

Abstract
T cell activation leads to engagement of cellular metabolic pathways necessary to support cell proliferation and function. However, our understanding of the signal transduction pathways that regulate metabolism and their impact on T cell function remains limited. The Liver Kinase B1 (LKB1) is a serine/threonine kinase that links cellular metabolism with cell growth and proliferation. Here we demonstrate that LKB1 is a critical regulator of T cell development, viability, activation, and metabolism. T cell-specific ablation of the gene that encodes LKB1 resulted in blocked thymocyte development and a reduction in peripheral T cells. LKB1-deficient T cells exhibited defects in cell proliferation and viability, and altered glycolytic and lipid metabolism. Interestingly, loss of LKB1 promoted increased T cell activation and inflammatory cytokine production by both CD4+ and CD8+ T cells. Activation of the AMP-activated protein kinase (AMPK) was decreased in LKB1-deficient T cells. AMPK was found to mediate a subset of LKB1 functions in T lymphocytes, as mice lacking the α1 subunit of AMPK displayed similar defects in T cell activation, metabolism, and inflammatory cytokine production, but normal T cell development and peripheral T cell homeostasis. LKB1- and AMPKα1-deficient T cells each displayed elevated mTORC1 signaling and IFN-γ production that could be reversed by rapamycin treatment. Our data highlight a central role for LKB1 in T cell activation, viability, and metabolism, and suggest that LKB1-AMPK signaling negatively regulates T cell effector function through regulation of mTOR activity.
Keywords: LKB1, AMPK, Bcl-xL, homeostasis, T cell activation, glucose metabolism, metabolic stress, IFN-γ
Introduction
T cells play a critical role in the generation of immunity and inflammatory processes. Upon T cell receptor (TCR) stimulation, T cells initiate a coordinated program of rapid proliferation and differentiation, leading to the development of activated T cells with specific effector functions tailored towards pathogen clearance or control. As part of their program of activation, T cells dramatically alter their metabolic activity to meet the increased metabolic demands of cell growth, proliferation, and effector function (1). Activated T cells generate ATP in large part by increasing their rate of glucose uptake and glycolysis (2, 3). Glucose availability is essential for the survival, proliferation, and effector function of activated T cells (4, 5). In contrast, quiescent T cells and Treg cells rely primarily on mitochondrial oxidative metabolism fueled by lipids and other metabolic substrates to maintain bioenergetic homeostasis and suppressor function (6-8). In addition, a switch from aerobic glycolysis to mitochondrial oxidative pathways is required for the establishment of CD8 T cell memory (9, 10). Thus, metabolic regulation is a crucial factor in T cell function, differentiation, and fate (11, 12).
The signal transduction pathways that regulate T cell metabolism remain poorly characterized. The phosphatidylinositol 3’-kinase (PI3K)/Akt signal transduction pathway is a central regulator of glycolytic metabolism in the immune system (13, 14). TCR- and CD28-dependent Akt activation results in increased Glut1-dependent glucose uptake and stimulation of glycolytic activity (13, 15). Akt also stimulates the mammalian target of rapamycin (mTOR), which regulates multiple metabolic pathways including protein translation, lipid synthesis, autophagy, and mitochondrial biogenesis and function in response to growth factor signals (16). mTOR has emerged as an important regulator of T cell differentiation and effector function (17). mTOR activity is required for the full effector function of both CD4+ (18) and CD8+ (19) T cells, and inhibition of mTOR signaling blocks the differentiation of CD4+ T cells towards inflammatory Th cell lineages (18, 20) while stimulating the development of regulatory T (Treg) cells (18, 21, 22). Similarly, inhibition of mTOR by rapamycin can promote the development of CD8+ T cell memory (9, 10). Identifying the signal transduction pathways that regulate T cell metabolism, and in particular modulators of mTOR activity, may help us understand how metabolic regulation impacts T cell function.
The liver kinase B1 (LKB1) is a serine/threonine kinase that functions as part of an evolutionarily conserved energy-sensing pathway to couple cellular bioenergetics to metabolic pathways important for cell growth control, proliferation, polarity, and stress resistance (23). LKB1 was first identified as the tumor suppressor responsible for Peutz-Jeghers syndrome, an autosomal dominant disorder that leads to intestinal hamartomas, mucocutaneous lesions, and an increased risk of spontaneous epithelial carcinomas (24, 25). LKB1 was recently found to have prominent roles in hematopoietic stem cell (HSC) viability and renewal (26-28), suggesting a prominent function for LKB1 in hematopoietic cells. One of the targets that links LKB1 to cellular metabolism is the serine/threonine kinase AMP-activated protein kinase (AMPK), which can act to suppress mTOR activity (29). LKB1-dependent phosphorylation of the α subunit of AMPK at Thr-172 stimulates AMPK activity, and LKB1 is essential for AMPK activation under conditions of bioenergetic stress (30-32). From a metabolic standpoint, AMPK promotes ATP conservation by activating pathways of catabolic metabolism and inhibiting anabolic processes that consume ATP (33). AMPK itself has been linked to stress resistance in lymphocytes (34).
Lymphocyte metabolism is a highly regulated process that plays a key role in the determination of T cell fate and function, and LKB1 is an established regulator of cell metabolism. However, while LKB1 has been shown to be important for thymocyte development (35, 36), its role in peripheral T cell metabolism, effector function, and mTOR regulation is not well defined. Here we show that LKB1 is a key regulator of glucose and lipid metabolism in T cells, and is essential for both normal T cell development and peripheral T cell function. T cells lacking LKB1 displayed defects in cellular proliferation and viability in response to metabolic stress, yet showed increased activation and inflammatory cytokine production upon TCR stimulation. Moreover, loss of LKB1 in the T cell compartment led to an accumulation of IFN-γ-producing CD8+ and CD4+ T cells in peripheral lymphoid organs. AMPK mediated a subset of LKB1 functions in peripheral T cells; AMPKα1-deficient mice displayed normal thymocyte development and peripheral T cell homeostasis, but AMPKα1-/- CD8+ T cells displayed an activated phenotype with alterations in metabolism and cytokine production similar to LKB1-deficient T cells. Moreover, mTOR Complex 1 (mTORC1) activity was elevated in LKB1- and AMPKα1-deficient T cells, and elevated IFN-γ production by these T cells could be reversed by rapamycin. Together these data demonstrate a critical role for LKB1 in the regulation of peripheral T cell function and metabolism through regulation of AMPK-dependent and independent pathways and mTORC1 signaling.
Materials and Methods
Mice
LKB1 floxed (stk11-fl/fl) mice, which carry an LKB1 allele with LoxP sites flanking exons 3 through 6 (37) were obtained from the National Cancer Institute (Frederick, MD). Rag1-/- mice, Lck-Cre mice, CD45.1 congenic mice, and Ub-Cre-ERT2 mice were purchased from the Jackson Laboratories (Bar Harbor, MA), and Bcl-xL transgenic mice have been described (4). Mice deficient for AMPKα1 (34) or floxed for AMPKα2 (38) have been described previously. Lck-Cre mice were crossed with LKB1 floxed mice to generate LKB1-fl/fl Lck-cre+ mutants or LKB1-fl/+ Lck-cre+, LKB1-fl/fl Lck-cre-, and LKB1-fl/+ Lck-cre- controls. Bcl-xL transgenic mice were crossed to the Lck-Cre LKB1 strains to generate LKB1-fl/fl Lck-cre+ mice that express the Bcl-xL transgene. Mice were bred and maintained under specific pathogen-free conditions at Duke University and McGill University under approved protocols. Experiments were performed using mice between 8 – 20 weeks of age.
T cell purification and culture
T cells were purified from spleen and mesenteric lymph nodes by negative selection (StemCell Technologies, Vancouver, British Columbia, Canada) and cells were cultured in IMDM or RPMI 1640 (Mediatech, Washington, DC) supplemented with 10% FBS (Gemini Bio-Products, West Sacramento, CA), L-glutamine (Invitrogen, Chicago, IL), penicillin-streptomycin (Invitrogen), and β-mercaptoethanol (Sigma-Aldrich, St. Louis, MO). CD44lo cells were isolated by negative selection via the addition of biotinylated anti-CD44 added to the enrichment cocktail. Where indicated, IL-7 was supplemented into media at a concentration of 10 ng/mL (eBioscience, San Diego, CA). T cell stimulation was induced by culturing purified T cells on anti-CD3ε (clone 145-2C11) and anti-CD28 (clone 37.51) coated plates (eBioscience), and supplemented with IL-2 (Preprotech, Rocky Hill, NJ). CD4+ T cell differentiation was performed as described previously (7). To activate Cre-ER and excise the floxed LKB1 transgene, isolated T cells were treated with 4-hydroxytamoxifen (4-OHT, 0.5 μM in ethanol, Sigma-Aldrich). 2-deoxyglucose, AICAR, and rapamycin were obtained from Sigma-Aldrich. T cell proliferation was measured by 3[H]-thymidine incorporation (1 μCi/ml) 48 hours post-activation as previously described (39) or via flow cytometry using CFSE (Molecular Probes, Eugene, OR).
Flow cytometry, viability, and cell size measurements
Single cell suspensions were stained with fluorescently conjugated Abs against murine CD4, CD8, CD3, Thy1.2, B220, CD19, CD25, CD44, CD45.1, CD62L, or CD69 (BD Biosciences, San Jose, CA). Cell viability was assessed via 7-aminoactinomycin D (Sigma-Aldrich) or propidium iodide (Molecular Probes) exclusion as previously described (39). Flow cytometry was performed on a FACScan or LSR II (BD Biosciences) or Gallios (Beckman Coulter, Fullerton, CA) flow cytometer and analyzed with FlowJo software (Tree Star, Ashland, OR). Cell size was determined using a Coulter Z2 particle counter (Beckman Coulter). Bax activation was measured by fixing cells for 5 minutes with 0.25% paraformaldehyde in PBS, followed by incubation with an antibody specific to amino acids 12-24 (BD Biosciences, clone 6A7) as previously described (40).
Cytokine measurements
Intracellular cytokine staining (ICS) was performed using specific fluorochrome labeled mAbs (eBioscience) and flow cytometry as described (41). In brief, single cell suspensions of spleen or lymph node cells were stimulated immediately ex vivo with PMA and ionomycin (Sigma-Aldrich) for 4 hours. Brefeldin A was added to cultures after 2 hours of stimulation. ICS was carried out using a Cytofix/Cytoperm kit (BD Biosciences) following manufacturer’s protocols. Cytokines in T cell supernatants were measured by capture ELISAs (eBioscience) using paired mAbs and recombinant standards, as previously described (15).
Quantitative Real-Time PCR
Total mRNA was isolated from mouse T cells using Trizol (Invitrogen), and cDNA was synthesized from 100ng of total RNA using the Superscript® VILO™ cDNA Synthesis Kit (Invitrogen). Quantitative PCR was performed using SYBR Green qPCR SuperMix (Invitrogen) and an Mx3005 qPCR machine (Agilent) using the following primers: glut1 forward 5’-CTG GAC CTC AAA CTT CAT TGT GGG-3’, glut1 reverse 5’-GGG TGT CTT GTC ACT TTG GCT GG-3’; hk2 forward: 5’-CCG TGG TGG ACA AGA TAA GAG AGA ACC-3’, hk2 reverse: 5’-GGA CAC GTC ACA TTT CGG AGC CAG-3’. All samples were normalized to β-actin mRNA levels.
