

Abstract
Chronic renal failure (CRF) is associated with malnutrition and renal tissue accumulation of lipids, which can contribute to progression of renal disease. This study was designed to explore the effect of a high-calorie diet on pathways involved in lipid metabolism in the remnant kidney of rats with CRF. 5/6 nephrectomized rats were randomized to receive a regular diet (3.0 kcal/g) or a high-calorie diet (4.5 kcal/g) for 12 weeks. Renal lipid contents and abundance of molecules involved in cholesterol and fatty acid metabolism were studied. The CRF group consuming a regular diet exhibited growth retardation; azotemia; proteinuria; glomerulosclerosis; tubulointerstitial injury; heavy lipid accumulation in the remnant kidney; up-regulation of lectin-like oxidized low-density lipoprotein receptor-1 (LOX-1), ATP-binding cassette transporter-1 (ABCA1), liver X receptor (LXR) α/β, carbohydrate-responsive element binding protein (ChREBP) and acyl-CoA carboxylase (ACC); and down-regulation of peroxisome proliferator-activated receptor-α (PPAR-α), carnitine palmitoyltransferase-1 (CPT1) and liver-type fatty acid binding protein (L-FABP). The high-calorie diet restored growth; reduced the severity of tubulointerstitial injury, proteinuria and azotemia; partially lowered renal tissue lipid contents; attenuated the up-regulation of mediators of lipid influx (LOX-1), lipid efflux (LXR-α/β and ABCA1) and fatty acid biosynthesis (ChREBP and ACC); and reversed the down-regulation of factors involved in fatty acid oxidation (PPAR-α, CPT1 and L-FABP). In conclusion, a high-calorie diet restores growth, improves renal function and structure, and lowers lipid burden in the remnant kidney. The latter is associated with and most likely due to reduction in lipid influx and enhancement of fatty acid oxidation.
Keywords: Chronic kidney disease, Renal lipid metabolism, Malnutrition, Inflammation, Progression of renal disease, Reverse cholesterol transport
1. Introduction
Malnutrition is a common feature of advanced chronic kidney disease (CKD) and a strong predictor of morbidity and mortality in patients with end-stage renal disease (ESRD) [1-3]. In fact, observational studies have shown that markers of overnutrition are strongly associated with decreased morbidity and mortality, including a high risk of cardiovascular events and death in ESRD patients [4-6]. Malnutrition in CKD patients is associated with accelerated whole-body and skeletal muscle proteolysis and increased energy expenditure, resulting in a net negative proteinenergy balance [7-9]. Parenteral intradialytic calorie and amino acid supplementation has been shown to acutely reverse the net negative whole body-forearm muscle protein balance [10], increasing body weight, serum albumin and ApoA-I levels without altering plasma lipid concentration in ESRD patients maintained on hemodialysis. Moreover, trial of energy supplementation via addition of the glucose polymer Polycose to the usual diet for 6 months has been shown to significantly increase total and lean body mass, with no significant effect on plasma triglyceride level [11], in hemodialysis patients. Observational studies have shown that in patients with CKD Stages 3-5, the rate of decline in glomerular filtration rate is higher in patients with low energy intake than in patients with moderate or high energy intake [12]. While high protein intake is known to accelerate the progression of renal disease in experimental animals [13,14], the effect of a high-calorie diet on the progression of nondiabetic renal disease is uncertain.
Malnutrition syndrome in CKD is almost invariably associated with oxidative stress and inflammation [15-17]. The associated oxidative stress and inflammation lead to oxidation of lipids and lipoproteins, uptake of oxidized lipoproteins by macrophages and resident cells, and formation of foam cells – events that promote atherosclerosis, glomerulosclerosis and tubulointerstitial injury [18]. Accumulation of excess lipids in nonadipose tissues can lead to lipotoxicity and cellular dysfunction, which is caused by the direct toxic effects of fatty acids or byproducts of their interaction with reactive oxygen species, ATP depletion and fatty-acid-induced apoptosis [19]. Cellular lipid homeostasis is regulated by the influx, synthesis, catabolism and efflux of lipids. An imbalance in theseprocesses can result in the conversion of macrophages, mesangial cells and vascular smooth muscle cells into foam cells. The influx of lipid into macrophages is mediated by several independent pathways, including scavenger receptors, whereas cholesterol efflux is primarily mediated by liver X receptor (LXR) α/β, which serves as an intracellular cholesterol sensor and regulates the expression of its target genes ATP-binding cassette transporter-1 (ABCA1) and the scavenger receptor class B type I (SR-BI), among others [18,20,21]. Sterol-responsive element binding proteins (SREBPs) and carbohydrate-responsive element binding protein (ChREBP) serve as master regulators of cellular lipid synthesis [22,23], whereas peroxisome proliferator-activated receptor-α (PPAR-α) regulates the expression of genes involved in the uptake, binding, transport, cellular retention and catabolism of fatty acids [24-26].
There is mounting evidence pointing to accumulation of lipids in the renal tissue and its contribution to the progression of glomerular and tubulointerstitial lesions in metabolic syndrome [18,27], chronic glomerulopathy [28,29], nephrotic syndrome [30], chronic renal insufficiency [31], diabetic nephropathy [32-34], obesity-associated renal disease [35], aging nephrosclerosis [36,37] and acute kidney injury in animals with experimental rhabdomyolysis [38].
In a recent study, we found heavy lipid accumulation and marked dysregulation of lipid regulatory proteins, enzymes, receptors and transcription factors in the remnant kidney of 5/6 nephrectomized animals [31]. The present study was undertaken to determine the effect of the long-term consumption of a high-calorie diet on renal tissue lipid contents and lipid regulatory proteins in rats with CKD induced by 5/6 nephrectomy.
2. Materials and methods
2.1. Animals and diet
Male Sprague–Dawley rats weighing 225–250 g were purchased from Harlan Sprague Dawley (Indianapolis, IN). They were housed in a climate-controlled light-regulated facility with 12:12-h day–night cycles. The animals were fed regular rat chow (Purina Mills, Brentwood, MO) and water ad libitum and randomly assigned to the chronic renal failure (CRF) group and the normal control group. The animals assigned to the CRF group were subjected to 5/6 nephrectomy by surgical resection using dorsal incision, as described previously [39]. The animals assigned to the control group were subjected to sham operation. All surgical procedures were carried out under general anesthesia (Nembutal, 50 mg/kg ip). Strict hemostasis and aseptic techniques were observed. After a 1-week observation period, the animals in the CRF group were randomized to receive a regular diet (3.0 kcal/g; Purina Mills) or a high-calorie diet (4.5 kcal/g; Harlan Teklad, Madison, WI) for 12 weeks. The shamoperated control group was fed a regular diet. The composition of the diets is shown in Table 1. At the conclusion of the 12-week observation period, the animals were placed in metabolic cages for a 24-h urine collection. They were then anesthetized (pentobarbital, 50 mg/kg ip) and euthanized by exsanguination using cardiac puncture. The kidneys were immediately removed, frozen in liquid nitrogen and stored at −70°C until processed. All experiments were approved by the Institutional Committee for the Use and Care of Experimental Animals at University of California, Irvine. The concentrations of serum creatinine, total cholesterol, triglyceride, highdensity lipoprotein (HDL) cholesterol and low-density lipoprotein (LDL) cholesterol, as well as urinary protein excretion, were measured as described in our previous studies [39].
