

Abstract
Experimental allergic encephalomyelitis (EAE) is an animal model of multiple sclerosis (MS), the most common human demyelinating disease of the central nervous system. Sodium benzoate (NaB), a metabolite of cinnamon and a FDA-approved drug against urea cycle disorders in children, is a widely used food additive, which is long known for its microbicidal effect. However, recent studies reveal that apart from its microbicidal effects, NaB can also regulate many immune signaling pathways responsible for inflammation, glial cell activation, switching of T-helper cells, modulation of regulatory T cells, cell-to-cell contact, and migration. As a result, NaB alters the neuroimmunology of EAE and ameliorates the disease process of EAE. In this review, we have made an honest attempt to analyze these newly-discovered immunomodulatory activities of NaB and associated mechanisms that may help in considering this drug for various inflammatory human disorders including MS as primary or adjunct therapy.
Keywords: Sodium benzoate, EAE, innate immunity, adaptive immunity, neuroinflammation, glial activation, signal transduction
Introduction
Multiple sclerosis (MS) is a T cell-mediated autoimmune disease affecting the central nervous system (CNS), and experimental allergic encephalomyelitis (EAE) is an animal model of MS.(1–3) EAE is actively induced by the injection of whole spinal cord preparations or proteins derived from myelin (commonly myelin basic protein (MBP) or proteolipid protein (PLP)).(4–7) Activated T cells isolated from the spleen and lymph nodes of actively immunized animals can also be used to transfer disease to naive recipients. In both models, neuroantigen-specific autoimmune T cells first contact a naive intact BBB and are able to extravasate through the BBB due to their activated status. These cells are retained in the CNS due to presentation of appropriate antigen and undergo further activation.(8,9) This is followed by the recruitment of non-antigen-specific lymphocytes and activated macrophages from the blood into this site, accompanied by the activation of resident glial cells and further disruption of the BBB.
Although there is no effective drug against MS, studies on neuroimmunological regulations of EAE have led to the discovery of many pharmacological interventions in MS that include interferons, copaxone, tysabri, statins, glucocorticoids, mitoxantrone, cyclophosphamide, etc.(6) Sodium benzoate (NaB), a metabolite of cinnamon, is a commonly used food additive and an FDA-approved therapy for reducing plasma ammonia and glutamine in urea cycle disorders. Recent discoveries suggest that NaB is an important modulator of adaptive and innate immune responses of EAE leading to attenuation of inflammation and demyelination in EAE.
Sodium benzoate (NaB)
Cinnamon is a commonly used spice and flavoring material for desert, candies, chocolate etc. In addition to containing manganese, dietary fiber, iron, and calcium, cinnamon contains three major compounds—cinnam-aldehyde, cinnamyl acetate and cinnamyl alcohol. After intake, these three active compounds are converted into cinnamic acid by oxidation and hydrolysis, respectively. Then cinnamic acid is β-oxidized to benzoate in the liver that exists as sodium salt (NaB) or benzoyl-CoA (Figure 1). NaB is a component of Ucephan, a FDA-approved drug used in the treatment for hepatic metabolic defects associated with hyperammonemia such as urea cycle disorders involving deficiencies of carbamoyl phosphate synthetase, ornithine transcarbamylase, or argininosuccinic acid synthetase.(10,11) It is also widely used as a preservative in broad range of foods and cosmetic products.(12) It is nontoxic and can be administered as a solution in drinking water. It has been reported that 2% solution of NaB in drinking water is safe for lifelong treatment in mice without any noticeable side effects.(13) In accordance with the published reports,(11,12) minor amount of benzoic acid, a direct metabolite of cinnamic acid,(13) is also excreted in the urine of human.
Figure 1.

Regulation of p21ras and associated signaling events by cinnamon metabolite NaB. Different components of cinnamon are oxidized to cinnamon acid, which is further β-oxidized to benzoyl-CoA. This benzoyl-CoA may compete with acetyl-CoA, acetoacetyl-CoA and HMG-CoA for thiolase (1), HMG-CoA synthetase (2) and HMG-CoA reductase (3), respectively. Inhibition of these biochemical steps leads to decreased synthesis of farnesyl pyrophosphate and inhibition of p21ras farnesylation, resulting in decreased activation of NF-κB and attenuation of proinflammatory gene transcription. (See colour version of this figure online at www.informahealthcare.com/ipi)
Modulation of adaptive immunity by NaB
Although not mutually exclusive, the immune system is broadly divided into two categories—adaptive and innate. During an immune attack, adaptive immunity utilizes the learning immunity to recognize a pathogen’s unique antigen to build antigen-specific immune response for its neutralization. Recent studies suggest that NaB is capable of modulating this learning immunity, which we delineate in following sections.
