quinn MT, Magder SA. Superoxide production in the vasculature of lipopolysaccharide treated rats and pigs. Shock. 2003;19:48–493. doi: 10.1097/01.shk.0000054374.88889.37. [DOI] [PubMed] [Google Scholar]
- 117.Peng T, Lu X, Feng Q. Pivotal role of gp91phox-containing NADH oxidase in lipopolysaccharide-induced tumor necrosis factor-alpha expression and myocardial depression. Circulation. 2005;111:1637–1644. doi: 10.1161/01.CIR.0000160366.50210.E9. [DOI] [PubMed] [Google Scholar]
- 118.Li JM, Shah AM. Intracellular localization and preassembly of the NADPH oxidase complex in cultured endothelial cells. J Biol Chem. 2002;277:19952–19960. doi: 10.1074/jbc.M110073200. [DOI] [PubMed] [Google Scholar]
- 119.Suh YA, Arnold RS, Lassegue B, Shi J, Xu X, Sorescu D, Chung AB, Griendling KK, Lambeth JD. Cell transformation by the superoxide-generating oxidase Mox1. Nature. 1999;401:79–82. doi: 10.1038/43459. [DOI] [PubMed] [Google Scholar]
- 120.Banfi B, Maturana A, Jaconi S, Arnaudeau S, Laforge T, Sinha B, Ligeti E, Demaurex N, Krause KH. A mammalian H+ channel generated through alternative splicing of the NADPH oxidase homolog NOH-1. Science. 2000;287:138–142. doi: 10.1126/science.287.5450.138. [DOI] [PubMed] [Google Scholar]
- 121.Cui XL, Brockman D, Campos B, Myatt L. Expression of NADPH oxidase isoform 1 (Nox1) in human placenta: involvement in preeclampsia. Placenta. 2006;27:422–431. doi: 10.1016/j.placenta.2005.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Lee NK, Choi YG, Baik JY, Han SY, Jeong DW, Bae YS, Kim N, Lee SY. A crucial role for reactive oxygen species in RANKL-induced osteoclast differentiation. Blood. 2005;106:852–859. doi: 10.1182/blood-2004-09-3662. [DOI] [PubMed] [Google Scholar]
- 123.Hilenski LL, Clempus RE, Quinn MT, Lambeth JD, Griendling KK. Distinct subcellular localizations of Nox1 and Nox4 in vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 2004;24:677–683. doi: 10.1161/01.ATV.0000112024.13727.2c. [DOI] [PubMed] [Google Scholar]
- 124.Helmcke I, Heumuller S, Tikkanen R, Schroder K, Brandes RP. Identification of structural elements in Nox1 and Nox4 controlling localization and activity. Antioxid Redox Signal. 2009;11:1279–1287. doi: 10.1089/ars.2008.2383. [DOI] [PubMed] [Google Scholar]
- 125.Miller FJ, Jr, Filali M, Huss GJ, Stanic B, Chamseddine A, Barna TJ, Lamb FS. Cytokine activation of nuclear factor kappa B in vascular smooth muscle cells requires signaling endosomes containing Nox1 and ClC-3. Circ Res. 2007;101:663–671. doi: 10.1161/CIRCRESAHA.107.151076. [DOI] [PubMed] [Google Scholar]
- 126.Kikuchi H, Hikage M, Miyashita H, Fukumoto M. NADPH oxidase subunit, gp91(phox) homologue, preferentially expressed in human colon epithelial cells. Gene. 2000;254:237–243. doi: 10.1016/s0378-1119(00)00258-4. [DOI] [PubMed] [Google Scholar]
- 127.Geiszt M, Kopp JB, Varnai P, Leto TL. Identification of renox, an NAD(P)H oxidase in kidney. Proc Natl Acad Sci U S A. 2000;97:8010–8014. doi: 10.1073/pnas.130135897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Shiose A, Kuroda J, Tsuruya K, Hirai M, Hirakata H, Naito S, Hattori M, Sakaki Y, Sumimoto H. A novel superoxide-producing NAD(P)H oxidase in kidney. J Biol Chem. 2001;276:1417–1423. doi: 10.1074/jbc.M007597200. [DOI] [PubMed] [Google Scholar]
- 129.Van Buul JD, Fernandez-Borja M, Anthony EC, Hordijk PL. Expression and localization of NOX2 and NOX4 in primary human endothelial cells. Antioxid Redox Signal. 2005;7:308–317. doi: 10.1089/ars.2005.7.308. [DOI] [PubMed] [Google Scholar]
- 130.Yang S, Madyastha P, Bingel S, Ries W, Key L. A new superoxide-generating oxidase in murine osteoclasts. Journal of Biological Chemistry. 2001;276:5452–5458. doi: 10.1074/jbc.M001004200. [DOI] [PubMed] [Google Scholar]
- 131.Ago T, Kitazono T, Ooboshi H, Iyama T, Han YH, Takada J, Wakisaka M, Ibayashi S, Utsumi H, Iida M. Nox4 as the major catalytic component of an endothelial NAD(P)H oxidase. Circulation. 2004;109:227–233. doi: 10.1161/01.CIR.0000105680.92873.70. [DOI] [PubMed] [Google Scholar]
- 132.Cucoranu I, Clempus R, Dikalova A, Phelan PJ, Ariyan S, Dikalov S, Sorescu D. NAD(P)H oxidase 4 mediates transforming growth factor-beta1-induced differentiation of cardiac fibroblasts into myofibroblasts. Circ Res. 2005;97:900–907. doi: 10.1161/01.RES.0000187457.24338.3D. [DOI] [PubMed] [Google Scholar]
- 133.Vallet P, Charnay Y, Steger K, Ogier-Denis E, Kovari E, Herrmann F, Michel JP, Szanto I. Neuronal expression of the NADPH oxidase NOX4, and its regulation in mouse experimental brain ischemia. Neuroscience. 2005;132:233–238. doi: 10.1016/j.neuroscience.2004.12.038. [DOI] [PubMed] [Google Scholar]
- 134.Kuroda J, Nakagawa K, Yamasaki T, Nakamura K, Takeya R, Kuribayashi F, Imajoh-Ohmi S, Igarashi K, Shibata Y, Sueishi K, Sumimoto H. The superoxide-producing NAD(P)H oxidase Nox4 in the nucleus of human vascular endothelial cells. Genes Cells. 2005;10:1139–1151. doi: 10.1111/j.1365-2443.2005.00907.x. [DOI] [PubMed] [Google Scholar]
- 135.Block K, Gorin Y, Abboud HE. Subcellular localization of Nox4 and regulation in diabetes. Proc Natl Acad Sci U S A. 2009;106:14385–14390. doi: 10.1073/pnas.0906805106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Ago T, Kuroda J, Pain J, Fu C, Li H, Sadoshima J. Upregulation of Nox4 by hypertrophic stimuli promotes apoptosis and mitochondrial dysfunction in cardiac myocytes. Circ Res. 106:1253–1264. doi: 10.1161/CIRCRESAHA.109.213116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Banfi B, Tirone F, Durussel I, Knisz J, Moskwa P, Molnar GZ, Krause KH, Cox JA. Mechanism of Ca2+ activation of the NADPH oxidase 5 (NOX5) J Biol Chem. 2004;279:18583–18591. doi: 10.1074/jbc.M310268200. [DOI] [PubMed] [Google Scholar]
- 138.BelAiba RS, Djordjevic T, Petry A, Diemer K, Bonello S, Banfi B, Hess J, Pogrebniak A, Bickel C, Gorlach A. NOX5 variants are functionally active in endothelial cells. Free Radic Biol Med. 2007;42:446–459. doi: 10.1016/j.freeradbiomed.2006.10.054. [DOI] [PubMed] [Google Scholar]
- 139.Jay DB, Papaharalambus CA, Seidel-Rogol B, Dikalova AE, Lassegue B, Griendling KK. Nox5 mediates PDGF-induced proliferation in human aortic smooth muscle cells. Free Radic Biol Med. 2008;45:329–335. doi: 10.1016/j.freeradbiomed.2008.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Dupuy C, Ohayon R, Valent A, Noel-Hudson MS, Deme D, Virion A. Purification of a novel flavoprotein involved in the thyroid NADPH oxidase. Cloning of the porcine and human cdnas. J Biol Chem. 1999;274:37265–37269. doi: 10.1074/jbc.274.52.37265. [DOI] [PubMed] [Google Scholar]
- 141.Luxen S, Noack D, Frausto M, Davanture S, Torbett BE, Knaus UG. Heterodimerization controls localization of Duox-DuoxA NADPH oxidases in airway cells. J Cell Sci. 2009;122:1238–1247. doi: 10.1242/jcs.044123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Zamocky M, Jakopitsch C, Furtmuller PG, Dunand C, Obinger C. The peroxidase-cyclooxygenase superfamily: Reconstructed evolution of critical enzymes of the innate immune system. Proteins. 2008;72:589–605. doi: 10.1002/prot.21950. [DOI] [PubMed] [Google Scholar]