Immunoblotting
Cell lysate preparation, SDS-PAGE, electrophoretic transfer, immunoblotting, and development using enhanced chemiluminescence were conducted as described previously (1). To probe for Glut1, cells were lysed for 1 hour on ice in PBS plus 1% Triton X-100 and 0.1% SDS containing protease inhibitors (BD Pharmingen). Otherwise, cells were lysed in radioimmunoprecipitation assay (RIPA) buffer or AMPK Lysis Buffer (42) supplemented with protease and phosphatase inhibitors (Sigma-Aldrich). Primary antibodies to AMPK (pT172-specific and total), phospho-Acetyl-CoA-carboxylase (pS79), p70 S6-kinase (pT389-specific and total), S6 ribosomal protein (pS235/236-specific and total), 4E-BP1 (pT37/46-specific and total), Bcl-xL, HKII, LDHA, and actin, as well as anti-rabbit and mouse HRP secondary antibodies were obtained from Cell Signaling Technology (Danvers, MA). Anti-LKB1 mouse monoclonal antibody (Ley 37D/G6) was obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Glut1 rabbit antibody was obtained from Abcam (Cambridge, MA).
Metabolic assays
Assays for glucose uptake, glycolytic flux, and mitochondrial-dependent β-oxidation of fatty acids were performed as previously described (9, 43, 44).
Statistical analysis
Data are presented as mean ± standard deviation and were analyzed using paired Student’s t-test. Unless stated otherwise, a confidence level of p < 0.05 was considered statistically significant for all data.
Results
LKB1 is required for T cell development and homeostasis
To understand the role of the serine/threonine kinase LKB1 in T cell function, animals lacking expression of LKB1 in the T cell lineage were generated by crossing mice with LKB1 floxed (LKB1-fl/fl) (37) with mice expressing a Cre recombinase transgene under the control of the proximal Lck promoter (Lck-cre). Cre-expressing LKB1-fl/fl thymocytes and peripheral T cells displayed decreased expression of LKB1 protein as demonstrated by Western blotting (Fig. 1A). Lck-cre+, LKB1-fl/fl mice displayed dramatic reductions in both thymocyte (Fig. 1B) and peripheral T cell (Fig. 1C) numbers compared to control animals that retained endogenous LKB1 expression. Targeted T cell deletion of LKB1 also resulted in an increased proportion of CD4-CD8- double negative thymocytes with accumulation in the DN3 (CD25hi, CD44lo) stage, and decreased numbers of double positive (CD4+CD8+) and CD4+ and CD8+ single positive thymocytes, suggesting incomplete thymic selection (Fig. 1D). Lck-cre+, LKB1-fl/fl LKB1-deficient mice displayed a decrease in both the proportion (Fig. 1E) and total number (Fig. 1F) of CD4+ and CD8+ peripheral T cells. LKB1-deficient T cells also displayed an increase in T cell size relative to Cre-negative controls (Fig. 1G).
FIGURE 1.
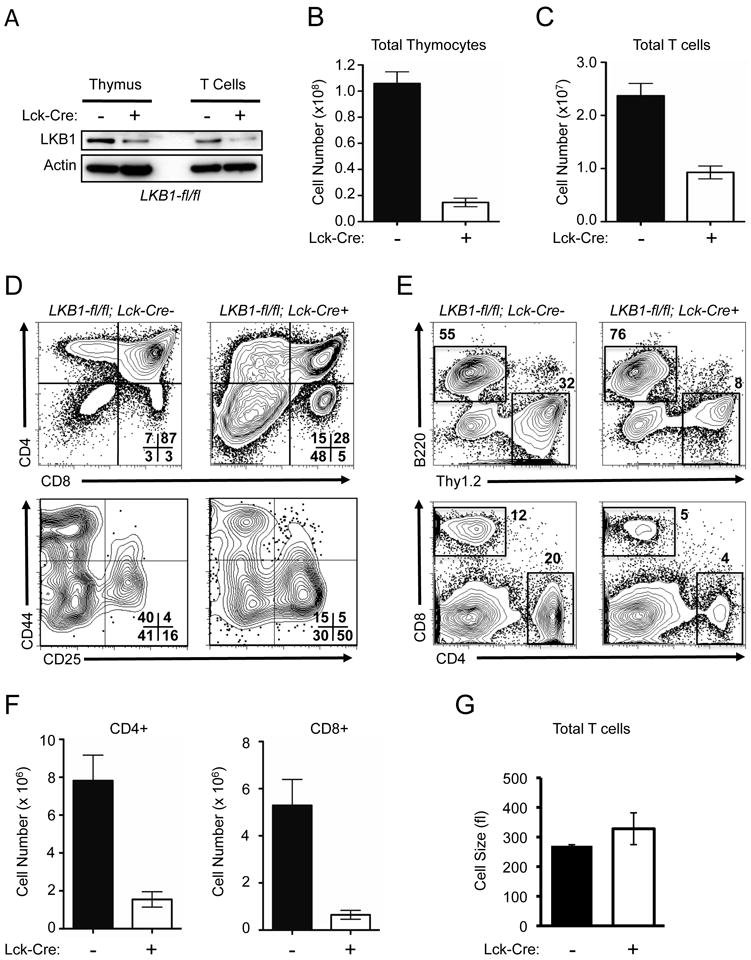
LKB1 is required for T cell development and peripheral homeostasis. A, LKB1 protein expression in LKB1-expressing (Lck-cre-) or LKB1-deficient (Lck-cre+) thymocytes and T cells. Total Thymocytes and purified peripheral T cells were lysed and immunoblotted with antibodies against LKB1 and actin. B-C, Summary of thymocyte (B) and peripheral T cell (C) counts in control (Lck-cre-, black bars) and LKB1 mutant (Lck-cre+, open bars) mice. Data are presented as the mean ± SEM for a minimum of 10 mice per genotype. D, Representative flow plots for expression of CD4 and CD8 on total thymocytes (top panel), and CD44 and CD25 expression on CD4/CD8-double negative (DN) thymocytes (lower panel) from control (LKB1-fl/fl, Lck-cre-) or LKB1 mutant (LKB1-fl/fl, Lck-cre+) animals. Numbers represent the percentage of cells in the indicated quadrant. E, Representative flow plots for B220 versus Thy1.2 expression (upper panel), and CD4 versus CD8 expression on total splenocytes (lower panel) from animals of the indicated genotypes. F, Summary of peripheral CD4 and CD8 T cell numbers from control (black bars) or LKB1 mutant (open bars) animals. G, Resting T cell size as measured by Coulter counter. Black bars represent control T cells, open bars represent LKB1-deficient T cells.
LKB1-deficient T cells display defects in T cell proliferation and viability
Despite an overall defect in thymic development, limited numbers of peripheral CD4+ and CD8+ T cells were observed in T cell-specific LKB1 mutant animals. To assess their functional status, LKB1-deficient T cells were purified from spleen, activated in vitro using anti-CD3 and anti-CD28 antibodies, and T cell proliferation rates measured by thymidine incorporation. As seen in Fig. 2A, T cells from Lck-cre+, LKB1-fl/fl mice displayed decreased proliferative capacity when activated with anti-CD3 antibodies alone or in combination with CD28 costimulation. The addition of recombinant IL-2 led to a partial rescue in proliferation of LKB1-deficient (LKB1-fl/fl; Lck-Cre+) T cells (Supplemental Fig. 1A), but did not fully rescue T cell proliferation. To assess whether enhanced apoptosis could underlie the defective proliferation of LKB1-deficient T cells, we assessed levels of T cell division and apoptosis using carboxyfluorescein succinimidyl ester (CFSE) dye dilution and viability dye exclusion using propidium iodide (PI). CFSE dilution profiles demonstrated that the majority of viable LKB1-deficient cells remained CFSEbright (Fig. 2B). However, LKB1-null T cells also displayed reduced viability relative to control T cells following activation (Fig. 2C). While we cannot exclude cell death that occurred at the time of cell division, these data indicate that viable LKB1-deficient T cells exhibited delays in initiating cell division in response to TCR stimulation.
FIGURE 2.
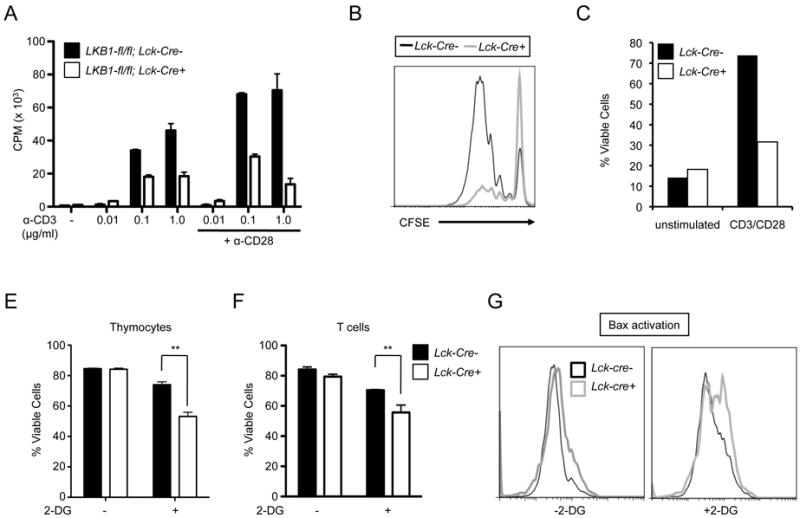
LKB1-deficient T cells display defects in proliferation and viability. A-C, Proliferation of LKB1-deficient T cells. A, Peripheral T cells from control (black bars) or LKB1 mutant (open bars) mice were cultured with various concentrations of anti-CD3 and anti-CD28 antibodies and proliferation measured by thymidine incorporation after 48 hours. Data represent the mean ± s.d. for samples in triplicate. B, Total T cells were activated with anti-CD3 (5 μg/mL) and anti-CD28 (5 μg/mL) for 72 hours and proliferation was measured by CFSE dilution. C, Viability of T cells in (B) was determined by flow cytometry using propidium iodide exclusion. E-F, Measurements of T cell apoptosis induced by metabolic stress. Thymocytes (E) or peripheral T cells (F) from control (black bars) or LKB1-mutant (open bars) animals were cultured with 2-deoxyglucose (2-DG, 10 mM) and cell viability measured 24 hours later by 7-AAD exclusion. Data are expressed as mean ± s.d. for triplicate samples. G, Activated T cells were cultured with 2-deoxyglucose (2-DG, 10 mM) for 18 hours and Bax activation was measured by flow cytometry.
