

Abstract
MnSOD plays a critical role in the survival of aerobic life, and its aberrant expression has been implicated in carcinogenesis and tumor resistance to therapy. However, despite extensive studies in MnSOD regulation and its role in cancer, when and how the alteration of MnSOD expression occurs during the process of tumor development in vivo are unknown. Here, we generated transgenic mice expressing a luciferase reporter gene under the control of human MnSOD promoter/enhancer elements and investigated the changes of MnSOD transcription using the 7–12-dimethylbenz-(α)-anthracene (DMBA)/TPA multistage skin carcinogenesis model. The results demonstrate that MnSOD expression was suppressed at a very early stage but increased at late stages of skin carcinogenesis. The suppression and subsequent restoration of MnSOD expression were mediated by two transcription-factors, Sp1 and p53. Exposure to DMBA and TPA activated p53 and decreased MnSOD expression via p53-mediated suppression of Sp1 binding to the MnSOD promoter in normal appearing skin and benign papillomas. In squamous cell carcinomas, Sp1 binding increased, due to the loss of functional p53. We used chromatin immunoprecipitation, EMSA, and both knockdown and overexpression of Sp1 and p53 to verify their roles in the expression of MnSOD at each stage of cancer development. The results identify MnSOD as a p53-regulated gene that switches between early and advanced stages of cancer. These findings also provide strong support for the development of means to reactivate p53 for the prevention of tumor progression.
INTRODUCTION
Manganese superoxide dismutase (MnSOD) is a nuclear-encoded mitochondrial antioxidant enzyme that is essential for the removal of superoxide radicals. Under normal physiological conditions, superoxide radicals in mitochondria are rapidly converted by MnSOD to hydrogen peroxide, which is further detoxified by catalase and/or glutathione peroxidase to form water and molecular oxygen (1). If not removed efficiently, superoxide radicals may accumulate and be converted to highly toxic species, such as hydroxyl radicals, that can damage DNA and the mitochondrial membrane, leading to defective mitochondria, which have been implicated in cancer (2, 3).
Due to its strategic location in mitochondria, MnSOD is essential for the survival of aerobic life (4). It has been demonstrated that homozygous MnSOD knockout mice often die from dilated cardiomyopathy and neurodegenerative disease within 2 to 3 weeks after birth (5, 6), and those that manage to survive longer than a week exhibit extensive mitochondrial injury (6). Lack of MnSOD has also been associated with an increased frequency of radiation- induced neoplastic transformation (7) and MnSOD overexpression can suppress tumor incidence and tumor multiplicity (8).
While many studies have shown that MnSOD is down-regulated in cancer cells (9), it has also been reported that MnSOD expression is increased in some cancer cells and human tissues (10). Thus, it is not clear whether and how the change in MnSOD expression correlates with stages of tumorigenesis and cancer progression. We and others have previously shown that MnSOD transcription is regulated by specificity protein 1 (Sp1), nuclear factor-kappa B (NF-κB), and p53, which are required for the basal and induced expression of the MnSOD gene, respectively (11–17).
p53 is a transcriptional regulator with tumor suppression capability. p53 is mutated in at least 50% of tumors, and the wild- type p53 tumor suppressor is completely lost in many tumor tissues (18,19). Here, we evaluated changes in MnSOD expression during the course of tumorigenesis and demonstrate that MnSOD expression was suppressed in early tumorigenic stages but was restored later in advanced carcinoma. Using siRNA knockdown approaches coupled with in vitro tumor invasion assay, we also demonstrate that suppression of p53 alone led to increased invasion. The loss of Sp1 function exacerbates p53 siRNA mediated cell transformation, but not invasion. The data support a model in which activation of p53 by carcinogens leads to a reduction in MnSOD that precedes the appearance of cancer. When a tumor begins to develop, the modulation of stress sensitive transcription factors, including p53, creates an environment of high oxidative stress achieved by the suppression of MnSOD. As the tumor progresses and p53 activity is lost, MnSOD levels rise again, creating conditions in which cancer cells can survive under oxidative stress.
Materials and Methods
Generation of transgenic mice
The human MnSOD promoter–enhancer-driven reporter gene construct (−555 to +24/I2E/pGL3) was restriction digested with KpnI and SalI to remove the bacterial sequences in the vector. Purified DNA was introduced by microinjection into the pronuclei of mouse fertilized eggs, as described previously (20, 21).
Treatment of animals for tumor initiation and promotion
All procedures using animals were performed according to the protocols approved by the University of Kentucky Animal Care and Use Committee. Female mice (6–8 weeks) bearing the human MnSOD reporter gene were used for the study when they were in the resting phase of the hair cycle. A single dose of DMBA (20 nmol) was applied to the shaved skin area (18–20 cm2) on the mouse back for initiation. Two weeks after the DMBA treatment, TPA(4 μg/mouse/day) was painted on the same area of the skin 5 days a week for 25 weeks. Control animals were treated with DMSO only.
In vivo bioluminescence
An IVIS® imaging system 100 (Xenogen, Hopkinton, MA) with live image acquisition software (version2.20, Xenogen) was used to detect the bioluminescent signals on mouse skin according to the protocol as described by Dhar et al. (22).
MnSOD activity assay
Mouse skin tissues were harvested and homogenized. MnSOD activity was measured by nitroblue tetrazolium reduction using xanthine and xanthine oxidase for superoxide generation as described by Spitz et al. (23).
Electrophoretic mobility shift assay
The consensus double-stranded oligonucleotides of NF-κB sequence5′-AGTTGAGGGGACTTTCCCAGGC-3′, Sp1 sequence 5′-ATTCGATCGGGGCGGGGCGAGC-3′ (Promega, Madison, WI) and p53 sequence 5′-. AGACATGCCT-AGACATGCCT-3′ were radioactively labeled with [32P]ATP and T4 polynucleotide kinases and the electrophoretic mobility assays were performed according to protocol as described previously (15, 16).