- 143.Amaya Y, Yamazaki K, Sato M, Noda K, Nishino T. Proteolytic conversion of xanthine dehydrogenase from the NAD-dependent type to the O2-dependent type. Amino acid sequence of rat liver xanthine dehydrogenase and identification of the cleavage sites of the enzyme protein during irreversible conversion by trypsin. J Biol Chem. 1990;265:14170–14175. [PubMed] [Google Scholar]
- 144.Waud WR, Rajagopalan KV. The mechanism of conversion of rat liver xanthine dehydrogenase from an NAD+-dependent form (type D) to an O2-dependent form (type O) Arch Biochem Biophys. 1976;172:365–379. doi: 10.1016/0003-9861(76)90088-6. [DOI] [PubMed] [Google Scholar]
- 145.Enroth C, Eger BT, Okamoto K, Nishino T, Pai EF. Crystal structures of bovine milk xanthine dehydrogenase and xanthine oxidase: structure-based mechanism of conversion. Proc Natl Acad Sci U S A. 2000;97:10723–10728. doi: 10.1073/pnas.97.20.10723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Harris CM, Massey V. The reaction of reduced xanthine dehydrogenase with molecular oxygen. Reaction kinetics and measurement of superoxide radical. J Biol Chem. 1997;272:8370–8379. doi: 10.1074/jbc.272.13.8370. [DOI] [PubMed] [Google Scholar]
- 147.Butler R, Morris AD, Belch JJ, Hill A, Struthers AD. Allopurinol normalizes endothelial dysfunction in type 2 diabetics with mild hypertension. Hypertension. 2000;35:746–751. doi: 10.1161/01.hyp.35.3.746. [DOI] [PubMed] [Google Scholar]
- 148.Desco MC, Asensi M, Marquez R, Martinez-Valls J, Vento M, Pallardo FV, Sastre J, Vina J. Xanthine oxidase is involved in free radical production in type 1 diabetes: protection by allopurinol. Diabetes. 2002;51:1118–1124. doi: 10.2337/diabetes.51.4.1118. [DOI] [PubMed] [Google Scholar]
- 149.Farquharson CA, Butler R, Hill A, Belch JJ, Struthers AD. Allopurinol improves endothelial dysfunction in chronic heart failure. Circulation. 2002;106:221–226. doi: 10.1161/01.cir.0000022140.61460.1d. [DOI] [PubMed] [Google Scholar]
- 150.Aslan M, Ryan TM, Adler B, Townes TM, Parks DA, Thompson JA, Tousson A, Gladwin MT, Patel RP, Tarpey MM, Batinic-Haberle I, White CR, Freeman BA. Oxygen radical inhibition of nitric oxide-dependent vascular function in sickle cell disease. Proc Natl Acad Sci U S A. 2001;98:15215–15220. doi: 10.1073/pnas.221292098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Ryter SW, Morse D, Choi AM. Carbon monoxide and bilirubin: potential therapies for pulmonary/vascular injury and disease. Am J Respir Cell Mol Biol. 2007;36:175–182. doi: 10.1165/rcmb.2006-0333TR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Idriss NK, Blann AD, Lip GY. Hemoxygenase-1 in cardiovascular disease. J Am Coll Cardiol. 2008;52:971–978. doi: 10.1016/j.jacc.2008.06.019. [DOI] [PubMed] [Google Scholar]
- 153.Pae HO, Lee YC, Chung HT. Heme oxygenase-1 and carbon monoxide: emerging therapeutic targets in inflammation and allergy. Recent Pat Inflamm Allergy Drug Discov. 2008;2:159–165. doi: 10.2174/187221308786241929. [DOI] [PubMed] [Google Scholar]
- 154.Ryter SW, Alam J, Choi AM. Heme oxygenase-1/carbon monoxide: from basic science to therapeutic applications. Physiol Rev. 2006;86:583–650. doi: 10.1152/physrev.00011.2005. [DOI] [PubMed] [Google Scholar]
- 155.Hamanaka RB, Chandel NS. Mitochondrial reactive oxygen species regulate cellular signaling and dictate biological outcomes. Trends Biochem Sci. 35:505–513. doi: 10.1016/j.tibs.2010.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Ramachandran A, Levonen AL, Brookes PS, Ceaser E, Shiva S, Barone MC, Darley-Usmar V. Mitochondria, nitric oxide, and cardiovascular dysfunction. Free Radic Biol Med. 2002;33:1465–1474. doi: 10.1016/s0891-5849(02)01142-5. [DOI] [PubMed] [Google Scholar]
- 157.Brookes PS, Levonen AL, Shiva S, Sarti P, Darley-Usmar VM. Mitochondria: regulators of signal transduction by reactive oxygen and nitrogen species. Free Radic Biol Med. 2002;33:755–764. doi: 10.1016/s0891-5849(02)00901-2. [DOI] [PubMed] [Google Scholar]
- 158.Brookes P, Darley-Usmar VM. Hypothesis: the mitochondrial NO(*) signaling pathway, and the transduction of nitrosative to oxidative cell signals: an alternative function for cytochrome C oxidase. Free Radic Biol Med. 2002;32:370–374. doi: 10.1016/s0891-5849(01)00805-x. [DOI] [PubMed] [Google Scholar]
- 159.Boveris A, Cadenas E. Mitochondrial production of hydrogen peroxide regulation by nitric oxide and the role of ubisemiquinone. IUBMB Life. 2000;50:245–250. doi: 10.1080/713803732. [DOI] [PubMed] [Google Scholar]
- 160.Cadenas E, Davies KJ. Mitochondrial free radical generation, oxidative stress, and aging. Free Radic Biol Med. 2000;29:222–230. doi: 10.1016/s0891-5849(00)00317-8. [DOI] [PubMed] [Google Scholar]
- 161.Lin TK, Hughes G, Muratovska A, Blaikie FH, Brookes PS, Darley-Usmar V, Smith RA, Murphy MP. Specific modification of mitochondrial protein thiols in response to oxidative stress: a proteomics approach. J Biol Chem. 2002;277:17048–17056. doi: 10.1074/jbc.M110797200. [DOI] [PubMed] [Google Scholar]
- 162.Guzy RD, Schumacker PT. Oxygen sensing by mitochondria at complex III: the paradox of increased reactive oxygen species during hypoxia. Exp Physiol. 2006;91:807–819. doi: 10.1113/expphysiol.2006.033506. [DOI] [PubMed] [Google Scholar]
- 163.Carreras MC, Franco MC, Peralta JG, Poderoso JJ. Nitric oxide, complex I, and the modulation of mitochondrial reactive species in biology and disease. Mol Aspects Med. 2004;25:125–139. doi: 10.1016/j.mam.2004.02.014. [DOI] [PubMed] [Google Scholar]
- 164.Morand S, Ueyama T, Tsujibe S, Saito N, Korzeniowska A, Leto TL. Duox maturation factors form cell surface complexes with Duox affecting the specificity of reactive oxygen species generation. FASEB J. 2009;23:1205–1218. doi: 10.1096/fj.08-120006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Lyle AN, Deshpande NN, Taniyama Y, Seidel-Rogol B, Pounkova L, Du P, Papaharalambus C, Lassegue B, Griendling KK. Poldip2, a novel regulator of Nox4 and cytoskeletal integrity in vascular smooth muscle cells. Circ Res. 2009;105:249–259. doi: 10.1161/CIRCRESAHA.109.193722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Janiszewski M, Lopes LR, Carmo AO, Pedro MA, Brandes RP, Santos CX, Laurindo FR. Regulation of NAD(P)H oxidase by associated protein disulfide isomerase in vascular smooth muscle cells. J Biol Chem. 2005;280:40813–40819. doi: 10.1074/jbc.M509255200. [DOI] [PubMed] [Google Scholar]
- 167.Santos CX, Tanaka LY, Wosniak J, Laurindo FR. Mechanisms and implications of reactive oxygen species generation during the unfolded protein response: roles of endoplasmic reticulum oxidoreductases, mitochondrial electron transport, and NADPH oxidase. Antioxid Redox Signal. 2009;11:2409–2427. doi: 10.1089/ars.2009.2625. [DOI] [PubMed] [Google Scholar]
- 168.Santos CX, Stolf BS, Takemoto PV, Amanso AM, Lopes LR, Souza EB, Goto H, Laurindo FR. Protein disulfide isomerase (PDI) associates with NADPH oxidase and is required for phagocytosis of Leishmania chagasi promastigotes by macrophages. J Leukoc Biol. 2009;86:989–998. doi: 10.1189/jlb.0608354. [DOI] [PubMed] [Google Scholar]
