

Abstract
Introduction
Microparticles (MPs) are submicron vesicles shed by activated or apoptotic cells, including platelets and monocytes. Increased circulating MPs are associated with thrombosis; however, their role in thrombogenesis is poorly understood.
Objective
To determine how MPs promote thrombin generation and modulate fibrin density and stability.
Methods
Platelets and monocytes were isolated from healthy donors. Platelets were stimulated with calcium ionophore, thrombin receptor agonist peptide (TRAP), or TRAP/convulxin. Monocytes and human monocytic THP-1 cells were stimulated with lipopolysaccharide. MPs were isolated, washed by high-speed centrifugation, and assessed by transmission electron microscopy, nanoparticle tracking analysis, flow cytometry, tissue factor (TF) activity, prothrombinase activity, thrombin generation, and clot formation, density, and stability.
Results
MPs from monocytes (M-MPs) and platelets (PMPs) had similar shapes and diameters (100–300 nm). M-MPs had TF activity (16.7±2.4 pM TF/106 MP), supported prothrombinase activity, and triggered shorter thrombin generation lag times than buffer controls (5.4±0.5 versus 84.2±4.8 min, respectively). Compared to controls, M-MPs supported faster fibrin formation (0.24±0.24 versus 76.7±15.1 mOD/min, respectively), 38% higher fibrin network density, and higher clot stability (3.8-fold higher turbidity in the presence of tissue plasminogen activator). In contrast, PMPs did not have TF activity and supported 2.8-fold lower prothrombinase activity than M-MPs. PMPs supported contact-dependent thrombin generation, but did not independently increase fibrin network density or stability. Interestingly, PMPs increased rates of thrombin generation and fibrin formation (1.7- and 1.3-fold, respectively) when mixed with THP-1-derived MPs.
Conclusion
MPs from platelets and monocytes differentially modulate clot formation, structure and stability, suggesting unique contributions to thrombosis.
Keywords: fibrinogen, microparticle, phosphatidylserine, thrombosis, tissue factor
Introduction
Once considered simply cell debris, microparticles (MPs) are bioactive sub-micron (0.1–1 μm) membrane vesicles shed from activated and apoptotic cells in culture and in vivo. While detectable in healthy controls[1], MPs levels are greatly elevated in patients with diseases including venous thromboembolism[2], hypertension[3], diabetes mellitus[4], and cancer[5, 6]. Platelet-derived MPs (PMPs) comprise the largest fraction of MPs in healthy controls[1] and patients[5, 7]. PMP concentrations in patients range from ~3000–11,000/μL.[2, 4, 8] Monocyte-derived MPs (M-MPs) are low or undetectable in healthy controls, but circulate at levels from ~300–1300/μL in sickle cell disease, cancer, and other diseases.[2, 4, 6, 9]
Despite studies demonstrating circulating MPs in healthy individuals and patients, MP-mediated mechanisms contributing to hemostasis or thrombosis are not understood. MPs bear surface antigens from their parent cells and anionic phospholipids such as phosphatidylserine (PS).[10] These properties are thought to define MP function. Observations that MPs support thrombin generation[1, 11] suggest MPs contribute to hemostasis via “idling” of the coagulation system, evidenced by the presence of activation peptides and prothrombin fragment 1.2 in healthy individuals[12]. However, it is difficult to confirm the role of MP procoagulant activity in hemostasis in vivo because MPs cannot be depleted from circulation. Increased levels of MPs in thrombotic disease suggest MP procoagulant activity tips coagulation “idling” towards full-fledged activation and promotes thrombosis. TF-bearing MPs, in particular, may independently initiate or propagate coagulation via recruitment to developing thrombi.[13–15] Human M-MPs promote fibrin accumulation in a murine carotid artery ligation model[16], and injection of MPs isolated from mice after inferior vena cava thrombosis into new mice prior to inferior vena cava ligation increases thrombus weight at early time points.[17] The mechanism(s) by which MP procoagulant activity increases fibrin deposition, thrombus growth, and/or weight are not known.
Thrombin generation promotes clotting and clot stability by modulating fibrin properties, including its network structure and resistance to fibrinolysis.[18, 19] These studies suggest MP procoagulant activity directly increases fibrin formation and stability. However, although procoagulant activity of heterogeneous MP pools isolated from plasma has been examined, to our knowledge, no study has examined how MPs from different parent cells promote procoagulant activity or fibrin production or quality. This information gap limits clinical interpretation of MP function in healthy individuals or pathologic contributions of elevated levels of certain MP subtypes in patients.
The goal of this study was to determine the specific contributions of monocyte- and platelet-derived MPs to thrombin generation and fibrin formation, structure, and stability. We prepared MPs from isolated human platelets, monocytes, and a monocytic cell line (THP-1) and compared their physical and biochemical properties. Our data show monocyte-derived MPs initiate thrombin generation and fibrin formation via their TF activity, whereas platelet-derived MPs propagate thrombin and fibrin production in TF- or contact-triggered plasma. These findings suggest MPs from different parent cells uniquely contribute to coagulation, and that the relative concentration of circulating MPs from different cell types influences thrombosis risk.
Materials & Methods
Materials
Monocyte Negative and Positive Selection Kits were from Miltenyi BioTec (Auburn, CA). Accu-Prep lymphocyte gradient medium was from Accurate Chemical Co. (Westbury, NY). Prostacyclin I2 was from Cayman Chemical Co. (Ann Arbor, MI). Thrombin receptor agonist peptide (TRAP) was from Bachem Inc. (Torrence, CA). Convulxin was from Centerchem Inc. (Norwalk, CT). Calcium Ionophore (A23187), lipopolysaccharide (LPS), control IgG (MOPC-1), and bovine serum albumin (BSA) were from Sigma Aldrich Corp. (St. Louis, MO). Fluorescein isothiocyanate (FITC)-Annexin V antibody and Annexin V Binding Buffer were from Beckman Coulter (Brea, CA). Phycoerythrin (PE)-anti-CD41 antibody and Megamix beads were from BioCytex (Marseille, France). Allophycocyanin (APC)-anti-CD14 antibody was from R&D Systems (Minneapolis, MN). Innovin was from Siemens Healthcare Diagnostics (Deerfield, IL). Mouse anti-human TF antibody (HTF-1, “anti-TF”)[20] was provided by Dr. Ronald Bach (Minneapolis VA Medical Center, MN), and recombinant factor VIIa by Novo Nordisk (Denmark). Human factor X was from Enzyme Research Laboratories (South Bend, IN). Human factors Xa, Va, prothrombin, and corn trypsin inhibitor (CTI) were from Haematologic Technologies, Inc. (Essex Junction, VT). Thrombin fluorogenic substrate (Z-Gly-Gly-Arg-AMC) and calibrator (α2-macroglobulin/thrombin) were from Diagnostica Stago (Parsippany, NJ). Tissue plasminogen activator (tPA) was from Calbiochem (La Jolla, CA). Citrated, contact-inhibited (18.6 μg/mL CTI[21]), normal-pooled, platelet-free plasma[18] was prepared from 27 individuals, with final centrifugation (20,000×g, 20 minutes) to prepare MP-depleted plasma (MDP).