Cell line
Normal mouse skin epithelial cell line (JB6 clone 41, P+), was obtained from Dr. Nancy H. Colburn at the National Cancer Institute (Frederick, MD). This cell line was initially characterized and described in Nature 1979, vol. 281, p. 589–91 (24). The cells used were from passage 23 of the original clone. The cells were characterized by measuring the Ap1 binding activity following TPA treatment. The results show that TPA induces Ap1 binding activity by 2–3 folds, which is consistent with the data from the original clone. These cells were tested routinely for cellular transformation by performing a soft agar transformation assay and they showed transformation negativity without any treatments.
Transformation assay in soft agar
For transformation assays, following transfection of siRNAs with or without Sp1 expression vector in JB6 cells for 48 h, 5,000 cells were mixed with 0.33% agar in MEM and overlaid on top of the 5% agar medium. After solidification, 2–3 ml of culture media were added over the soft agar layer and incubated at 37° C in a 5% CO2 air-humidified incubator for 14 days. Colonies consisting of at least 50 cells were counted under light microscope.
Matrigel invasion assay
Cellular invasion into matrigel was performed using the BD BioCoat Matrigel Invasion Chamber (BD Biosciences, Bedford, MA). Briefly, following transfection, 5×104 cells were added to the upper chamber containing a polycarbonate membrane filter of 8 μM pore size with rehydrated matrigel. The bottom chamber was filled with MEM supplemented with 10% FBA. The same number of cells was also seeded in the control chamber containing an identical polycarbonate membrane filter without matrigel, for free migration and viability testing. The chambers were then incubated for 24 h at 37° C, in 5% CO2. Non-invading or non-migrating cells were removed from the upper side of the matrigel chamber by gently but firmly swabbing with cotton tips. The cells that migrated to the lower side of the filter were then fixed with 100% methanol for 2 min and stained with 0.1% crystal violet. The stained nuclei on six randomly selected microscopic fields for each sample were counted and the quantification of migrated cells was performed in three independent experiments. The data are expressed as the percent of invasion.
Statistical analysis
Data were analyzed using one-way analysis of variance (ANOVA) followed by Bonferroni’s post-test multiple comparison. Each data point is represented as mean ± SD.
RESULTS
Generation of human MnSOD promoter-enhancer mice
The regulation of human MnSOD during tumorigenesis was evaluated using a transgenic mouse model carrying a human MnSOD promoter-enhancer reporter gene construct. To create this model, a 922 bp DNA fragment containing the MnSOD basal promoter (−555 to+24) and a DNA fragment containing enhancer (Intron 2 [I2E], +1742 to +2083) were separately amplified by PCR and ligated to a luciferase reporter vector (Fig. 1A) to generate transgenic mice. The DNA sequence of the gene construct was verified, and the stable integration of the human MnSOD promoter/enhancer elements in the transgenic mice was confirmed by Southern blot analysis of mouse genomic DNA using the intronic fragments of the human MnSOD gene as a probe (Fig. 1B). As shown previously, the transgenic mice did not show any difference in MnSOD protein or activity levels as compared with non-transgenic littermates (22).
Figure 1. Human MnSOD transgenic mouse model and MnSOD regulation in vivo.

Human MnSOD promoter-enhancer-driven luciferase gene construct used to generate transgenic mice (A). Transgenic mice showing a band for the intronic fragment (I2E) were obtained by restriction digestion of genomic DNA by kpn1 and bglII (B). Schematic diagram outlining the method of DMBA and TPA treatment and non-invasive imaging (C). Bioluminescence images of mice treated with DMBA followed by TPA were acquired by CCD camera (bottom panel). Photon counts were estimated within the defined gated area on the image (top panel) (D). After 25 weeks, the animals were humanely euthanized and skin and tumor tissues were harvested and the luciferase activity (E), MnSOD mRNA (F), MnSOD protein (G) and MSOD activity (H) were measured. Data are presented as the mean ± SD, with significant differences from control indicated by *p<0.05 and **p<0.01.
Non-invasive in vivo monitoring of transgene expression
Transgenic mice expressing the luciferase reporter gene under the control of the MnSOD promoter/enhancer were used to monitor MnSOD expression in vivo after DMBA treatment with or without subsequent daily treatment with TPA (4 μg/mouse/day) for 25 weeks (Fig. 1C). Prior to each measurement, D-luciferin was painted on the back of the mouse skin and the mouse was placed under a CCD camera. The enzyme-substrate reaction was allowed to stabilize for 20 min and then bioluminescence signals were collected. Our previous studies have shown that a single treatment with TPA alone increases MnSOD expression (22). In the present study, treatment with DMBA alone also significantly increased the level of MnSOD reporter gene activity. However, treatment with DMBA in combination with subsequent TPA treatment consistently decreased the MnSOD reporter gene activity during the entire course of treatment (Fig. 1D). To confirm this finding, the luciferase activity was measured in isolated skin tissues after 25 weeks of TPA treatment. We observed that the reporter gene activity was significantly decreased in isolated normal appearing skin and tumor tissues (Fig. 1E), consistent with the live imaging data.
Loss of endogenous MnSOD in early stages of tumorigenesis
To confirm the effects of DMBA and TPA treatment on MnSOD-reporter activity in vivo, we determined the endogenous MnSOD mRNA and protein levels and enzymatic activity following 25 weeks of repeated TPA treatment. Consistent with the reporter gene activity measured by the live imaging data, endogenous MnSOD mRNA (Fig. 1F) and protein (Fig. 1G) levels and enzymatic activity (Fig. 1H) all decreased significantly in isolated tumor tissues and surrounding skin tissues as compared to vehicle treated control. These results conclusively demonstrate that the loss of MnSOD expression occurred very early after treatment with DMBA and TPA in the two-stage tumorigenesis model.