Table 1.
Chow and high-calorie diet composition
| Composition | Chowa | High-calorie dietb |
|---|---|---|
| Energy as carbohydrate (%) | 59.1 | 42.7 |
| Energy as fat (%) | 12.1 | 42.0 |
| Energy as protein (%) | 28.8 | 15.2 |
| Carbohydrate (g/100 g) | 49.3 | 49.1 |
| Starch | 21.5 | 15 |
| Sucrose | 3.4 | 34.1 |
| Protein (g/100 g) | 24 | 19.5 |
| Fat (g/100 g) | 10.5 | 21.0 |
| Vitamins (g/100 g) | 4 | 1 |
| Minerals (g/100 g) | 6.9 | 3.9 |
| Fiber (g/100 g) | 5.3 | 5 |
| Digestible energy (kcal/g) | 3.04 | 4.5 |
Standard laboratory rat chow (Prolab RMH 2500).
Adjusted high-calorie diet: protein is casein, vitamin mixture is Teklad no. 40060, mineral mixture is AIN-76 (170915) and fat is supplied in the form of anhydrous milk fat.
2.2. Tissue lipid contents
Total lipids were extracted from 100 mg of tissue by the method of Folch et al. [40]. Briefly, samples were homogenized in 6 ml of chloroform–methanol (2:1). The mixture stood for 1 h, after which 1.5 ml of water was added, and the mixture was centrifuged for 10 min at 2000×g. The organic phase was evaporated to dryness under nitrogen stream and taken up in chloroform. Fifty-microliter aliquots of this organic phase were solubilized by adding a drop of Triton X-100, and total cholesterol and triglyceride contents were determined using enzymatic kits from Stanbio Laboratories (Boerne, TX). Data were expressed as the amount of the given lipids per gram of original kidney mass.
2.3. Lipid staining
After euthanasia of the animals, the kidneys were rapidly removed, sliced longitudinally and immersed in 4% paraformaldehyde/phosphate-buffered saline (at 4°C) overnight. Subsequently, the tissue was cryoprotected in 30% sucrose at 4°C and then frozen using liquid nitrogen. Frozen sections were then cut using Leica CM 1900 UV (Leica, Germany) at 10 μm. Each section was air dried for 1 h and fixed in 10% formalin for 10 min. Subsequently, the tissue was rinsed with distilled water, and the sections were stained with Oil Red O (Sigma Aldrich, St. Louis, MO) in accordance with the manufacturer’s protocol.
2.4. Preparation of kidney homogenates and nuclear extracts
All solutions, tubes and centrifuges were maintained at 0-4°C. Nuclear extract was prepared as described previously [41]. Briefly, 100 mg of kidney cortex was homogenized in 0.5 ml of buffer A containing 10 mM HEPES (pH 7.8), 10 mM KCl, 2 mM MgCl2, 1 mM dithiothreitol (DTT), 0.1 mM EDTA, 0.1 mM phenylmethanesulphonylfluoride (PMSF), 1 μM pepstatin and 1 mM P-aminobenzamidine using a tissue homogenizer for 20 s. Homogenates were kept on ice for 15 min, 125 μl of a 10% Nonidet P40 (NP-40) solution was added and mixed for 15 s, and the mixture was centrifuged for 2 min at 12,000 rpm. The supernatant containing cytosolic proteins was collected. Pelleted nuclei were washed once with 200 μl of buffer A plus 25 μl of 10% NP-40, centrifuged, suspended in 50 μl of buffer B [50 mM HEPES (pH 7.8), 50 mM KCl, 300 mM NaCl, 0.1 mM EDTA, 1 mM DTT, 0.1 mM PMSF and 10% (vol/vol) glycerol], mixed for 20 min and centrifuged for 5 min at 12,000 rpm. The supernatant containing nuclear proteins was stored at −80°C. The protein concentration in tissue homogenates and nuclear extracts was determined by Bio-Rad protein assay (Bio-Rad Laboratories, Hercules, CA).
2.5. Western blot analyses
Target proteins in the cytoplasmic and/or nuclear fractions of the kidney tissue were measured by Western blot analysis using the following antibodies. Rabbit antibodies against rat SREBP-1, SREBP-2, SREBP cleavage-activating protein (SCAP), insulin-induced gene-1 (Insig-1) and insulin-induced gene-2 (Insig-2), PPAR-α, liver-type fatty acid binding protein (L-FABP) and LXR-α/β antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Antibodies against ChREBP, ABCA1 and carnitine palmitoyltransferase-1 (CPT1) were obtained from Novus Biologicals (Littleton, CO). Antibodies against acyl-CoA:cholesterol acyltransferase (ACAT)-1 were obtained from GenScript (Piscataway, NJ). Antibodies against 3-hydroxy-3-methylglutaryl coenzyme A reductase (HMG-CoA reductase) were purchased from Upstate (Billerica, MA). Anti-low-density lipoprotein receptor-1 (LOX-1) was purchased from Abcam (Cambridge, MA). These antibodies were used in Western blot analyses for the corresponding proteins. Antibodies to histone H1 (Santa CruzBiotechnology) and β-actin (Sigma, St. Louis, MO) were used for measurements of histone and β-actin, which served as the housekeeping proteins for nuclear and cytosolic target proteins, respectively.
Briefly, aliquots containing 50 μg of proteins were fractionated on 8% and 4-20% Tris-glycine gel (Novex, San Diego, CA) at 120 V for 2 h and transferred to Hybond-ECL membrane (Amersham Life Science, Arlington Heights, IL). The membrane was incubated for 1 h in blocking buffer [1× Tris-buffered saline (TBS), 0.05% Tween-20 and 5% nonfat milk] and then overnight in the same buffer containing the given antibodies. The membrane was washed three times for 5 min in 1× TBS and 0.05% Tween-20 prior to 2-h incubation in a buffer (1× TBS, 0.05% Tween-20 and 3% nonfat milk) containing horseradish-peroxidase-linked anti-rabbit IgG and anti-mouse IgG (Amersham Life Science) at 1:1000 dilution. The membrane was washed four times and developed by autoluminography using ECL chemiluminescent agents (Amersham Life Science).
2.6. Statistics
Analysis of variance, multiple range test and regression analysis were used in statistical analysis of the data. Data are presented as mean±S.D. P values less than .05 were considered significant.