- 169.Rigutto S, Hoste C, Grasberger H, Milenkovic M, Communi D, Dumont JE, Corvilain B, Miot F, De Deken X. Activation of dual oxidases Duox1 and Duox2: differential regulation mediated by camp-dependent protein kinase and protein kinase C-dependent phosphorylation. J Biol Chem. 2009;284:6725–6734. doi: 10.1074/jbc.M806893200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Jackson HM, Kawahara T, Nisimoto Y, Smith SM, Lambeth JD. Nox4 B-loop creates an interface between the transmembrane and dehydrogenase domains. J Biol Chem. 285:10281–10290. doi: 10.1074/jbc.M109.084939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Heyworth PG, Cross AR, Curnutte JT. Chronic granulomatous disease. Curr Opin Immunol. 2003;15:578–584. doi: 10.1016/s0952-7915(03)00109-2. [DOI] [PubMed] [Google Scholar]
- 172.Debeurme F, Picciocchi A, Dagher MC, Grunwald D, Beaumel S, Fieschi F, Stasia MJ. Regulation of NADPH oxidase activity in phagocytes: relationship between FAD/NADPH binding and oxidase complex assembly. J Biol Chem. 285:33197–33208. doi: 10.1074/jbc.M110.151555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Biberstine-Kinkade KJ, Yu L, Dinauer MC. Mutagenesis of an arginine- and lysine-rich domain in the gp91(phox) subunit of the phagocyte NADPH-oxidase flavocytochrome b558. J Biol Chem. 1999;274:10451–10457. doi: 10.1074/jbc.274.15.10451. [DOI] [PubMed] [Google Scholar]
- 174.Li XJ, Grunwald D, Mathieu J, Morel F, Stasia MJ. Crucial role of two potential cytosolic regions of Nox2, 191 TSSTKTIRRS 200 and 484 DESQANHFAVHHDEEKD 500, on NADPH oxidase activation. J Biol Chem. 2005;280:14962–14973. doi: 10.1074/jbc.M500226200. [DOI] [PubMed] [Google Scholar]
- 175.Nisimoto Y, Jackson HM, Ogawa H, Kawahara T, Lambeth JD. Constitutive NADPH-dependent electron transferase activity of the Nox4 dehydrogenase domain. Biochemistry. 49:2433–2442. doi: 10.1021/bi9022285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Carrichon L, Picciocchi A, Debeurme F, Defendi F, Beaumel S, Jesaitis AJ, Dagher MC, Stasia MJ. Characterization of superoxide overproduction by the D-Loop(Nox4)-Nox2 cytochrome b(558) in phagocytes-Differential sensitivity to calcium and phosphorylation events. Biochim Biophys Acta. 1808:78–90. doi: 10.1016/j.bbamem.2010.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.von Lohneysen K, Noack D, Jesaitis AJ, Dinauer MC, Knaus UG. Mutational analysis reveals distinct features of the Nox4-p22 phox complex. J Biol Chem. 2008;283:35273–35282. doi: 10.1074/jbc.M804200200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Zhu Y, Marchal CC, Casbon AJ, Stull N, von Lohneysen K, Knaus UG, Jesaitis AJ, McCormick S, Nauseef WM, Dinauer MC. Deletion mutagenesis of p22phox subunit of flavocytochrome b558: identification of regions critical for gp91phox maturation and NADPH oxidase activity. J Biol Chem. 2006;281:30336–30346. doi: 10.1074/jbc.M607191200. [DOI] [PubMed] [Google Scholar]
- 179.Nakano Y, Longo-Guess CM, Bergstrom DE, Nauseef WM, Jones SM, Banfi B. Mutation of the Cyba gene encoding p22phox causes vestibular and immune defects in mice. J Clin Invest. 2008;118:1176–1185. doi: 10.1172/JCI33835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Niu XL, Madamanchi NR, Vendrov AE, Tchivilev I, Rojas M, Madamanchi C, Brandes RP, Krause KH, Humphries J, Smith A, Burnand KG, Runge MS. Nox activator 1: a potential target for modulation of vascular reactive oxygen species in atherosclerotic arteries. Circulation. 121:549–559. doi: 10.1161/CIRCULATIONAHA.109.908319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Weber DS, Taniyama Y, Rocic P, Seshiah PN, Dechert MA, Gerthoffer WT, Griendling KK. Phosphoinositide-dependent kinase 1 and p21-activated protein kinase mediate reactive oxygen species-dependent regulation of platelet-derived growth factor-induced smooth muscle cell migration. Circ Res. 2004;94:1219–1226. doi: 10.1161/01.RES.0000126848.54740.4A. [DOI] [PubMed] [Google Scholar]
- 182.Katsuyama M, Fan C, Yabe-Nishimura C. NADPH oxidase is involved in prostaglandin F2alpha-induced hypertrophy of vascular smooth muscle cells: induction of NOX1 by PGF2alpha. J Biol Chem. 2002;277:13438–13442. doi: 10.1074/jbc.M111634200. [DOI] [PubMed] [Google Scholar]
- 183.Dikalov SI, Dikalova AE, Bikineyeva AT, Schmidt HH, Harrison DG, Griendling KK. Distinct roles of Nox1 and Nox4 in basal and angiotensin II-stimulated superoxide and hydrogen peroxide production. Free Radic Biol Med. 2008;45:1340–1351. doi: 10.1016/j.freeradbiomed.2008.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Frey RS, Ushio-Fukai M, Malik AB. NADPH oxidase-dependent signaling in endothelial cells: role in physiology and pathophysiology. Antioxid Redox Signal. 2009;11:791–810. doi: 10.1089/ars.2008.2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Fisher AB. Redox signaling across cell membranes. Antioxid Redox Signal. 2009;11:1349–1356. doi: 10.1089/ars.2008.2378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Peshavariya HM, Dusting GJ, Selemidis S. Analysis of dihydroethidium fluorescence for the detection of intracellular and extracellular superoxide produced by NADPH oxidase. Free Radic Res. 2007;41:699–712. doi: 10.1080/10715760701297354. [DOI] [PubMed] [Google Scholar]
- 187.Souchard JP, Barbacanne MA, Margeat E, Maret A, Nepveu F, Arnal JF. Electron spin resonance detection of extracellular superoxide anion released by cultured endothelial cells. Free Radic Res. 1998;29:441–449. doi: 10.1080/10715769800300491. [DOI] [PubMed] [Google Scholar]
- 188.Kaplan-Albuquerque N, Bogaert YE, Van Putten V, Weiser-Evans MC, Nemenoff RA. Patterns of gene expression differentially regulated by platelet-derived growth factor and hypertrophic stimuli in vascular smooth muscle cells: markers for phenotypic modulation and response to injury. J Biol Chem. 2005;280:19966–19976. doi: 10.1074/jbc.M500917200. [DOI] [PubMed] [Google Scholar]
- 189.Sturrock A, Cahill B, Norman K, Huecksteadt TP, Hill K, Sanders K, Karwande SV, Stringham JC, Bull DA, Gleich M, Kennedy TP, Hoidal JR. Transforming growth factor-beta1 induces Nox4 NAD(P)H oxidase and reactive oxygen species-dependent proliferation in human pulmonary artery smooth muscle cells. Am J Physiol Lung Cell Mol Physiol. 2006;290:L661–L673. doi: 10.1152/ajplung.00269.2005. [DOI] [PubMed] [Google Scholar]
- 190.Lassegue B, Sorescu D, Szocs K, Yin Q, Akers M, Zhang Y, Grant SL, Lambeth JD, Griendling KK. Novel gp91(phox) homologues in vascular smooth muscle cells: nox1 mediates angiotensin II-induced superoxide formation and redox-sensitive signaling pathways. Circ Res. 2001;88:888–894. doi: 10.1161/hh0901.090299. [DOI] [PubMed] [Google Scholar]
- 191.Carmona-Cuenca I, Herrera B, Ventura JJ, Roncero C, Fernandez M, Fabregat I. EGF blocks NADPH oxidase activation byTGF-beta in fetal rat hepatocytes, impairing oxidative stress, and cell death. J Cell Physiol. 2006;207:322–330. doi: 10.1002/jcp.20568. [DOI] [PubMed] [Google Scholar]
- 192.Petry A, Weitnauer M, Gorlach A. Receptor activation of NADPH oxidases. Antioxid Redox Signal. 13:467–487. doi: 10.1089/ars.2009.3026. [DOI] [PubMed] [Google Scholar]
- 193.Zhang L, Sheppard OR, Shah AM, Brewer AC. Positive regulation of the NADPH oxidase NOX4 promoter in vascular smooth muscle cells by E2F. Free Radic Biol Med. 2008;45:679–685. doi: 10.1016/j.freeradbiomed.2008.05.019. [DOI] [PubMed] [Google Scholar]