Identification of transcription factors involved in MnSOD regulation during tumorigenesis
To identify the transcription factors involved in regulation of MnSOD transcription after DMBA and TPA treatment, we prepared nuclear extracts from the epithelium isolated from the mouse skin and tumor tissue. These samples were analyzed by electrophoretic mobility shift assay (EMSA) to measure the levels of transcription factors that bind to the promoter and enhancer regions of the MnSOD gene. We have previously shown that the NF-κB site in the intron of MnSOD is essential for the induction of MnSOD by cytokines (11). The electrophoretic supershift shows that the level of NF-κB-DNA complexes was higher in tumor tissues than in control skin tissues or normal appearing skin tissues treated with DMBA and TPA. In addition, the amount of NF-κB-DNA complexes supershifted by anti-p50 antibody was disproportionately higher than the amount of the DNA binding complexes supershifted by the anti-p65 antibody (Fig. 2A). Consistent with the binding activity, p50 was significantly increased, whereas p65 was significantly decreased in tumor tissues as compared to the control or normal appearing skin (Fig. 2C). Collectively, these results indicate that increased NF-κB DNA binding activity in DMBA-TPA induced tumor tissue was primarily due to the increased p50/p50 homodimer complex, with little or no contribution from the p50/p65 heterodimer. On the other hand the level of Sp1 DNA-binding activity, which is essential for basal levels of MnSOD transcription, decreased significantly in tumors as well as in skin tissues treated with DMBA and TPA (Fig. 2B).
Figure 2. Transcription factor binding to the MnSOD promoter and enhancer in vivo.
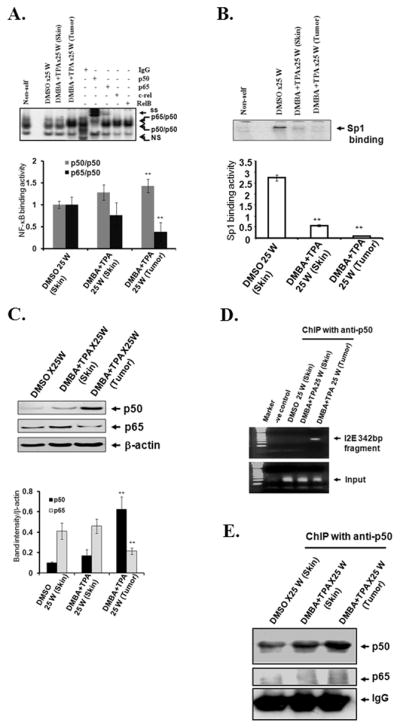
EMSA was performed in purified nuclear extract from skin and tumor tissues. For super-shift experiments, the EMSA reaction mixture was incubated with 1 μg antibody or IgG alone. The arrows point to the protein-DNA complex and super-shifted protein-antibody complexes (top panel). NF-κB-DNA binding complex was densitometrically scanned and expressed as a relative quantity (bottom panel) (A), Sp1 binding activity (top panel). The Sp1-DNA binding complexes were quantified (bottom panel) (B). Western analysis of p50 and p65 was performed in the purified nuclear extracts. Data were quantified and the relative levels of NF-κB proteins were estimated (C). Association of transcription factors with the enhancer region of the MnSOD gene was evaluated by ChIP assay in isolated skin and tumor tissues (D). The immunoprecipitated proteins were detected by Western analysis (E). Data shown are representative of three independent experiments. Significantly different from control group, **p<0.01.
To confirm that the binding of p50 to the MnSOD enhancer region is involved in the modulation of MnSOD expression after treatment, we cross-linked the proteins and DNA in vivo using formaldehyde. The cross-linked DNA–protein complexes were subjected to ChIP analysis using an antibody specific to p50, and the immunoprecipitation product was analyzed by Western analysis. We decross-linked the immunoprecipitation product and purified the DNA for PCR amplification. Using the purified DNA as a template, the intronic enhancer (I2E) fragments were amplified following ChIP assay. Results show that the amount of I2E pulled down with p50 was remarkably higher in tumor tissues as compared to normal skin (Fig. 2D). ChIP and subsequent Western blotting analyses also demonstrate higher levels of p50 protein in cross-linked DNA–protein complexes prepared from tumor tissues as compared to normal appearing skin tissues (Fig. 2E). These results indicate that the level of NF-κB family members was indeed increased after DMBA and TPA treatment and that p50 was the predominant NF-κB family member involved in the tumor tissues.
Tumor tissue pathology
Based on pathological examination, the histology of the tumors was characteristic of papillomas, and no pure carcinoma was observed after 25 weeks of TPA treatment (Supplementary Table 1). We therefore decided to extend the observation until 48–60 weeks to allow for progression following the cessation of TPA treatment. Thereafter, skin and tumor samples were collected for pathological examination. The results reveal that at later stages of tumorigenesis, papilloma progressed to squamous cell carcinoma (Supplementary Table 2). In two sets of experiments, at least ten papillomas and ten squamous cell carcinomas (SCC) were collected.
Transition of MnSOD expression between benign tumor and squamous cell carcinoma (SCC)
SCC tissues were separated from benign papilloma based on tissue pathology, and tissue lysates were prepared for measurement of luciferase reporter gene activity, endogenous MnSOD protein levels and MnSOD activity. As shown in Fig. 3A, luciferase activity decreased significantly in normal appearing skin tissues after DMBA and TPA treatment and further decreased in papilloma. Surprisingly, luciferase activity increased in SCC several-fold as compared to papilloma or normal skin (Fig. 3A). The endogenous MnSOD protein levels and enzymatic activity were decreased significantly in papilloma and skin tissues treated with DMBA and TPA (Fig. 3B, C), but increased significantly in SCC (Fig. 3B, C). These results demonstrate a transition from reduced MnSOD gene expression in early-stage tumorigenesis (papilloma) to increased MnSOD gene expression in late-stage tumorigenesis (SCC).
Figure 3. Increased MnSOD transcription, activity and protein in late-stage tumorigenesis.

MnSOD reporter gene activity, protein levels and activity were evaluated in normal skin tissues and tissues bearing papillomas or SCC. Luciferase activity (A), protein levels (B), and MnSOD activity (C). Ap1 binding activity was measured by EMSA. The Ap1 DNA-protein complex bands were densitometrically scanned and the relative levels were determined (bottom panel) (D). PCNA levels were detected in nuclear extracts by Western blotting. The protein bands were densitometrically scanned and the relative levels were determined after normalization to lamin C (E). Each group of tissues was collected from 10 individual animals. Data presented are the mean ± SD, and significant differences from control are indicated by *p<0.05 and **p<0.01.