3. Results
3.1. General data
Data are shown in Tables 2 and 3. As expected, caloric intake was significantly higher in the CRF group fed a high-calorie diet than in the CRF group fed a regular diet. However, there was no significant difference in protein intake among the study groups (Table 2). The CRF group exhibited azotemia; proteinuria; hypertension; marked elevation of plasma total cholesterol, LDL cholesterol and triglycerides; growth retardation; reduced visceral fat mass; and marked remnant kidney hypertrophy. Consumptionof the high-calorie diet resulted in attenuation of azotemia, hypercholesterolemia, proteinuria and remnant kidney hypertrophy; prevented growth retardation; and increased visceral fat mass, but did not significantly change arterial pressure.
Table 2.
Food and energy intakes
| Group | Control | CRF | CRF+high-calorie diet |
|---|---|---|---|
| Food intake (g/day) | 14.6±2.8 | 15.8±4.9 | 15.3±1.6 |
| Carbohydrate intake (g/day) | 5.7±0.9 | 6.8±2.1 | 7.4±0.7 |
| Protein intake (g/day) | 3.4±0.7 | 3.8±1.2 | 2.9±0.3 |
| Fat intake (g/day) | 0.5±0.1 | 0.8±0.3 | 3.2±0.3### |
| Energy intake (kcal/day) | 43.8±8.5 | 47.4±14.8 | 67.7±7.2# |
Values are given as mean±S.D.
P<.05 versus 5/6 nephrectomized CRF groups.
P<.001 versus 5/6 nephrectomized CRF groups
Table 3.
Biochemical parameters in CRF rats fed a regular diet or a high-calorie diet
| Group | Control | CRF | CRF+high-calorie diet |
|---|---|---|---|
| Total cholesterol (mg/dl) | 71.2±9.8 | 221.2±20.5 *** | 186.4±29.9 *** |
| LDL cholesterol (mg/dl) | 19.1±6.0 | 95.9±14.1 *** | 66.7±10.7***,# |
| HDL cholesterol (mg/dl) | 45.3±8.6 | 100.9±10.4 *** | 61.6±19.0*,# |
| Triglycerides (mg/dl) | 45.8±18.3 | 99.7±3.6 * | 192.8±63.6***,# |
| Free fatty acid (mEq/L) | 0.192±0.074 | 0.220±0.068 | 0.382±0.052***,## |
| Epididymal fat weight (g) | 4.6±0.6 | 3.6±0.6 * | 6.2±1.7*,## |
| Abdominal fat weight (g) | 2.6±0.7 | 1.8±0.7 | 5.6±1.1***,### |
| Urine protein (mg/24 h) | 6.7±1.3 | 80.3±7.3 *** | 31.1±12.8***,### |
| Creatinine clearance (ml/min) |
5.62±1.18 | 1.74±0.38 *** | 1.44±0.33 *** |
| Blood urea nitrogen (mg/dl) | 25.4±2.1 | 53.6±5.0 *** | 42.1±9.9**,# |
| Serum creatinine (mg/dl) | 0.50±0.1 | 1.56±0.5 *** | 1.43±0.2 *** |
| Systolic blood pressure (mmHg) |
123.5±13.4 | 161.4±10.0 ** | 150.6±14.2 ** |
| Diastolic blood pressure (mmHg) |
87.5±7.4 | 117.0±4.5 | 111.0±13.1 |
| Weight gain (g) | 146.0±13.4 | 119.3±12.0 * | 148.7±14.6## |
| Left kidney weight (g) | 1.45±0.16 | 2.38±0.38 *** | 1.65±0.13*,## |
Values are given as mean±S.D.
P<.05 versus control groups.
P<.01 versus control groups.
P<.001 versus control groups.
P<.05 versus 5/6 nephrectomized CRF groups.
P<.01 versus 5/6 nephrectomized CRF groups.
P<.001 versus 5/6 nephrectomized CRF groups.
3.2. Tissue lipid contents
Data are shown in Fig. 1. Oil Red O staining of the frozen kidney tissue revealed a marked accumulation of neutral lipids in the glomerular and tubulointerstitial regions of the remnant kidney in the CRF groups compared with that found in the kidneys of normal control rats. Consumption of the high-calorie diet reduced, but did not prevent, lipid accumulation in the remnant kidney. Similarly, the total cholesterol and triglyceride contents of the kidney tissue were markedly increased in the CRF rats consuming the regular diet and were partially reduced in rats fed the high-calorie diet.
Fig. 1.

Top: Representative photomicrographs of the Oil-Red-O-stained glomerular and tubulointerstitial regions of the kidney in the control (CTL; A and D), CRF (B and E) and CRF groups fed the high-calorie diet (CRF+HC; C and F). Bottom: Bar graphs depicting tissue cholesterol and triglyceride contents expressed per gram of the original kidney mass in the CRF groups fed the high-calorie diet. ***P<.001 versus control groups; ##P<.01 versus 5/6 nephrectomized CRF groups.
3.3. LOX-1 data
Data are shown in Fig. 2. Compared to the control group, the CRF group exhibited a marked increase in LOX-1 protein abundance in the remnant kidney. Consumption of the high-calorie diet attenuated CRF-induced up-regulation of LOX-1 in the remnant kidney.
Fig. 2.
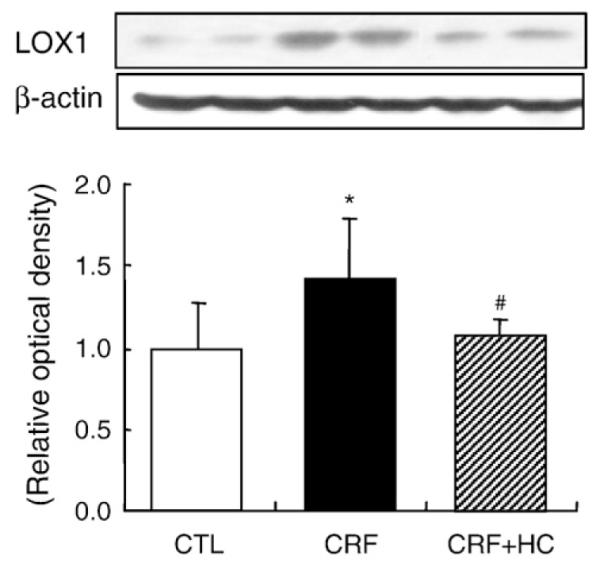
Representative Western blot analyses and group data depicting the protein abundance of LOX-1 in the renal tissues of CRF groups fed the high-calorie diet. *P<.05 versus control groups; #P<.05 versus 5/6 nephrectomized CRF groups.
3.4. SREBP-2, ACAT1 and HMG-CoA reductase data
Data are illustrated in Fig. 3. The 68-kDa active SREBP-2 fragment was significantly reduced in the nuclear fraction of the renal tissue in the CRF rats fed the regular diet as compared to that found in the control group. This was associated with a significant reduction in HMG-CoA reductase and a marked increase in ACAT1 abundance. Consumption ofthe high-calorie diet partially reversed the down-regulation of HMG-CoA reductase and the up-regulation of ACAT1 in the renal tissues without significantly affecting nuclear SREBP-2 abundance.
Fig. 3.