- 194.Yamagishi S, Nakamura K, Ueda S, Kato S, Imaizumi T. Pigment epithelium-derived factor (PEDF) blocks angiotensin II signaling in endothelial cells via suppression of NADPH oxidase: a novel anti-oxidative mechanism of PEDF. Cell Tissue Res. 2005;320:437–445. doi: 10.1007/s00441-005-1094-8. [DOI] [PubMed] [Google Scholar]
- 195.Adachi T, Pimentel DR, Heibeck T, Hou X, Lee YJ, Jiang B, Ido Y, Cohen RA. S-glutathiolation of Ras mediates redox-sensitive signaling by angiotensin II in vascular smooth muscle cells. J Biol Chem. 2004;279:29857–29862. doi: 10.1074/jbc.M313320200. [DOI] [PubMed] [Google Scholar]
- 196.Ushio-Fukai M, Griendling KK, Becker PL, Hilenski H, Halleran S, Alexander RW. Epidermal growth factor receptor transactivation by angiotensin II requires reactive oxygen species in vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 2001;21:489–495. doi: 10.1161/01.atv.21.4.489. [DOI] [PubMed] [Google Scholar]
- 197.Ushio-Fukai M, Alexander RW, Akers M, Griendling KK. p38 mitogen-activated protein kinase is a critical component of the redox-sensitive signaling pathways activated by angiotensin II. Role in vascular smooth muscle cell hypertrophy. J Biol Chem. 1998;273:1502–5029. doi: 10.1074/jbc.273.24.15022. [DOI] [PubMed] [Google Scholar]
- 198.Touyz RM, Yao G, Viel E, Amiri F, Schiffrin EL. Angiotensin II and endothelin-1 regulate MAP kinases through different redox-dependent mechanisms in human vascular smooth muscle cells. J Hypertens. 2004;22:114–149. doi: 10.1097/00004872-200406000-00015. [DOI] [PubMed] [Google Scholar]
- 199.Peshavariya H, Dusting GJ, Jiang F, Halmos LR, Sobey CG, Drummond GR, Selemidis S. NADPH oxidase isoform selective regulation of endothelial cell proliferation and survival. Naunyn Schmiedebergs Arch Pharmacol. 2009;380:193–204. doi: 10.1007/s00210-009-0413-0. [DOI] [PubMed] [Google Scholar]
- 200.Heneberg P, Draber P. Regulation of cys-based protein tyrosine phosphatases via reactive oxygen and nitrogen species in mast cells and basophils. Curr Med Chem. 2005;12:1859–1871. doi: 10.2174/0929867054546636. [DOI] [PubMed] [Google Scholar]
- 201.Fan J, Frey RS, Rahman A, Malik AB. Role of neutrophil NADPH oxidase in the mechanism of tumor necrosis factor-a-induced NF-kB activation and intercellular adhesion molecule-1 expression in endothelial cells. Journal of Biological Chemistry. 2002;277:3404–3411. doi: 10.1074/jbc.M110054200. [DOI] [PubMed] [Google Scholar]
- 202.Zhang M, Kho AL, Anilkumar N, Chibber R, Pagano PJ, Shah AM, Cave AC. Glycated proteins stimulate reactive oxygen species production in cardiac myocytes: involvement of Nox2 (gp91phox)-containing NADPH oxidase. Circulation. 2006;113:1235–1243. doi: 10.1161/CIRCULATIONAHA.105.581397. [DOI] [PubMed] [Google Scholar]
- 203.Clempus RE, Sorescu D, Dikalova AE, Pounkova L, Jo P, Sorescu GP, Schmidt HH, Lassegue B, Griendling KK. Nox4 is required for maintenance of the differentiated vascular smooth muscle cell phenotype. Arterioscler Thromb Vasc Biol. 2007;27:42–48. doi: 10.1161/01.ATV.0000251500.94478.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Menshikov M, Plekhanova O, Cai H, Chalupsky K, Parfyonova Y, Bashtrikov P, Tkachuk V, Berk BC. Urokinase plasminogen activator stimulates vascular smooth muscle cell proliferation via redox-dependent pathways. Arterioscler Thromb Vasc Biol. 2006;26:801–807. doi: 10.1161/01.ATV.0000207277.27432.15. [DOI] [PubMed] [Google Scholar]
- 205.Chen K, Kirber MT, Xiao H, Yang Y, Keaney JF., Jr Regulation of ROS signal transduction by NADPH oxidase 4 localization. J Cell Biol. 2008;181:1129–1139. doi: 10.1083/jcb.200709049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Martin-Garrido A, Brown DI, Lyle AN, Dikalova A, Seidel-Rogol B, Lassegue B, San Martin A, Griendling KK. NADPH oxidase 4 mediates TGF-beta-induced smooth muscle alpha-actin via p38MAPK and serum response factor. Free Radic Biol Med. 50:354–362. doi: 10.1016/j.freeradbiomed.2010.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Lee MY, San Martin A, Mehta PK, Dikalova AE, Garrido AM, Datla SR, Lyons E, Krause KH, Banfi B, Lambeth JD, Lassegue B, Griendling KK. Mechanisms of vascular smooth muscle NADPH oxidase 1 (Nox1) contribution to injury-induced neointimal formation. Arterioscler Thromb Vasc Biol. 2009;29:480–487. doi: 10.1161/ATVBAHA.108.181925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Harrison DG, Gongora MC, Guzik TJ, Widder J. Oxidative stress and hypertension. J Am Soc Hypertens. 2007;1:30–44. doi: 10.1016/j.jash.2006.11.006. [DOI] [PubMed] [Google Scholar]
- 209.Sedeek M, Hebert RL, Kennedy CR, Burns KD, Touyz RM. Molecular mechanisms of hypertension: role of Nox family NADPH oxidases. Curr Opin Nephrol Hypertens. 2009;18:122–127. doi: 10.1097/MNH.0b013e32832923c3. [DOI] [PubMed] [Google Scholar]
- 210.Addabbo F, Montagnani M, Goligorsky MS. Mitochondria and reactive oxygen species. Hypertension. 2009;53:885–892. doi: 10.1161/HYPERTENSIONAHA.109.130054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Modlinger P, Chabrashvili T, Gill PS, Mendonca M, Harrison DG, Griendling KK, Li M, Raggio J, Wellstein A, Chen Y, Welch WJ, Wilcox CS. RNA silencing in vivo reveals role of p22phox in rat angiotensin slow pressor response. Hypertension. 2006;47:238–244. doi: 10.1161/01.HYP.0000200023.02195.73. [DOI] [PubMed] [Google Scholar]
- 212.Rajagopalan S, Kurz S, Münzel T, Tarpey M, Freeman BA, Griendling KK, Harrison DG. Angiotensin II-mediated hypertension in the rat increases vascular superoxide production via membrane NADH/NADPH oxidase activation. Contribution to alterations of vasomotor tone. J Clin Invest. 1996;97:1916–1923. doi: 10.1172/JCI118623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Liu J, Ormsby A, Oja-Tebbe N, Pagano PJ. Gene transfer of NAD(P)H oxidase inhibitor to the vascular adventitia attenuates medial smooth muscle hypertrophy. Circ Res. 2004;95:587–594. doi: 10.1161/01.RES.0000142317.88591.e6. [DOI] [PubMed] [Google Scholar]
- 214.Chen X, Touyz RM, Park JB, Schiffrin EL. Antioxidant effects of vitamins C and E are associated with altered activation of vascular NADPH oxidase and superoxide dismutase in stroke-prone SHR. Hypertension. 2001;38:606–611. doi: 10.1161/hy09t1.094005. [DOI] [PubMed] [Google Scholar]
- 215.Duffy SJ, Gokce N, Holbrook M, Huang A, Frei B, Keaney JF, Jr, Vita JA. Treatment of hypertension with ascorbic acid. Lancet. 1999;354:2048–2049. doi: 10.1016/s0140-6736(99)04410-4. [DOI] [PubMed] [Google Scholar]
- 216.Lacy F, Kailasam MT, O’Connor DT, Schmid-Sch”nbein GW, Parmer RJ. Plasma hydrogen peroxide production in human essential hypertension. Role of heredity, gender, and ethnicity. Hypertension. 2000;36:878–884. doi: 10.1161/01.hyp.36.5.878. [DOI] [PubMed] [Google Scholar]
- 217.Chen J, He J, Hamm L, Batuman V, Whelton PK. Serum antioxidant vitamins and blood pressure in the United States population. Hypertension. 2002;40:810–816. doi: 10.1161/01.hyp.0000039962.68332.59. [DOI] [PubMed] [Google Scholar]
- 218.Hasnain BI, Mooradian AD. Recent trials of antioxidant therapy: what should we be telling our patients? Cleve Clin J Med. 2004;71:327–334. doi: 10.3949/ccjm.71.4.327. [DOI] [PubMed] [Google Scholar]