Activation of proliferation in tumor tissues
It has been demonstrated that induction of Ap1 activity leads to activation of several genes required for cell proliferation (8, 25). To monitor cell proliferation in the skin and tumor tissues, we performed EMSAs to detect the level of Ap1 binding activity. The results show that Ap1 binding activity was significantly increased both in papilloma and SCC tissues; however, Ap1 binding activity did not further increase in SCC, as compared to papilloma (Fig. 3D). We also measured the level of proliferating cell nuclear antigen (PCNA) in the nuclear extract of the skin and tumor tissues by Western blotting using anti-PCNA antibody, and found that the nuclear PCNA levels increased both in papilloma and SCC after treatment (Fig. 3E), which is consistent with the observed increase in Ap1 binding activity. These results indicate that increased cell proliferation in papilloma and SCC developed after DMBA and TPA treatment. Interestingly, the level of PCNA was also slightly increased in the histologically normal skin tissue treated with DMBA and TPA. This result is consistent with the observed decline in MnSOD levels at the very early stages of cancer development.
Regulation of MnSOD transcription by p53 and Sp1
To identify the mechanism underlying the increased MnSOD expression in SCC, we further investigated the DNA binding activity of NF-κB and Sp1 to the MnSOD enhancer and promoter region. We found that the total NF-κB DNA binding activity was decreased in SCC as compared to the papilloma or normal skin (Fig. 4A), indicating that the increased MnSOD transcription in SCC is unlikely to be mediated by the binding of NF-κB to the MnSOD enhancer element. Consistent with the results of the above studies on papilloma tissues collected after 25 weeks of TPA treatment, the promoter binding activity of transcription factor Sp1 in papilloma remained suppressed through week 48. In contrast, the Sp1 binding activity increased significantly in SCC, as compared with papilloma (Fig. 4B). Since Sp1 is the critical basal transcription factor known to interact with p53 (15, 26), we measured p53 DNA binding activity in the nuclear extracts by EMSA analysis. The p53 DNA binding activity was increased in papilloma. However, there was a significant decrease of p53 binding activity in SCC as compared to skin tissues treated with either DMSO or DMBA and TPA (Fig. 4C). Since p53 and Sp1 compete to bind to the MnSOD promoter (15, 16), our results suggest that the increase in Sp1 binding resulting from the absence of p53 contributed to the restoration of MnSOD transcription in SCC.
Figure 4. Alteration of transcription factor binding activity and nuclear p53 levels.
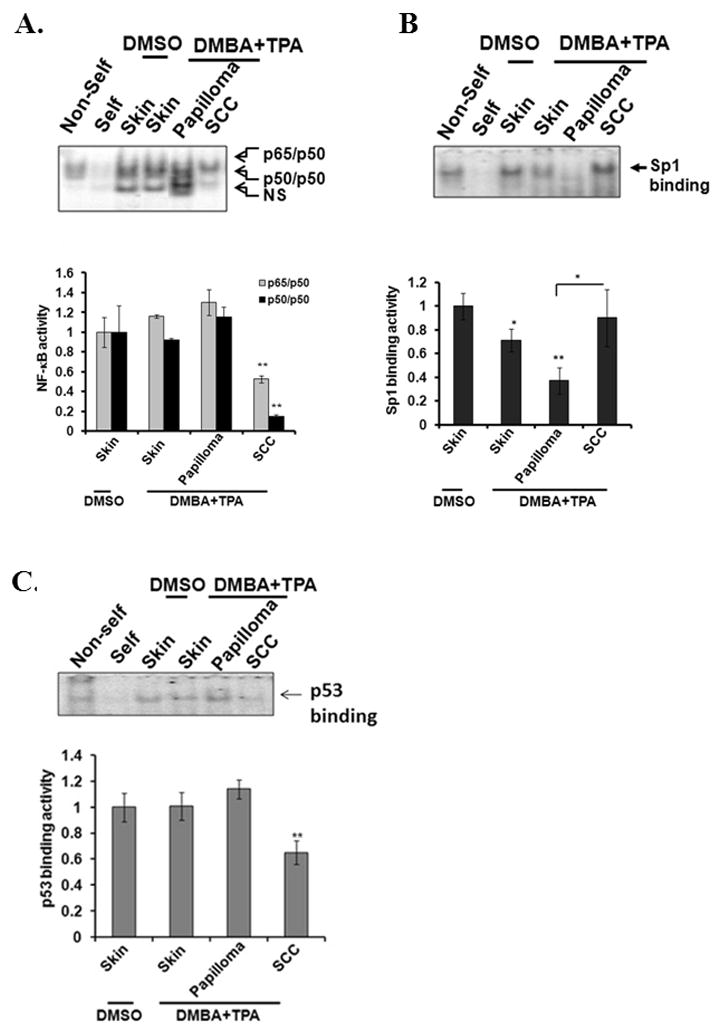
DNA binding activity of each transcription factor was evaluated by EMSA (top panel) and quantified (bottom panel) (A) NF-κB, (B) Sp1. (C) p53. The quantification of p53-DNA complexes is shown (bottom panel) (C). Data presented are the mean ± SD, and significant differences from control, *p<0.05 and **p<0.01.