Because altered development may have influenced activation of peripheral LKB1-deficient T cells, we examined proliferation following inducible deletion of LKB1 in mature peripheral T cells. T cells from LKB1-fl/fl, Ub-Cre-ER+ or LKB1-fl/fl, Ub-Cre-ER- animals were cultured with rIL-7 and 4-OHT for 4 days to promote deletion of the floxed LKB1 alleles in Ub-Cre-ER+ T cells, and then T cells were labeled with CFSE and stimulated with anti-CD3 and anti-CD28 antibodies for 48 hours. CFSE dilution profiles showed that T cells with acute deletion of LKB1 displayed reduced levels of cell division (Supplemental Fig. 1B).
LKB1 is an important regulator of cellular stress responses (45), and plays an essential role in the maintenance of hematopoietic stem cell viability (27). To test whether loss of LKB1 could influence T cell viability, LKB1-deficient or control thymocytes and peripheral T cells were cultured in the presence of the glycolytic inhibitor 2-deoxyglucose (2-DG), and cell viability was assessed. While the viability of control cells was largely unaffected by 2-DG treatment (10 mM), LKB1-deficient thymocytes (Fig. 2E) and T cells (Fig. 2F) displayed increased levels of cell death. Withdrawal of IL-2, which reduces glucose metabolism similar to the direct inhibition of glycolysis (9), promoted increased rates of cell death of LKB1-deficient T cells compared to controls (Supplemental Fig. 1C). In addition, treatment of activated LKB1-null T cells with 2-DG (Fig. 2G), or withdrawal from IL-2 (Supplemental Fig. 1D), promoted increased activation of the pro-apoptotic Bax protein, demonstrating an increase in apoptosis following metabolic stress in LKB1-null T cells.
LKB1 regulates T cell metabolism
Activated effector T cells rely heavily on glucose metabolism to survive, proliferate, and support specific effector functions (4, 5, 46). To evaluate the influence of LKB1 loss on T cell metabolism, the rates of glucose uptake and glycolysis were assessed in LKB1-deficient T cells. T cells lacking LKB1 displayed increased rates of glucose uptake (Fig. 3A) and glycolytic flux (Fig. 3B) relative to control T cells, consistent with findings suggesting that LKB1 is required to regulate glucose metabolism in metabolic tissues (47). LKB1-deficient T cells displayed elevated expression of both glut1 and hk2 mRNA (Fig. 3C), as well as elevated Glut1 and hexokinase 2 protein levels (Figs. 3D,E). Glut1 protein expression was increased both at baseline and following TCR stimulation in the LKB1-null T cells as compared to controls (Fig. 3D).
FIGURE 3.
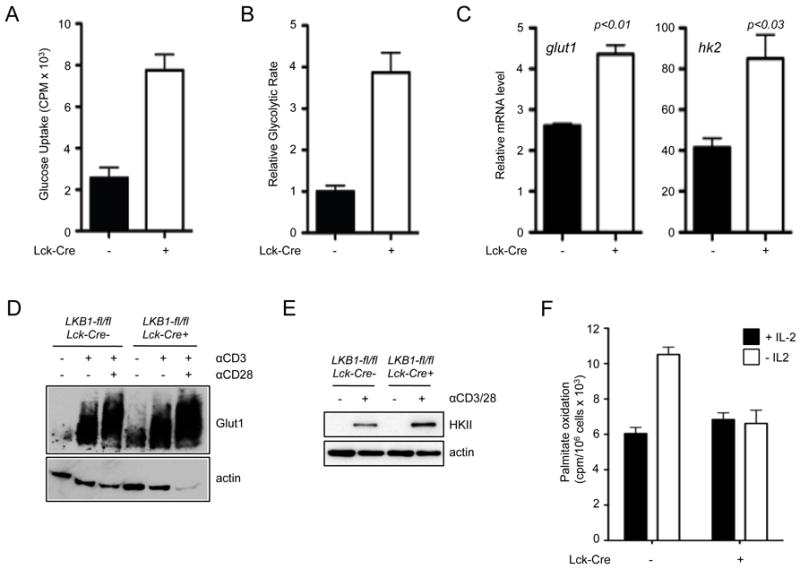
LKB1 regulates T cell metabolism. A, Measurement of LKB1-dependent glucose uptake by T cells. Peripheral T cells from LKB1-expressing (black bars) or LKB1-mutant (open bars) mice were isolated from spleen and lymph nodes, and glucose uptake assessed by 3[H]-2-DG uptake. B, Glycolytic flux in LKB1-deficient T cells. Control (black bars) or LKB1-null (open bars) T cells were isolated and glycolytic flux determined by the generation of tritiated water by enolase. Data are expressed relative to the glycolytic rate (nmol glucose/106 cells/hour) of control T cells. C, Control (black bars) or LKB1-deficient (open bars) T cells were stimulated with anti-CD3 and anti-CD28 antibodies for 24 hours and Glut 1 and hexokinase 2 mRNA expression determined by qPCR. Data are expressed relative to actin mRNA levels, and normalized to mRNA levels in unstimulated T cells. D, Purified T cells from LKB1-expressing (LKB1-fl/fl, Lck-cre-) or LKB1-mutant (LKB1-fl/fl, Lck-cre+) mice were activated with anti-CD3 (5 μg/mL) and anti-CD28 (5 μg/mL) antibodies for 24 hours. Lysates were resolved by SDS-PAGE and immunoblotted for Glut1 and actin. E, Western blot of Hexokinase II expression in control (LKB1-fl/fl, Lck-cre-) or LKB1-deficient (LKB1-fl/fl, Lck-cre+) T cells 24 hours following anti-CD3 and anti-CD28 treatment. F, Measurement of lipid oxidation in LKB1-deficient T cells. Activated T cells were cultured in the presence (black bars) or absence (open bars) of rIL-2 for 14 hours, and the liberation of tritiated water from 3[H]-palmitate (CPM per 106 cells) was measured 6 hours later. Data are presented as the mean ± s.d. for samples in triplicate.
We have previously shown that lipid oxidation can function as an alternate metabolic pathway in resting and memory T cells when glucose uptake is low or reduced or when cytokine availability is limiting (9). Thus, we assessed whether IL-2 dependent control of lipid metabolism was altered in LKB1-deficient T cells. Following cytokine withdrawal, which leads to downregulation of the glucose transporter Glut1 and reduced glycolysis (44), control T cells displayed an increase in their rate of lipid oxidation; however, the induction of lipid oxidation induced by IL-2 withdrawal was impaired in LKB1-deficient T cells (Fig. 3F). Collectively these data suggest that LKB1 functions to coordinate glucose and lipid metabolism in T cells, and loss of LKB1 leads to deregulated metabolic function.
Loss of LKB1 results in the accumulation of activated inflammatory T cells
Given the disrupted metabolic and proliferative capacity of LKB1-deficient T cells, we next examined the effect of LKB1 loss on T cell effector function. Despite possessing fewer T cells overall, Lck-cre+, LKB1-fl/fl animals displayed increased numbers of CD4+ and CD8+ T cells with an activated phenotype (CD44hiCD62Llo) (Fig. 4A). Both the CD4+ and CD8+ T cell populations from Lck-cre+, LKB1-fl/fl mice displayed a higher proportion of CD44-expressing cells (Supplemental Fig. 2A). Moreover, both conditional and inducible deletion of LKB1 resulted in a pronounced increase in the surface expression of activation markers CD25, CD44, and CD69 following TCR ligation (Supplemental Fig. 2B,C).
FIGURE 4.
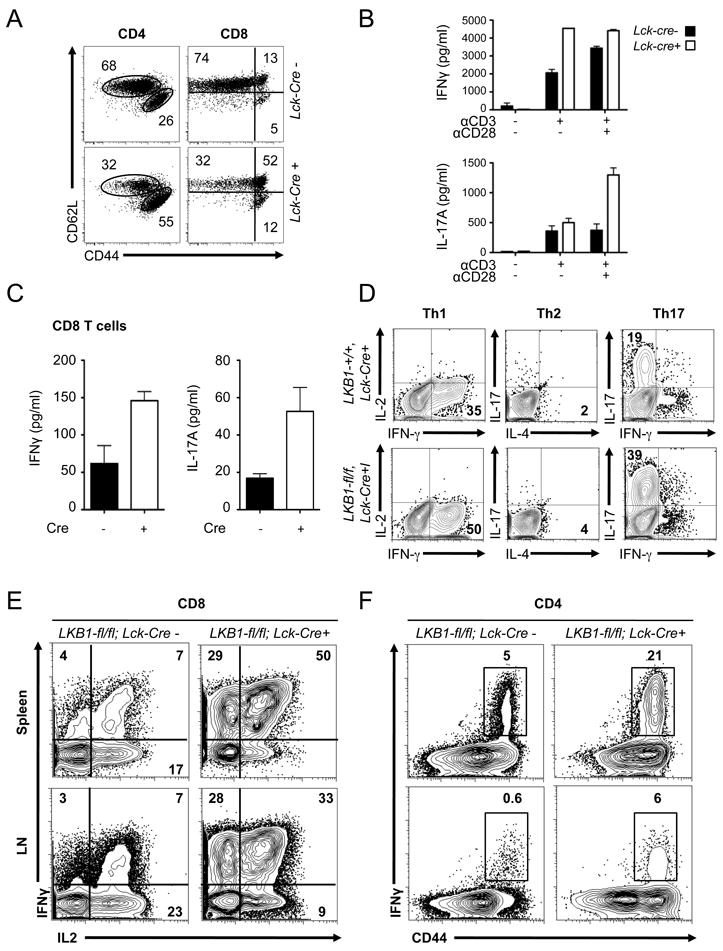
LKB1-deficient T cells display a hyperactivated phenotype. A, CD4+ and CD8+ splenocytes from LKB1-expressing (LKB1-fl/fl, Lck-cre-) or LKB1-deficient (LKB1-fl/fl, Lckcre+) mice were analyzed for CD44 and CD62L expression by flow cytometry. B, IFN-γ and IL-17A production by LKB1-null T cells. Control (black bars) or LKB1-deficient (open bars) T cells were cultured with anti-CD3 antibodies in the presence or absence of CD28 costimulation for 24 hours, and IFN-γ and IL-17A in culture supernatants was measured by ELISA. C, IFN-γ (left panel) and IL-17A (right panel) production by anti-CD3/CD28-activated CD44loCD8+ T cells from LKB1-fl/fl, Lck-cre- or LKB1-fl/fl, Lck-cre+ mice as measured by ELISA. D, CD44loCD4+ T cells from LKB1-expressing (LKB1-wt/wt, Lck-cre+) or LKB1-deficient (LKB1-fl/fl, Lck-cre+) mice were cultured in Th1, Th2, or Th17 skewing conditions and examined for effector function by intracellular staining for IL-2, IFN-γ, IL-4, and IL-17. E-F, Frequency of IFN-γ and IL-2 producing T cells in LKB1-mutant mice. Splenocytes (Spleen) or lymph node (LN) cells from LKB1-expressing (LKB1-fl/fl, Lck-cre-) or LKB1-deficient (LKB1-fl/fl, Lck-cre+) animals were stimulated with PMA and ionomycin for 4 hours, and cytokine producing cells were identified by ICS and flow cytometry. CD8+ T cells were analyzed for IFN-γ versus IL-2 expression (E), while CD4+ cells were analyzed for surface expression of CD44 and intracellular IFN-γ (F). The gated population in (F) refers to the percentage of endogenous IFN-γ-producing CD4+ T cells.