Th1-Th2 switching: turning on anti-inflammatory T cells
It is known that MS is a Th1-mediated autoimmune disease where an autoreactive and inflammatory population of Th1 cells participates in the demyelination of CNS white matter.(6,14–16) Accordingly, Th1 cells produce proinflammatory cytokines like IFN-γ, IL-2, TNF-α etc., which ultimately aggravate the disease process. There is another subtype of helper T cells in MS patients, known as Th2 that is less in number and anti-inflammatory in nature because they secrete anti-inflammatory cytokines like IL-4, IL-10 and IL-13.(14–17) The stimulation of Th2 cell-driven response and suppression of Th1 cell-mediated response at the same time has been confirmed to ameliorate the symptoms of EAE and MS. We have demonstrated that NaB efficiently inhibits the production of IFN-γ while stimulating the release of IL-4 and IL-10 from MBP-immunized splenocytes.(18) However, under similar condition, sodium formate (NaFO), having a similar structure but without the benzene ring, does not exhibit any effect on either Th1 or Th2 cytokines, suggesting the specificity of the effect. Therefore, NaB is capable of switching off the differentiation of Th1 cells and turning on the differentiation of Th2 cells (Figure 2).
Figure 2.
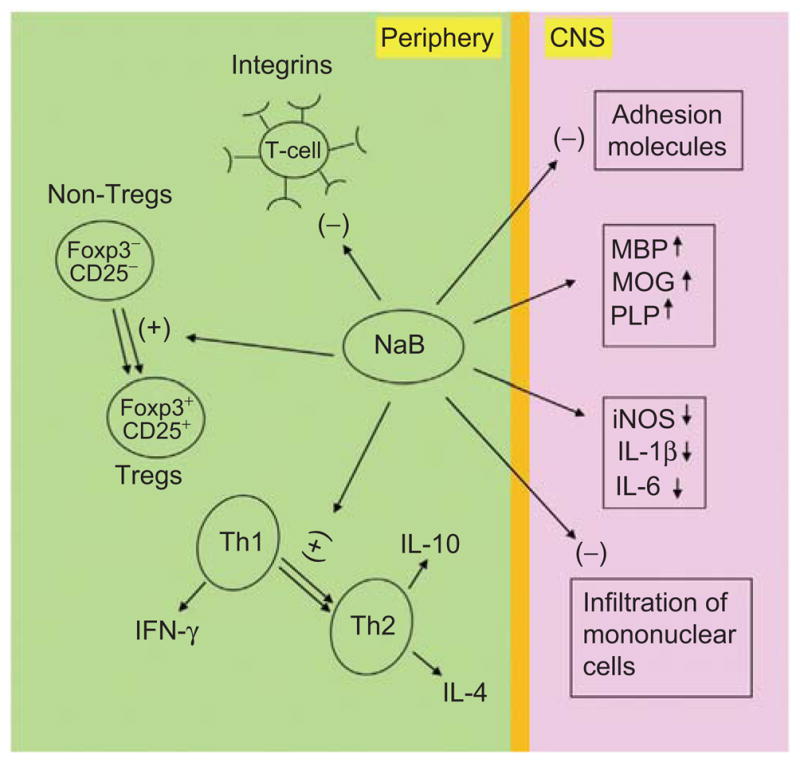
Alteration of EAE-related multiple immune responses by NaB. During NaB treatment of EAE mice, NaB inhibits several integrins on T cells, enriches Tregs and causes Th1 to Th2 shift of the adaptive T cells response in the periphery. On the other hand, NaB treatment leads to suppression of adhesion molecules, attenuation of mononuclear cell infiltration, decrease in proinflammatory molecules, and normalization and/or up-regulation of myelin genes in the CNS compartment. (See colour version of this figure online at www.informahealthcare.com/ipi)
While mechanisms underlying Th1-Th2 switching are poorly understood, earlier we have demonstrated that NO plays a critical role in Th1 to Th2 switching.(17) Interestingly, scavenging of NO favored the induction of GATA3 expression and the production of IL-10 from MBP-primed splenocytes.(17) On the other hand, excess NO stimulated the expression of T-bet and the release of IFN-γ.(17) Because NaB is capable of attenuating the induction of NO production,(19) it is possible that NaB switches the differentiation from Th1 to Th2 via reducing the level of NO.
Enriching regulatory T cells (Tregs): a sword against autoimmunity
Although autoreactive lymphocytes are normally deleted in the thymus, yet some self-reactive T cells are found in the periphery of normal healthy individuals.(20) However, these T cells usually do not attack organs that produce self-antigens. Therefore, a state of self-tolerance is maintained in normal individual. Recently, this has been shown to be mediated by regulatory T cells (Tregs) that are characterized by the expression of Foxp3, a transcription factor, and CD25 (IL-2 receptor α-chain). Although there are many types of Tregs, CD4+CD25+Foxp3+ Tregs are known to be the key regulatory cell type that prevents activation and differentiation of non-Tregs including autoreactive T cells.(21,22) In autoimmune diseases, autoreactive T cells somehow overcome the resistance provided by the Tregs and therefore undergo activation and proliferation. Accordingly, we have demonstrated that after MBP-priming, the expression of Foxp3 and CD25 goes down leading to the suppression of Tregs.(23) Interestingly, NaB treatment significantly up-regulates the expression Foxp3 and CD25 and enriches the level of CD4+CD25+ Tregs in MBP-primed splenocytes.(23) Similarly, treatment of donor mice with NaB during MBP-priming reveals significant up-regulation of both Foxp3 and CD25 in vivo in the spleen.(18) However, NaB remains unable to change the level of CD4 in donor spleen. Stimulation of Foxp3 and CD25 by NaB strongly suggest that this compound may exhibit its protective action in EAE by enriching CD4+CD25+ Tregs.(18,23) Therefore, NaB is capable of increasing the abundance of Tregs in cultured splenocytes and in vivo in donor mice.