Representative Western blot analyses and group data depicting the protein abundance of precursor SREBP-2 (pSREBP-2), mature SREBP-2 (nSREBP-2), ACAT1 and HMG-CoA reductase in the renal tissues of CRF groups fed the high-calorie diet. *P<.05, **P<.01, ***P<.001 versus control groups; #P<.05, ###P<.001 versus 5/6 nephrectomized CRF groups.
3.5. SCAP, Insig-1 and Insig-2 data
Data are depicted in Fig. 4. SCAP and Insig-2 protein abundance was significantly lower in the remnant kidney of the CRF group fed the regular diet than in the control kidneys. Consumption of the high-calorie diet partially reversed the down-regulation of SCAP, but did not significantly affect Insig-1 or Insig-2 abundance.
Fig. 4.

Representative Western blot analyses and group data depicting SCAP, Insig-1 and Insig-2 protein abundance in the renal tissues of CRF groups fed the high-calorie diet. *P<.05, **P<.01 versus control groups; #P<.05 versus 5/6 nephrectomized CRF groups.
3.6. ChREBP, SREBP-1, and acyl-CoA carboxylase (ACC) data
Data are shown in Fig. 5. ChREBP and ACC protein abundance was significantly higher and SREBP-1 protein abundance was significantly lower in the kidney of the CRF rats fed the regular diet as comparedwith the values found in the control group. Consumption of the high-calorie diet reversed CRF-induced up-regulation of ChREBP and ACC, but did not alter SREBP-1 abundance in the remnant kidney.
Fig. 5.

Representative Western blot analyses and group data depicting the protein abundance of precursor SREBP-1 (pSREBP-1), mature SREBP-1 (nSREBP-1), ChREBP and ACC in the renal tissues of CRF groups fed the high-calorie diet. *P<.05, **P<.01 versus control groups; #P<.05, ##P<.01 versus 5/6 nephrectomized CRF groups.
3.7. PPAR-α, CPT1 and L-FABP data
Data are illustrated in Fig. 6. PPAR-α, CPT1 and L-FABP abundance was significantly reduced in the remnant kidneys of the CRF group consuming the regular diet when compared with those found in the control group. Consumption of the high-calorie diet partially reversed these abnormalities.
Fig. 6.

Representative Western blot analyses and group data depicting renal PPAR-α, CPT1 and L-FABP protein abundance in CRF groups fed the high-calorie diet. *P<.05, ***P<.001 versus control groups; #P<.05, ##P<.01 versus 5/6 nephrectomized CRF groups.
3.8. LXR-α/β and ABCA1 data
Data are shown in Fig. 7. LXR-α/β and ABCA1 abundance was significantly increased in the kidney tissues of the CRF group fed the regular diet. Consumption of the high-calorie diet partially reversed the CRF-induced up-regulation of ABCA1 and LXR-α/β in the remnant kidney.
Fig. 7.

Representative Western blot analyses and group data depicting the protein abundance of renal tissue ABCA1 and nuclear LXR-α/β in CRF groups fed the high-calorie diet. ***P<.001 versus control groups; ##P<.01 versus 5/6 nephrectomized CRF groups.
4. Discussion
Malnutrition is highly prevalent among patients with advanced CKD, and indices of malnutrition are powerful predictors of morbidity and mortality in this population [14]. Inflammation, oxidative stress, uremic toxins, metabolic and hormonal derangements, and comorbid conditions work in concert to raise catabolic rate and to promote anorexia in patients with advanced CRF [42]. The prevailing anorexia and catabolic state, in turn, lead to malnutrition, proteolysis, muscle atrophy and depletion of body fat stores in patients with advanced CRF. However, the effect of malnutrition on the progression of renal disease is not known.
Despite a similar daily food intake, body weight and visceral fat mass were significantly lower in our CRF rats consuming a regular diet when compared with the control group. These findings point to a catabolic state marked by heightened energy expenditure in the CRF animals. Increased delivery of energy with the high-calorie diet reversed CRF-induced growth retardation, prevented depletion of fat stores and appeared to have attenuated whole-body and skeletal muscle protein catabolism, as evidenced by a lower urea nitrogen concentration. Retrospective studies have shown that the rate of decline in glomerular filtration rate in patients with CKD Stages 3-5 is higher in those with low energy intake than in those with moderate or high energy intake [43]. However, prospective studies of the effect of a high-calorie diet on the progression of renal disease are lacking, and its potential impact on the cellular and molecular mechanisms involved in the progressive deterioration of renal function and structure are unknown.
Accumulation of lipids in the renal tissue plays an important part in the progression of renal disease in metabolic syndrome [18,27], chronic glomerulopathy [28,29], nephrotic syndrome [30], chronicrenal insufficiency [31], diabetic nephropathy [32-34], obesity-associated renal disease [35], aging nephrosclerosis [36,37] and acute kidney injury in experimental rhabdomyolysis [38]. Cellular lipid homeostasis is regulated by the influx, synthesis, catabolism and efflux of lipids. An imbalance in these processes can result in the conversion of macrophages, mesangial cells and vascular smooth muscle cells into foam cells. Influx of lipid into macrophages is mediated by several independent pathways, including scavenger receptors, whereas cholesterol efflux is primarily mediated by LXR-α/β, which serves as an intracellular cholesterol sensor and regulates the expression of its target genes ABCA1 and SR-BI, among others [18,20,21]. SREBPs and ChREBP serve as master regulators of cellular lipid synthesis. For instance, SREBP-1c and ChREBP independently regulate fatty acid synthesis, whereas SREBP-2 regulates cholesterol synthesis [22,23]. Proteolytic activation of SREBPs is inhibited by elevated intracellular free cholesterol concentration, thereby limiting the production of cholesterol through feedback repression [44,45]. Sterols induce the binding of SCAP, which has an escort function and a sterol-sensing function, to Insig-1 and facilitates retention of the SCAP-SREBP complex in the endoplasmic reticulum (ER). In sterol-depleted cells, SCAP escorts SREBPs from the ER to the Golgi apparatus for proteolytic processing, thereby allowing SREBP-2 to stimulate cholesterol synthesis [46]. There is growing evidence that dysregulation of SREBP contributes to the pathogenesis of nephropathy in diabetes and obesity, aging nephrosclerosis, nephrotic syndrome and CRF [28-37]. PPAR-α regulates the expression of genes involved in the uptake, binding, transport, cellular retention and catabolism of fatty acids [24-26]. In fact, PPAR-α deficiency has been shown to accelerate dyslipidemia and to cause glomerular matrix expansion, inflammatory cell infiltration and proteinuria in animals with experimental diabetic nephropathy [47,48]. The present study was undertaken to determine the effect of the long-term consumption of a high-calorie diet on renal tissue lipid content and lipid regulatory proteins in rats with CKD induced by 5/6 nephrectomy.
In a recent study [31], we found heavy lipid accumulation in the remnant kidney of 5/6 nephrectomized animals fed a regular diet. This was associated with a marked up-regulation of proteins involved in lipid influx, lipid efflux and fatty acid synthesis, and a down-regulation of factors involved in cholesterol synthesis and fatty acid catabolism. In confirmation of the above study [31], the CRF rats fed the regular diet exhibited heavy lipid accumulation in the glomeruli and tubulointerstitial regions of the remnant kidney. Consumption of the high-calorie diet significantly reduced, but did not normalize, the kidney tissue lipid burden in the study animals.