Primary cell isolation
Peripheral blood mononuclear cells (PBMCs), monocytes, and platelets were freshly-isolated from whole blood from healthy donors in a protocol approved by the University of North Carolina (UNC) Institutional Review Board. Blood was collected into 3.2% sodium citrate and centrifuged (150×g, 20 minutes). The platelet-rich plasma was treated with 50 ng/mL prostacyclin I2 to prevent nonspecific activation of platelets during preparation and centrifuged (400×g, 20 minutes) to pellet platelets. Platelets were re-suspended in warm (37 °C) Tyrode’s buffer [1.5 mM N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid, 0.33 mM NaH2PO4 (pH 7.4), 13.8 mM NaCl, 0.27 mM KCl, 0.1 mM MgCl2, 0.55 mM dextrose, 1 mg/mL BSA] with 50 ng/mL prostacyclin I2 and incubated for 30 minutes at 37 °C before stimulation. To isolate PBMCs and monocytes, the buffy coat and erythrocytes were diluted with Hank’s balanced salt solution containing 5 mM ethylenediamine tetraaceticacid (HBSS/EDTA), layered over Accu-prep Lymphocyte gradient medium, and centrifuged (400×g, 30 minutes). The PBMC buffy coat fraction was removed, washed twice by centrifugation at 250×g, and re-suspended in HBSS/EDTA. For negative and positive monocyte selection, the PBMC buffy coat was washed twice and re-suspended in ice-cold MACS sorting buffer (phosphate-buffered saline/0.5% BSA/2 mM EDTA). Negative and positive monocyte selection kits were used according to the manufacturer’s instructions. Final enriched monocyte populations were suspended in Macrophage-Serum-Free Media (Gibco, Grand Island, NY).
Cell culture
Human monocytic leukemia cell line (THP-1) was obtained from Dr. Nigel Mackman (UNC), and cultured in RPMI 1640 media with 10% fetal bovine serum.
Microparticle isolation
Platelets were stimulated for 30 minutes at room temperature with 6 or 50 μg/mL TRAP, 6 or 50 μg/mL TRAP plus 500 ng/mL convulxin (TRAP/convulxin), or 10 μM Ca2+ ionophore (A23187). There were no differences in peak thrombin generation between PMPs generated with 6 or 50 μg/mL TRAP. Samples were centrifuged (1,500×g, 15 minutes) and MP-containing supernatants were collected. PBMCs, monocytes, and THP-1 cells were stimulated with 100 ng/mL LPS (6 hours, 37 °C) in Macrophage Serum-Free Media; this condition supports cell stimulation in the absence of LPS-binding protein.[22] Trypan blue (HyClone, Logan, UT) exclusion experiments indicated less than 10% of monocytes and less than 15% of THP-1 cells were dead following the 6-hour incubation. Cells were removed by sequential centrifugation (500×g for 10 minutes, 1,500×g for 15 minutes, 13,000×g for 2 minutes). MPs were pelleted (20,000×g, 15 minutes), re-suspended in HEPES-buffered saline with 0.1% BSA (HBS/BSA), and re-pelleted (20,000×g, 15 minutes). MP pellets were re-suspended in HBS/BSA and stored at 4 °C up to one week. Control experiments showed no difference in thrombin generation in fresh and 1 week-old MPs (9.6±2.7 versus 12.0±0.9 nM peak thrombin, respectively).
Microparticle enumeration
Flow cytometry of washed MPs was performed on an LSR-II flow cytometer (BD Biosciences, San Jose, CA), using Megamix polystyrene beads (0.5, 0.9, and 3 μm) to gate the MP region by forward- and side-scatter.[23] MPs were incubated with antibodies in Annexin V Binding Buffer. FITC-Annexin V was used to detect PS. PE-anti-CD41 was used as a cell marker for PMPs and APC-anti-CD14 as a marker for PBMC-MPs and M-MPs. Fluorospheres (10 μm) were added and samples were analyzed for PS-positive/cell marker-positive (dual positive) events. MP concentration was determined per: [(Dual positive Events/Fluorosphere Bead Events)* Fluorosphere Bead Concentration]*Dilution Factor.
Microparticle physical characterization
Transmission electron microscopy (TEM) was performed by depositing MPs onto carbon-coated Formvar copper grids treated with 0.01% poly-L-lysine. MPs were negatively stained with 3% ammonium molybdate (pH 7.0)/0.1% trehalose and imaged on a LEO EM910 transmission electron microscope (Carl Zeiss SMT, Peabody, MA) operating at 80 kV. Digital images were recorded using a Gatan Orius CCD Digital Camera and Digital Micrograph 3.11.0 (Gatan INC, Pleasanton, CA). MP diameters were measured using Adobe Photoshop 8.0.
Nanoparticle Tracking Analysis (NTA) was performed using the Nanosight NS500 system (NanoSight, Amesbury, UK), which focuses a laser beam through a suspension of the particles of interest, visualizes particles resident within the beam using a conventional optical microscope aligned normally to the beam axis, and collects light scattered from all particles in the field of view. Videos (60 seconds) were recorded using an electron multiplying charge-coupled device, and particle movement was analyzed by NTA software. Each particle was identified and its Brownian movement tracked and measured frame-to-frame. The particle movement velocity was used to calculate particle size by applying the Stokes-Einstein equation.
TF and prothrombinase activity
MPs were pre-incubated with anti-TF antibody (10 μg/mL, final) or isotype-matched control IgG (10 minutes, 37 °C) and then with factor VIIa (100 pM, final, 5 minutes) and factor X (135 nM, final, 30 minutes, 37 °C), in calcium (5 mM, final). Factor Xa generation per MP (per number of cell-marker-and Annexin V-positive events) was measured by chromogenic substrate cleavage[24] and referenced to a standard curve of lipidated recombinant human TF (Innovin). Prothrombinase activity (per Annexin V-positive events) was measured as described.[18]
Thrombin generation by calibrated automated thrombography
MPs (10 μL) and 20 μL HBS/BSA were spiked into 70 μL contact-inhibited MDP with anti-TF or control IgG (10 μg/mL, final) (58% MDP, final). Thrombin generation was initiated by automatically dispensing fluorogenic substrate (Z-Gly-Gly-Arg-AMC) in CaCl2 (416 μM and 16 mM, final, respectively), calibrated against wells containing α2-macroglobulin/thrombin complex and plasma, and analyzed with Thrombinoscope software v3.0.0.29 (Thrombinoscope BV, Maastricht, Netherlands).