Mechanisms by which Tregs are regulated are poorly understood. Because immune stimulation is always associated with the generation of NO, recently we examined the role of NO in the modulation of Foxp3 and Tregs.(23,24) We have described that NO plays a pivotal role in the regulation of Foxp3 in MBP-primed T cells. Although decreasing the level of NO by inhibitors of inducible NO synthase (iNOS), knockdown of the iNOS gene or scavengers of NO stimulates the expression of Foxp3, increasing the level of NO by an NO donor decreases the expression of Foxp3. In addition to MBP-primed T cells, T cell priming with myelin oligodendrocyte glycoprotein (MOG), another target neuroantigen in MS, as well as collagen, a target autoantigen in rheumatoid arthritis (RA), also reduces the expression of Foxp3 via NO.(23) Accordingly, the prevention of Foxp3 and CD25 expression by pharmacological compounds is abrogated by GSNO, an NO donor, suggesting that NaB also restores Foxp3 in MBP-primed T cells via inhibiting synthesis of NO.(23)
Modulation of ligands on T cell surface: altering T cell infiltration
T cell trafficking into the CNS is an important event in the pathogenesis of MS and EAE. It is believed that disease-causing autoreactive T cells interact with the BBB endothelium prior to their entry into the CNS parenchyma.(14,25) It has been found that integrins like α4β1 (very late antigen-4 (VLA-4)) and αLβ2 (lymphocyte function-associated antigen-1 (LFA-1)) on T cells bind to counter-receptors on the BBB endothelial cells, which probably facilitates the extravasation of lymphocytes.(25–27) Furthermore, these integrins also play an active role in T cell—APC interaction and provides co-stimulatory signals for T cell receptor (TCR)-induced T cell activation. Accordingly, the level of these integrins increases on autoreactive T cells.(25,26,28) However, NaB treatment markedly inhibits the expression of α4, β1, αL, and β2 in neuroantigen-primed T cells.(18) On the other hand, NaFO, an inactive analog of NaB, does not modulate any of these integrins in T cells suggesting the specificity of NaB effect. Spleen is the major organ responding to various immune responses. Accordingly, spleens of MBP-immunized donor mice have markedly elevated level of α4, β1, αL, and β2 as compared to normal mice. However, NaB treatment of mice via drinking water results in attenuation of α4, β1, αL, and β2 in spleens of donor mice suggesting that NaB is capable of suppressing VLA-4 and LFA-1 integrins in cultured T cells as well as in vivo in spleen.(18)
Modulation of innate immunity by NaB
The innate immunity uses the inborn memory of germline-encoded proteins to read through the molecular patterns of common pathogens or insults. This is the immunity one is born with and is the initial response by the body to eliminate microbes and prevent infection. In following lines, we describe how NaB modulates such immune response.
Knocking down proinflammatory molecules in microglia
Microglia are considered as CNS-resident professional macrophages and sensor cells that function as the principal immune effector cells of the CNS responding to any pathological event.(29) It has been found that activated microglia accumulate at and around plaques in the CNS of MS patients.(29) Although proinflammatory molecules released from microglia may help in the elimination of microbes from the CNS, excessive proinflammatory molecules attack host cells. Therefore, once microglia are activated in neurodegenerating microenvironment, it always goes beyond control and eventually detrimental effects override beneficial effects. Accordingly, wide variety of proinflammatory molecules, such as proinflammatory cytokines (IL-1β, IFN-γ, IL-6, and TNF-α), proinflammatory chemokines (MCP-1/CCL2, IP-10/CXCL10 and IL-8), proinflammatory enzymes (inducible nitric oxide synthase, cyclooxygenase-2) and proinflammatory transcription factors (NF-κB, C/EBP) have been detected in microglia in CNS lesions of MS patients.(1,2,29,30)
NaB has been found to inhibit LPS-induced production of NO and the expression of iNOS in both CNS microglia and peritoneal macrophages.(19) Similarly, NaB also attenuates the expression of proinflammatory cytokines (TNF-α and IL-1β) in microglia. Although LPS requires TLR4 to transduce its signal and subsequently to induce iNOS, NaB has no effect on the expression of TLR4 in LPS-treated microglia, suggesting that NaB inhibits LPS-induced expression of iNOS without modulating its receptor TLR4.(19) Similarly, etiological reagents of various neurological disorders such as fibrillar Aβ peptides (etiological reagent for Alzheimer’s disease), fibrillar PrP peptides (etiological reagent for prion diseases), dsRNA in the form of poly IC (one of the etiological reagents for viral encephalopathy), HIV-1 gp120 (one of the etiological reagents for HIV-associated dementia), IL-12 p402 (one of the etiological reagent for MS), and MPP+ (a Parkinsonian toxin) also induce the expression of iNOS in microglia via NaB-sensitive pathway.(19)
Mechanisms for NaB-mediated inhibition of microglial proinflammatory molecules
Microglial proinflammatory cytokines are up-regulated in response to a wide range of inducers. Broadly, these inducers can be categorized by their ability to either elicit innate or adaptive immune responses. The first group consists of products of bacterial and viral origin, while the second group is primarily composed of proinflammatory cytokines. Signaling mechanisms by which proinflammatory molecules are induced in microglia by both group of inducers are becoming clear. In following paragraphs, we delineate how NaB modulates such signaling processes to contain proinflammatory molecules in microglia.