CRF is associated with oxidative stress, inflammation, elevation of proinflammatory cytokines and formation of oxidized lipids and lipoproteins [15,17,49], which can collectively stimulate the expression of scavenger receptors, heighten lipid influx in macrophages and mesangial cells, and contribute to interstitial injury and glomerulosclerosis [50-52]. In fact, in confirmation of earlier studies [31,53], LOX-1 was up-regulated in the remnant kidneys of our CRF rats consuming the regular diet. Consumption of the high-calorie diet reversed the CRF-induced up-regulation of LOX-1 in the remnant kidney. This phenomenon can, in part, contribute to the observed reduction in lipid burden in the remnant kidneys of CRF animals fed the high-calorie diet.
In confirmation of our previous findings [31], HMG-CoA reductase, the rate-limiting enzyme in cholesterol biosynthesis, and the nuclear content of its master regulator SREBP-2 were significantly reduced in the CRF rats fed the regular diet. This was associated with significant down-regulations of SCAP and Insig-2 and a mild reduction in Insig-1. In the presence of an elevated cellular sterol level, SREBP-SCAP complexes are anchored by Insigs to the ER membrane as transcriptionally inactive precursor proteins. This process prevents the migration of SREBP to the Golgi apparatus for proteolytic activation and eventual translocation to the nucleus [54]. In addition to blocking the activation of SREBPs, Insigs bind and facilitate the ubiquitination of HMG-CoA reductase in response to elevated cellular sterols concentrations [55]. Down-regulation of SCAP, Insig-1 and Insig-2 in the remnant kidney is consistent with general suppression of the sterol-regulated lipogenic pathway occasioned by heavy lipid influx from the extracellular compartment in the CRF animals. Consumption of a high-calorie diet did not alter SREBP abundance or activity in the remnant kidney of the CRF rats.
The remnant kidneys of the CRF rats fed the regular diet showed a marked up-regulation of ACAT1, which catalyzes the esterification of cholesterol for sequestration in intracellular vesicles – events that play a central role in foam cell formation [56]. Together, down-regulation of HMG-CoA reductase and up-regulation of ACAT1 in the remnant kidney represent a coordinated response to a heightened influx of cholesterol. The observed up-regulation of ACAT expression in the remnant kidney is also found in the liver in this model [57]. The contribution of elevated ACAT to the pathogenesis of lipid disorders and the progression of renal disease in this model is supported by amelioration of proteinuria and preservation of residual renal function in response to pharmacological inhibition of ACAT in this model [58]. Consumption of a high-calorie diet partially reversed these changes, reflecting a lower influx of cholesterol occasioned by partial reduction in plasma LDL and LOX-1 in the remnant kidney.
Genes involved in fatty acid synthesis are independently regulated by SREBP-1c and ChREBP, which are activated by reduced cellular sterol and increased cellular glucose loads, respectively [23,45]. Expression of ACC, the key enzyme in fatty acid synthesis, was significantly higher in the kidneys of the CRF rats fed the regular diet than that in the kidneys of the rats in the control group. This was associated with a marked increase in the nuclear abundance of ChREBP and a significant reduction in SREBP-1 in the remnant kidney tissue of the CRF rats fed the regular diet. The observed up-regulation of the lipogenic enzyme ACC in the face of divergent regulatory pathways that are independently driven by cellular sterol and glucose loads points to the dominant influence of the latter in the diseased kidney. We believe that the observed up-regulation of ChREBP and the consequent up-regulation of ACC in the remnant kidney are driven by increased filtered glucose load in the remaining nephrons undergoing compensatory hypertrophy and hyperfiltration. Consumption of the high-calorie diet attenuated CRF-induced remnant kidney hypertrophy and reversed the up-regulation of ChREBP and ACC in the remnant kidney.
PPAR-α, CPT1 and L-FABP abundance was significantly reduced in the remnant kidneys of the CRF group consuming the regular diet when compared with that found in the control group. Consumption of the high-calorie diet partially reversed these abnormalities. PPAR-α is predominantly expressed in tissues with high fatty acid catabolic rates such as the liver, kidney, heart and muscle. In the kidney, PPAR-α is highly abundant in the proximal tubules and medullary thick ascending limb of loop of Henle, with much lower levels in glomerular mesangial cells [26]. PPAR-α promotes fatty acid catabolism via up-regulation of L-FABP and CPT1. L-FABP and CPT1 serve as vehicles for the intracellular transport of long-chain acyl-CoAs for mitochondrial and peroxisomal β-oxidation [59] and, as such, promote a shift in fatty acid metabolism from reesterification and storage to oxidation. The CRF animals consuming a regular diet exhibited a significant down-regulation of PPAR-α, L-FABP and CPT1 in the remnant kidneys. Consumption of the high-calorie diet reversed the down-regulation of PPAR-α and its target genes and reduced fat storage in the remnant/diseased kidney. In addition to causing lipotoxicity, impaired fatty acid oxidation can contribute to cellular injury and dysfunction by limiting lipid-derived energy production in the remnant kidney, which faces increased energy demand by the remaining nephrons occasioned by compensatory hypertrophy and hyperfiltration. Thus,restoration of fatty acid utilization by the high-calorie diet may have, in part, contributed to the observed attenuation of tubulointerstitial injury in these animals.
ABCA1 transporter is the gatekeeper of the reverse cholesterol transport pathway, which mediates the transfer of cellular cholesterol and phospholipids to lipid-poor HDL for disposal in the liver [60]. Expression of ABCA1 and a number of other genes involved in cholesterol absorption, efflux, transport and excretion is regulated by LXR-α/β, which serves as a cholesterol sensor [61]. The CRF rats employed in the present study exhibited a marked up-regulation of ABCA1 in the remnant kidney. This was associated with an increased nuclear translocation of LXR-α/β. Up-regulation of ABCA1, which is the primary pathway of cellular cholesterol and phospholipid efflux, and activation of its master regulator LXR point to the cellular response to increased cholesterol burden in the remnant kidney. Activation of LXR in macrophages leads to the coordinate induction of multiple genes potentially involved in cholesterol efflux, including ABCA1, ABCG1 and ApoE. Induction of these genes in response to lipid loading may serve to limit the accumulation of lipid and thus protect against the development of fatty lesions and atherosclerosis [62,63]. The CRF rats fed the high-calorie diet exhibited a significant reversal of the CRF-induced up-regulation of ABCA1 in the remnant kidney. This was associated with a decreased nuclear translocation of LXR-α/β in the remnant kidney. It should be noted, however, that despite the up-regulation of cellular efflux pathways, HDL-mediated reverse cholesterol and phospholipid transport may be impaired in CRF. Several factors can contribute to defective reverse cholesterol transport in CRF. First, CRF significantly lowers the hepatic production of ApoA-I, which is the principal constituent of HDL [64]. Second, CRF significantly lowers hepatic production and reduces the plasma concentration of lecithin:cholesterol acyltransferase [57], which is essential for efficient HDL-mediated cholesterol uptake and maturation of HDL. Finally, CRF results in oxidative modification of HDL [65], impairs the anti-inflammatory properties of HDL [66] and can thereby interfere with HDL binding to ABCA1 transporter [60].