Fibrin formation, network structure and clot stability
MPs were added to re-calcified (10 mM, final) MDP (88% MDP, final) in the absence or presence of 0.5 μg/mL tPA. Fibrin formation and lysis were measured by turbidity as described.[19] Fibrin networks were imaged in 10 μm z-stacks (4 stacks/clot) using laser scanning confocal microscopy in the presence of Alexa488-conjugated fibrinogen, as described.[18, 19] Fibrin density was measured by counting fibers intersecting a randomly-applied grid, as described.[18, 19]
Statistical analysis
Data are described using descriptive statistics [mean (±standard error of the mean) or mean (range), as indicated]. TF-dependent activity (anti-TF versus control IgG) and PMP-dependent activity (±PMPs) were analyzed by paired student’s t test. For all other data, Analysis of Variance (ANOVA) with Dunnett’s post hoc test using buffer as the index group was used to control type I error. P<0.05 was considered significant.
Results
MPs from platelets and monocytes have a similar size distribution
We first characterized MPs from TRAP, TRAP/convulxin, and A23187-treated platelets, and LPS-treated PBMCs, monocytes, and THP-1 cells using flow cytometry with Annexin V- and cell-specific marker positivity, the gold standard for MP characterization and enumeration.[23] Figure 1A shows the MP gate (determined by 500 nm and 900 nm Megamix fluorescent beads) from representative PMPs from TRAP-stimulated platelets; MP from TRAP/convulxin- and A23187-treated platelets appeared similar (data not shown). M-MP contamination in PMP preparations was <5%. PMP contamination in MP preparations from LPS-treated PBMCs, negatively-selected monocytes, and positively-selected monocytes was 45.4±7.4%, 33.4±8.7%, and 16.6±2.9% of Annexin V-positive events, respectively, likely from platelets contaminating monocyte preparations. M-MPs derived from positively-selected monocytes (Figure 1B) were used for subsequent experiments. We also used THP-1-derived MPs (THP-MPs) as a platelet-free source of monocytic MPs, as indicated.
Figure 1. M-MPs and PMPs have similar size distributions.
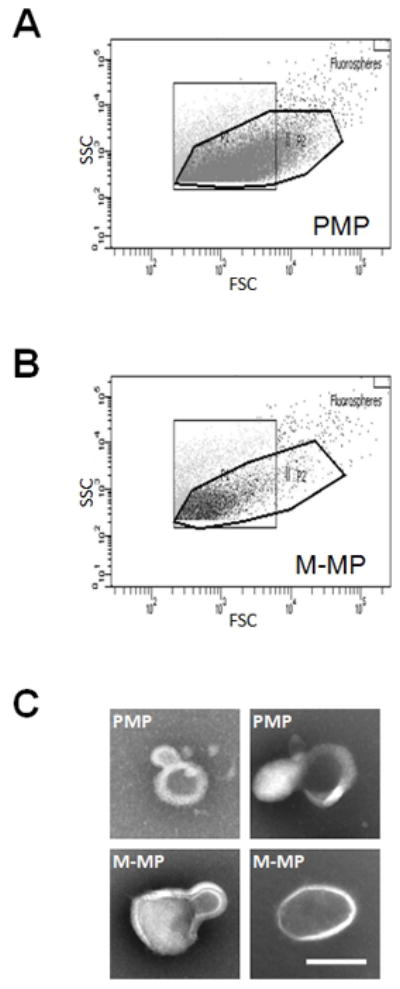
Representative bivariate flow cytometry plots (forward and side scatter) of (A) TRAP-derived PMPs and (B) M-MPs. (C) TEM of representative TRAP-derived PMPs and M-MPs. Scale bar is 200 nm.
Although flow cytometry detects particles as small as 300 nm, studies indicate MPs may be smaller (100–300 nm)[25]. To explicitly determine MP size we utilized two independent techniques: TEM and NTA. TEM enables imaging and measurement of individual MP (Figure 1C), while NTA enables enumeration and size analysis of fully hydrated MPs (from 20–1000 nm) in suspension[26]. Both methodologies indicated MPs have similar diameters (150–300 nm), regardless of the parent cell (monocytes or platelets), or stimulation method (LPS, TRAP, TRAP/convulxin, or A23187) (Table 1). NTA indicated LPS-stimulated monocytes generated approximately 60 M-MP/monocyte, and platelets stimulated with TRAP, TRAP/convulxin, or A23187 produced approximately 1.0 PMP/platelet; both were approximately 85-fold higher than that suggested by flow cytometry, consistent with reports that flow cytometry detects only a modest proportion of MPs[26]. However, although NTA can detect fluorescently-labeled placental MPs[26], it has not been validated for fluorescently detecting M-MPs or PMPs. Since MP enumeration in prior studies of human plasmas utilized flow cytometry, we used flow cytometry-determined enumeration in all subsequent assays.
Table 1.
Comparison of MP sizes by TEM and NTA.
| TEM (nm, n=3–6) | NTA (nm, n=2) | |||
|---|---|---|---|---|
| Mean | Range | Mean | Range | |
| M-MP | 270 | 32 | 227 | 46 |
| PMP (TRAP) | 220 | 199 | 246 | 69 |
| PMP (TRAP/convulxin) | 290 | 182 | 184 | 4.3 |
| PMP (A23187) | 267 | 480 | 233 | 61 |
M-MPs, but not PMPs, promote thrombin generation in a TF-dependent manner
To compare inherent functional properties of MPs from platelets and monocytes, we measured both TF- and PS-dependent procoagulant activity. Figure 2A shows TF activity on M-MPs (16.7±2.4 pM TF/106 MP) and THP-MPs (2.88±0.8 pM TF/106 MP). However, PMPs, regardless of the agonist used for their derivation, did not support factor Xa generation (Figure 2A), suggesting PMPs do not express TF.
Figure 2. M-MPs, but not PMPs, promote thrombin generation in a TF-dependent manner.
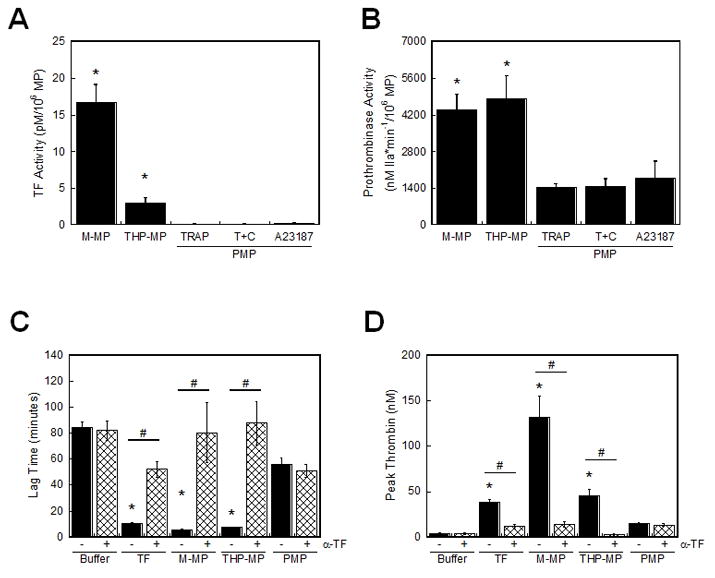
(A) TF activity (±SEM) was determined by factor Xa chromogenic substrate cleavage (n=5). *P<0.02 by ANOVA versus PMPs. (B) Prothrombinase activity (±SEM) was determined by thrombin chromogenic substrate cleavage (n=3–4). MP counts were determined by Annexin V-positive events in flow cytometry. *P<0.01 by ANOVA versus all PMPs. (C–D) Thrombin generation supported by 5000 PMP/μL or 1000 M-MP or THP-MP/μL was measured in the presence of anti-TF or control IgG. (C) Thrombin lag time and (D) peak (±SEM, n=3–6). Data from PMP from TRAP-stimulated platelets are shown; PMP from TRAP/convulxin- or A23187-stimulated platelets were similar. “Buffer” is HBS/BSA. TF is 1 pM (Innovin). *P<0.05 by ANOVA versus buffer controls. #P<0.05 by paired Student’s t test between anti-TF and IgG controls.