- 219.Van der Zwan LP, Scheffer PG, Dekker JM, Stehouwer CD, Heine RJ, Teerlink T. Hyperglycemia and oxidative stress strengthen the association between myeloperoxidase and blood pressure. Hypertension. 55:1366–1372. doi: 10.1161/HYPERTENSIONAHA.109.147231. [DOI] [PubMed] [Google Scholar]
- 220.Fortuno A, Bidegain J, Baltanas A, Moreno MU, Montero L, Landecho MF, Beloqui O, Diez J, Zalba G. Is leptin involved in phagocytic NADPH oxidase overactivity in obesity? Potential clinical implications. J Hypertens. 28:194–950. doi: 10.1097/HJH.0b013e32833c21af. [DOI] [PubMed] [Google Scholar]
- 221.Gutierrez J, Ballinger SW, Darley-Usmar VM, Landar A. Free radicals, mitochondria, and oxidized lipids: the emerging role in signal transduction in vascular cells. Circ Res. 2006;99:924–932. doi: 10.1161/01.RES.0000248212.86638.e9. [DOI] [PubMed] [Google Scholar]
- 222.Houston MC. Nutrition and nutraceutical supplements in the treatment of hypertension. Expert Rev Cardiovasc Ther. 8:821–833. doi: 10.1586/erc.10.63. [DOI] [PubMed] [Google Scholar]
- 223.Kelly RP, Poo Yeo K, Isaac HB, Lee CY, Huang SH, Teng L, Halliwell B, Wise SD. Lack of effect of acute oral ingestion of vitamin C on oxidative stress, arterial stiffness or blood pressure in healthy subjects. Free Radic Res. 2008;42:514–522. doi: 10.1080/10715760802087431. [DOI] [PubMed] [Google Scholar]
- 224.Cave A. Selective targeting of NADPH oxidase for cardiovascular protection. Curr Opin Pharmacol. 2009;9:208–213. doi: 10.1016/j.coph.2008.10.001. [DOI] [PubMed] [Google Scholar]
- 225.Weseler AR, Bast A. Oxidative stress and vascular function: implications for pharmacologic treatments. Curr Hypertens Rep. 12:154–161. doi: 10.1007/s11906-010-0103-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Goettsch C, Goettsch W, Arsov A, Hofbauer LC, Bornstein SR, Morawietz H. Long-term cyclic strain downregulates endothelial Nox4. Antioxid Redox Signal. 2009;11:2385–2397. doi: 10.1089/ars.2009.2561. [DOI] [PubMed] [Google Scholar]
- 227.Datla SR, Peshavariya H, Dusting GJ, Jiang F. Important Role of Nox4 Type NADPH Oxidase in Angiogenic Responses in Human Microvascular Endothelial Cells In Vitro. Arteriosclerosis, Thrombosis, and Vascular Biology:ATVBAHA. 2007 doi: 10.1161/ATVBAHA.107.149450. [DOI] [PubMed] [Google Scholar]
- 228.Lener B, Koziel R, Pircher H, Hutter E, Greussing R, Herndler-Brandstetter D, Hermann M, Unterluggauer H, Jansen-Durr P. The NADPH oxidase Nox4 restricts the replicative lifespan of human endothelial cells. Biochem J. 2009;423:363–374. doi: 10.1042/BJ20090666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Kleinschnitz C, Grund H, Wingler K, Armitage ME, Jones E, Mittal M, Barit D, Schwarz T, Geis C, Kraft P, Barthel K, Schuhmann MK, Herrmann AM, Meuth SG, Stoll G, Meurer S, Schrewe A, Becker L, Gailus-Durner V, Fuchs H, Klopstock T, de Angelis MH, Jandeleit-Dahm K, Shah AM, Weissmann N, Schmidt HH. Post-stroke inhibition of induced NADPH oxidase type 4 prevents oxidative stress and neurodegeneration. PLoS Biol. :8. doi: 10.1371/journal.pbio.1000479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Zhang M, Brewer AC, Schroder K, Santos CX, Grieve DJ, Wang M, Anilkumar N, Yu B, Dong X, Walker SJ, Brandes RP, Shah AM. NADPH oxidase-4 mediates protection against chronic load-induced stress in mouse hearts by enhancing angiogenesis. Proc Natl Acad Sci U S A. 2010;107:18121–18126. doi: 10.1073/pnas.1009700107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Tirone F, Radu L, Craescu CT, Cox JA. Identification of the binding site for the regulatory calcium-binding domain in the catalytic domain of NOX5. Biochemistry. 49:761–771. doi: 10.1021/bi901846y. [DOI] [PubMed] [Google Scholar]
- 232.Guzik TJ, Chen W, Gongora MC, Guzik B, Lob HE, Mangalat D, Hoch N, Dikalov S, Rudzinski P, Kapelak B, Sadowski J, Harrison DG. Calcium-dependent NOX5 nicotinamide adenine dinucleotide phosphate oxidase contributes to vascular oxidative stress in human coronary artery disease. J Am Coll Cardiol. 2008;52:1803–1809. doi: 10.1016/j.jacc.2008.07.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Fu X, Beer DG, Behar J, Wands J, Lambeth D, Cao W. cAMP-response element-binding protein mediates acid-induced NADPH oxidase NOX5-S expression in Barrett esophageal adenocarcinoma cells. J Biol Chem. 2006;281:20368–20382. doi: 10.1074/jbc.M603353200. [DOI] [PubMed] [Google Scholar]
- 234.Montezano AC, Burger D, Paravicini TM, Chignalia AZ, Yusuf H, Almasri M, He Y, Callera GE, He G, Krause KH, Lambeth D, Quinn MT, Touyz RM. Nicotinamide adenine dinucleotide phosphate reduced oxidase 5 (Nox5) regulation by angiotensin II and endothelin-1 is mediated via calcium/calmodulin-dependent, rac-1-independent pathways in human endothelial cells. Circ Res. 106:1363–1373. doi: 10.1161/CIRCRESAHA.109.216036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Thannickal VJ, Toews GB, White ES, Lynch JP, 3rd, Martinez FJ. Mechanisms of pulmonary fibrosis. Annu Rev Med. 2004;55:395–417. doi: 10.1146/annurev.med.55.091902.103810. [DOI] [PubMed] [Google Scholar]
- 236.American Thoracic Society. Idiopathic pulmonary fibrosis: diagnosis and treatment. International consensus statement. American Thoracic Society (ATS), and the European Respiratory Society (ERS) Am J Respir Crit Care Med. 2000;161:646–664. doi: 10.1164/ajrccm.161.2.ats3-00. [DOI] [PubMed] [Google Scholar]
- 237.Kinnula VL, Fattman CL, Tan RJ, Oury TD. Oxidative stress in pulmonary fibrosis: a possible role for redox modulatory therapy. Am J Respir Crit Care Med. 2005;172:417–422. doi: 10.1164/rccm.200501-017PP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Manoury B, Nenan S, Leclerc O, Guenon I, Boichot E, Planquois JM, Bertrand CP, Lagente V. The absence of reactive oxygen species production protects mice against bleomycin-induced pulmonary fibrosis. Respir Res. 2005;6:11. doi: 10.1186/1465-9921-6-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Shvedova AA, Kisin ER, Murray AR, Kommineni C, Castranova V, Fadeel B, Kagan VE. Increased accumulation of neutrophils and decreased fibrosis in the lung of NADPH oxidase-deficient C57BL/6 mice exposed to carbon nanotubes. Toxicol Appl Pharmacol. 2008;231:235–240. doi: 10.1016/j.taap.2008.04.018. [DOI] [PubMed] [Google Scholar]
- 240.Cantin AM, North SL, Fells GA, Hubbard RC, Crystal RG. Oxidant-mediated epithelial cell injury in idiopathic pulmonary fibrosis. J Clin Invest. 1987;79:1665–1673. doi: 10.1172/JCI113005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Strausz J, Muller-Quernheim J, Steppling H, Ferlinz R. Oxygen radical production by alveolar inflammatory cells in idiopathic pulmonary fibrosis. Am Rev Respir Dis. 1990;141:124–128. doi: 10.1164/ajrccm/141.1.124. [DOI] [PubMed] [Google Scholar]