In conclusion, CRF results in heavy lipid accumulation in the remnant kidney, which is mediated by increased cellular influx of oxidized lipoproteins, heightened fatty acid synthesis pathway and down-regulation of fatty acid oxidation system. Consumption of a high-calorie diet attenuates lipid accumulation by lowering key molecules involved in lipid influx and fatty acid synthesis and by restoring pathways involved in fatty acid oxidation in the diseased kidney.
Acknowledgments
This study was supported, in part, by National Institutes of Health grant 5 U54 RR0119234.
References
- [1].Acchiardo SR, Moore LW, Latour PA. Malnutrition as the main factor in morbidity and mortality of hemodialysis patients. Kidney Int Suppl. 1983;16:S199–203. [PubMed] [Google Scholar]
- [2].Kopple JD. Pathophysiology of protein-energy wasting in chronic renal failure. J Nutr. 1999;129:247S–51S. doi: 10.1093/jn/129.1.247S. [DOI] [PubMed] [Google Scholar]
- [3].Lindholm B, Bergström J. Nutritional aspects on peritoneal dialysis. Kidney Int Suppl. 1992;38:S165–71. [PubMed] [Google Scholar]
- [4].Kalantar-Zadeh K, Block G, Humphreys MH, Kopple JD. Reverse epidemiology of cardiovascular risk factors in maintenance dialysis patients. Kidney Int. 2003;63:793–808. doi: 10.1046/j.1523-1755.2003.00803.x. [DOI] [PubMed] [Google Scholar]
- [5].Kalantar-Zadeh K, Abbott KC, Salahudeen AK, Kilpatrick RD, Horwich TB. Survival advantages of obesity in dialysis patients. Am J Clin Nutr. 2005;81:543–54. doi: 10.1093/ajcn/81.3.543. [DOI] [PubMed] [Google Scholar]
- [6].Kalantar-Zadeh K. Causes and consequences of the reverse epidemiology of body mass index in dialysis patients. J Renal Nutr. 2005;15:142–7. doi: 10.1053/j.jrn.2004.09.020. [DOI] [PubMed] [Google Scholar]
- [7].Kopple JD. McCollum Award Lecture, 1996: protein-energy malnutrition in maintenance dialysis patients. Am J Clin Nutr. 1997;65:1544–57. doi: 10.1093/ajcn/65.5.1544. [DOI] [PubMed] [Google Scholar]
- [8].Schneeweiss B, Graninger W, Stockenhuber F, Druml W, Ferenci P, Eichinger S, et al. Energy metabolism in acute and chronic renal failure. Am J Clin Nutr. 1990;52:596–601. doi: 10.1093/ajcn/52.4.596. [DOI] [PubMed] [Google Scholar]
- [9].Mitch WE. Mechanisms causing loss of lean body mass in kidney disease. Am J Clin Nutr. 1998;67:359–66. doi: 10.1093/ajcn/67.3.359. [DOI] [PubMed] [Google Scholar]
- [10].Pupim LB, Flakoll PJ, Brouillette JR, Levenhagen DK, Hakim RM, Ikizler TA. Intradialytic parenteral nutrition improves protein and energy homeostasis in chronic hemodialysis patients. J Clin Invest. 2002;110:483–92. doi: 10.1172/JCI15449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Allman MA, Stewart PM, Tiller DJ, Horvath JS, Duggin GG, Truswell AS. Energy supplementation and the nutritional status of hemodialysis patients. Am J Clin Nutr. 1990;51:558–62. doi: 10.1093/ajcn/51.4.558. [DOI] [PubMed] [Google Scholar]
- [12].Hung SC, Tung TY, Yang CS, Tarng DC. High-calories supplementation increases serum leptin levels and improves response to rHuEPO in long-term hemodialysis patients. Am J Kidney Dis. 2005;45:1073–83. doi: 10.1053/j.ajkd.2005.02.020. [DOI] [PubMed] [Google Scholar]
- [13].Meireles CL, Price SR, Pereira AM, Carvalhaes JT, Mitch WE. Nutrition and chronic renal failure in rats: what is an optimal dietary protein? J Am Soc Nephrol. 1999;10:2367–73. doi: 10.1681/ASN.V10112367. [DOI] [PubMed] [Google Scholar]
- [14].Wolfson M. Management of protein and energy intake in dialysis patients. J Am Soc Nephrol. 1999;10:2244–7. doi: 10.1681/ASN.V10102244. [DOI] [PubMed] [Google Scholar]
- [15].Vaziri ND. Oxidative stress in chronic renal failure: the nature, mechanism and consequences. Semin Nephrol. 2004;24:469–73. doi: 10.1016/j.semnephrol.2004.06.026. [DOI] [PubMed] [Google Scholar]
- [16].Kaysen GA. Inflammation and oxidative stress in end-stage renal disease. Adv Nephrol Necker Hosp. 2000;30:201–14. [PubMed] [Google Scholar]
- [17].Himmelfarb J, Stenvinkel P, Ikizler TA, Hakim R. The elephant in uremia: oxidant stress as a unifying concept of cardiovascular disease in uremia. Kidney Int. 2002;62:1524–38. doi: 10.1046/j.1523-1755.2002.00600.x. [DOI] [PubMed] [Google Scholar]
- [18].Abrass CK. Cellular lipid metabolism and the role of lipids in progressive renal disease. Am J Nephrol. 2004;24:46–53. doi: 10.1159/000075925. [DOI] [PubMed] [Google Scholar]
- [19].Schaffer JE. Lipotoxicity: when tissues overeat. Curr Opin Lipidol. 2003;14:281–7. doi: 10.1097/00041433-200306000-00008. [DOI] [PubMed] [Google Scholar]
- [20].Glass CK, Witztum JL. Atherosclerosis: the road ahead. Cell. 2001;104:503–16. doi: 10.1016/s0092-8674(01)00238-0. [DOI] [PubMed] [Google Scholar]
- [21].Lusis AJ. Atherosclerosis. Nature. 2000;407:233–41. doi: 10.1038/35025203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Brown MS, Goldstein JL. The SREBP pathway: regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell. 1997;89:331–40. doi: 10.1016/s0092-8674(00)80213-5. [DOI] [PubMed] [Google Scholar]
- [23].Ishii S, Iizuka K, Miller BC, Uyeda K. Carbohydrate response element binding protein directly promotes lipogenic enzyme gene transcription. Proc Natl Acad Sci USA. 2004;101:15597–602. doi: 10.1073/pnas.0405238101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Barbier O, Torra P, Duguay Y, Blanquart C, Fruchart JC, Glineur C, et al. Pleiotropic actions of peroxisome proliferator-activated receptors in lipid metabolism and atherosclerosis. Arterioscler Thromb Vasc Biol. 2002;22:717–26. doi: 10.1161/01.atv.0000015598.86369.04. [DOI] [PubMed] [Google Scholar]