Role of mevalonate pathway
Isoprenoids are a major class of nonsaponifiable lipids which is polymeric derivative of C5 compound isopentenyl pyrophosphate (IPP). Isoprenoids like farnesyl- and geranylgeranyl-pyrophosphate, are biosynthesized in animals from acetyl-CoA via the mevalonate pathway, as is depicted in Figure 1. These isoprenoids covalently modify and thus modulate the biological activity of important signal transduction components like small G proteins Ras, Rac and Rho.(31) Upon isoprenylation, these G proteins become membrane-bound and transduce several intracellular signaling pathways that lead to the expression of proinflammatory molecules like iNOS, TNF-α, IL-1β, IL-6 etc. The experimental evidence for this statement has been provided by a pioneering study by Pahan et al.,(32) where lovastatin, a blocker of the mevalonate pathway and also a FDA-approved drug for hypercholesterolemia, inhibits the expression of iNOS and proinflammatory cytokines in activated glial cells.
The requirement of at least 6h of preincubation of cells with NaB to see its anti-inflammatory effect suggests that metabolite(s) sensitive to NaB may be involved in the process.(19) Accordingly, it has been found that intermediates of the mevalonate pathway are capable of reversing the anti-inflammatory effect of NaB. For example, HMG-CoA, mevalonate and farnesyl pyrophosphate abrogate the inhibitory effect of NaB on the expression of iNOS in microglial cells.(19) End product of the mevalonate pathway is cholesterol. It is interesting because NaB alone is capable of reducing the level of cholesterol in serum of mice at a level that is comparable to that of pravastatin-treated mice suggesting that NaB may be considered for cholesterol lowering. However, cholesterol and coenzyme Q has no effect on NaB-mediated inhibition of iNOS in microglia. These results suggest that depletion of intermediary products rather than end products of the mevalonate pathway is responsible for the observed anti-inflammatory effect of NaB.(19)
Suppression of p21ras activation
Suppression of LPS-induced activation of NF-κB and expression of iNOS in rat astrocytes by farnesyltransferase inhibitor(32) suggests an important role of farnesylation reaction in the regulation of iNOS gene. Consistent to a role of farnesylation in the activation of p21ras, a dominant-negative mutant of p21ras (S17N) also attenuates the expression of iNOS in rat and human primary astrocytes.(33) Accordingly, expression of a constitutively active mutant of p21ras (RasV12) alone induces the production of NO and the expression of iNOS in microglial cells(19) suggesting that signal(s) provided by the activation of p21ras alone is sufficient to induce the expression of iNOS. Therefore, there is evidence to suggest an important role of p21ras signaling in the expression of iNOS in glial cells. Consistent to the reversal of NaB-mediated inhibition of iNOS in microglia by intermediates of the mevalonate pathway, NaB suppresses the activation of p21ras in microglial cells suggesting that NaB attenuates the expression of proinflammatory molecules in glial cells probably by suppressing the activation of p21ras.
Inhibition of proinflammatory transcription factor NF-κB
The signaling events required for the transcription of iNOS and proinflammatory cytokines are becoming clear. Although many transcription factors such as NF-κB, C/EBPβ, AP-1, STAT, IRF-1 etc. play a role in the expression of various proinflammatory molecules, activation of NF-κB seems essential for the transcription of most of the proinflammatory molecules.(19,31,32,34) Therefore, for a drug to exhibit anti-inflammatory effect, it is almost mandatory to attenuate the activation of NF-κB. Although NaB does not abrogate NF-κB activation completely, NaB markedly inhibits the activation of NF-κB in microglia.(19) It has been found that NaB suppresses the activation of p21ras and thereby inhibits the activation of NF-κB in microglia. This conclusion is based on the following observations. First, FPT inhibitor II, capable of inhibiting farnesylation of p21ras, inhibits the activation of NF-κB. Second, NaB, but not NaFO, attenuates the activation of p21ras in LPS-stimulated microglia. Third, activation of p21ras alone was sufficient for the activation of NF-κB in microglial cells.(19)
Inhibition of NF-κB activation by NaB suggests that NaB exhibits anti-inflammatory efficacy via NF-κB inhibition. To further confirm this, the effect of NaB on the expression of IFNγ-induced expression of iNOS is also investigated in microglial cells because IFN-γ induces the expression of iNOS via STATs, but not NF-κB. Although NaB inhibits the expression of iNOS in microglia stimulated by various proinflammatory insults, NaB does not exhibit any inhibitory effect on IFNγ-induced expression of iNOS in microglial cells suggesting that NaB is only able to inhibit iNOS if it is mediated by the activation of NF-κB.(19)
Attenuation of microglial surface markers and co-stimulatory molecules
Microglial activation is a hallmark of various neurodegenerative disorders and during severe activation, microglia not only secretes various neurotoxic molecules but also expresses different proteins and surface markers.(35,36) Among different surface markers, CD11b is the most potential one with immense biological significance. It acts as a binding protein for intracellular cell adhesion molecule-1 and complement C3bi.(35,36) It is reported that in various neuroinflammatory diseases, the increased CD11b expression corresponds to the severity of microglial activation.(35–37) Morphologically, microglial activation is associated with intense ramification and cytoskeletal rearrangement in which changes in shape and motility correlate with increased expression of CD11b. During this activation process, the cytoplasmic domain of CD11b is believed to interact increasingly with cytoskeletal proteins.(36) As expected, LPS increased the expression of CD11b, CD11c and CD68 in microglial cells. However, it has been found that NaB abrogates increased expression of CD11b, CD11c and CD68 in LPS-activated microglia.(19) MPP+ (a Parkinsonian neurotoxin) also stimulate CD11b in microglia via NaB-sensitive pathways.(19)
How might NaB suppress increased expression of CD11b in microglia? We have found that NO plays a critical role in the up-regulation of CD11b. For example, either a scavenger of NO (PTIO) or an inhibitor of inducible nitric oxide synthase (L-NIL) blocks the increased expression of CD11b in activated microglia.(37) On the other hand, GSNO (a NO donor) alone increases the level of CD11b in microglia.(37) Because NaB is capable of suppressing the expression of iNOS and the production of NO in microglia,(37) it is possible that NaB decreases the levels of microglial surface markers via attenuation of NO production. Activated microglia also express class-II MHC and co-stimulatory molecules like B7-1 and B7-2, which are critical for antigen presentation. NaB also knocks down the increased the expression of class-II MHC and the co-stimulatory molecules B7-1 and B7-2 in activated microglia.(19) Taken together, NaB is capable of attenuating microglial surface markers and co-stimulatory molecules in activated microglia.