To verify the in vivo finding that increased Sp1 binding to the MnSOD promoter is associated with the loss of p53, we co-transfected mouse skin epithelial cells with p53 siRNA and/or Sp1 expression vector and measured the MnSOD reporter gene activity, Sp1 binding activity and endogenous MnSOD expression. Transfection with either p53 siRNA or Sp1 expression vector alone significantly induced MnSOD promoter activity (Fig. 5A) and increased endogenous MnSOD protein levels (Fig. 5B), and co-transfection with p53 siRNA and Sp1 expression vector further increased the reporter gene activity and endogenous MnSOD levels. The level of p21, another known p53 target gene, was also suppressed following overexpression of pool of p53 siRNA. The level of p53 is suppressed by individual p53 siRNAs were also verified by Western blotting (Supplementary Fig. S1). Overexpression of Sp1 did not change the p53 siRNA mediated suppressed level of p21 (Fig. 5B). These results confirm that suppression of p53 increases Sp1-mediated MnSOD transcription. To further investigate the association between p53 levels and Sp1 DNA binding, we performed EMSAs using the nuclear extract prepared following transfection. The results also show that Sp1 binding activity increased after p53 siRNA transfection and further increased upon Sp1 overexpression (Fig. 5C). These results suggest that p53 suppression and Sp1 over-expression acted in concert to up-regulate MnSOD transcription in squamous cell carcinoma developed after DMBA and TPA treatment.
Figure 5. Knockdown of p53 alone or overexpression of Sp1 with subsequent p53 knockdown enhances Sp1-DNA binding activity and MnSOD transcription in vitro.
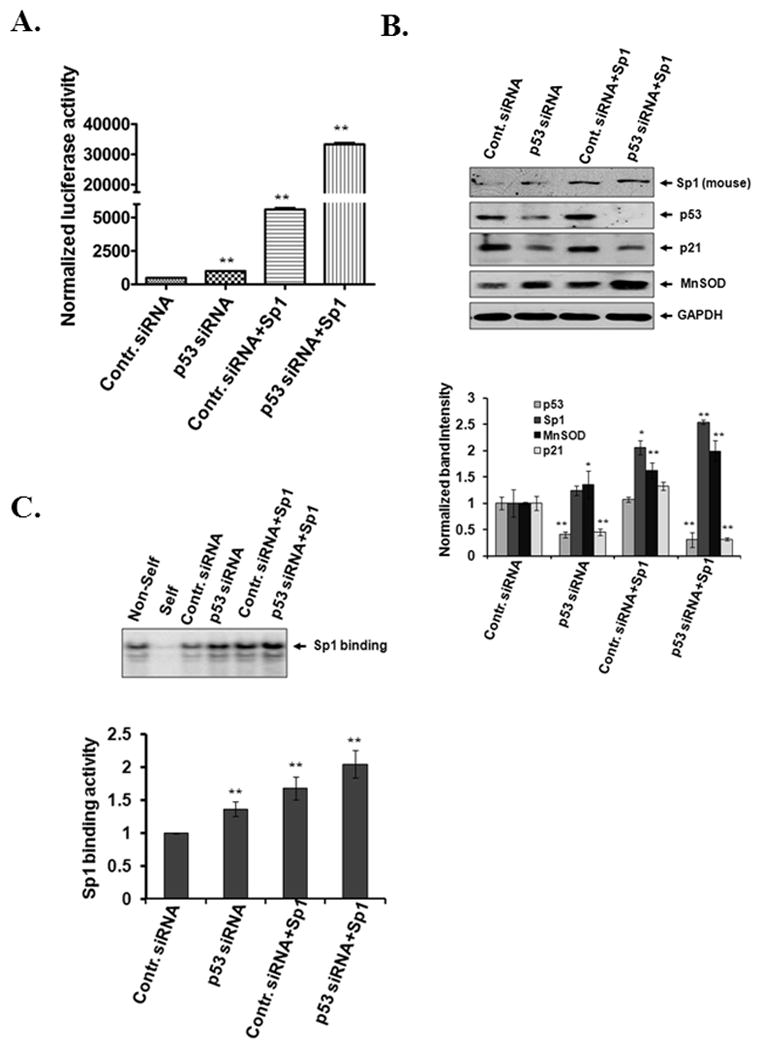
Mouse epithelial cells (JB6) were co-transfected with control siRNA or p53 siRNA with or without Sp1 expression vector and with MnSOD reporter vector. Luciferase activity was normalized to beta-galactosidase activity and represented as a measure of MnSOD transcription (A). Suppression of p53 protein and increase of Sp1 expression upon transfection of p53 siRNA and Sp1 expression vector, respectively, were verified by Western blotting. Endogenous MnSOD and p21 protein levels were verified by reprobing the membrane with MnSOD or p21 antibody (top panel). The gels were densitometrically scanned and the relative levels were normalized to GAPDH as an internal control (bottom panel) (B). Sp1 binding activity was evaluated by EMSA following transfection of p53 siRNA with or without Sp1 expression vector (top panel). The binding complexes were densitometrically scanned and quantified (bottom panel) (C). Data consist of three representative experiments and are expressed as the mean ± SD. Significantly different from control, *p<0.05 and **p<0.01.
Effects of p53 and Sp1 on cell transformation and invasiveness
To further determine whether the loss of p53 and Sp1 is associated with cell transformation and invasion, we suppressed p53 and/or Sp1 expression in mouse epithelial cells by siRNA transfection and performed a soft agar colony formation assay. The results show that p53 siRNA transfection significantly increased the yield of anchorage-independent colonies in soft agar (Fig. 6A, B, C), which was further increased after co-transfection with Sp1 siRNA and p53 siRNA. We also performed a matrigel invasion assay using JB6 cells transfected with p53 siRNA and/or Sp1 siRNA. Similar to the results observed in the soft agar colony formation assay, the matrigel assay results show that p53 siRNA transfection significantly increased the cell invasion capability, but co-transfection with Sp1 siRNA and p53 siRNA did not further increase the matrigel invasion in the transfected cells (Fig. 6D, E). Overexpression of Sp1 alone did not decrease the yield of the number of anchorage-independent colonies in soft agar or the invasion capability in matrigel, as compared to the control (Fig. 6A–E). These results suggest that suppression of p53 alone led to cell invasion as measured by the matrigel invasion. The loss of Sp1 function further enhanced the p53 siRNA mediated cell transformation but not invasion.
Figure 6. Knockdown of p53 enhances transformation phenotype and increases matrigel invasiveness in JB6 cells.