We next compared the ability of these MPs to support prothrombinase activity. Regardless of the agonist used for their derivation, PMPs had similar prothrombinase activity (Figure 2B). PMPs did not support thrombin generation in the absence of exogenous factor Va (data not shown), indicating functional, platelet-derived factor Va was not present on PMPs. Similarly, M-MPs and THP-MPs did not support thrombin generation in the absence of exogenous factor Va (data not shown). Flow cytometry indicated M-MPs had higher Annexin V mean fluorescence intensity than PMPs (711.0±71.0 versus 418.7±43.8 arbitrary units, p<0.002). Accordingly, M-MPs showed 2.8-fold higher prothrombinase activity than PMPs (4382±584 nM thrombin*min−1/106 M-MP versus 1556±189 nM thrombin*min−1/106 PMP, Figure 2B]. Although THP-MPs had lower Annexin V mean fluorescence intensity than PMPs (111.8±2.7 arbitrary units, P<0.001), they demonstrated higher prothrombinase activity (4807±879 nM thrombin*min−1/106 THP-MP, Figure 2B) than PMPs. These data suggest functional prothrombinase activity is not necessarily predicted by Annexin V fluorescence intensity, and confirm that M-MP prothrombinase activity was not due to PMP contamination in M-MP preparations.
Given these inherent differences in the nature of procoagulant activity of monocyte (TF and PS)-and platelet (only PS)-derived MPs, we then compared their ability to support thrombin generation in plasma. We spiked MPs at levels associated with thrombosis in humans (1000 M-MP/μL and 5000 PMP/μL)[2, 4, 6, 9] into re-calcified MDP and measured thrombin generation by calibrated automated thrombography. PMPs from TRAP-, TRAP/convulxin-, or A23187-stimulated platelets slightly but non-significantly shortened the lag time and increased the peak over buffer controls (Figures 2C–D, data not shown). These activities were not blocked by anti-TF antibody, indicating this activity was TF-independent. Although MDP was treated with CTI to inhibit contact activation, studies have reported time-dependent loss of CTI-mediated contact inhibition.[27] Accordingly, PMPs did not trigger clotting in factor XI- or IX-deficient plasma (data not shown). These data are consistent with observations that PMP-enriched plasma from healthy donors supports contact pathway-driven thrombin generation[1], and attribute this finding specifically to the PMP fraction.
In contrast to PMPs, compared to buffer controls, both M-MPs and THP-MPs significantly (P<0.001) shortened the thrombin lag time (5.4±0.5 min and 7.5±0.4 min, respectively, versus 84.2±4.8 min, Figure 2C) and increased the peak (131.5±23.4 and 45.6±6.9 nM, respectively, versus 3.6±0.8 nM, Figure 2D) in a TF-dependent manner. These data indicate monocyte-derived MPs initiate thrombin generation in a TF-dependent manner, while PMPs support thrombin generation only in the presence of an intact contact pathway.
M-MPs promote fibrin formation
Given differences in PMP and M-MP procoagulant activity, we then compared the ability of MPs to support fibrin formation by spiking MPs into re-calcified MDP and following fibrin formation by turbidity. PMPs from TRAP- (Figures 3A, B), TRAP/convulxin- (data not shown) or A23187-(data not shown) stimulated platelets inconsistently triggered fibrin formation after 60 minutes only at the highest concentrations tested, and in a TF-independent mechanism. As in thrombin generation assays (Figure 2), PMPs did not trigger clotting in contact (factor XII)-deficient plasma for up to 120 minutes (data not shown), indicating PMP activity in MDP reflected time-dependent loss of contact inhibition[27]. In contrast, M-MPs and THP-MPs significantly (P<0.0001) shortened the onset (Figures 3A, B) and increased the rate (Figures 3A, C) of fibrin formation in a concentration-dependent manner that was completely blocked by anti-TF antibody (data not shown). These data indicate M-MPs and THP-MPs trigger TF-dependent fibrin production, while PMPs support fibrin formation only in the presence of an intact contact pathway.
Figure 3. M-MPs initiate fibrin formation.

MPs were spiked into re-calcified MDP and fibrin formation was followed by turbidity. (A) Representative fibrin formation curves. (B) Onset and (C) rate of fibrin formation. For conditions in which clots did not form, onsets were censored at 120 minutes and rates at 0 mOD/min. Data show mean (±SEM, n=4–6). TF is 1 pM (Innovin). *P<0.0001 by ANOVA versus buffer controls.
M-MPs, but not PMPs, increase fibrin network density
The fibrin formation rate is correlated with fibrin network density.[18, 19] We used laser scanning confocal microscopy to compare effects of PMP and M-MPs on network density versus networks produced by the background level of contact activation in MDP. Addition of TRAP-derived PMPs to clotting reactions did not significantly alter network density compared to buffer controls (Figure 4), although fibers appeared slightly more branched in samples clotted in the presence of 5000 PMP/μL. TRAP/convulxin- and A23187-derived PMPs similarly had no significant effect on network density (data not shown). Since platelets increase fibrin density via interactions between their integrins and the fibrin network[19, 28], this observation suggests integrin density on PMP is lower than on intact platelets and does not sufficiently interact with the fibrin network to alter structure. In contrast to PMPs, M-MPs significantly increased fiber density (P=0.001) over controls (Figure 4).
Figure 4. M-MPs, but not PMPs, increase fibrin network density.