Modulation of microglial adhesion molecules
Adhesion molecules are crucial for cell-to-cell contact, attachment and cell migration.(38) For example, during lymphocyte migration into the CNS, the initial contact between a lymphocyte and an endothelial cell is referred to as “tethering”, while the immediate subsequent interaction is described as “rolling”.(39) These steps, which allow the lymphocyte to slow down and roll along the vascular wall, are mediated predominantly by selectins (E-selectin and P-selectin). Importance of E- and P-selectins in the extravasation of T lymphocytes into the CNS has been documented when complete inhibition of rolling and arrest of Th1 cells in inflamed brain is observed with anti-E- and P-selectin antibodies.(40) Interestingly, NaB treatment via drinking water strongly inhibits the expression of E- and P-selectins in the cerebellum as well as in the spinal cord of EAE mice.(18)
In addition to E-selectin and P-selectin, adhesion molecules like ICAM-1 and VCAM-1 expressing in the endothelium of BBB as well as in glial cells in CNS parenchyma play an important role in the infiltration of T cells.(38–40) There are various ligands for these adhesion molecules and interestingly, T cells express these ligands. For example, VLA-4 and LFA-1 (ligands) on T cell surface interact directly with endothelial or glial VCAM-1 and ICAM-1, respectively. Accordingly, up-regulation of ICAM-1 and VCAM-1 has been reported on cerebral vessels during EAE preceding the perivascular infiltration by lymphocytes and the onset of disease.(40) Interestingly, NaB treatment effectively inhibits the expression of these adhesion molecules in both cerebellum and spinal cord of EAE mice.(18) Consistent to the suppression of various contact molecules, NaB treatment also inhibits the infiltration of inflammatory cells into the CNS of EAE mice.(18)
Controlling astroglial activation
Astrocytes are the major glial cells present in CNS. In counting, they greatly outnumber neurons, microglia or oligodendrocytes. More often than not, the dogma has held these cells as mere bystanders during brain pathophysiology. However, as suggested by growing number of literatures, astrocytes form the bulk of all resident brain cells with an ability to initiate and amplify immune response.(41,42) Furthermore, according to several lines of evidences, astrocytes express various proinflammatory molecules and all three isoforms of NOS,(42,43) including iNOS that is expressed prolifically by these cells in response to various stimuli. Therefore, it is also important to understand mechanisms by which astroglia are activated and to delineate ways to attenuate astroglial activation. It has been shown that NaB inhibits IL-1β-induced production of NO and the expression of iNOS protein in primary human astroglia.(19) Inhibition of iNOS promoter-driven luciferase activity in IL-1β-stimulated astroglia by NaB suggests that NaB inhibits the production of NO and the expression of iNOS by inhibiting the activation of iNOS promoter.(19)
Increased expression of glial fibrillary acidic protein (GFAP) represents astroglial activation and gliosis during neurodegeneration. Senile plaques, a pathologic hallmark of Alzheimer’s disease, are associated with GFAP-positive activated astrocytes.(44) It is reported that in various neuroinflammatory diseases, the increased GFAP expression corresponds to the severity of astroglial activation.(41) Consistent to the inhibition of iNOS, NaB also decreases IL-1β-induced expression of GFAP in human astroglia(19) suggesting that NaB is capable of attenuating astroglial activation and astrogliosis. Although mechanism by which astroglial expression of GFAP is increased in neurodegenerative CNS remains unclear, recently we have delineated that NO is an important regulator GFAP in which NO uses the guanylate cyclase (GC)–cGMP–cGMP-activated protein kinase (PKG) signaling pathway, but not NF-κB, to induce the expression of GFAP in astrocytes.(45) Because NaB decreases NO production, it is possible that NaB suppresses astroglial GFAP induction via reduction of NO.