JB6 cells were plated on a soft-agar dish and cultured for 14 days for colony formation. For each treatment, 6–10 dishes were used and a representative picture is shown (A). Microscopic images of transformed colonies were taken from six randomly selected fields in each dish (6–10 dishes per group). A representative microscopic view is shown (B). The number of transformed colonies was counted and quantified (C). Cells were seeded onto three-dimensional migration chamber inserts and the matrigel invasion assays were performed as described in materials and methods (D). Matrigel invasion was quantified using six different random fields per insert (4 inserts per group). Migrated cells were counted in the matrigel and control chambers following staining with crystal violet. Data were normalized to the cells that migrated from the control chamber (E). Each bar represents the average of the mean ± SD of 6 different randomly selected fields per dish (6 dishes per group). Significantly different from control, *p<0.05 and **p<0.01.
DISCUSSION
Accumulating data suggest that MnSOD constitutes one of the major cellular defense mechanisms against the toxic effects of agents that cause oxidative stress and that MnSOD functions as a tumor suppressor gene in several experimental systems (7, 27). Many types of transformed and cancer cells have been shown to have altered levels of MnSOD activity (28). However, when and how the alteration of MnSOD occurs in vivo are unknown. The present study is the first to directly demonstrate when alteration of MnSOD expression occurs during the process of cancer development. The results indicate that the loss of MnSOD transcription occurred very early and persisted prior to SCC formation. This result suggests the role of MnSOD in the early stage of tumor development, which is consistent with our previous tumor promotion studies in which we have shown that the increased cell proliferation resulting from MnSOD deficiency is essential for TPA-mediated skin tumor promotion (8, 29).
Although studies to determine the mechanisms responsible for the induction of MnSOD in established cultured cells have yielded useful information concerning the regulation of MnSOD in vitro, there is little information concerning the regulation of MnSOD during carcinogenesis in vivo. In this study, we examined MnSOD expression and activity in papillomas formed after 25 weeks of TPA treatment following DMBA initiation, and in squamous cell carcinomas developed between 26 and 48–64 weeks. The results demonstrate that MnSOD transcription is regulated by Sp1 and p53, which bind to the MnSOD promoter, and members of the NF-κB family of transcription factors, which bind to the enhancer region of the MnSOD gene (16, 30). The finding that suppressed MnSOD transcription in the early stages of tumorigenesis following DMBA and TPA treatment is associated with decreased Sp1 binding to the MnSOD promoter is consistent with previous observations that Sp1 is essential for the basal expression of MnSOD (13,16). The suppression of MnSOD expression in papillomas in the presence of increased NF-κB binding activity indicates that NF-κB alone is not sufficient to increase MnSOD transcription in vivo. Co-transfection of mouse skin epithelial cells with Sp1 siRNA and NF-κB family members p65 and p50 demonstrates that Sp1 siRNA-mediated suppression of MnSOD transcription could not be reversed by over-expressed NF-κB (data not shown). This finding further establishes the vital role of Sp1 in the regulation of basal MnSOD transcription. Although the members of the NF-κB family are known to up-regulate MnSOD transcription (13), the relative and differential abundance of homodimers and heterodimers may determine their collective effectiveness. The NF-κB p50-p50 homodimer is a known negative regulator of gene transcription (31), which could negate the augmentative effect of NF-κB (both p65 and p50) on MnSOD transcription. NF-κB is a ubiquitous transcription factor overexpressed in human hematopoietic malignancies and some solid tumors. We and others have demonstrated that NF-κB interacts with Sp1 and enhances its target gene transcription (15, 32). Down-regulation of Sp1 is expected to decrease the amount of Sp1 available for the formation of Sp1/NF-κB complexes, which could be responsible for the suppressed MnSOD expression observed in the papillomas described here.
In contrast to the increase of NF-κB binding activity in the papillomas, NF-κB binding activity was significantly suppressed in SCC. Although it is possible that a higher p65/p50 to p50/p50 ratio, as was observed in the SCC, could enhance MnSOD transcription despite an overall decrease of NF-κB proteins, such an effect is unlikely to occur in cells with abundant Sp1. It is conceivable that the binding of p53 to its target site hinders the access of Sp1 to the Sp1 binding site and that the loss of p53 during development of the SCC may allow Sp1 to bind more efficiently to the MnSOD promoter. This hypothesis is supported by the siRNA transfection results showing enhanced Sp1 binding activity as well as increased MnSOD transcription and endogenous MnSOD expression after p53 siRNA transfection. Our finding that the presence of p53 plays an important role in neoplastic transformation and invasion and that the loss of Sp1 function further enhances p53 siRNA mediated cell transformation but not invasion is consistent with the bi-directional role of p53 in the regulation of MnSOD and the Janus face of p53. In addition to p53, two p53 homologs, p63 and p73, also act as transactivators of many genes, including p21, Bax and redox-related genes such as PIGs (33, 34). Both p63 and p73 possess structural similarities with p53 in their DNA-binding, transactivation and oligomerization domain (35, 36). Similar to p53, transient transfection of p63 also is capable of activating or repressing transcription of the reporter gene downstream of an optimal p53 DNA binding site (37). Our results demonstrate that loss of p53 DNA binding activity leads to the increase of MnSOD expression in SCC and suggest that the mechanism of p63- mediated MnSOD expression, if any, is different from p53- mediated MnSOD induction in SCC. Interestingly, p73 is a potential tumor suppressor gene that possesses very high sequence similarity with p53, suggesting that the two may have a similar ability to activate or repress transcription. Indeed, Steven and Jonathan have reported that expression of 73α and p73β suppresses the transcription of cyclin B1 and is dependent on functional Sp1 binding sites in the promoter (38). Our results clearly demonstrate that induction of MnSOD transcription, in the absence of p53, in SCC is a Sp1 dependent phenomenon. However, we could not rule out the possibility that the loss of p73 in SCC that has been reported (39) may also contribute to the availability of Sp1 for MnSOD transcription in SCC.