- [25].Guan Y, Breyer MD. Peroxisome proliferator-activated receptors (PPARs): novel therapeutic targets in renal disease. Kidney Int. 2001;60:14–30. doi: 10.1046/j.1523-1755.2001.00766.x. [DOI] [PubMed] [Google Scholar]
- [26].Guan Y. Peroxisome proliferator-activated receptor family and its relationship to renal complications of the metabolic syndrome. J Am Soc Nephrol. 2004;15:2801–15. doi: 10.1097/01.ASN.0000139067.83419.46. [DOI] [PubMed] [Google Scholar]
- [27].Moorhead JF, Chan MK, El-Nahas M, Varghese Z. Lipid nephrotoxicity in chronic progressive glomerular and tubulo-interstitial disease. Lancet. 1982;2:1309–11. doi: 10.1016/s0140-6736(82)91513-6. [DOI] [PubMed] [Google Scholar]
- [28].Johnson ACM, Yabu JM, Hanson S, Shah VO, Zager RA. Experimental glomerulopathy alters renal cortical cholesterol, SR-B1, ABCA1, and HMG CoA reductase expression. Am J Pathol. 2003;163:313–20. doi: 10.1016/S0002-9440(10)63819-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Sun L, Halaihel N, Zhang W, Rogers T, Levi M. Role of sterol regulatory element-binding protein 1 in regulation of renal lipid metabolism and glomerulosclerosis in diabetes mellitus. J Biol Chem. 2002;277:18919–27. doi: 10.1074/jbc.M110650200. [DOI] [PubMed] [Google Scholar]
- [30].Kim HJ, Vaziri ND. Sterol regulatory element-binding proteins, liver X receptor, ABCA1 transporter, CD36, scavenger receptors A1 and B1 in nephrotic kidney. Am J Nephrol. 2009;29:607–14. doi: 10.1159/000193631. [DOI] [PubMed] [Google Scholar]
- [31].Kim HJ, Moradi H, Yuan J, Norris K, Vaziri ND. Renal mass reduction results in accumulation of lipids and dysregulation of lipid regulatory proteins in the remnant kidney. Am J Physiol Renal Physiol. 2009;296:F1297–306. doi: 10.1152/ajprenal.90761.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Ishigaki N, Yamamoto T, Shimizu Y, Kobayashi K, Yatoh S, Sone H, et al. Involvement of glomerular SREBP-1c in diabetic nephropathy. Biochem Biophys Res Commun. 2007;364:502–8. doi: 10.1016/j.bbrc.2007.10.038. [DOI] [PubMed] [Google Scholar]
- [33].Wang Z, Jiang T, Li J, Proctor G, McManaman JL, Lucia S, et al. Regulation of renal lipid metabolism, lipid accumulation, and glomerulosclerosis in FVBdb/db mice with type 2 diabetes. Diabetes. 2005;54:2328–35. doi: 10.2337/diabetes.54.8.2328. [DOI] [PubMed] [Google Scholar]
- [34].Proctor G, Jiang T, Iwahashi M, Wang Z, Li J, Levi M. Regulation of renal fatty acid and cholesterol metabolism, inflammation, and fibrosis in Akita and OVE26 mice with type 1 diabetes. Diabetes. 2006;55:2502–9. doi: 10.2337/db05-0603. [DOI] [PubMed] [Google Scholar]
- [35].Jiang T, Wang Z, Proctor G, Moskowitz S, Liebman SE, Rogers T, et al. Diet-induced obesity in C57BL/6J mice causes increased renal lipid accumulation and glomerulosclerosis via a sterol regulatory element-binding protein-1c-dependent pathway. J Biol Chem. 2005;280:32317–25. doi: 10.1074/jbc.M500801200. [DOI] [PubMed] [Google Scholar]
- [36].Jiang T, Liebman SE, Lucia S, Li J, Levi M. Role of altered lipid metabolism and the sterol regulatory element binding proteins in the pathogenesis of age-related renal disease. Kidney Int. 2005;68:2608–20. doi: 10.1111/j.1523-1755.2005.00733.x. [DOI] [PubMed] [Google Scholar]
- [37].Jiang T, Liebman SE, Scott Lucia M, Phillips CL, Levi M. Calorie restriction modulates renal expression of sterol regulatory element binding proteins, lipid accumulation, and age-related renal disease. J Am Soc Nephrol. 2005;16:2385–94. doi: 10.1681/ASN.2004080701. [DOI] [PubMed] [Google Scholar]
- [38].Zager RA, Johnson ACM, Hanson SY, Shah VO. Acute tubular injury causes dysregulation of cellular cholesterol transport proteins. Am J Pathol. 2003;163:313–20. doi: 10.1016/S0002-9440(10)63655-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Vaziri ND, Dang B, Zhan CD, Liang K. Downregulation of hepatic acyl-CoA: diglycerol acyltransferase (DGAT) in chronic renal failure. Am J Physiol Renal Physiol. 2004;287:F90–4. doi: 10.1152/ajprenal.00358.2003. [DOI] [PubMed] [Google Scholar]
- [40].Folch J, Lee M, Stanley GH. A simple method for the isolation and purification of total lipids from animal tissues. J Biol Chem. 1957;226:479–509. [PubMed] [Google Scholar]
- [41].Sakurai H, Hisada Y, Ueno M, Sugiura M, Kawashima K, Sugita T. Activation of transcription factor NF-κB in experimental glomerulonephritis in rats. Biochim Biophys Acta. 1996;1316:132–8. doi: 10.1016/0925-4439(96)00022-1. [DOI] [PubMed] [Google Scholar]
- [42].Mitch WE, Maroni BJ. Factors causing malnutrition in patients with chronic uremia. Am J Kidney Dis. 1999;33:176–9. doi: 10.1016/s0272-6386(99)70279-9. [DOI] [PubMed] [Google Scholar]
- [43].Huang MC, Chen ME, Hung HC, Chen HC, Chang WT, Lee CH, et al. Inadequate energy and excess protein intakes may be associated with worsening renal function in chronic kidney disease. J Renal Nutr. 2008;18:187–94. doi: 10.1053/j.jrn.2007.08.003. [DOI] [PubMed] [Google Scholar]
- [44].Brown MS, Goldstein JL. A proteolytic pathway that controls the cholesterol content of membranes, cells, and blood. Proc Natl Acad Sci USA. 1999;96:11041–8. doi: 10.1073/pnas.96.20.11041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Wang X, Sato R, Brown MS, Hua X, Goldstein JL. SREBP-1, a membrane-bound transcription factor released by sterol-regulated proteolysis. Cell. 1994;77:53–62. doi: 10.1016/0092-8674(94)90234-8. [DOI] [PubMed] [Google Scholar]