NaB in EAE
EAE is particularly important for testing different therapeutic approaches against MS and adoptively-transferred EAE mimics the relapsing-remitting MS, the most common form of MS found in patients. Because NaB is highly water soluble, mice with relapsing-remitting EAE (RR-EAE) and acute EAE were allowed to drink NaB-containing water. According to Brahmachari and Pahan,(18) NaB markedly inhibits the clinical symptoms and disease severity of RR-EAE and acute EAE in mice. On the other hand, NaFO is unable to inhibit the clinical symptoms of EAE suggesting the specificity of the effect.(18)
Suppression of disease progression in mice with RR-EAE
Although MS could be monophasic, majority of MS patients (>80%) experience relapsing-remitting symptoms. Relapsing-remitting MS (RR-MS) is characterized by relapses when new symptoms may appear and old ones resurface or worsen. The relapses are followed by periods of remission, during which the person recovers either fully or partially from the deficits acquired during the relapse. These characteristics are modeled in adoptively-transferred RR-EAE mice in which the acute phase is followed by several relapses that are separated by periods of remission. When mice with established RR-EAE receive NaB via drinking water from the onset of acute phase, NaB exhibits inhibitory effect on the clinical symptoms within 4 days of treatment followed by marked inhibition on subsequent days of treatment that is maintained throughout the duration of the experiment.(18) Similarly when NaB treatment began from the onset of relapsing phase, NaB also halts disease progression displaying its inhibitory effect within only 5 days of treatment. It is believed that infiltration of blood mononuclear cells and associated neuroinflammation are critical for CNS demyelination observed in MS patients and EAE animals. However, NaB treatment of established RR-EAE mice remarkably inhibits infiltration of mononuclear cells and associated inflammation and restores myelin level in the CNS of RR-EAE mice.(18)
NaB and encephalitogenicity of T cells
Neuroantigen-specific T cells play an important role in the disease process of MS and EAE. After transfer, these cells first contact a naive intact BBB and are able to extravasate through the BBB due to their activated status. Therefore, these neuroantigen-specific T cells are called encephalitogenic T cells because in susceptible animals, these cells alone can cause EAE. Blocking the mechanisms by which neuroantigen-specific T cells are activated before adoptive transfer may help characterize the immune arm of MS and offer novel targets for therapy. Interestingly, incubation of MBP-specific T cells with NaB, but not NaFO, before adoptive transfer markedly suppressed the ability of these T cells to induce clinical symptoms of RR-EAE suggesting that NaB inhibits the encephalitogenicity of T cells.(18) Similarly, NaB treatment in donor mice also inhibits the generation of encephalitogenic T cells in vivo. For example, mice that receive MBP-primed T cells isolated from NaB-treated donor mice exhibit dramatically less clinical symptoms and disease severity compared to the control EAE group receiving untreated MBP-primed T cells.(18) Taken together, NaB has the capability of suppressing the EAE-inducing activity of T cells.
Conclusion
Unraveling the mechanisms involved in normalizing aberrant immune responses and rescuing and sustenance of oligodendrocytes and myelin in MS patients(46) is of high priority for developing effective therapies. Here we discuss how NaB, a commonly used food additive, a metabolite of cinnamon and a FDA-approved drug for urea cycle disorders, suppresses inflammation, switches the differentiation of Th cells, enriches Treg population (Figure 2), and blocks the disease process of EAE, ultimately suggesting that this drug may be used for therapeutic intervention in MS. Although there is no effective therapy against MS, different forms of interferon-β (IFN-β) have been used to treat this disease. However, NaB has several advantages over IFN-β. First, IFN-β has a number of side effects including flu-like symptoms, menstrual disorders in women, decrease in neutrophil count and white blood cell count, increase in AST and ALT levels, and development of neutralizing antibodies to IFN-β(47) However, NaB is fairly nontoxic and a widely used food preservative having a long clinical history as a treatment for conditions associated with hyperammonemia such as urea cycle disorders in children. In addition, it scavenges glycine. NaB reacts with glycine to form hippurate, which is excreted through the urine. It has been reported that the level of glycine is increased in vivo in the CNS and plasma of MS patients.(48) Therefore, this glycine-scavenging property of NaB could be a blessing for MS patients because glycine is mainly responsible for inhibiting motor neurons. When impaired, glycinergic inhibition leads to spastic and hypertonic disorders such as featured in MS. Second, MS patients are treated with IFN-β through painful injections which often lead to injection site reactions such as skin necrosis. However, NaB can be taken through food and drinking water or milk, the least painful route. Third, IFN-β is costly, whereas NaB is very economical. Fourth, IFN-β should not cross the intact blood-brain barrier (BBB). However, NaB is an amphipathic molecule and in hyperammonemia, this drug is believed to scavenge excessive glycine. After treatment of patients with urea cycle disorders, NaB combines with glycine to produce hippurate, a compound that is readily excreted in the urine. Simultaneous serum and CSF sampling in those patients showed comparable levels of NaB and hippurate in the CSF,(49–51) suggesting that NaB is capable of crossing the BBB. Therefore, NaB may function at multiple steps in both CNS and periphery (Figure 2).
Footnotes
Declaration of interest
This study was supported by grants from NIH (AT6681 and NS39940).