- 242.Hecker L, Vittal R, Jones T, Jagirdar R, Luckhardt TR, Horowitz JC, Pennathur S, Martinez FJ, Thannickal VJ. NADPH oxidase-4 mediates myofibroblast activation and fibrogenic responses to lung injury. Nat Med. 2009;15:1077–1081. doi: 10.1038/nm.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Amara N, Goven D, Prost F, Muloway R, Crestani B, Boczkowski J. NOX4/NADPH oxidase expression is increased in pulmonary fibroblasts from patients with idiopathic pulmonary fibrosis and mediates TGFbeta1-induced fibroblast differentiation into myofibroblasts. Thorax. 65:733–738. doi: 10.1136/thx.2009.113456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Lu X, Murphy TC, Nanes MS, Hart CM. PPAR{gamma} regulates hypoxia-induced Nox4 expression in human pulmonary artery smooth muscle cells through NF-{kappa}B. Am J Physiol Lung Cell Mol Physiol. 299:L559–566. doi: 10.1152/ajplung.00090.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Pache JC, Carnesecchi S, Deffert C, Donati Y, Herrmann FR, Barazzone-Argiroffo C, Krause KH. NOX-4 is expressed in thickened pulmonary arteries in idiopathic pulmonary fibrosis. Nat Med. 17:31–32. doi: 10.1038/nm0111-31. author reply 3–3. [DOI] [PubMed] [Google Scholar]
- 246.Hecker L, Crowe DR, Thannickal VJ. Reply to: “NOX-4 is expressed in thickened pulmonary arteries in idiopathic pulmonary fibrosis”. Nat Med. 2011;17:32–33. doi: 10.1038/nm0111-31. [DOI] [PubMed] [Google Scholar]
- 247.Mittal M, Roth M, Konig P, Hofmann S, Dony E, Goyal P, Selbitz AC, Schermuly RT, Ghofrani HA, Kwapiszewska G, Kummer W, Klepetko W, Hoda MA, Fink L, Hanze J, Seeger W, Grimminger F, Schmidt HH, Weissmann N. Hypoxia-dependent regulation of nonphagocytic NADPH oxidase subunit NOX4 in the pulmonary vasculature. Circ Res. 2007;101:258–267. doi: 10.1161/CIRCRESAHA.107.148015. [DOI] [PubMed] [Google Scholar]
- 248.States JC, Srivastava S, Chen Y, Barchowsky A. Arsenic and cardiovascular disease. Toxicol Sci. 2009;107:312–323. doi: 10.1093/toxsci/kfn236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Smith KR, Klei LR, Barchowsky A. Arsenite stimulates plasma membrane NADPH oxidase in vascular endothelial cells. Am J Physiol Lung Cell Mol Physiol. 2001;280:L442–L449. doi: 10.1152/ajplung.2001.280.3.L442. [DOI] [PubMed] [Google Scholar]
- 250.Lynn S, Gurr JR, Lai HT, Jan KY. NADH oxidase activation is involved in arsenite-induced oxidative DNA damage in human vascular smooth muscle cells. Circ Res. 2000;86:514–519. doi: 10.1161/01.res.86.5.514. [DOI] [PubMed] [Google Scholar]
- 251.Barchowsky A, Klei LR, Dudek EJ, Swartz HM, James PE. Stimulation of reactive oxygen, but not reactive nitrogen species, in vascular endothelial cells exposed to low levels of arsenite. Free Radic Biol Med. 1999;27:1405–1412. doi: 10.1016/s0891-5849(99)00186-0. [DOI] [PubMed] [Google Scholar]
- 252.Bunderson M, Coffin JD, Beall HD. Arsenic induces peroxynitrite generation and cyclooxygenase-2 protein expression in aortic endothelial cells: possible role in atherosclerosis. Toxicol Appl Pharmacol. 2002;184:11–18. [PubMed] [Google Scholar]
- 253.Straub AC, Clark KA, Ross MA, Chandra AG, Li S, Gao X, Pagano PJ, Stolz DB, Barchowsky A. Arsenic-stimulated liver sinusoidal capillarization in mice requires NADPH oxidase-generated superoxide. J Clin Invest. 2008;118:3980–3989. doi: 10.1172/JCI35092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Straub AC, Stolz DB, Ross MA, Hernandez-Zavala A, Soucy NV, Klei LR, Barchowsky A. Arsenic stimulates sinusoidal endothelial cell capillarization and vessel remodeling in mouse liver. Hepatology. 2007;45:205–212. doi: 10.1002/hep.21444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Straub AC, Klei LR, Stolz DB, Barchowsky A. Arsenic requires sphingosine-1-phosphate type 1 receptors to induce angiogenic genes and endothelial cell remodeling. Am J Pathol. 2009;174:1949–1958. doi: 10.2353/ajpath.2009.081016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Faurschou M, Borregaard N. Neutrophil granules and secretory vesicles in inflammation. Microbes Infect. 2003;5:1317–1327. doi: 10.1016/j.micinf.2003.09.008. [DOI] [PubMed] [Google Scholar]
- 257.Nauseef WM. How human neutrophils kill and degrade microbes: an integrated view. Immunol Rev. 2007;219:88–102. doi: 10.1111/j.1600-065X.2007.00550.x. [DOI] [PubMed] [Google Scholar]
- 258.Schultz J, Kaminker K. Myeloperoxidase of the leucocyte of normal human blood. I. Content and localization. Arch Biochem Biophys. 1962;96:465–467. doi: 10.1016/0003-9861(62)90321-1. [DOI] [PubMed] [Google Scholar]
- 259.Winterbourn CC, Hampton MB, Livesey JH, Kettle AJ. Modeling the reactions of superoxide and myeloperoxidase in the neutrophil phagosome: implications for microbial killing. J Biol Chem. 2006;281:39860–39869. doi: 10.1074/jbc.M605898200. [DOI] [PubMed] [Google Scholar]
- 260.Klebanoff SJ. Myeloperoxidase: friend and foe. J Leukoc Biol. 2005;77:598–625. doi: 10.1189/jlb.1204697. [DOI] [PubMed] [Google Scholar]
- 261.Painter RG, Wang G. Direct measurement of free chloride concentrations in the phagolysosomes of human neutrophils. Anal Chem. 2006;78:3133–3137. doi: 10.1021/ac0521706. [DOI] [PubMed] [Google Scholar]
- 262.Painter RG, Marrero L, Lombard GA, Valentine VG, Nauseef WM, Wang G. CFTR-mediated halide transport in phagosomes of human neutrophils. J Leukoc Biol. 87:933–942. doi: 10.1189/jlb.1009655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Painter RG, Bonvillain RW, Valentine VG, Lombard GA, LaPlace SG, Nauseef WM, Wang G. The role of chloride anion and CFTR in killing of Pseudomonas aeruginosa by normal and CF neutrophils. J Leukoc Biol. 2008;83:1345–1353. doi: 10.1189/jlb.0907658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Thomas EL, Grisham MB, Jefferson MM. Myeloperoxidase-dependent effect of amines on functions of isolated neutrophils. J Clin Invest. 1983;72:441–454. doi: 10.1172/JCI110992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Chapman AL, Senthilmohan R, Winterbourn CC, Kettle AJ. Comparison of mono- and dichlorinated tyrosines with carbonyls for detection of hypochlorous acid modified proteins. Arch Biochem Biophys. 2000;377:95–100. doi: 10.1006/abbi.2000.1744. [DOI] [PubMed] [Google Scholar]
- 266.Rosen H, Crowley JR, Heinecke JW. Human neutrophils use the myeloperoxidase-hydrogen peroxide-chloride system to chlorinate but not nitrate bacterial proteins during phagocytosis. J Biol Chem. 2002;277:30463–30468. doi: 10.1074/jbc.M202331200. [DOI] [PubMed] [Google Scholar]
- 267.Gattas MV, Forteza R, Fragoso MA, Fregien N, Salas P, Salathe M, Conner GE. Oxidative epithelial host defense is regulated by infectious and inflammatory stimuli. Free Radic Biol Med. 2009;47:1450–1458. doi: 10.1016/j.freeradbiomed.2009.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Moskwa P, Lorentzen D, Excoffon KJ, Zabner J, McCray PB, Jr, Nauseef WM, Dupuy C, Banfi B. A novel host defense system of airways is defective in cystic fibrosis. Am J Respir Crit Care Med. 2007;175:174–183. doi: 10.1164/rccm.200607-1029OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Forteza R, Salathe M, Miot F, Conner GE. Regulated hydrogen peroxide production by Duox in human airway epithelial cells. Am J Respir Cell Mol Biol. 2005;32:462–469. doi: 10.1165/rcmb.2004-0302OC. [DOI] [PubMed] [Google Scholar]
- 270.Wijkstrom-Frei C, El-Chemaly S, Ali-Rachedi R, Gerson C, Cobas MA, Forteza R, Salathe M, Conner GE. Lactoperoxidase and human airway host defense. Am J Respir Cell Mol Biol. 2003;29:206–212. doi: 10.1165/rcmb.2002-0152OC. [DOI] [PubMed] [Google Scholar]
- 271.Salathe M, Holderby M, Forteza R, Abraham WM, Wanner A, Conner GE. Isolation and characterization of a peroxidase from the airway. Am J Respir Cell Mol Biol. 1997;17:97–105. doi: 10.1165/ajrcmb.17.1.2719. [DOI] [PubMed] [Google Scholar]