Our results show that suppression of Sp1 binding to the MnSOD promoter is responsible for DMBA- and TPA-mediated decrease in MnSOD expression in early stages of tumorigenesis, whereas reduced p53 activity is responsible for restoration of MnSOD at later stages of tumorigenesis. Only then does tumor progression reach the aggressively malignant stage represented by the formation of SCC. Due to the critical function of MnSOD in aerobic respiration, it is conceivable that in the early stages of tumorigenesis the suppressed MnSOD may create a cellular environment conducive to the increase of ROS in mitochondria and subsequent mitochondrial injury resulting in the increased glycolysis observed in cancer cells. However, the restoration of MnSOD expression in fully malignant cells may allow them to more efficiently combat increased oxidative stress, thereby conferring a sustained growth advantage.
Supplementary Material
Acknowledgments
We thank Dr. Jonathan M. Horowitz, North Carolina State University, for the generous gift of Sp1 expression vector and Dr. Ravikumar Rangaswamy Rao for assisting in image acquisition by microscope following matrigel assay. This work was supported, in part, by National Institute of Health Grants CA 49797 and CA 73599 to Daret K. St. Clair.
Footnotes
References
- 1.Gregory EM, Fridovich I. Oxygen toxicity the superoxide dismutase. J Becteriol. 1973;114:1193–1197. doi: 10.1128/jb.114.3.1193-1197.1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kowald A, Kirkwood TB. Accumulation of defective mitochondria through delayed degradation of damaged organelles and its possible role in the ageing of post-mitotic and dividing cells. J Theor Biol. 2000;202:145–160. doi: 10.1006/jtbi.1999.1046. [DOI] [PubMed] [Google Scholar]
- 3.Singh KK. Mitochondria damage checkpoint, aging, and cancer. Annals of the New York Academy of Sciences. 2006;1067:182–190. doi: 10.1196/annals.1354.022. [DOI] [PubMed] [Google Scholar]
- 4.Carlioz A, Touati D. Isolation of superoxide dismutase mutants in Escherichia coli: is superoxide dismutase necessary for aerobic life? EMBO Journa. 1986;5:623–630. doi: 10.1002/j.1460-2075.1986.tb04256.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lebovitz RM, Zhang H, Vogel H, Cartwright J, Jr, Dionne L, Lu N, Huang S, et al. Neurodegeneration, myocardial injury, and perinatal death in mitochondrial superoxide dismutase-deficient mice. Proc Natl Acad Sci USA. 1996;93:9782–9787. doi: 10.1073/pnas.93.18.9782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li Y, Huang TT, Carlson EJ, Melov S, Ursell PC, Olson JL, et al. Dilated cardiomyopathy and neonatal lethality in mutant mice lacking manganese superoxide dismutase. Nature Genetics. 1995;11:376–381. doi: 10.1038/ng1295-376. [DOI] [PubMed] [Google Scholar]
- 7.St Clair D, Zhao Y, Chaiswing L, Oberley T. Modulation of skin tumorigenesis by SOD. Biomedicine & Pharmacotherapy. 2005;59:209–214. doi: 10.1016/j.biopha.2005.03.004. [DOI] [PubMed] [Google Scholar]
- 8.Zhao Y, Xue Y, Oberley TD, Kiningham KK, Lin SM, Yen HC, et al. Overexpression of manganese superoxide dismutase suppresses tumor formation by modulation of activator protein-1 signaling in a multistage skin carcinogenesis model. Cancer Res. 2001;61:6082–6088. [PubMed] [Google Scholar]
- 9.Oberley LW, Buettner GR. Role of superoxide dismutase in cancer: a review. Cancer Res. 1979;39:1141–1149. [PubMed] [Google Scholar]
- 10.Izutani R, Asano S, Imano M, Kuroda D, Kato M, Ohyanagi H. Expression of manganese superoxide dismutase in esophageal and gastric cancers. J Gastroenterol. 1998;33:816–822. doi: 10.1007/s005350050181. [DOI] [PubMed] [Google Scholar]
- 11.Xu Y, Kiningham KK, Devalaraja MN, Yeh CC, Majima H, Kasarskis EJ, et al. An intronic NF-kappaB element is essential for induction of the human manganese superoxide dismutase gene by tumor necrosis factor-alpha and interleukin-1beta. DNA and Cell Biol. 1999;18:709–722. doi: 10.1089/104454999314999. [DOI] [PubMed] [Google Scholar]
- 12.Porntadavity S, Xu Y, Kiningham K, Rangnekar VM, Prachayasittikul V, St Clair DK. TPA-activated transcription of the human MnSOD gene: role of transcription factors Sp-1 and Egr-1. DNA and Cell Biol. 2001;20:473–481. doi: 10.1089/104454901316976109. [DOI] [PubMed] [Google Scholar]
- 13.Xu Y, Porntadavity S, St Clair DK. Transcriptional regulation of the human manganese superoxide dismutase gene: the role of specificity protein 1 (Sp1) and activating protein-2 (AP-2) Biochem J. 2002;362:401–412. doi: 10.1042/0264-6021:3620401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hussain SP, Amstad P, He P, Robles A, Lupold S, Kaneko I, et al. p53-induced up-regulation of MnSOD and GPx but not catalase increases oxidative stress and apoptosis. Cancer Res. 2004;64:2350–2356. doi: 10.1158/0008-5472.can-2287-2. [DOI] [PubMed] [Google Scholar]
- 15.Dhar SK, Xu Y, Chen Y, St Clair DK. Specificity protein 1-dependent p53-mediated suppression of human manganese superoxide dismutase gene expression. J Biol Chem. 2006;281:21698–21709. doi: 10.1074/jbc.M601083200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dhar SK, Xu Y, St Clair DK. Nuclear factor kappa B- and specificity protein 1-dependent p53-mediated bi-directional regulation of the human manganese superoxide dismutase gene. J Biol Chem. 2010;285:9835–9846. doi: 10.1074/jbc.M109.060715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Drane P, Bravard A, Bouvard V, May E. Reciprocal down-regulation of p53 and SOD2 gene expression-implication in p53 mediated apoptosis. Oncogene. 2001;20:430–439. doi: 10.1038/sj.onc.1204101. [DOI] [PubMed] [Google Scholar]