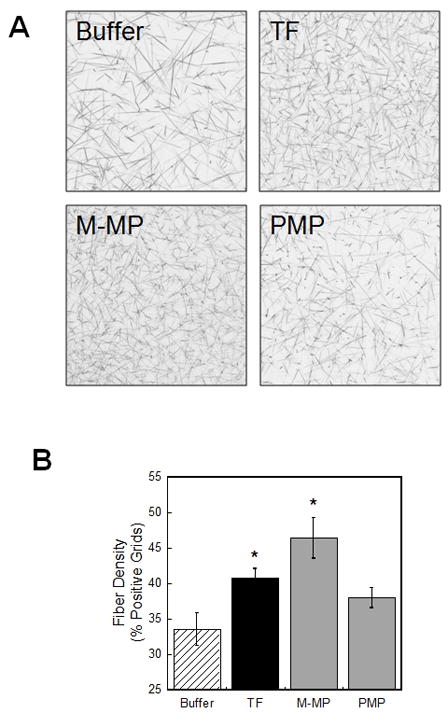
Clots were formed by incubating MPs (5000 PMP/μL and 1000 M-MP/μL, final) in re-calcified MDP containing Alexa488-conjugated fibrinogen, and imaged by confocal microscopy. (A) Micrographs of fibrin networks after clots were fully-formed (180 minutes). (B) Fibrin fiber density shown as mean percent positive grids (±SEM, n=3–5). TF is 1 pM (Innovin). *P<0.05 by ANOVA versus buffer controls.
M-MPs increase clot resistance to fibrinolysis
Fibrin network density is positively associated with network stability.[18, 19] To determine the ability of MPs to promote clot stability, we spiked MPs into re-calcified MDP in the presence of tPA, and followed fibrin formation and lysis by turbidity (Figure 5A). The time to peak and peak turbidity reflect the time to maximum fibrin formation and peak incorporation of fibrin into the clot, respectively.[19] Both M-MPs and THP-MPs shortened the time to peak (P<0.0001, Figure 5B) and increased the peak (P<0.03, Figure 5C) turbidity versus control (buffer), whereas PMPs did not enhance fibrin formation over buffer (contact activation), alone. These data suggest monocyte-derived MPs trigger rapid accumulation of fibrin, even in the presence of fibrinolytic activity.
Figure 5. M-MPs increase clot resistance to fibrinolysis.
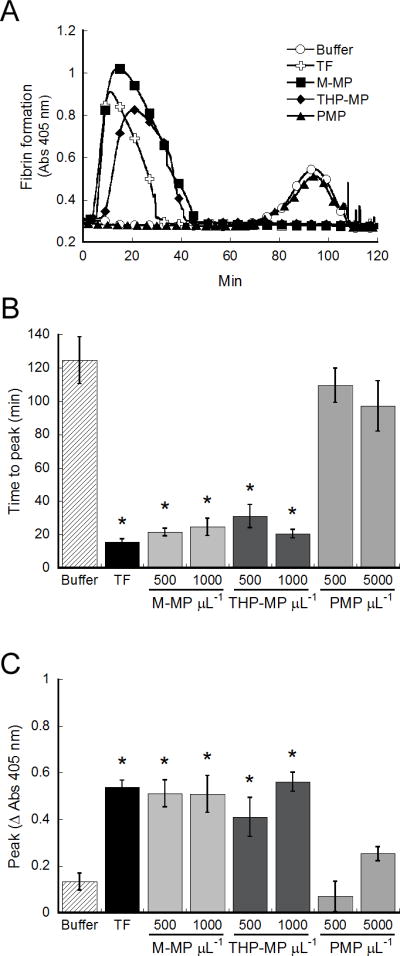
MPs (1000 M-MP or THP-MP/μL and 5000 PMP/μL) were spiked into re-calcified MDP in the presence of tPA and fibrin formation and lysis were followed by turbidity. (A) Representative turbidity curves. (B) Time to peak and (C) peak turbidity change. Data show mean (±SEM, n=4–7). TF is 1 pM (Innovin). *P<0.05 by ANOVA versus buffer controls.
PMPs increase thrombin generation and the fibrin formation rate during TF-initiated clotting
Our data indicated PMPs supported little to no fibrin generation in the absence of a procoagulant stimulus (contact activation), consistent with their apparently non-pathogenic presence in healthy individuals[1]. However, since PMP supported procoagulant activity (Figure 2) and fibrin formation following contact activation (Figures 2–5), and since MPs are recruited to sites of vascular injury (TF exposure)[13–15], we tested the hypothesis that PMPs augment coagulation in reactions triggered by TF. We spiked PMPs into re-calcified MDP, triggered clotting with TF (1 pM Innovin or THP-MPs as a model monocyte TF-bearing MP to avoid any influence of contaminating PMPs), and followed thrombin generation and fibrin formation. Addition of PMPs from TRAP-stimulated platelets to Innovin-triggered reactions shortened the thrombin lag time from 10.4±0.8 to 8.7±0.5 minutes (P<0.01). PMPs also shortened the lag time in 500/uL and 1000/uL THP-MP-triggered reactions from 4.6±0.2 to 4.0±0.1 minutes, and 3.7±0.6 to 3.2±0.5 minutes, respectively (P<0.04). PMPs increased the thrombin peak (P<0.05, Figure 6A). These data reflect the ability of PMPs to support thrombin propagation, as well positively feedback on the initiation phase. Consequently, addition of PMPs to Innovin- or THP-MP-triggered clotting assays consistently increased the fibrin formation rate over Innovin or THP-MP, alone (P<0.01, Figure 6B), and slightly, but non-significantly, shortened the time to peak (Figure 6C) and increased the peak (Figure 6D) turbidity in fibrinolysis assays. These findings suggest that following initiation, PMPs enhance clotting propagation and promote faster fibrin growth.
Figure 6. PMPs increase thrombin generation and the rate of fibrin formation during TF-initiated clotting.
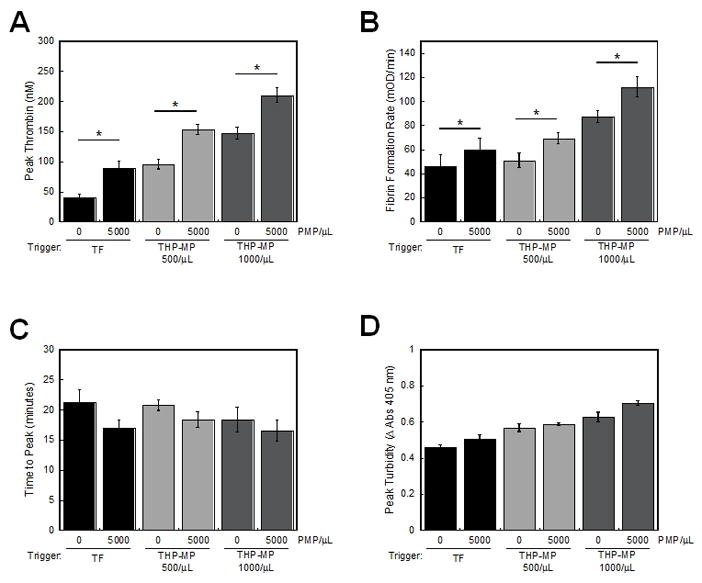
PMPs were mixed with THP-MPs or Innovin and spiked into re-calcified MDP, and thrombin generation and fibrin formation were measured. (A) Peak thrombin and (B) fibrin formation rate in the absence of tPA. (C) Time to peak and (D) peak turbidity in the presence of tPA. Data show mean (±SEM, n=3–4). TF is 1 pM (Innovin). *P<0.02 by Student’s t test versus 0 PMP/μL.