- 272.Conner GE, Salathe M, Forteza R. Lactoperoxidase and hydrogen peroxide metabolism in the airway. Am J Respir Crit Care Med. 2002;166:S57–61. doi: 10.1164/rccm.2206018. [DOI] [PubMed] [Google Scholar]
- 273.Conner GE, Wijkstrom-Frei C, Randell SH, Fernandez VE, Salathe M. The lactoperoxidase system links anion transport to host defense in cystic fibrosis. FEBS Lett. 2007;581:271–278. doi: 10.1016/j.febslet.2006.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Klebanoff SJ, Clem WH, Luebke RG. The peroxidase-thiocyanate-hydrogen peroxide antimicrobial system. Biochim Biophys Acta. 1966;117:63–72. doi: 10.1016/0304-4165(66)90152-8. [DOI] [PubMed] [Google Scholar]
- 275.Thomas EL, Aune TM. Lactoperoxidase, peroxide, thiocyanate antimicrobial system: correlation of sulfhydryl oxidation with antimicrobial action. Infect Immun. 1978;20:456–463. doi: 10.1128/iai.20.2.456-463.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Pattison DI, Davies MJ. Absolute rate constants for the reaction of hypochlorous acid with protein side chains and peptide bonds. Chem Res Toxicol. 2001;14:1453–1464. doi: 10.1021/tx0155451. [DOI] [PubMed] [Google Scholar]
- 277.Winterbourn CC, Hampton MB. Thiol chemistry and specificity in redox signaling. Free Radic Biol Med. 2008;45:549–561. doi: 10.1016/j.freeradbiomed.2008.05.004. [DOI] [PubMed] [Google Scholar]
- 278.Imlay JA. Pathways of oxidative damage. Annu Rev Microbiol. 2003;57:395–418. doi: 10.1146/annurev.micro.57.030502.090938. [DOI] [PubMed] [Google Scholar]
- 279.Arlandson M, Decker T, Roongta VA, Bonilla L, Mayo KH, MacPherson JC, Hazen SL, Slungaard A. Eosinophil peroxidase oxidation of thiocyanate. Characterization of major reaction products and a potential sulfhydryl-targeted cytotoxicity system. J Biol Chem. 2001;276:215–224. doi: 10.1074/jbc.M004881200. [DOI] [PubMed] [Google Scholar]
- 280.Grisham MB, Ryan EM. Cytotoxic properties of salivary oxidants. Am J Physiol. 1990;258:C115–121. doi: 10.1152/ajpcell.1990.258.1.C115. [DOI] [PubMed] [Google Scholar]
- 281.Hawkins CL. The role of hypothiocyanous acid (HOSCN) in biological systems. Free Radic Res. 2009;43:1147–1158. doi: 10.3109/10715760903214462. [DOI] [PubMed] [Google Scholar]
- 282.Xu Y, Szep S, Lu Z. The antioxidant role of thiocyanate in the pathogenesis of cystic fibrosis and other inflammation-related diseases. Proc Natl Acad Sci U S A. 2009;106:20515–20519. doi: 10.1073/pnas.0911412106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Ashby MT, Carlson AC, Scott MJ. Redox buffering of hypochlorous acid by thiocyanate in physiologic fluids. J Am Chem Soc. 2004;126:15976–15977. doi: 10.1021/ja0438361. [DOI] [PubMed] [Google Scholar]
- 284.Fridovich I. Quantitative aspects of the production of superoxide anion radical by milk xanthine oxidase. J Biol Chem. 1970;245:4053–4057. [PubMed] [Google Scholar]
- 285.Kelley EE, Khoo NK, Hundley NJ, Malik UZ, Freeman BA, Tarpey MM. Hydrogen peroxide is the major oxidant product of xanthine oxidase. Free Radic Biol Med. 48:493–498. doi: 10.1016/j.freeradbiomed.2009.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Cai H. Hydrogen peroxide regulation of endothelial function: origins, mechanisms, and consequences. Cardiovasc Res. 2005;68:26–36. doi: 10.1016/j.cardiores.2005.06.021. [DOI] [PubMed] [Google Scholar]
- 287.Godber BL, Doel JJ, Sapkota GP, Blake DR, Stevens CR, Eisenthal R, Harrison R. Reduction of nitrite to nitric oxide catalyzed by xanthine oxidoreductase. J Biol Chem. 2000;275:7757–7763. doi: 10.1074/jbc.275.11.7757. [DOI] [PubMed] [Google Scholar]
- 288.Millar TM, Stevens CR, Benjamin N, Eisenthal R, Harrison R, Blake DR. Xanthine oxidoreductase catalyses the reduction of nitrates and nitrite to nitric oxide under hypoxic conditions. FEBS Lett. 1998;427:225–228. doi: 10.1016/s0014-5793(98)00430-x. [DOI] [PubMed] [Google Scholar]
- 289.Li H, Samouilov A, Liu X, Zweier JL. Characterization of the magnitude and kinetics of xanthine oxidase-catalyzed nitrite reduction. Evaluation of its role in nitric oxide generation in anoxic tissues. J Biol Chem. 2001;276:24482–24489. doi: 10.1074/jbc.M011648200. [DOI] [PubMed] [Google Scholar]
- 290.Kleinbongard P, Dejam A, Lauer T, Rassaf T, Schindler A, Picker O, Scheeren T, Godecke A, Schrader J, Schulz R, Heusch G, Schaub GA, Bryan NS, Feelisch M, Kelm M. Plasma nitrite reflects constitutive nitric oxide synthase activity in mammals. Free Radic Biol Med. 2003;35:790–796. doi: 10.1016/s0891-5849(03)00406-4. [DOI] [PubMed] [Google Scholar]
- 291.Taille C, El-Benna J, Lanone S, Boczkowski J, Motterlini R. Mitochondrial respiratory chain and NAD(P)H oxidase are targets for the antiproliferative effect of carbon monoxide in human airway smooth muscle. J Biol Chem. 2005;280:25350–25360. doi: 10.1074/jbc.M503512200. [DOI] [PubMed] [Google Scholar]
- 292.Kwak JY, Takeshige K, Cheung BS, Minakami S. Bilirubin inhibits the activation of superoxide-producing NADPH oxidase in a neutrophil cell-free system. Biochim Biophys Acta. 1991;1076:369–373. doi: 10.1016/0167-4838(91)90478-i. [DOI] [PubMed] [Google Scholar]
- 293.Nakahira K, Kim HP, Geng XH, Nakao A, Wang X, Murase N, Drain PF, Sasidhar M, Nabel EG, Takahashi T, Lukacs NW, Ryter SW, Morita K, Choi AM. Carbon monoxide differentially inhibits TLR signaling pathways by regulating ROS-induced trafficking of TLRs to lipid rafts. J Exp Med. 2006;203:2377–2389. doi: 10.1084/jem.20060845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Rodriguez AI, Gangopadhyay A, Kelley EE, Pagano PJ, Zuckerbraun BS, Bauer PM. HO-1 and CO decrease platelet-derived growth factor-induced vascular smooth muscle cell migration via inhibition of Nox1. Arterioscler Thromb Vasc Biol. 30:98–104. doi: 10.1161/ATVBAHA.109.197822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Clowes AW, Clowes MM, Fingerle J, Reidy MA. Regulation of smooth muscle cell growth in injured artery. J Cardiovasc Pharmacol. 1989;14(Suppl 6):S12–15. [PubMed] [Google Scholar]
- 296.Santos JH, Meyer JN, Van Houten B. Mitochondrial localization of telomerase as a determinant for hydrogen peroxide-induced mitochondrial DNA damage and apoptosis. Hum Mol Genet. 2006;15:1757–1768. doi: 10.1093/hmg/ddl098. [DOI] [PubMed] [Google Scholar]
- 297.Dai DF, Rabinovitch PS. Cardiac aging in mice and humans: the role of mitochondrial oxidative stress. Trends Cardiovasc Med. 2009;19:213–220. doi: 10.1016/j.tcm.2009.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Scolletta S, Biagioli B. Energetic myocardial metabolism and oxidative stress: let’s make them our friends in the fight against heart failure. Biomed Pharmacother. 64:203–207. doi: 10.1016/j.biopha.2009.10.002. [DOI] [PubMed] [Google Scholar]