Given the heightened activation state of LKB1-null T cells, we next examined cytokine production by T cells lacking LKB1. Peripheral T cells from LKB1-fl/fl; Lck-Cre+ or LKB1-fl/fl; Lck-Cre- control animals were isolated, stimulated with anti-CD3 and anti-CD28 antibodies, and IFN-γ and IL-17A in culture supernatants was measured. Despite lower overall levels of proliferation (Fig. 2A), LKB1-deficient T cells displayed elevated IFN-γ and IL-17A production relative to control T cells (Fig. 4B). We next isolated naïve (CD44lo) CD4+ and CD8+ T cells from LKB1 mutant and control mice and examined TCR-induced cytokine production by individual T cell subsets. LKB1-deficient CD8+ T cells produced increased amounts of IFN-γ and IL-17A upon activation relative to LKB1-expressing T cells (Fig. 4C). LKB1-deficient CD4+ T cells also displayed a slight enhancement in IFN-γ production (Supplemental Fig. 2D). Finally, we assessed the capacity of naïve CD4+ LKB1-null T cells to differentiate into Th1, Th2, or Th17 effector cells in vitro. T cells lacking LKB1 displayed enhanced differentiation towards Th1 and Th17 CD4+ T cell lineages (Fig. 4D).
We next analyzed whether LKB1 deficiency could alter the number of cytokine producing T cells in vivo. Total splenocytes or lymph node cells were isolated from Lck-cre+, LKB1-fl/fl animals or littermate controls, stimulated immediately ex vivo with PMA and ionomycin, and the presence of IFN-γ and IL-2-producing cells was measured by intracellular cytokine staining (ICS). Peripheral CD8+ (Fig. 4E) and CD4+ (Fig. 4F) T cells from T cell-specific LKB1-null animals were enriched for IFN-γ-producing cells as determined by ICS. In particular, a significant increase in the level of IFN-γ/IL-2-double positive CD8+ T cells was observed in LKB1 T cell-deficient animals (Fig. 4E), suggesting that LKB1 functions as a negative regulator of IFN-γ-producing T cells in vivo.
Bcl-xL rescues decreased thymocyte and peripheral T lymphocyte cellularity caused by LKB1 deficiency
In principle, deletion of LKB1 at the DN stage of thymocyte development may promote low T lymphocyte cellularity (Fig. 1) due to either inefficient thymocyte differentiation or reduced viability. The anti-apoptotic Bcl-2 family protein Bcl-xL is a key regulator of thymocyte viability (48), and reduced Bcl-xL levels have been linked to LKB1 deficiency (35). Indeed, thymocytes with deletion of both alleles of LKB1 gene displayed decreased levels of Bcl-xL protein (Fig. 5A). To assess whether restoring Bcl-xL levels could rescue the defect in T cell development in T cell LKB1-deficient mice, we generated Lck-cre+, LKB1-fl/fl mice that also expressed a T cell-specific Bcl-xL transgene, and analyzed the central and peripheral T cell compartments of these mice by flow cytometry. Transgenic expression of Bcl-xL rescued total thymocyte cellularity in T cell-specific LKB1-deficient mice to wild-type levels, and increased splenic T cell cellularity above wild-type levels regardless of LKB1 status (Fig. 5B). Expression of Bcl-xL also restored the positive selection of CD4+ and CD8+ T cells in the thymus (Fig. 5C), and circumvented the developmental block at the DN3 (CD25hiCD44lo) stage of thymocyte development caused by targeted deletion of LKB1 in thymocytes (Fig. 5D).
FIGURE 5.
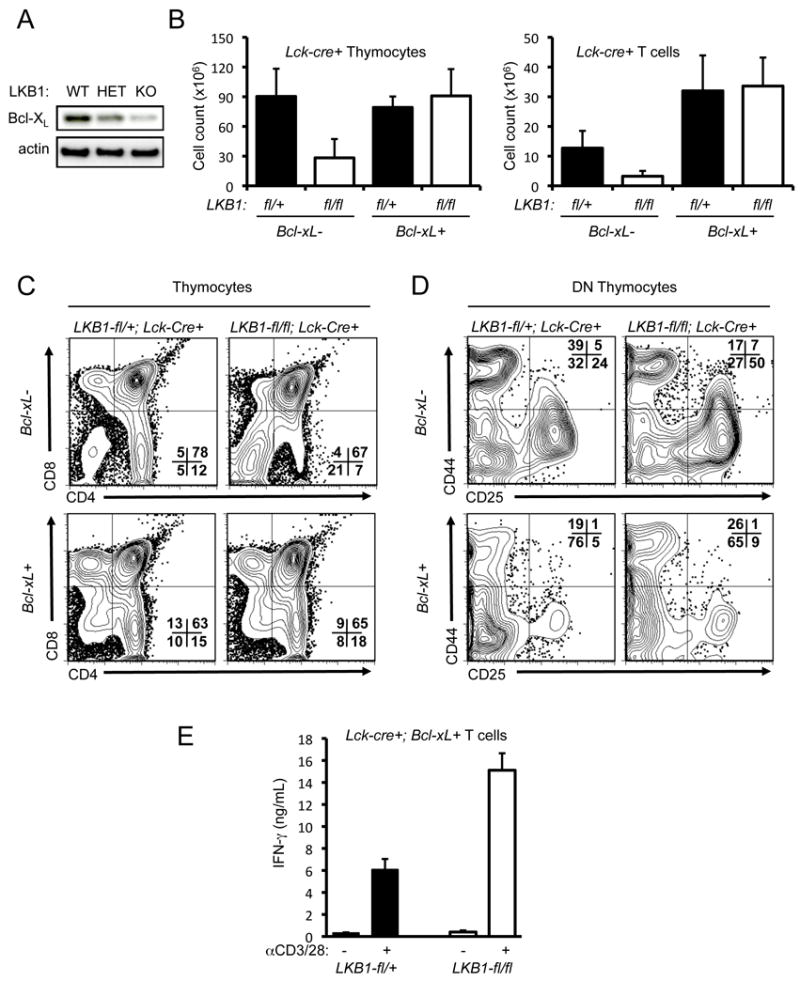
Transgenic expression of Bcl-xL rescues thymocyte development but not aberrant T cell activation and cytokine production in LKB1-mutant animals. A, Bcl-xL protein expression in LKB1-deficient thymocytes. Lysates from homozygous LKB1-deficient (KO), LKB1 heterozygous (HET), or wild-type (WT) thymocytes were resolved by SDS-PAGE and immunoblotted using antibodies to Bcl-xL and actin. B, Thymocyte and T cell numbers from LKB1-expressing (LKB1-fl/wt, Lck-cre+) or LKB1-deficient (LKB1-fl/fl, Lck-cre+) mice with or without expression of a Bcl-xL transgene. Data are expressed as mean ± SEM for a minimum of 5 animals per genotype. C-D, Expression profiling of thymocytes from LKB1-mutant animals expressing a Bcl-xL transgene. Thymocytes from LKB1-expressing (LKB1-fl/wt, Lck-cre+) or LKB1-deficient (LKB1-fl/fl, Lck-cre+) animals with (+) or without (-) expression of a Bcl-xL transgene were analyzed for CD4 versus CD8 expression on total thymocytes (C) or CD25 versus CD44 expression on CD4-CD8- double negative (DN) thymocytes (D) by flow cytometry. E, IFN-γ production of LKB1-null T cells overexpressing Bcl-xL. Bcl-xL expressing T cells from LKB1-expressing (black bars) or LKB1-deficient (open bars) animals were stimulated with anti-CD3 and anti-CD28 antibodies, and IFN-γ in supernatants was measured at 48 hours by ELISA. Data are expressed as the mean ± s.d. for triplicate cultures.
One explanation for the accumulation of activated T cells in T cell-specific LKB1-null animals is increased homeostatic T cell proliferation brought on by the reduced T cell compartment in these mice. To test this possibility, CFSE-labeled CD45.1 congenic donor T cells were adoptively transferred into either LKB1-deficient hosts or various control mice, and the level of homeostatic proliferation was assessed by CFSE dye dilution one week post-transfer. Minimal levels of homeostatic proliferation by the CD45.1 donor T cell population were detected in LKB1-fl/fl, Lck-Cre+, Bcl-xL-Tg mice, with significant levels of T cell division observed only in Rag1-/- hosts (Supplemental Fig. 3A). Moreover, LKB1-dependent thymic cellularity was restored by expression of a Bcl-xL transgene (Fig. 5B), which permitted the direct examination of LKB1-deficient peripheral T cell function in the absence of lymphopenia. Despite equivalent cell numbers as control Bcl-xL transgenic animals, LKB1-deficient T cells expressing Bcl-xL continued to display elevated CD44 expression at baseline, as well as increased CD44, CD25 and CD69 expression upon stimulation (Supplemental Fig. 3B). Similarly, LKB1-deficient T cells expressing Bcl-xL displayed increased IFN-γ production upon T cell activation relative to control Bcl-xL transgenic T cells (Fig. 5E). Thus, transgenic expression of Bcl-xL reversed the developmental and homeostatic defects, but not the activated T cell phenotype, in T cell-specific LKB1-deficient mice (Fig. 5), suggesting a cell-intrinsic role for LKB1 in the regulation of T cell activation.
AMPKα1 regulates T cell viability and metabolism, but not TCR-mediated proliferation
The most well-defined physiological target of LKB1 is the serine/threonine kinase AMPK (45). Phosphorylation of AMPKα at Thr-172 and phosphorylation of its downstream target Acetyl-CoA Carboxylase (ACC) occurs rapidly following TCR stimulation (Fig. 6A). As seen in Fig. 6B, LKB1-deficient T cells (LKB1-fl/fl, Lck-cre+) displayed impaired AMPKα and ACC phosphorylation following stimulation with the AMPK agonist aminoimidazole carboxamide ribonucleotide (AICAR), in agreement with previous work suggesting that T cells lacking LKB1 are defective for AMPKα phosphorylation at Thr-172 (36). To determine if the phenotype of LKB1-deficient T cells could be attributed to the loss of AMPK signaling, we examined T lymphocyte development and function in animals lacking prkaa1, the gene that encodes AMPKα1. AICAR-induced AMPK activation was completely abolished in thymocytes lacking the α1 subunit of AMPK (Supplemental Fig. 4A), suggesting that T cells, like other hematopoietic cells, predominantly express the α1 catalytic subunit of the trimeric AMPK complex (49). In agreement with previous work (34), AMPKα1-deficient mice displayed no overt defects in thymocyte development or peripheral T cell subsets (Supplemental Fig. 4B), and displayed normal proliferative responses to TCR and CD28 stimulation (Fig. 6C).