Discussion
The presence of circulating, cell-derived MPs in healthy individuals and their increased numbers in disease is well-documented; however, their potential mechanistic role(s) in hemostasis and/or thrombosis are poorly-defined. This information gap results, in part, from the lack of studies investigating independent procoagulant contributions of pure MP populations. We specifically characterized properties of MPs from isolated platelets and monocytes—MP subtypes most frequently implicated in procoagulant/prothrombotic disorders. Our data show TF-bearing MPs (M-MPs, THP-MPs) initiated plasma thrombin generation, promoted fibrin formation, and increased fibrin network density and resistance to lysis. In contrast, PMPs did not support plasma thrombin generation or clotting in the absence of a procoagulant trigger (contact or TF). However, following initiation, PMPs significantly increased thrombin generation and the fibrin formation rate. These data support findings demonstrating MP-enriched plasma supports thrombin generation[1, 11], but extend these observations to show consequential, functional effects of MPs from specific parent cells on fibrin formation and stability.
Fueled by reports that MPs are smaller (<0.5 μm) than initially reported (≤1 μm)[25], use of flow cytometry to characterize and enumerate MPs is controversial; the refractive index of cellular vesicles appears lower than that of polystyrene beads used to gate MP populations by flow cytometry, leading to underestimation of size.[29] Our data are consistent with reports that NTA detects more MPs than flow cytometry[26] due to increased detection of small (<0.5 μm) particles. However, using NTA and TEM in addition to flow cytometry to assess physical characteristics of platelet and monocyte-derived MPs, our data show MPs from both cell types, regardless of the agonist used for their derivation, are ~150–300 nm in diameter. These findings support observations that MPs from LPS- and P-selectin-activated monocytes are similar in size[25], and extend the observations to include similarity with PMPs, suggesting cellular mechanisms producing MPs are common between cell types.[10] Nonetheless, MP characterization may be enhanced by continued refinement of methods that detect smaller vesicles.[29]
Whereas PMPs circulate in healthy individuals, M-MPs are present only in prothrombotic disease. Our findings provide a rationale for different roles of these MPs in hemostasis and thrombosis. Consistent with the role of activated platelets in coagulation propagation[30], PMPs augmented thrombin generation and fibrin formation only following a procoagulant trigger. Although it is well-accepted that such a trigger may arise in vivo from vascular injury that exposes subendothelial collagen or TF, the contact pathway contributes to thrombosis in murine models.[31, 32] Thus, PMPs may promote thrombus propagation following TF- or contact-initiated clotting in humans. In contrast to PMPs, M-MPs independently initiated thrombin generation and fibrin formation, and increased fibrin network density and stability, stemming from their ability to support both TF and prothrombinase activities. These findings are consistent with previous findings that leukocyte-derived MPs promote thrombogenesis, whereas PMPs are a marker of on-going thrombosis.[17] Together, these data suggest MPs derived from different parent cells uniquely contribute TF-dependent and-independent activities that promote thrombus formation and growth.
Given reports of platelet TF expression, it is notable that we did not detect TF activity in PMPs. Discrepancies between findings of TF mRNA processing and de novo protein synthesis in PAR- or A23187-stimulated platelets in some[33, 34] but not other[35, 36] studies have been potentially explained by the transient nature of TF expression following platelet activation[37]. Given the time required for MP production and isolation following platelet stimulation (~30 minutes in our study), it is unclear whether the lack of PMP TF reflects a lack of platelet TF expression, or that TF is not packaged into PMPs. Further studies are required to resolve this controversy. Similarly interesting was our finding that M-MPs and THP-MPs had greater prothrombinase activity than PMPs. Since NTA indicated not all MPs were detected by flow cytometry, the higher prothrombinase activity of M-MPs and THP-MPs could indicate higher numbers of small MPs were generated by monocytic cells and were therefore present in the prothrombinase assay. However, NTA and TEM indicated MPs generated by monocytes and platelets had similar physical characteristics. Although high lipid concentrations cause an inhibitory effect in prothrombinase assays, testing additional MP dilutions confirmed that MP concentrations used in these assays were below concentrations that caused this effect (data not shown). Tracy et al. previously observed 15-fold higher prothrombinase activity on monocytes than platelets.[38] Our findings support and extend this observation by demonstrating these differences are not due to different cellular surface area.
Our study has potential limitations. First, MP function was analyzed from isolated parent cells; MP produced in vivo may possess unique properties. However, our findings, as well as those of others[25] indicate similar physical and procoagulant properties of MPs isolated from given cell types, suggesting MPs have defined characteristics whether generated in vivo or in vitro. Second, while we focused on MPs from monocytes and platelets, MPs from other parent cells may possess other procoagulant or anticoagulant/profibrinolytic properties.[39] Since reports suggest circulating CD41-positive MPs derive from megakaryocytes[40], caution is advised when extrapolating our findings with PMPs to CD41-positive MPs in vivo. Third, we examined M-MP and PMP procoagulant activity; however, their impact depends on their ability to be recruited to and retained in, clots. Accumulation of TF-bearing MPs in thrombi but not hemostatic clots[41] suggests not all circulating MPs contribute procoagulant activity to all clots. Fourth, even by positive selection, we were unable to reduce the level of platelet contamination in M-MP preparations to less than 17%. Therefore, at least some procoagulant activity exhibited by M-MPs may derive from contaminating PMPs. However, we were able to recapitulate activities observed in M-MPs with THP-MPs, supporting our conclusion that monocyte-derived MPs exhibited both TF and prothrombinase activity in the assays. Finally, given 10–15% cell death that occurred during monocyte/THP-MPs generation, it is possible that a minor population of MPs were generated from dying cells. However, the recent study by Boles et al. (2011)[42] demonstrates apoptosis-derived THP-MPs have procoagulant properties (TF and PS activity) similar to those produced by LPS-stimulated cells, and therefore would be expected to exhibit similar behavior as LPS-derived MP in our thrombin generation and fibrin formation assays.
In conclusion, MPs from monocytes and platelets exhibit unique procoagulant activities; M-MPs initiated the extrinsic pathway, PMPs supported intrinsic pathway-dependent clotting. Both MP types contributed to propagation of clotting. These data imply a pathogenic role for M-MPs, and suggest PMPs exhibit prothrombotic activity once initiation has occurred. It will be important to compare the contributions of MP from different parent cells in in vivo thrombosis models.
Acknowledgments
Funding: This study was supported by research funding from the NIH (R01HL094740 to ASW and T32ES007017 to MMA). The Nanoparticle Tracking Analysis work was supported by the Wellcome Trust (GR087730).
Footnotes
Disclosure of Conflicts of Interest
No relevant conflicts of interest to disclose.
Authorship
M.M.A. performed experiments, analyzed data, and wrote the manuscript; C.G. performed experiments, analyzed data, and wrote the manuscript; P.H. analyzed data and wrote the manuscript; A.S.W. designed and supervised the study, analyzed data, and wrote the manuscript.