References
- 1.Parkinson JF, Mitrovic B, Merrill JE. The role of nitric oxide in multiple sclerosis. J Mol Med. 1997;75:174–186. doi: 10.1007/s001090050102. [DOI] [PubMed] [Google Scholar]
- 2.Brück W, Stadelmann C. Inflammation and degeneration in multiple sclerosis. Neurol Sci. 2003;24 (Suppl 5):S265–S267. doi: 10.1007/s10072-003-0170-7. [DOI] [PubMed] [Google Scholar]
- 3.Martin R, McFarland HF, McFarlin DE. Immunological aspects of demyelinating diseases. Annu Rev Immunol. 1992;10:153–187. doi: 10.1146/annurev.iy.10.040192.001101. [DOI] [PubMed] [Google Scholar]
- 4.Swanborg RH. Experimental autoimmune encephalomyelitis in rodents as a model for human demyelinating disease. Clin Immunol Immunopathol. 1995;77:4–13. doi: 10.1016/0090-1229(95)90130-2. [DOI] [PubMed] [Google Scholar]
- 5.Tuohy VK, Sobel RA, Lees MB. Myelin proteolipid protein-induced experimental allergic encephalomyelitis. Variations of disease expression in different strains of mice. J Immunol. 1988;140:1868–1873. [PubMed] [Google Scholar]
- 6.Pahan K. Neuroimmune pharmacological control of EAE. J Neuroimmune Pharmacol. 2010;5:165–167. doi: 10.1007/s11481-010-9219-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Libbey JE, Tsunoda I, Fujinami RS. Studies in the modulation of experimental autoimmune encephalomyelitis. J Neuroimmune Pharmacol. 2010;5:168–175. doi: 10.1007/s11481-010-9215-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sabatino JJ, Jr, Rosenthal KM, Evavold BD. Manipulating antigenic ligand strength to selectively target myelin-reactive CD4+ T cells in EAE. J Neuroimmune Pharmacol. 2010;5:176–188. doi: 10.1007/s11481-009-9181-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Becher B, Bechmann I, Greter M. Antigen presentation in autoimmunity and CNS inflammation: how T lymphocytes recognize the brain. J Mol Med. 2006;84:532–543. doi: 10.1007/s00109-006-0065-1. [DOI] [PubMed] [Google Scholar]
- 10.Toth B. Lack of tumorigenicity of sodium benzoate in mice. Fundam Appl Toxicol. 1984;4:494–496. doi: 10.1016/0272-0590(84)90208-2. [DOI] [PubMed] [Google Scholar]
- 11.Bridges JW, French MR, Smith RL, Williams RT. The fate of benzoic acid in various species. Biochem J. 1970;118:47–51. doi: 10.1042/bj1180047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kubota K, Ishizaki T. Dose-dependent pharmacokinetics of benzoic acid following oral administration of sodium benzoate to humans. Eur J Clin Pharmacol. 1991;41:363–368. doi: 10.1007/BF00314969. [DOI] [PubMed] [Google Scholar]
- 13.Abd El-Mawla AM, Schmidt W, Beerhues L. Cinnamic acid is a precursor of benzoic acids in cell cultures of Hypericum androsaemum L. but not in cell cultures of Centaurium erythraea RAFN. Planta. 2001;212:288–293. doi: 10.1007/s004250000394. [DOI] [PubMed] [Google Scholar]
- 14.Jana A, Pahan K. Sphingolipids in multiple sclerosis. Neuromolecular Med. 2010;12:351–361. doi: 10.1007/s12017-010-8128-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.El-behi M, Rostami A, Ciric B. Current views on the roles of Th1 and Th17 cells in experimental autoimmune encephalomyelitis. J Neuroimmune Pharmacol. 2010;5:189–197. doi: 10.1007/s11481-009-9188-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen Z, Freedman MS. Gammadelta T cells and multiple sclerosis: Friends, foes, or both? Autoimmun Rev. 2010 Dec 30; doi: 10.1016/j.autrev.2010.12.005. [DOI] [PubMed] [Google Scholar]
- 17.Dasgupta S, Roy A, Jana M, Hartley DM, Pahan K. Gemfibrozil ameliorates relapsing-remitting experimental autoimmune encephalomyelitis independent of peroxisome proliferator-activated receptor-alpha. Mol Pharmacol. 2007;72:934–946. doi: 10.1124/mol.106.033787. [DOI] [PubMed] [Google Scholar]
- 18.Brahmachari S, Pahan K. Sodium benzoate, a food additive and a metabolite of cinnamon, modifies T cells at multiple steps and inhibits adoptive transfer of experimental allergic encephalomyelitis. J Immunol. 2007;179:275–283. doi: 10.4049/jimmunol.179.1.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brahmachari S, Jana A, Pahan K. Sodium benzoate, a metabolite of cinnamon and a food additive, reduces microglial and astroglial inflammatory responses. J Immunol. 2009;183:5917–5927. doi: 10.4049/jimmunol.0803336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Coffer PJ, Burgering BM. Forkhead-box transcription factors and their role in the immune system. Nat Rev Immunol. 2004;4:889–899. doi: 10.1038/nri1488. [DOI] [PubMed] [Google Scholar]