FIGURE 6.
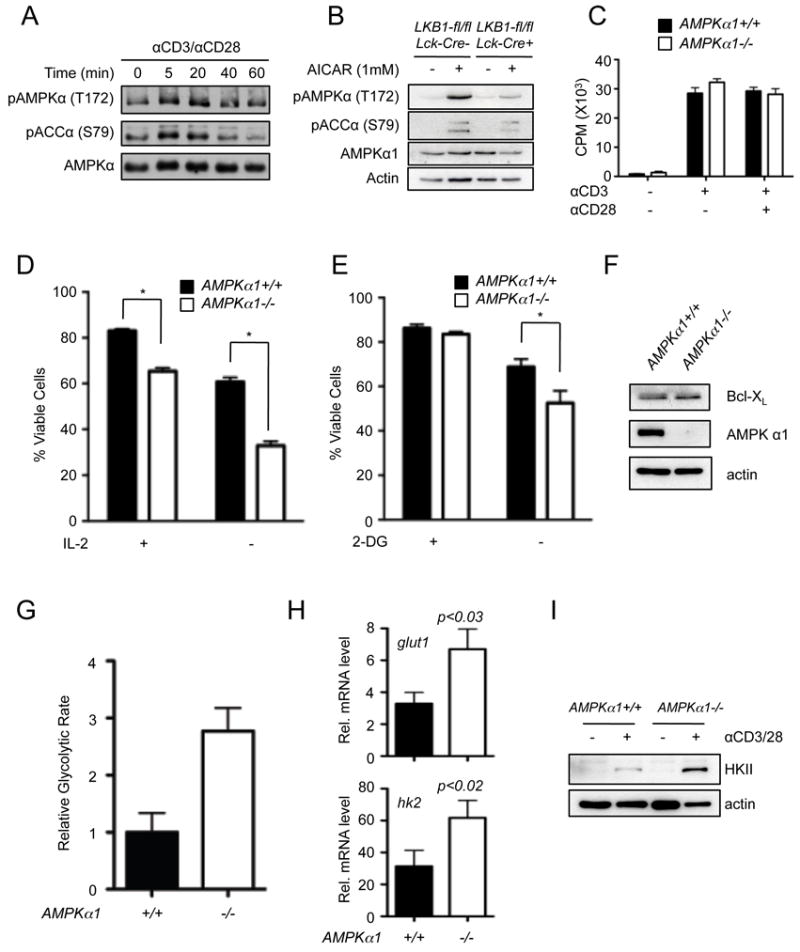
AMPK regulates T cell viability and metabolism, but not proliferation. A, Timecourse of AMPK activation following TCR and CD28 stimulation. Purified T cells were activated with anti-CD3 and anti-CD28 antibodies for the indicated times, and lysates resolved by SDS-PAGE were immunoblotted for phospho-AMPKα (pAMPK T172), phospho-ACC (pACC S79) and total AMPKα. B, Western Blot of phospho-AMPKα (T172) and phospho-ACC (S79) levels in control (Lck-Cre-) or LKB1-deficient (Lck-Cre+) T cells following AICAR treatment (1 mM, 1h). C, Peripheral T cells from wild-type (AMPKα1+/+, black bars) or AMPKα1-deficient (AMPKα1-/-, open bars) mice were stimulated with anti-CD3 antibodies (1 μg/mL) with or without anti-CD28 costimulation (0.5 μg/mL). Proliferation was measured by thymidine incorporation after 48 hours. Data represent the mean ± s.d. for triplicate samples. D-E, Measurements of apoptosis resistance of AMPK-deficient T cells. Peripheral T cells from control (black bars) or AMPKα1-/- (open bars) animals were cultured in the presence (+) or absence (-) of rIL-2 (D) or treated with 10 mM 2-deoxyglucose (E), and cell viability was measured 24 hours later by 7-AAD exclusion. Data are expressed as mean ± s.d. for triplicate samples. F, Western Blot of Bcl-xL expression in AMPKα1+/+ and AMPKα1-/- thymocytes. G, Measurement of relative glycolytic flux in AMPK-deficient T cells. Control (black bars) or AMPKα1-/- (open bars) T cells were isolated by magnetic sorting and glycolytic flux determined as in Figure 3. H, Relative Glut 1 and hexokinase 2 mRNA levels in AMPKα1+/+ (black bars) and AMPKα1-/- (open bars) T cells 24 hours following anti-CD3/CD28 treatment as determined by qPCR. Data are expressed relative to actin mRNA levels, and normalized to mRNA levels in unstimulated T cells. I, Western Blot of Hexokinase II expression in AMPKα1+/+ and AMPKα1-/- T cells 24 hours following anti-CD3/CD28 treatment.
We next examined the effects of AMPKα1 loss on T cell viability and metabolism. Activated T cells from AMPKα1-deficient or control animals were cultured in the absence of IL-2 (Fig. 6D) or subjected to metabolic stress by 2-DG treatment (Fig. 6E), and cell viability measured. Similar to LKB1-deficiency, AMPKα1-deficient T cells displayed reduced viability relative to control cells in response to these stresses (Fig. 6D,E). However, the expression of Bcl-xL protein was equivalent in both AMPKα1-/- and control T cells (Fig. 6F). Similar to LKB1-deficient T cells, AMPKα1-/- T cells displayed a 3-fold increase in the basal glycolytic rate of resting T cells ex vivo (Fig. 6G) and increased levels of glut1 and hk2 mRNA (Fig. 6H). AMPKα1-deficient T cells also displayed elevated expression of hexokinase 2 protein (Fig. 6I). Thus, loss of AMPK activity appears to phenocopy the effect of LKB1 deficiency on T cell metabolism (Fig. 3).
AMPKα1 regulates CD8+ T cell activation and cytokine production
To assess the effect of AMPKα1 loss on T cell effector function, we measured the level of activation and cytokine production of AMPKα1-deficient T cells following TCR stimulation. CD8+ T cells from AMPKα1-/- mice displayed elevated CD44 expression both prior to and following anti-CD3 and anti-CD28 stimulation (Fig. 7A). Similar to LKB1-deficient T cells, increased levels of secreted IFN-γ and IL-17A (Fig. 7B) were detected in AMPKα1-/- cultures following TCR stimulation. To ensure that these effects were not due to the presence of previously activated cells, naïve (CD44lo) CD4+ and CD8+ T cells were isolated, activated with anti-CD3 and anti-CD28 antibodies, and supernatants were analyzed for cytokine production by ELISA. Interestingly, AMPKα1-/- CD8+ T cells displayed elevated IFN-γ and IL-17 production (Fig. 7C), while IFN-γ production by AMPKα1-/- CD4+ T cells did not differ significantly from control cells (Supplemental Fig. 4C). Elevated production of IFN-γ by AMPKα1-/- CD8+ T cells was also detected by intracellular cytokine staining (Fig. 7D). We next induced naïve AMPKα1-/- CD4+ T cells to differentiate into Th1, Th2, and Th17 effector cells, and observed no discernible difference in cytokine production between AMPKα1-deficient or control CD4+ T cells (Fig. 7E). Altogether these data suggest that, in contrast to LKB1 (Fig. 4), AMPKα1 specifically regulates the production of inflammatory cytokines by CD8+ but not CD4+ T cells.
FIGURE 7.
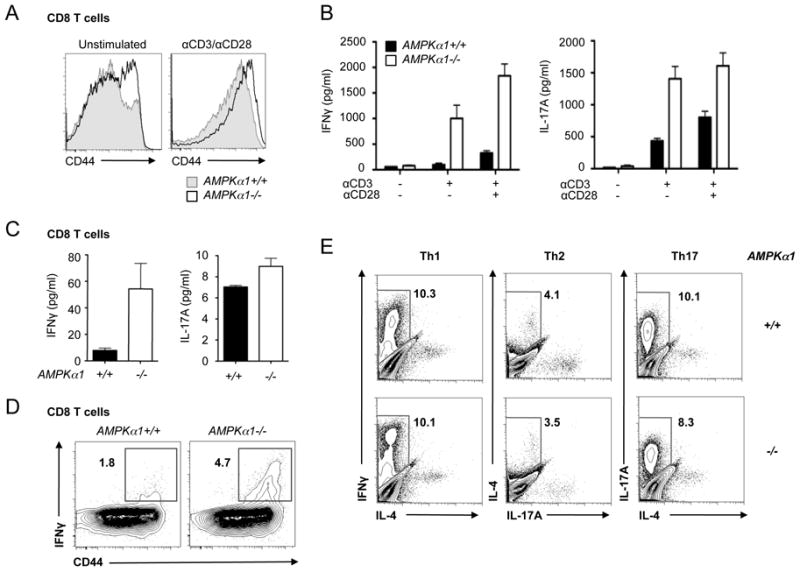
Loss of AMPKα1 promotes hyperactivation of CD8+ T cells. A, CD44 surface expression on AMPKα1-deficient T cells. T cells isolated from control (grey histogram) or AMPKα1-/- (open histogram) mice were left unstimulated (left panel) or anti-CD3/CD28 treated (right panel) for 24 hours. CD44 surface expression on CD8+ T cells was analyzed by flow cytometry. B, Inflammatory cytokine production by activated AMPKα1-deficient T cells. Wild type (black bars) or AMPKα1-null (open bars) T cells were activated with anti-CD3 antibodies in the presence or absence of anti-CD28 costimulation (0.5 μg/ml). Culture supernatants were harvested and analyzed for IFN-γ or IL-17A levels by ELISA. C, IFN-γ (left panel) and IL-17A (right panel) production by anti-CD3/CD28-activated CD44loCD8+ T cells from AMPKα1+/+ (black bars) or AMPKα1-/- (open bars) mice. Cytokines were measured by ELISA 24 hours post-stimulation. D, IFN-γ production by AMPK-deficient T cells. Splenocytes from AMPKα1+/+ or AMPKα1-/- animals were activated with anti-CD3 antibodies, and IFN-γ-producing cells were identified by ICS and flow cytometry. Surface expression of CD44 versus intracellular IFN-γ is displayed for CD8+ cells. E, T helper differentiation of AMPKα1-/- T cells in vitro. AMPKα1+/+ or AMPKα1-/- CD44loCD4+ T cells were cultured in Th1, Th2, or Th17 skewing conditions and examined for effector function after 4 days of culture by intracellular staining for IFN-γ, IL-4, and IL-17.