References
- 1.Berckmans RJ, Neiuwland R, Boing AN, Romijn FP, Hack CE, Sturk A. Cell-derived microparticles circulate in healthy humans and support low grade thrombin generation. Thromb Haemost. 2001;85:639–46. [PubMed] [Google Scholar]
- 2.Chirinos JA, Heresi GA, Velasquez H, Jy W, Jimenez JJ, Ahn E, Horstman LL, Soriano AO, Zambrano JP, Ahn YS. Elevation of endothelial microparticles, platelets, and leukocyte activation in patients with venous thromboembolism. J Am Coll Cardiol. 2005;45:1467–71. doi: 10.1016/j.jacc.2004.12.075. [DOI] [PubMed] [Google Scholar]
- 3.Preston RA, Jy W, Jimenez JJ, Mauro LM, Horstman LL, Valle M, Aime G, Ahn YS. Effects of severe hypertension on endothelial and platelet microparticles. Hypertension. 2003;41:211–7. doi: 10.1161/01.hyp.0000049760.15764.2d. [DOI] [PubMed] [Google Scholar]
- 4.Tripodi A, Branchi A, Chantarangkul V, Clerici M, Merati G, Artoni A, Mannucci PM. Hypercoagulability in patients with type 2 diabetes mellitus detected by a thrombin generation assay. J Thromb Thrombolysis. 2010 doi: 10.1007/s11239-010-0506-0. [DOI] [PubMed] [Google Scholar]
- 5.Hron G, Kollars M, Weber H, Sagaster V, Quehenberger P, Eichinger S, Kyrle PA, Weltermann A. Tissue factor-positive microparticles: cellular origin and association with coagulation activation in patients with colorectal cancer. Thromb Haemost. 2007;97:119–23. [PubMed] [Google Scholar]
- 6.Kanazawa S, Nomura S, Kuwana M, Muramatsu M, Yamaguchi K, Fukuhara S. Monocyte-derived microparticles may be a sign of vascular complication in patients with lung cancer. Lung Cancer. 2003;39:145–9. doi: 10.1016/s0169-5002(02)00441-5. [DOI] [PubMed] [Google Scholar]
- 7.Joop K, Berckmans RJ, Nieuwland R, Berkhout J, Romijn FP, Hack CE, Sturk A. Microparticles from patients with multiple organ dysfunction syndrome and sepsis support coagulation through multiple mechanisms. Thromb Haemost. 2001;85:810–20. [PubMed] [Google Scholar]
- 8.Tesselaar ME, Romijn FP, Van Der Linden IK, Prins FA, Bertina RM, Osanto S. Microparticle-associated tissue factor activity: a link between cancer and thrombosis? J Thromb Haemost. 2007;5:520–7. doi: 10.1111/j.1538-7836.2007.02369.x. [DOI] [PubMed] [Google Scholar]
- 9.Shet AS, Aras O, Gupta K, Hass MJ, Rausch DJ, Saba N, Koopmeiners L, Key NS, Hebbel RP. Sickle blood contains tissue factor-positive microparticles derived from endothelial cells and monocytes. Blood. 2003;102:2678–83. doi: 10.1182/blood-2003-03-0693. [DOI] [PubMed] [Google Scholar]
- 10.Morel O, Jesel L, Freyssinet JM, Toti F. Cellular mechanisms underlying the formation of circulating microparticles. Arterioscler Thromb Vasc Biol. 2011;31:15–26. doi: 10.1161/ATVBAHA.109.200956. [DOI] [PubMed] [Google Scholar]
- 11.Bidot L, Jy W, Bidot C, Jr, Jimenez JJ, Fontana V, Horstman LL, Ahn YS. Microparticle-mediated thrombin generation assay: increased activity in patients with recurrent thrombosis. J Thromb Haemost. 2008;6:913–9. doi: 10.1111/j.1538-7836.2008.02963.x. [DOI] [PubMed] [Google Scholar]
- 12.Bauer KA, Kass BL, ten Cate H, Bednarek MA, Hawiger JJ, Rosenberg RD. Detection of factor X activation in humans. Blood. 1989;74:2007–15. [PubMed] [Google Scholar]
- 13.Falati S, Liu Q, Gross P, Merrill-Skoloff G, Chou J, Vandendries E, Celi A, Croce K, Furie BC, Furie B. Accumulation of tissue factor into developing thrombi in vivo is dependent upon microparticle P-selectin glycoprotein ligand 1 and platelet P-selectin. J Exp Med. 2003;197:1585–98. doi: 10.1084/jem.20021868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gross PL, Furie BC, Merrill-Skoloff G, Chou J, Furie B. Leukocyte-versus microparticle-mediated tissue factor transfer during arteriolar thrombus development. J Leukoc Biol. 2005;78:1318–26. doi: 10.1189/jlb.0405193. [DOI] [PubMed] [Google Scholar]
- 15.Thomas GM, Panicot-Dubois L, Lacroix R, Dignat-George F, Lombardo D, Dubois C. Cancer cell-derived microparticles bearing P-selectin glycoprotein ligand 1 accelerate thrombus formation in vivo. J Exp Med. 2009;206:1913–27. doi: 10.1084/jem.20082297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Reinhardt C, von Bruhl ML, Manukyan D, Grahl L, Lorenz M, Altmann B, Dlugai S, Hess S, Konrad I, Orschiedt L, Mackman N, Ruddock L, Massberg S, Engelmann B. Protein disulfide isomerase acts as an injury response signal that enhances fibrin generation via tissue factor activation. J Clin Invest. 2008;118:1110–22. doi: 10.1172/JCI32376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ramacciotti E, Hawley AE, Farris DM, Ballard NE, Wrobleski SK, Myers DD, Jr, Henke PK, Wakefield TW. Leukocyte- and platelet-derived microparticles correlate with thrombus weight and tissue factor activity in an experimental mouse model of venous thrombosis. Thromb Haemost. 2009;101:748–54. [PMC free article] [PubMed] [Google Scholar]
- 18.Campbell RA, Overmyer KA, Bagnell CR, Wolberg AS. Cellular procoagulant activity dictates clot structure and stability as a function of distance from the cell surface. Arterioscler Thromb Vasc Biol. 2008;28:2247–54. doi: 10.1161/ATVBAHA.108.176008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Campbell RA, Overmyer KA, Selzman CH, Sheridan BC, Wolberg AS. Contributions of extravascular and intravascular cells to fibrin network formation, structure, and stability. Blood. 2009;114:4886–96. doi: 10.1182/blood-2009-06-228940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Carson SD, Ross SE, Bach R, Guha A. An inhibitory monoclonal antibody against human tissue factor. Blood. 1987;70:490–3. [PubMed] [Google Scholar]
- 21.Luddington R, Baglin T. Clinical measurement of thrombin generation by calibrated automated thrombography requires contact factor inhibition. J Thromb Haemost. 2004;2:1954–9. doi: 10.1111/j.1538-7836.2004.00964.x. [DOI] [PubMed] [Google Scholar]