- 299.Dranka BP, Hill BG, Darley-Usmar VM. Mitochondrial reserve capacity in endothelial cells: The impact of nitric oxide and reactive oxygen species. Free Radic Biol Med. :905–914. doi: 10.1016/j.freeradbiomed.2010.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Hill BG, Dranka BP, Zou L, Chatham JC, Darley-Usmar VM. Importance of the bioenergetic reserve capacity in response to cardiomyocyte stress induced by 4-hydroxynonenal. Biochem J. 2009;424:99–107. doi: 10.1042/BJ20090934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Belikova NA, Vladimirov YA, Osipov AN, Kapralov AA, Tyurin VA, Potapovich MV, Basova LV, Peterson J, Kurnikov IV, Kagan VE. Peroxidase activity and structural transitions of cytochrome c bound to cardiolipin-containing membranes. Biochemistry. 2006;45:4998–5009. doi: 10.1021/bi0525573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Vlasova, Tyurin VA, Kapralov AA, Kurnikov IV, Osipov AN, Potapovich MV, Stoyanovsky DA, Kagan VE. Nitric oxide inhibits peroxidase activity of cytochrome c.cardiolipin complex and blocks cardiolipin oxidation. J Biol Chem. 2006;281:14554–14562. doi: 10.1074/jbc.M509507200. [DOI] [PubMed] [Google Scholar]
- 303.Groeger AL, Cipollina C, Cole MP, Woodcock SR, Bonacci G, Rudolph TK, Rudolph V, Freeman BA, Schopfer FJ. Cyclooxygenase-2 generates anti-inflammatory mediators from omega-3 fatty acids. Nat Chem Biol. 2010;6:433–441. doi: 10.1038/nchembio.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.Freeman BA, Baker PR, Schopfer FJ, Woodcock SR, Napolitano A, d’Ischia M. Nitro-fatty acid formation and signaling. J Biol Chem. 2008;283:15515–15519. doi: 10.1074/jbc.R800004200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305.Villacorta L, Zhang J, Garcia-Barrio MT, Chen XL, Freeman BA, Chen YE, Cui T. Nitro-linoleic acid inhibits vascular smooth muscle cell proliferation via the Keap1/Nrf2 signaling pathway. Am J Physiol Heart Circ Physiol. 2007;293:H770–H776. doi: 10.1152/ajpheart.00261.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Cui T, Schopfer FJ, Zhang J, Chen K, Ichikawa T, Baker PR, Batthyany C, Chacko BK, Feng X, Patel RP, Agarwal A, Freeman BA, Chen YE. Nitrated fatty acids: Endogenous anti-inflammatory signaling mediators. J Biol Chem. 2006;281:35686–35698. doi: 10.1074/jbc.M603357200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Schopfer FJ, Lin Y, Baker PR, Cui T, Garcia-Barrio M, Zhang J, Chen K, Chen YE, Freeman BA. Nitrolinoleic acid: an endogenous peroxisome proliferator-activated receptor gamma ligand. Proc Natl Acad Sci USA. 2005;102:2340–2345. doi: 10.1073/pnas.0408384102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.Kansanen E, Jyrkkanen HK, Volger OL, Leinonen H, Kivela AM, Hakkinen SK, Woodcock SR, Schopfer FJ, Horrevoets AJ, Yla-Herttuala S, Freeman BA, Levonen AL. Nrf2-dependent and -independent responses to nitro-fatty acids in human endothelial cells: identification of heat shock response as the major pathway activated by nitro-oleic acid. J Biol Chem. 2009;284:33233–33241. doi: 10.1074/jbc.M109.064873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309.Khoo NKH, Rudolph V, Cole MP, Golin-Bisllo F, Schopfer FJ, Woodcock SR, Batthyany C, Freeman BA. Activation of Vascular Endothelial Nitric Oxide Synthase and Heme Oxygenase-1 Expression by Electrophilc Nitro-Fatty Acids. Free Radic Biol Med. 2010;48:230–239. doi: 10.1016/j.freeradbiomed.2009.10.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310.Wright MM, Schopfer FJ, Baker PR, Vidyasagar V, Powell P, Chumley P, Iles KE, Freeman BA, Agarwal A. Fatty acid transduction of nitric oxide signaling: nitrolinoleic acid potently activates endothelial heme oxygenase 1 expression. Proc Natl Acad Sci USA. 2006;103:4299–4304. doi: 10.1073/pnas.0506541103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311.Khoo NKH, Freeman BA. Electrophilic nitro-fatty acids: anti-inflammatory mediators in the vascular compartment. Curr Opin Pharmacol. 2010;10:179–184. doi: 10.1016/j.coph.2009.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312.Li Y, Zhang J, Schopfer FJ, Martynowski D, Garcia-Barrio MT, Kovach A, Suino-Powell K, Baker PR, Freeman BA, Chen YE, Xu HE. Molecular recognition of nitrated fatty acids by PPAR gamma. Nat Struct Mol Biol. 2008;15:865–867. doi: 10.1038/nsmb.1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313.Rudolph TK, Rudolph V, Edreira MM, Cole MP, Bonacci G, Schopfer FJ, Woodcock SR, Franek A, Pekarova M, Khoo NK, Hasty AH, Baldus S, Freeman BA. Nitro-Fatty Acids Reduce Atherosclerosis in Apolipoprotein E-Deficient Mice. Arterioscler Thromb Vasc Biol. 2010;30:938–945. doi: 10.1161/ATVBAHA.109.201582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314.Cole MP, Rudolph TK, Khoo NKH, Motanya UN, Golin-Bisllo F, Wertz JW, Schopfer FJ, Woodcock SR, Ali MS, Rudolph V, Zhang J, Chen YE, Agarwal A, Freeman BA, Bauer PM. Nitro-Fatty Acid Inhibition of Neointima Formation After Endoluminal Vessel Injury. Circ Res. 2009;105:965–972. doi: 10.1161/CIRCRESAHA.109.199075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315.Rudolph V, Rudolph TK, Schopfer FJ, Bonacci G, Woodcock SR, Cole MP, Baker PR, Ramani R, Freeman BA. Endogenous generation and protective effects of nitro-fatty acids in a murine model of focal cardiac ischaemia and reperfusion. Cardiovasc Res. 2010;85:155–166. doi: 10.1093/cvr/cvp275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316.Schopfer FJ, Cole MP, Groeger AL, Chen CS, Khoo NK, Woodcock SR, Golin-Bisello F, Motanya UN, Li Y, Zhang J, Garcia-Barrio MT, Rudolph TK, Rudolph V, Bonacci G, Baker PR, Xu HE, Batthyany CI, Chen YE, Hallis TM, Freeman BA. Covalent peroxisome proliferator-activated receptor g binding by nitro-fatty acids: Endogenous ligands act as selective modulators. J Biol Chem. 2010;285:12321–12333. doi: 10.1074/jbc.M109.091512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317.Ueyama T, Geiszt M, Leto TL. Involvement of Rac1 in activation of multicomponent Nox1- and Nox3-based NADPH oxidases. MolCell Biol. 2006;26:2160–2174. doi: 10.1128/MCB.26.6.2160-2174.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318.Leto TL, Morand S, Hurt D, Ueyama T. Targeting and regulation of reactive oxygen species generation by Nox family NADPH oxidases. Antioxid Redox Signal. 2009;11:2607–2619. doi: 10.1089/ars.2009.2637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319.Serrander L, Jaquet V, Bedard K, Plastre O, Hartley O, Arnaudeau S, Demaurex N, Schlegel W, Krause KH. NOX5 is expressed at the plasma membrane and generates superoxide in response to protein kinase C activation. Biochimie. 2007;89:1159–1167. doi: 10.1016/j.biochi.2007.05.004. [DOI] [PubMed] [Google Scholar]
- 320.Kawahara T, Lambeth JD. Phosphatidylinositol (4,5)-bisphosphate modulates Nox5 localization via an N-terminal polybasic region. Mol Biol Cell. 2008;19:4020–4031. doi: 10.1091/mbc.E07-12-1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321.Brar SS, Corbin Z, Kennedy TP, Hemendinger R, Thornton L, Bommarius B, Arnold RS, Whorton AR, Sturrock AB, Huecksteadt TP, Quinn MT, Krenitsky K, Ardie KG, Lambeth JD, Hoidal JR. NOX5 NAD(P)H oxidase regulates growth and apoptosis in DU 145 prostate cancer cells. Am J Physiol Cell Physiol. 2003;285:C353–369. doi: 10.1152/ajpcell.00525.2002. [DOI] [PubMed] [Google Scholar]
- 322.Straub AC, Stolz DB, Vin H, Ross MA, Soucy NV, Klei LR, Barchowsky A. Low level arsenic promotes progressive inflammatory angiogenesis and liver blood vessel remodeling in mice. Toxicol Appl Pharmacol. 2007;222:327–336. doi: 10.1016/j.taap.2006.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]