LKB1-AMPKα1-dependent control of mTORC1 regulates IFN-γ production
A major downstream effect of LKB1-AMPK signaling is the inactivation of the mTOR pathway (45). To determine the influence of reduced LKB1 signaling on mTORC1 activity LKB1-null T cells were stimulated with anti-CD3 and anti-CD28 for 24 hours, and phosphorylation of the downstream mTORC1 targets S6 kinase (S6K), rS6, and 4EBP1 were assessed by Western Blot. T cells lacking LKB1 expression displayed elevated S6K, rS6, and 4EBP1 phosphorylation following activation (Fig. 8A). Similar increases in S6K, rS6, and 4EBP1 phosphorylation were observed in T cells lacking AMPKα1 (Fig. 8B). Baseline phosphorylation of rS6 and 4EBP1 was also elevated in unstimulated AMPKα1-/- T cells (Fig. 8B). Inhibition of mTORC1 signaling by the addition of rapamycin (Rapa) to T cell cultures ablated the effect of LKB1- or AMPKα1-deficiency on enhanced IFN-γ production (Fig. 8C,D). Thus, LKB1 and AMPKα1 appear to play a key role in suppressing mTORC1 signaling, and elevated mTORC1 activity appears to promote the enhanced cytokine production by LKB1 and AMPKα1-deficient T cells.
FIGURE 8.
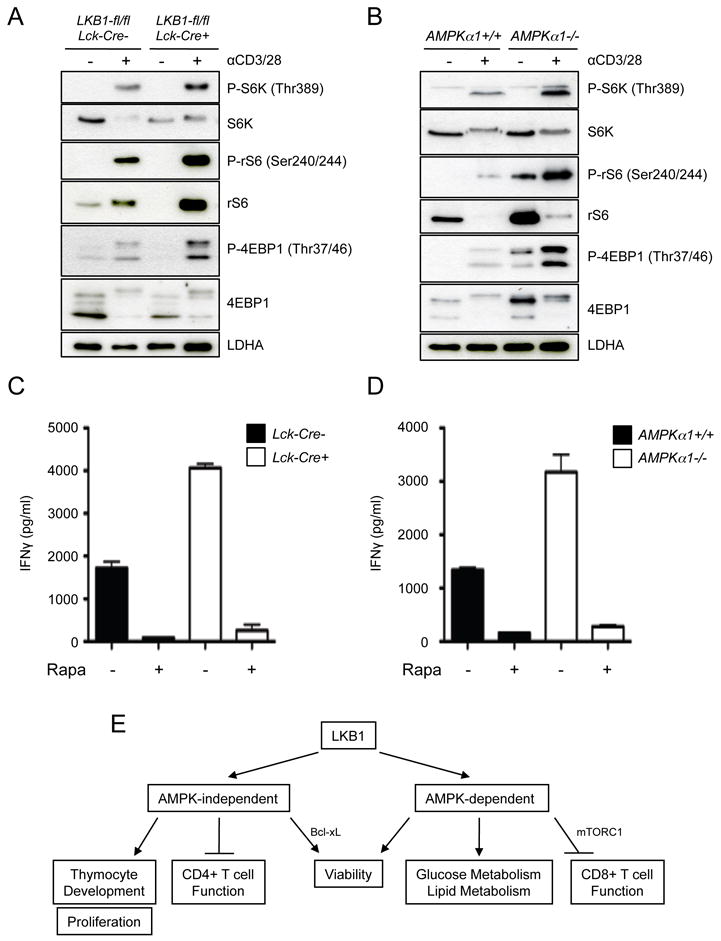
mTORC1 activity is deregulated in LKB1 and AMPKα1-deficient T cells. A, Examination of mTORC1 signaling in LKB1-deficient T cells by Western Blot. T cells were isolated from control (Lck-cre-) or LKB1-deficient (Lck-cre+) mice, left unstimulated (-) or stimulated (+) with anti-CD3 and anti-CD28 antibodies for 24 hours, and lysates resolved by SDS-PAGE. Phosphorylated and total levels of S6K, rS6, and 4E-BP were measured by immunoblotting. Protein loading was assessed by measuring total LDHA levels. B, mTORC1 signaling in AMPKα1-deficient T cells. T cells from AMPKα1+/+ or AMPKα1-/- mice were activated and lysates analyzed for mTORC1 signaling as in (A). C-D, Analysis of LKB1- and AMPK-dependent IFN-γ production in the presence of rapamycin. LKB1-deficient (C) or AMPKα1-deficient (D) T cells were stimulated in the presence (+) or absence (-) of rapamycin (Rapa, 25 nM), and IFN-γ in culture supernatants was measured by ELISA at 24 hours. Black bars represent control T cell cultures for each experiment. Data are presented as the mean ± s.d. for samples in triplicate. E, Integrated model for the function of LKB1 in T cell biology.
Discussion
LKB1 functions at the center of a signaling axis that couples cellular bioenergetics and environmental stimuli with cell growth control and metabolism. Here we demonstrate that LKB1 plays a critical role in regulating T cell development and peripheral T cell function. Animals lacking LKB1 in the T cell compartment displayed a block in thymocyte development at the DN3 stage, resulting in decreased thymic cellularity and a reduced peripheral T cell compartment. LKB1-deficient T cells displayed defective proliferation in response to TCR and costimulatory signals, were increasingly susceptible to apoptosis induced by metabolic stressors or cytokine withdrawal, and displayed perturbations in glucose and lipid metabolism. Surprisingly, LKB1-deficient T cells that did reach the periphery, either naturally or through forced expression of Bcl-xL to block aberrant cell death due to the lack of LKB1, displayed an activated phenotype characterized by increased expression of activation markers and elevated production of inflammatory cytokines. In particular, loss of LKB1 enhanced IFN-γ and IL-17 production by CD8+ T cells, and promoted enhanced differentiation of CD4+ T cells towards Th1 and Th17 effector subtypes. In contrast, loss of AMPKα1, a prominent downstream target of LKB1, only partially recapitulated the phenotype of LKB1-deficient T cells. AMPKα1-/- T cells displayed similar defects in T cell viability and metabolism, while T cell development and proliferative responses were normal. While IFN-γ and IL-17 production by AMPKα1-/- T cells was also enhanced, this was largely due to deregulated CD8+ T cell function in the absence of AMPKα1. The mTORC1 pathway was hyperactivated in both LKB1- and AMPKα1-deficient T cells, and inhibition of this pathway with rapamycin was sufficient to block elevated IFN-γ production by these cells. Together our results establish the LKB1-AMPK signaling axis as a critical regulator of peripheral T cell viability and metabolism, and a negative regulator of T cell inflammatory cytokine production through regulation of mTORC1 activity (Fig. 8E).
The present study, coupled with lines of evidence from other groups, indicate that LKB1 plays a multifaceted role in T cell development and peripheral function. Our data support previous work focused largely on the role of LKB1 in thymocyte development (36) and thymocyte survival (35), and suggests that LKB1 is a cell-autonomous factor essential for T cell development. We also demonstrate that LKB1 functions largely to regulate thymocyte viability, as both thymocyte cellularity and the impaired DN to DP transition are restored when apoptosis is blocked in T cell-specific LKB1-null mice by transgenic overexpression of Bcl-xL. This suggests that LKB1 is not essential for the T cell differentiation process itself, but rather may function to support cell survival during thymocyte development. Indeed, thymocytes and T cells lacking LKB1 display increased susceptibility to apoptosis induced by metabolic stress or cytokine withdrawal. Collectively our data suggest that loss of LKB1 in T cells decreases overall T cell fitness leading to decreased viability, similar to that observed in hematopoietic stem cells (26-28). It is important to note, however, that the LKB1 mouse strain used in this study, which targets exons 3 through 6 of the stk11 gene for LKB1 (37), is different from the animals used by Tamas et al., which targets exons 4 through 7 and results in the generation of a hypomorphic mutation that decreases LKB1 expression in the absence Cre recombinase (50). Consequently, one cannot rule out the possibility that hypomorphic LKB1 expression in other tissues, such as thymic epithelium, could contribute to the phenotypes previously described (36). Together, however, these data suggest that LKB1 functions to couple environmental cues to T cell survival either through the regulation of cellular metabolism and bioenergetics and/or Bcl-xL levels.
AMPK is one of the key physiological effectors downstream of LKB1. However, our data suggest that LKB1 exerts effects on T cell biology through both AMPK-dependent and AMPK-independent pathways (Fig. 8E). While AMPKα1 was largely dispensable for thymocyte development and T cell proliferation, both LKB1 and AMPKα1 mutant T cells displayed defects in glucose and lipid metabolism and stress-induced apoptosis, suggesting these biological processes are regulated through a common pathway. Similarly, LKB1 and AMPKα1 both function as negative regulators of inflammatory cytokine production. However, AMPK appears only to regulate CD8+ T cell activation and cytokine production, while LKB1 influences both CD4+ and CD8+ T cell function. AMPKα1 is just 1 of 13 enzymes of the AMPK-related family of kinases activated by LKB1 (51), and as such, may only represent a fraction of LKB1-dependent processes in T cells.
One striking aspect of the “activated” T cell phenotype of LKB1 mutant mice is the appearance of enhanced cytokine-producing CD4+ and CD8+ T cells with diminished proliferative capacity. LKB1 has previously been shown to be a negative regulator of cell cycle entry (52), so loss of proliferative capacity with LKB1 loss in T cells runs counter to other cellular systems. One possible explanation for the increased activation of LKB1-null T cells could be homeostatic proliferation driven by lymphopenia due to the reduced T cell compartment in Lck-Cre+, LKB1-fl/fl mice (53, 54). However, expression of a Bcl-xL transgene rescues T cell numbers in LKB1 mutant mice but does not affect the heightened activation profile of or cytokine production by LKB1-deficient T cells. Moreover, the observation that AMPKα1-deficient animals display normal T cell development but enhanced T cell activation and cytokine production upon stimulation, suggests that lymphopenia may not drive the activated phenotype of LKB1-deficient cells. Rather, altered LKB1/AMPK-dependent processes such as cellular metabolism or deregulated signaling downstream of LKB1 (i.e. increased mTORC1 activity) may drive T cell activation and cytokine production. Again, the complete scope of LKB1-dependent processes in T cells may only partially depend on AMPK, with AMPKα1 coupling LKB1 to the regulation of effector cytokine production while being dispensable for proliferation (Fig. 8E).
Our data suggest that regulation of mTORC1 signaling downstream of LKB1/AMPK is critical for inflammatory cytokine production. mTORC1 activity is elevated in LKB1- and AMPKα1-deficient T cells and rapamycin can reverse the elevated IFN-γ production by these T cells. Thus, LKB1-AMPK signaling represents a novel mechanism for regulation of mTORdependent effector functions in T cells. Increased mTORC1 activity appears to generally influence T cell viability and effector function. T cell-specific deletion of Tsc1, a tumor suppressor that lies downstream of LKB1-AMPK signaling, results in increased mTORC1 activity and is required for the maintenance of T cell viability and quiescence (55-57). Whether LKB1-AMPK-signaling influences T cell function through control of metabolism remains unclear. Increased glucose uptake, such as that observed in LKB1- and AMPKα1-deficient T cells, can enhance T cell activation and cytokine production (5). Thus, the increase in glucose metabolism associated with loss of LKB1-AMPK signaling may drive a feed-forward loop that promotes the development of proinflammatory effector T cells, resulting in the accumulation of activated IFN-γ producing cells. The engagement of alternate metabolic pathways such as lipid oxidation (9), which is defective in LKB1- and AMPKα1-deficient T cells, may also contribute to this process. The activation of mTORC1 signaling, either through PI3K/Akt-mediated signal transduction or suppression of LKB1-AMPK activity, as shown here, appears to favor the development of inflammatory effector T cells. Our data adds to the growing body of evidence that AMPK signaling can influence inflammatory processes (58, 59).