- 22.Hasty DL, Meron-Sudai S, Cox KH, Nagorna T, Ruiz-Bustos E, Losi E, Courtney HS, Mahrous EA, Lee R, Ofek I. Monocyte and macrophage activation by lipoteichoic Acid is independent of alanine and is potentiated by hemoglobin. J Immunol. 2006;176:5567–76. doi: 10.4049/jimmunol.176.9.5567. [DOI] [PubMed] [Google Scholar]
- 23.Robert S, Poncelet P, Lacroix R, Arnaud L, Giraudo L, Hauchard A, Sampol J, Dignat-George F. Standardization of platelet-derived microparticle counting using calibrated beads and a Cytomics FC500 routine flow cytometer: a first step towards multicenter studies? J Thromb Haemost. 2009;7:190–7. doi: 10.1111/j.1538-7836.2008.03200.x. [DOI] [PubMed] [Google Scholar]
- 24.Wang JG, Manly D, Kirchhofer D, Pawlinski R, Mackman N. Levels of microparticle tissue factor activity correlate with coagulation activation in endotoxemic mice. J Thromb Haemost. 2009;7:1092–8. doi: 10.1111/j.1538-7836.2009.03448.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bernimoulin M, Waters EK, Foy M, Steele BM, Sullivan M, Falet H, Walsh MT, Barteneva N, Geng JG, Hartwig JH, Maguire PB, Wagner DD. Differential stimulation of monocytic cells results in distinct populations of microparticles. J Thromb Haemost. 2009;7:1019–28. doi: 10.1111/j.1538-7836.2009.03434.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dragovic RA, Gardiner C, Brooks AS, Tannetta DS, Ferguson DJP, Hole P, Carr B, Redman CWG, Harris AL, Dobson PJ, Harrison P, Sargent IL. Sizing and phenotyping of cellular vesicles using Nanoparticle Tracking Analysis. Nanomedicine. 2011 doi: 10.1016/j.nano.2011.04.003. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Orfeo T, Brummel-Ziedins KE, Gissel M, Butenas S, Mann KG. The nature of the stable blood clot procoagulant activities. J Biol Chem. 2008;283:9776–86. doi: 10.1074/jbc.M707435200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Collet JP, Montalescot G, Lesty C, Weisel JW. A structural and dynamic investigation of the facilitating effect of glycoprotein IIb/IIIa inhibitors in dissolving platelet-rich clots. Circ Res. 2002;90:428–34. doi: 10.1161/hh0402.105095. [DOI] [PubMed] [Google Scholar]
- 29.Chandler WL, Yeung W, Tait JF. A new microparticle size calibration standard for use in measuring smaller microparticles using a new flow cytometer. J Thromb Haemost. 2011;9:1216–24. doi: 10.1111/j.1538-7836.2011.04283.x. [DOI] [PubMed] [Google Scholar]
- 30.Hoffman M, Monroe DM., 3rd A cell-based model of hemostasis. Thromb Haemost. 2001;85:958–65. [PubMed] [Google Scholar]
- 31.Renne T, Pozgajova M, Gruner S, Schuh K, Pauer HU, Burfeind P, Gailani D, Nieswandt B. Defective thrombus formation in mice lacking coagulation factor XII. J Exp Med. 2005;202:271–81. doi: 10.1084/jem.20050664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang X, Smith PL, Hsu MY, Gailani D, Schumacher WA, Ogletree ML, Seiffert DA. Effects of factor XI deficiency on ferric chloride-induced vena cava thrombosis in mice. J Thromb Haemost. 2006;4:1982–8. doi: 10.1111/j.1538-7836.2006.02093.x. [DOI] [PubMed] [Google Scholar]
- 33.Camera M, Frigerio M, Toschi V, Brambilla M, Rossi F, Cottell DC, Maderna P, Parolari A, Bonzi R, De Vincenti O, Tremoli E. Platelet activation induces cell-surface immunoreactive tissue factor expression, which is modulated differently by antiplatelet drugs. Arterioscler Thromb Vasc Biol. 2003;23:1690–6. doi: 10.1161/01.ATV.0000085629.23209.AA. [DOI] [PubMed] [Google Scholar]
- 34.Schwertz H, Tolley ND, Foulks JM, Denis MM, Risenmay BW, Buerke M, Tilley RE, Rondina MT, Harris EM, Kraiss LW, Mackman N, Zimmerman GA, Weyrich AS. Signal-dependent splicing of tissue factor pre-mRNA modulates the thrombogenicity of human platelets. J Exp Med. 2006;203:2433–40. doi: 10.1084/jem.20061302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bouchard BA, Mann KG, Butenas S. No evidence for tissue factor on platelets. Blood. 2010;116:854–5. doi: 10.1182/blood-2010-05-285627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Osterud B, Olsen JO, Bjorklid E. What is blood borne tissue factor? Thromb Res. 2009;124:640–1. doi: 10.1016/j.thromres.2009.06.027. [DOI] [PubMed] [Google Scholar]
- 37.Camera M, Brambilla M, Toschi V, Tremoli E. Tissue factor expression on platelets is a dynamic event. Blood. 2010;116:5076–7. doi: 10.1182/blood-2010-09-307306. [DOI] [PubMed] [Google Scholar]
- 38.Tracy PB, Rohrbach MS, Mann KG. Functional prothrombinase complex assembly on isolated monocytes and lymphocytes. J Biol Chem. 1983;258:7264–7. [PubMed] [Google Scholar]
- 39.Lacroix R, Sabatier F, Mialhe A, Basire A, Pannell R, Borghi H, Robert S, Lamy E, Plawinski L, Camoin-Jau L, Gurewich V, Angles-Cano E, Dignat-George F. Activation of plasminogen into plasmin at the surface of endothelial microparticles: a mechanism that modulates angiogenic properties of endothelial progenitor cells in vitro. Blood. 2007;110:2432–9. doi: 10.1182/blood-2007-02-069997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Flaumenhaft R, Dilks JR, Richardson J, Alden E, Patel-Hett SR, Battinelli E, Klement GL, Sola-Visner M, Italiano JE., Jr Megakaryocyte-derived microparticles: direct visualization and distinction from platelet-derived microparticles. Blood. 2009;113:1112–21. doi: 10.1182/blood-2008-06-163832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hoffman M, Whinna HC, Monroe DM. Circulating tissue factor accumulates in thrombi, but not in hemostatic plugs. J Thromb Haemost. 2006;4:2092–3. doi: 10.1111/j.1538-7836.2006.02085.x. [DOI] [PubMed] [Google Scholar]
- 42.Boles JC, Williams JC, Hollingsworth RM, Wang JG, Glover SL, Owens AP, 3rd, Barcel DA, Kasthuri RS, Key NS, Mackman N. Anthracycline treatment of the human monocytic leukemia cell line THP-1 increases phosphatidylserine exposure and tissue factor activity. Thromb Res. 2011 doi: 10.1016/j.thromres.2011.06.022. In Press. [DOI] [PubMed] [Google Scholar]