- 18.Hollstein M, Sidransky D, Vogelstein B, Harris CC. p53 mutations in human cancers. Science. 1991;253:49–53. doi: 10.1126/science.1905840. [DOI] [PubMed] [Google Scholar]
- 19.Soussi T, Lozano G. p53 mutation heterogeneity in cancer. Biochem Biophysic Res Comm. 2005;331:834–842. doi: 10.1016/j.bbrc.2005.03.190. [DOI] [PubMed] [Google Scholar]
- 20.Hogan B, Beddington R, Costantini R, Lacy E. Manipulating the mouse embryo. A Laboratory Manual. 2. Cold Spring Harbor, New York: Cold Spring Harbor Laboratory Press; 1994. [Google Scholar]
- 21.Yen HC, Oberley TD, Vichitbandha S, Ho YS, St Clair DK. The protective role of manganese superoxide dismutase against adriamycin-induced acute cardiac toxicity in transgenic mice. J Clinic Invest. 1996;98:1253–1260. doi: 10.1172/JCI118909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dhar SK, Xu Y, Noel T, St Clair DK. Chronic exposure to 12-O-tetradecanoylphorbol-13-acetate represses sod2 induction in vivo: the negative role of p50. Carcinogenesis. 2007;28:2605–2613. doi: 10.1093/carcin/bgm163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Spitz DR, Oberley LW. An assay for superoxide dismutase activity in mammalian tissue homogenates. Anal Biochem. 1989;179:8–18. doi: 10.1016/0003-2697(89)90192-9. [DOI] [PubMed] [Google Scholar]
- 24.Colburn NH, Former BF, Nelson KA, Yuspa SH. Tumor promoter induces anchorage independence irreversibly. Nature. 1979;281:589–591. doi: 10.1038/281589a0. [DOI] [PubMed] [Google Scholar]
- 25.Shaulian E, Karin M. AP-1 as a regulator of cell life and death. Nature Cell Biol. 2002;4:131–136. doi: 10.1038/ncb0502-e131. [DOI] [PubMed] [Google Scholar]
- 26.Koutsodontis G, Vasilaki E, Chou WC, Papakosta P, Kardassis D. Physical and functional interactions between members of the tumour suppressor p53 and the Sp families of transcription factors: importance for the regulation of genes involved in cell-cycle arrest and apoptosis. Biochem J. 2005;389:443–455. doi: 10.1042/BJ20041980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Oberley LW. Mechanism of the tumor suppressive effect on MnSOD overexpression. Biomed & Pharmacotherapy. 2005;59:143–148. doi: 10.1016/j.biopha.2005.03.006. [DOI] [PubMed] [Google Scholar]
- 28.Sun Y, Oberley LW, Oberley TD, Elwell JH, Sierra-Rivera E. Lowered antioxidant enzymes in spontaneously transformed embryonic mouse liver cells in culture. Carcinogenesis. 1993;14:1457–1463. doi: 10.1093/carcin/14.7.1457. [DOI] [PubMed] [Google Scholar]
- 29.Zhao Y, Oberley TD, Chaiswing L, Lin SM, Epstein CJ, Huang TT, St Clair DK. Manganese superoxide dismutase deficiency enhances cell turnover via tumor promoter-induced alterations in AP-1 and p53-mediated pathways in a skin cancer model. Oncogene. 2002;21:3836–3846. doi: 10.1038/sj.onc.1205477. [DOI] [PubMed] [Google Scholar]
- 30.Miao L, St Clair DK. Regulation of superoxide dismutase gene: implications in disease. Free Radic Biol Med. 2009;15:344–356. doi: 10.1016/j.freeradbiomed.2009.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Grundstrom S, Anderson P, Scheipers P, Sundstedt A. Bcl-3 and NFkappaB p50-p50 homodimers act as transcriptional repressors in tolerant CD4+ T cells. J Biol Chem. 2004;279:8460–8468. doi: 10.1074/jbc.M312398200. [DOI] [PubMed] [Google Scholar]
- 32.Hirano F, Tanaka H, Hirano Y, Hiramoto M, Handa H, Makino I, et al. Functional interference of Sp1 and NF-kappaB through the same DNA binding site. Mol Cell Biol. 1998;18:1266–1274. doi: 10.1128/mcb.18.3.1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Helton ES, Zhang J, Chen X. The proline-rich domain in p63 is necessary for the transcriptional and apoptosis-inducing activities of Tap63. Oncogene. 2008;27:2843–2850. doi: 10.1038/sj.onc.1210948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jost CA, Marin WG, Kaelin p73 is a simian [correction of human] p53-related protein that can induce apoptosis. Nature. 1997;389:191–194. doi: 10.1038/38298. [DOI] [PubMed] [Google Scholar]
- 35.Kaghad M, Bonnet H, Yang A, Creancier L, Biscan JC, Valent A, et al. Monoallelically expressed gene related to p53 at 1p36, a region frequently deleted in neuroblastoma and other cancers. Cell. 1997;90:809–819. doi: 10.1016/s0092-8674(00)80540-1. [DOI] [PubMed] [Google Scholar]
- 36.Zhu J, Jiang J, Zhou W, Chen X. The potential tumor suppressor p73 differentially regulates cellular p53 target genes. Cancer Res. 1998;58:5061–5065. [PubMed] [Google Scholar]
- 37.Westfall MD, Pietenpol JA. p63: molecular complexity in development and cancer. Carcinogenesis. 2004;25:857–864. doi: 10.1093/carcin/bgh148. [DOI] [PubMed] [Google Scholar]
- 38.Innocente SA, Lee JM. p73 is a p53-independent, Sp1-dependent repressor of cyclin B1 transcription. Biochem Biophys Res Comm. 2005;329:713–718. doi: 10.1016/j.bbrc.2005.02.028. [DOI] [PubMed] [Google Scholar]
- 39.Johnson J, Lagowski J, Lawson S, Liu Y, Kulesz-Martin M. p73 expression modulates p63 and Mdm2 protein presence in complex with p53 family-specific DNA target sequence in squamous cell carcinogenesis. Oncogene. 2008;27:2780–2787. doi: 10.1038/sj.onc.1210941. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.