The signal transduction pathways that regulate LKB1 and AMPK activity in T cells remain unclear. LKB1 is considered to have constitutive kinase activity, and may be limited by the availability and/or localization of its substrates (23). More recent data has suggested that LKB1 is phosphorylated by Lck following TCR stimulation and associates with downstream TCR signaling components LAT and PLCγ1 (60). AMPK itself can be activated by other kinases in addition to LKB1, including CaMKK2 (61, 62), that may promote AMPK activation downstream of TCR stimulation (49). TCR activation stimulates the PI3K/Akt/mTOR pathway to promote Glut1 cell surface trafficking and glycolysis, but upregulation of this metabolic pathway following TCR ligation takes several hours (5, 63). AMPK is rapidly activated following T cell receptor stimulation (49), and, thus, may function to promote rapid increases in ATP generation through alternative metabolic pathways prior to the induction of glycolysis. Alternatively, the increased metabolic demands of T cell activation may promote metabolic stress conditions that in turn activate AMPK via LKB1. Developing and resting T cells require extrinsic signals mediated through the TCR and IL-7R to maintain glucose metabolism (4, 64), and these receptors may signal through LKB1 to regulate basal T cell metabolism. Further work must be done to characterize the signaling pathways both upstream and downstream of LKB1.
Together, our data establish LKB1 as a critical regulator of T cell development and peripheral T cell function. Our data suggest that inhibition of the LKB1-AMPK signaling axis disrupts T cell metabolism and promotes the development of proinflammatory T cells through hyperactivation of mTORC1 signaling. Deficiency in LKB1 or AMPKα1 leads to a reciprocal phenotype to mTOR deficiency (17), supporting the model that these pathways act in opposition as critical regulators of T cell metabolism and function. Thus, manipulation of LKB1-AMPK signaling may provide a new means by which to modulate inflammation and autoimmunity.
Supplementary Material
Acknowledgments
We thank Benoit Viollet for his generous gift of AMPKα1- and AMPKα2-deficient mice, Erika Pearce and Pamela Ohashi for critical reading of the manuscript, and members of the Rathmell and Jones laboratories for helpful discussion and feedback.
Footnotes
Disclosures The authors have no financial conflicts of interest.
References
- 1.Jones RG, Thompson CB. Revving the engine: signal transduction fuels T cell activation. Immunity. 2007;27:173–178. doi: 10.1016/j.immuni.2007.07.008. [DOI] [PubMed] [Google Scholar]
- 2.Roos D, Loos JA. Changes in the carbohydrate metabolism of mitogenically stimulated human peripheral lymphocytes. II. Relative importance of glycolysis and oxidative phosphorylation on phytohaemagglutinin stimulation. Exp Cell Res. 1973;77:127–135. doi: 10.1016/0014-4827(73)90561-2. [DOI] [PubMed] [Google Scholar]
- 3.Bental M, Deutsch C. Metabolic changes in activated T cells: an NMR study of human peripheral blood lymphocytes. Magn Reson Med. 1993;29:317–326. doi: 10.1002/mrm.1910290307. [DOI] [PubMed] [Google Scholar]
- 4.Rathmell JC, Vander Heiden MG, Harris MH, Frauwirth KA, Thompson CB. In the absence of extrinsic signals, nutrient utilization by lymphocytes is insufficient to maintain either cell size or viability. Mol Cell. 2000;6:683–692. doi: 10.1016/s1097-2765(00)00066-6. [DOI] [PubMed] [Google Scholar]
- 5.Jacobs SR, Herman CE, Maciver NJ, Wofford JA, Wieman HL, Hammen JJ, Rathmell JC. Glucose uptake is limiting in T cell activation and requires CD28-mediated Akt-dependent and independent pathways. J Immunol. 2008;180:4476–4486. doi: 10.4049/jimmunol.180.7.4476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Krauss S, Brand MD, Buttgereit F. Signaling takes a breath--new quantitative perspectives on bioenergetics and signal transduction. Immunity. 2001;15:497–502. doi: 10.1016/s1074-7613(01)00205-9. [DOI] [PubMed] [Google Scholar]
- 7.Michalek RD, Gerriets VA, Jacobs SR, Macintyre AN, MacIver NJ, Mason EF, Sullivan SA, Nichols AG, Rathmell JC. Cutting edge: distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J Immunol. 2011;186:3299–3303. doi: 10.4049/jimmunol.1003613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shi LZ, Wang R, Huang G, Vogel P, Neale G, Green DR, Chi H. HIF1{alpha}-dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J Exp Med. 2011;208:1367–1376. doi: 10.1084/jem.20110278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pearce EL, Walsh MC, Cejas PJ, Harms GM, Shen H, Wang LS, Jones RG, Choi Y. Enhancing CD8 T-cell memory by modulating fatty acid metabolism. Nature. 2009;460:103–107. doi: 10.1038/nature08097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Araki K, Turner AP, Shaffer VO, Gangba S, Keller SA, Bachmann MF, Larsen CP, Ahmed R. mTOR regulates memory CD8 T-cell differentiation. Nature. 2009;460:108–112. doi: 10.1038/nature08155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Michalek RD, Rathmell JC. The metabolic life and times of a T-cell. Immunol Rev. 2010;236:190–202. doi: 10.1111/j.1600-065X.2010.00911.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Finlay D, Cantrell DA. Metabolism, migration and memory in cytotoxic T cells. Nat Rev Immunol. 2011;11:109–117. doi: 10.1038/nri2888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Frauwirth KA, Riley JL, Harris MH, Parry RV, Rathmell JC, Plas DR, Elstrom RL, June CH, Thompson CB. The CD28 signaling pathway regulates glucose metabolism. Immunity. 2002;16:769–777. doi: 10.1016/s1074-7613(02)00323-0. [DOI] [PubMed] [Google Scholar]
- 14.Krawczyk CM, Holowka T, Sun J, Blagih J, Amiel E, DeBerardinis RJ, Cross JR, Jung E, Thompson CB, Jones RG, Pearce EJ. Toll-like receptor-induced changes in glycolytic metabolism regulate dendritic cell activation. Blood. 2010;115:4742–4749. doi: 10.1182/blood-2009-10-249540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rathmell JC, Elstrom RL, Cinalli RM, Thompson CB. Activated Akt promotes increased resting T cell size, CD28-independent T cell growth, and development of autoimmunity and lymphoma. Eur J Immunol. 2003;33:2223–2232. doi: 10.1002/eji.200324048. [DOI] [PubMed] [Google Scholar]
- 16.Laplante M, Sabatini DM. mTOR signaling at a glance. J Cell Sci. 2009;122:3589–3594. doi: 10.1242/jcs.051011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Powell JD, Delgoffe GM. The mammalian target of rapamycin: linking T cell differentiation, function, and metabolism. Immunity. 2010;33:301–311. doi: 10.1016/j.immuni.2010.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Delgoffe GM, Kole TP, Zheng Y, Zarek PE, Matthews KL, Xiao B, Worley PF, Kozma SC, Powell JD. The mTOR kinase differentially regulates effector and regulatory T cell lineage commitment. Immunity. 2009;30:832–844. doi: 10.1016/j.immuni.2009.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rao RR, Li Q, Odunsi K, Shrikant PA. The mTOR kinase determines effector versus memory CD8+ T cell fate by regulating the expression of transcription factors T-bet and Eomesodermin. Immunity. 2010;32:67–78. doi: 10.1016/j.immuni.2009.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee K, Gudapati P, Dragovic S, Spencer C, Joyce S, Killeen N, Magnuson MA, Boothby M. Mammalian target of rapamycin protein complex 2 regulates differentiation of Th1 and Th2 cell subsets via distinct signaling pathways. Immunity. 2010;32:743–753. doi: 10.1016/j.immuni.2010.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Battaglia M, Stabilini A, Roncarolo MG. Rapamycin selectively expands CD4+CD25+FoxP3+ regulatory T cells. Blood. 2005;105:4743–4748. doi: 10.1182/blood-2004-10-3932. [DOI] [PubMed] [Google Scholar]
- 22.Kopf H, de la Rosa GM, Howard OM, Chen X. Rapamycin inhibits differentiation of Th17 cells and promotes generation of FoxP3+ T regulatory cells. Int Immunopharmacol. 2007;7:1819–1824. doi: 10.1016/j.intimp.2007.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Alessi DR, Sakamoto K, Bayascas JR. LKB1-dependent signaling pathways. Annu Rev Biochem. 2006;75:137–163. doi: 10.1146/annurev.biochem.75.103004.142702. [DOI] [PubMed] [Google Scholar]
- 24.Hemminki A, Markie D, Tomlinson I, Avizienyte E, Roth S, Loukola A, Bignell G, Warren W, Aminoff M, Hoglund P, Jarvinen H, Kristo P, Pelin K, Ridanpaa M, Salovaara R, Toro T, Bodmer W, Olschwang S, Olsen AS, Stratton MR, de la Chapelle A, Aaltonen LA. A serine/threonine kinase gene defective in Peutz-Jeghers syndrome. Nature. 1998;391:184–187. doi: 10.1038/34432. [DOI] [PubMed] [Google Scholar]
- 25.Jenne DE, Reimann H, Nezu J, Friedel W, Loff S, Jeschke R, Muller O, Back W, Zimmer M. Peutz-Jeghers syndrome is caused by mutations in a novel serine threonine kinase. Nat Genet. 1998;18:38–43. doi: 10.1038/ng0198-38. [DOI] [PubMed] [Google Scholar]
- 26.Gan B, Hu J, Jiang S, Liu Y, Sahin E, Zhuang L, Fletcher-Sananikone E, Colla S, Wang YA, Chin L, Depinho RA. Lkb1 regulates quiescence and metabolic homeostasis of haematopoietic stem cells. Nature. 2010;468:701–704. doi: 10.1038/nature09595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gurumurthy S, Xie SZ, Alagesan B, Kim J, Yusuf RZ, Saez B, Tzatsos A, Ozsolak F, Milos P, Ferrari F, Park PJ, Shirihai OS, Scadden DT, Bardeesy N. The Lkb1 metabolic sensor maintains haematopoietic stem cell survival. Nature. 2010;468:659–663. doi: 10.1038/nature09572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nakada D, Saunders TL, Morrison SJ. Lkb1 regulates cell cycle and energy metabolism in haematopoietic stem cells. Nature. 2010;468:653–658. doi: 10.1038/nature09571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Inoki K, Zhu T, Guan KL. TSC2 mediates cellular energy response to control cell growth and survival. Cell. 2003;115:577–590. doi: 10.1016/s0092-8674(03)00929-2. [DOI] [PubMed] [Google Scholar]