- [46].Yang T, Espenshade PJ, Wright ME, Yabe D, Gong Y, Aebersold R, et al. Crucial step in cholesterol homeostasis: sterols promote binding of SCAP to INSIG-1, a membrane protein that facilitates retention of SREBPs in ER. Cell. 2002;110:489–500. doi: 10.1016/s0092-8674(02)00872-3. [DOI] [PubMed] [Google Scholar]
- [47].Mori Y, Hirano T, Nagashima M, Shiraishi Y, Fukui T, Adachi M. Decreased peroxisome proliferator-activated receptor α gene expression is associated with dyslipidemia in a rat model of chronic renal failure. Metabol Clin Exp. 2007;56:1714–8. doi: 10.1016/j.metabol.2007.07.016. [DOI] [PubMed] [Google Scholar]
- [48].Park CW, Kim HW, Ko SH, Chung HW, Lim SW, Yang CW, et al. Accelerated diabetic nephropathy in mice lacking the peroxisome proliferator-activated receptor α. Diabetes. 2006;55:885–93. doi: 10.2337/diabetes.55.04.06.db05-1329. [DOI] [PubMed] [Google Scholar]
- [49].Vaziri ND, Bai Y, Ni Z, Quiroz Y, Rodriguez-Iturbe B. Intra-renal angiotensin II/AT1 receptor, oxidative stress, inflammation and progressive injury in renal mass reduction. J Pharmacol Exp Ther. 2007;323:85–93. doi: 10.1124/jpet.107.123638. [DOI] [PubMed] [Google Scholar]
- [50].Okamura DM, López-Guisa JM, Koelsch K, Collins S, Eddy AA. Atherogenic scavenger receptor modulation in the tubulointerstitium in response to chronic renal injury. Am J Physiol Renal Physiol. 2007;293:F575–85. doi: 10.1152/ajprenal.00063.2007. [DOI] [PubMed] [Google Scholar]
- [51].Kelly KJ, Wu P, Patterson CE, Temm C, Dominguez JH. LOX-1 and inflammation: a new mechanism for renal injury in obesity and diabetes. Am J Physiol Renal Physiol. 2008;294:F1136–45. doi: 10.1152/ajprenal.00396.2007. [DOI] [PubMed] [Google Scholar]
- [52].Ruan XZ, Moorhead JF, Varghese Z. Lipid redistribution in renal dysfunction. Kidney Int. 2008;74:407–9. doi: 10.1038/ki.2008.226. [DOI] [PubMed] [Google Scholar]
- [53].Ueno T, Kaname S, Takaichi K, Nagase M, Tojo A, Onozato ML, et al. Lox-1, an oxidized low-density lipoprotein receptor, was upregulated in the kidneys of chronic renal failure rats. Hypertens Res. 2003;26:117–22. doi: 10.1291/hypres.26.117. [DOI] [PubMed] [Google Scholar]
- [54].Adams CM, Reitz J, De Brabander JK, Feramisco JD, Li L, Brown MS, et al. Cholesterol and 25-hydroxycholesterol inhibit activation of SREBPs by different mechanisms, both involving SCAP and Insigs. J Biol Chem. 2004;279:52772–80. doi: 10.1074/jbc.M410302200. [DOI] [PubMed] [Google Scholar]
- [55].Goldstein JL, DeBose-Boyd RA, Brown MS. Protein sensors for membrane sterols. Cell. 2006;124:35–46. doi: 10.1016/j.cell.2005.12.022. [DOI] [PubMed] [Google Scholar]
- [56].Bocan TM, Krause BR, Rosebury WS, Mueller SB, Lu X, Dagle C, et al. The ACAT inhibitor avasimibe reduces macrophages and matrix metalloproteinase expression in atherosclerotic lesions of hypercholesterolemic rabbits. Arterioscler Thromb Vasc Biol. 2000;20:70–9. doi: 10.1161/01.atv.20.1.70. [DOI] [PubMed] [Google Scholar]
- [57].Vaziri ND, Liang K, Parks JS. Downregulation of lecithin:cholesterol acyltransferase (LCAT) in chronic renal failure. Kidney Int. 2001;59:2192–6. doi: 10.1046/j.1523-1755.2001.00734.x. [DOI] [PubMed] [Google Scholar]
- [58].Vaziri ND, Liang K. ACAT inhibition reverses LCAT deficiency and improves plasma HDL in chronic renal failure. Am J Physiol Renal Physiol. 2004;287:F1038–43. doi: 10.1152/ajprenal.00150.2004. [DOI] [PubMed] [Google Scholar]
- [59].Storch J, Thumser AEA. The fatty acid transport function of fatty acid-binding proteins. Biochim Biophys Acta. 2000;1486:28–44. doi: 10.1016/s1388-1981(00)00046-9. [DOI] [PubMed] [Google Scholar]
- [60].Oram JF, Vaughan AM. ATP-binding cassette cholesterol transporters and cardiovascular disease. Circ Res. 2006;99:1031–43. doi: 10.1161/01.RES.0000250171.54048.5c. [DOI] [PubMed] [Google Scholar]
- [61].Tontonoz P, Mangelsdorf DJ. Liver X receptor signaling pathways in cardiovascular disease. Mol Endocrinol. 2003;17:985–93. doi: 10.1210/me.2003-0061. [DOI] [PubMed] [Google Scholar]
- [62].Laffitte BA, Repa JJ, Joseph SB, Wilpitz DC, Kast HR, Mangelsdorf DJ, et al. LXRs control lipid-inducible expression of the apolipoprotein E gene in macrophages and adipocytes. Proc Natl Acad Sci USA. 2001;98:507–12. doi: 10.1073/pnas.021488798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Jiang XC, Beyer TP, Li Z, Liu J, Quan W, Schmidt RJ, et al. LXRs control lipidinducible expression of the apolipoprotein E gene in macrophages and adipocytes. J Biol Chem. 2003;278:49072–8. doi: 10.1074/jbc.M304274200. [DOI] [PubMed] [Google Scholar]
- [64].Vaziri ND, Deng G, Liang K. Hepatic HDL receptor, SR-B1 and Apo A-I expression in chronic renal failure. Nephrol Dial Transplant. 1999;14:1462–6. doi: 10.1093/ndt/14.6.1462. [DOI] [PubMed] [Google Scholar]
- [65].Moradi H, Pahl MV, Elahimehr R, Vaziri ND. Impaired antioxidant activity of HDL in chronic kidney disease. Translational Res. 2009;153:77–85. doi: 10.1016/j.trsl.2008.11.007. [DOI] [PubMed] [Google Scholar]
- [66].Vaziri ND, Moradi H, Pahl MV, Fogelman AM, Navab M. In vitro stimulation of HDL anti-inflammatory activity and inhibition of LDL pro-inflammatory activity in the plasma of patients with end-stage renal disease by an apoA-1 mimetic peptide. Kidney Int. 2009;76:437–44. doi: 10.1038/ki.2009.177. [DOI] [PMC free article] [PubMed] [Google Scholar]