- 30.Shaw RJ, Kosmatka M, Bardeesy N, Hurley RL, Witters LA, DePinho RA, Cantley LC. The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proc Natl Acad Sci U S A. 2004;101:3329–3335. doi: 10.1073/pnas.0308061100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hawley SA, Boudeau J, Reid JL, Mustard KJ, Udd L, Makela TP, Alessi DR, Hardie DG. Complexes between the LKB1 tumor suppressor, STRAD alpha/beta and MO25 alpha/beta are upstream kinases in the AMP-activated protein kinase cascade. J Biol. 2003;2:28. doi: 10.1186/1475-4924-2-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Woods A, Johnstone SR, Dickerson K, Leiper FC, Fryer LG, Neumann D, Schlattner U, Wallimann T, Carlson M, Carling D. LKB1 is the upstream kinase in the AMP-activated protein kinase cascade. Curr Biol. 2003;13:2004–2008. doi: 10.1016/j.cub.2003.10.031. [DOI] [PubMed] [Google Scholar]
- 33.Hardie DG. AMP-activated/SNF1 protein kinases: conserved guardians of cellular energy. Nat Rev Mol Cell Biol. 2007;8:774–785. doi: 10.1038/nrm2249. [DOI] [PubMed] [Google Scholar]
- 34.Mayer A, Denanglaire S, Viollet B, Leo O, Andris F. AMP-activated protein kinase regulates lymphocyte responses to metabolic stress but is largely dispensable for immune cell development and function. Eur J Immunol. 2008;38:948–956. doi: 10.1002/eji.200738045. [DOI] [PubMed] [Google Scholar]
- 35.Cao Y, Li H, Liu H, Zheng C, Ji H, Liu X. The serine/threonine kinase LKB1 controls thymocyte survival through regulation of AMPK activation and Bcl-XL expression. Cell Res. 2010;20:99–108. doi: 10.1038/cr.2009.141. [DOI] [PubMed] [Google Scholar]
- 36.Tamas P, Macintyre A, Finlay D, Clarke R, Feijoo-Carnero C, Ashworth A, Cantrell D. LKB1 is essential for the proliferation of T-cell progenitors and mature peripheral T cells. Eur J Immunol. 2010;40:242–253. doi: 10.1002/eji.200939677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bardeesy N, Sinha M, Hezel AF, Signoretti S, Hathaway NA, Sharpless NE, Loda M, Carrasco DR, DePinho RA. Loss of the Lkb1 tumour suppressor provokes intestinal polyposis but resistance to transformation. Nature. 2002;419:162–167. doi: 10.1038/nature01045. [DOI] [PubMed] [Google Scholar]
- 38.Viollet B, Andreelli F, Jorgensen SB, Perrin C, Geloen A, Flamez D, Mu J, Lenzner C, Baud O, Bennoun M, Gomas E, Nicolas G, Wojtaszewski JF, Kahn A, Carling D, Schuit FC, Birnbaum MJ, Richter EA, Burcelin R, Vaulont S. The AMP-activated protein kinase alpha2 catalytic subunit controls whole-body insulin sensitivity. J Clin Invest. 2003;111:91–98. doi: 10.1172/JCI16567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jones RG, Bui T, White C, Madesh M, Krawczyk CM, Lindsten T, Hawkins BJ, Kubek S, Frauwirth KA, Wang YL, Conway SJ, Roderick HL, Bootman MD, Shen H, Foskett JK, Thompson CB. The proapoptotic factors Bax and Bak regulate T Cell proliferation through control of endoplasmic reticulum Ca(2+) homeostasis. Immunity. 2007;27:268–280. doi: 10.1016/j.immuni.2007.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rathmell JC, Fox CJ, Plas DR, Hammerman PS, Cinalli RM, Thompson CB. Akt-directed glucose metabolism can prevent Bax conformation change and promote growth factor-independent survival. Mol Cell Biol. 2003;23:7315–7328. doi: 10.1128/MCB.23.20.7315-7328.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Krawczyk CM, Shen H, Pearce EJ. Functional plasticity in memory T helper cell responses. J Immunol. 2007;178:4080–4088. doi: 10.4049/jimmunol.178.7.4080. [DOI] [PubMed] [Google Scholar]
- 42.Bungard D, Fuerth BJ, Zeng PY, Faubert B, Maas NL, Viollet B, Carling D, Thompson CB, Jones RG, Berger SL. Signaling kinase AMPK activates stress-promoted transcription via histone H2B phosphorylation. Science. 2010;329:1201–1205. doi: 10.1126/science.1191241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wieman HL, Wofford JA, Rathmell JC. Cytokine stimulation promotes glucose uptake via phosphatidylinositol-3 kinase/Akt regulation of Glut1 activity and trafficking. Mol Biol Cell. 2007;18:1437–1446. doi: 10.1091/mbc.E06-07-0593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Plas DR, Talapatra S, Edinger AL, Rathmell JC, Thompson CB. Akt and Bcl-xL promote growth factor-independent survival through distinct effects on mitochondrial physiology. J Biol Chem. 2001;276:12041–12048. doi: 10.1074/jbc.M010551200. [DOI] [PubMed] [Google Scholar]
- 45.Shackelford DB, Shaw RJ. The LKB1-AMPK pathway: metabolism and growth control in tumour suppression. Nat Rev Cancer. 2009;9:563–575. doi: 10.1038/nrc2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cham CM, Driessens G, O’Keefe JP, Gajewski TF. Glucose deprivation inhibits multiple key gene expression events and effector functions in CD8+ T cells. Eur J Immunol. 2008;38:2438–2450. doi: 10.1002/eji.200838289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shaw RJ, Lamia KA, Vasquez D, Koo SH, Bardeesy N, Depinho RA, Montminy M, Cantley LC. The kinase LKB1 mediates glucose homeostasis in liver and therapeutic effects of metformin. Science. 2005;310:1642–1646. doi: 10.1126/science.1120781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Motoyama N, Wang F, Roth KA, Sawa H, Nakayama K, Negishi I, Senju S, Zhang Q, Fujii S, et al. Massive cell death of immature hematopoietic cells and neurons in Bcl-x-deficient mice. Science. 1995;267:1506–1510. doi: 10.1126/science.7878471. [DOI] [PubMed] [Google Scholar]
- 49.Tamas P, Hawley SA, Clarke RG, Mustard KJ, Green K, Hardie DG, Cantrell DA. Regulation of the energy sensor AMP-activated protein kinase by antigen receptor and Ca2+ in T lymphocytes. J Exp Med. 2006;203:1665–1670. doi: 10.1084/jem.20052469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Huang X, Wullschleger S, Shpiro N, McGuire VA, Sakamoto K, Woods YL, McBurnie W, Fleming S, Alessi DR. Important role of the LKB1-AMPK pathway in suppressing tumorigenesis in PTEN-deficient mice. Biochem J. 2008;412:211–221. doi: 10.1042/BJ20080557. [DOI] [PubMed] [Google Scholar]
- 51.Lizcano JM, Goransson O, Toth R, Deak M, Morrice NA, Boudeau J, Hawley SA, Udd L, Makela TP, Hardie DG, Alessi DR. LKB1 is a master kinase that activates 13 kinases of the AMPK subfamily, including MARK/PAR-1. Embo J. 2004;23:833–843. doi: 10.1038/sj.emboj.7600110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tiainen M, Vaahtomeri K, Ylikorkala A, Makela TP. Growth arrest by the LKB1 tumor suppressor: induction of p21(WAF1/CIP1) Hum Mol Genet. 2002;11:1497–1504. doi: 10.1093/hmg/11.13.1497. [DOI] [PubMed] [Google Scholar]
- 53.Boyman O, Letourneau S, Krieg C, Sprent J. Homeostatic proliferation and survival of naive and memory T cells. Eur J Immunol. 2009;39:2088–2094. doi: 10.1002/eji.200939444. [DOI] [PubMed] [Google Scholar]
- 54.Surh CD, Sprent J. Homeostasis of naive and memory T cells. Immunity. 2008;29:848–862. doi: 10.1016/j.immuni.2008.11.002. [DOI] [PubMed] [Google Scholar]
- 55.Yang K, Neale G, Green DR, He W, Chi H. The tumor suppressor Tsc1 enforces quiescence of naive T cells to promote immune homeostasis and function. Nat Immunol. 2011 doi: 10.1038/ni.2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Parkhitko A, Myachina F, Morrison TA, Hindi KM, Auricchio N, Karbowniczek M, Wu JJ, Finkel T, Kwiatkowski DJ, Yu JJ, Henske EP. Tumorigenesis in tuberous sclerosis complex is autophagy and p62/sequestosome 1 (SQSTM1)-dependent. Proc Natl Acad Sci U S A. 2011;108:12455–12460. doi: 10.1073/pnas.1104361108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.O’Brien TF, Gorentla BK, Xie D, Srivatsan S, McLeod IX, He YW, Zhong XP. Regulation of T cell survival and mitochondrial homeostasis by TSC1. Eur J Immunol. 2011 doi: 10.1002/eji.201141411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sag D, Carling D, Stout RD, Suttles J. Adenosine 5’-monophosphate-activated protein kinase promotes macrophage polarization to an anti-inflammatory functional phenotype. J Immunol. 2008;181:8633–8641. doi: 10.4049/jimmunol.181.12.8633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nath N, Giri S, Prasad R, Salem ML, Singh AK, Singh I. 5-aminoimidazole-4-carboxamide ribonucleoside: a novel immunomodulator with therapeutic efficacy in experimental autoimmune encephalomyelitis. J Immunol. 2005;175:566–574. doi: 10.4049/jimmunol.175.1.566. [DOI] [PubMed] [Google Scholar]
- 60.Cao Y, Li H, Liu H, Zhang M, Hua Z, Ji H, Liu X. LKB1 regulates TCR-mediated PLCgamma1 activation and thymocyte positive selection. Embo J. 2011;30:2083–2093. doi: 10.1038/emboj.2011.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hawley SA, Pan DA, Mustard KJ, Ross L, Bain J, Edelman AM, Frenguelli BG, Hardie DG. Calmodulin-dependent protein kinase kinase-beta is an alternative upstream kinase for AMP-activated protein kinase. Cell Metab. 2005;2:9–19. doi: 10.1016/j.cmet.2005.05.009. [DOI] [PubMed] [Google Scholar]
- 62.Woods A, Dickerson K, Heath R, Hong SP, Momcilovic M, Johnstone SR, Carlson M, Carling D. Ca2+/calmodulin-dependent protein kinase kinase-beta acts upstream of AMP-activated protein kinase in mammalian cells. Cell Metab. 2005;2:21–33. doi: 10.1016/j.cmet.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 63.Maciver NJ, Jacobs SR, Wieman HL, Wofford JA, Coloff JL, Rathmell JC. Glucose metabolism in lymphocytes is a regulated process with significant effects on immune cell function and survival. J Leukoc Biol. 2008;84:949–957. doi: 10.1189/jlb.0108024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jacobs SR, Michalek RD, Rathmell JC. IL-7 is essential for homeostatic control of T cell metabolism in vivo. J Immunol. 2010;184:3461–3469. doi: 10.4049/jimmunol.0902593. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.