- 21.O’Garra A, Vieira PL, Vieira P, Goldfeld AE. IL-10-producing and naturally occurring CD4+ Tregs: limiting collateral damage. J Clin Invest. 2004;114:1372–1378. doi: 10.1172/JCI23215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xu D, Liu H, Komai-Koma M, Campbell C, McSharry C, Alexander J, Liew FY. CD4+CD25+ regulatory T cells suppress differentiation and functions of Th1 and Th2 cells, Leishmania major infection, and colitis in mice. J Immunol. 2003;170:394–399. doi: 10.4049/jimmunol.170.1.394. [DOI] [PubMed] [Google Scholar]
- 23.Brahmachari S, Pahan K. Myelin basic protein priming reduces the expression of Foxp3 in T cells via nitric oxide. J Immunol. 2010;184:1799–1809. doi: 10.4049/jimmunol.0804394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brahmachari S, Pahan K. Suppression of regulatory T cells by IL-12p40 homodimer via nitric oxide. J Immunol. 2009;183:2045–2058. doi: 10.4049/jimmunol.0800276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Steinman L. A molecular trio in relapse and remission in multiple sclerosis. Nat Rev Immunol. 2009;9:440–447. doi: 10.1038/nri2548. [DOI] [PubMed] [Google Scholar]
- 26.Brahmachari S, Pahan K. Gender-specific expression of beta1 integrin of VLA-4 in myelin basic protein-primed T cells: implications for gender bias in multiple sclerosis. J Immunol. 2010;184:6103–6113. doi: 10.4049/jimmunol.0804356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dasgupta S, Jana M, Liu X, Pahan K. Role of very-late antigen-4 (VLA-4) in myelin basic protein-primed T cell contact-induced expression of proinflammatory cytokines in microglial cells. J Biol Chem. 2003;278:22424–22431. doi: 10.1074/jbc.M301789200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Roy A, Liu X, Pahan K. Myelin basic protein-primed T cells induce neurotrophins in glial cells via alphavbeta3 [corrected] integrin. J Biol Chem. 2007;282:32222–32232. doi: 10.1074/jbc.M702899200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Benveniste EN. Role of macrophages/microglia in multiple sclerosis and experimental allergic encephalomyelitis. J Mol Med. 1997;75:165–173. doi: 10.1007/s001090050101. [DOI] [PubMed] [Google Scholar]
- 30.Steinman L. Multiple sclerosis: a coordinated immunological attack against myelin in the central nervous system. Cell. 1996;85:299–302. doi: 10.1016/s0092-8674(00)81107-1. [DOI] [PubMed] [Google Scholar]
- 31.Saha RN, Pahan K. Regulation of inducible nitric oxide synthase gene in glial cells. Antioxid Redox Signal. 2006;8:929–947. doi: 10.1089/ars.2006.8.929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pahan K, Sheikh FG, Namboodiri AM, Singh I. Lovastatin and phenylacetate inhibit the induction of nitric oxide synthase and cytokines in rat primary astrocytes, microglia, and macrophages. J Clin Invest. 1997;100:2671–2679. doi: 10.1172/JCI119812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pahan K, Liu X, McKinney MJ, Wood C, Sheikh FG, Raymond JR. Expression of a dominant-negative mutant of p21(ras) inhibits induction of nitric oxide synthase and activation of nuclear factor-kappaB in primary astrocytes. J Neurochem. 2000;74:2288–2295. doi: 10.1046/j.1471-4159.2000.0742288.x. [DOI] [PubMed] [Google Scholar]
- 34.Ghosh S, Hayden MS. New regulators of NF-kappaB in inflammation. Nat Rev Immunol. 2008;8:837–848. doi: 10.1038/nri2423. [DOI] [PubMed] [Google Scholar]
- 35.Ling EA, Wong WC. The origin and nature of ramified and amoeboid microglia: a historical review and current concepts. Glia. 1993;7:9–18. doi: 10.1002/glia.440070105. [DOI] [PubMed] [Google Scholar]
- 36.González-Scarano F, Baltuch G. Microglia as mediators of inflammatory and degenerative diseases. Annu Rev Neurosci. 1999;22:219–240. doi: 10.1146/annurev.neuro.22.1.219. [DOI] [PubMed] [Google Scholar]
- 37.Roy A, Fung YK, Liu X, Pahan K. Up-regulation of microglial CD11b expression by nitric oxide. J Biol Chem. 2006;281:14971–14980. doi: 10.1074/jbc.M600236200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.von Andrian UH, Mackay CR. T-cell function and migration. Two sides of the same coin. N Engl J Med. 2000;343:1020–1034. doi: 10.1056/NEJM200010053431407. [DOI] [PubMed] [Google Scholar]
- 39.Vajkoczy P, Laschinger M, Engelhardt B. Alpha4-integrin-VCAM-1 binding mediates G protein-independent capture of encephalitogenic T cell blasts to CNS white matter microvessels. J Clin Invest. 2001;108:557–565. doi: 10.1172/JCI12440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Steffen BJ, Butcher EC, Engelhardt B. Evidence for involvement of ICAM-1 and VCAM-1 in lymphocyte interaction with endothelium in experimental autoimmune encephalomyelitis in the central nervous system in the SJL/J mouse. Am J Pathol. 1994;145:189–201. [PMC free article] [PubMed] [Google Scholar]
- 41.Eng LF, Ghirnikar RS. GFAP and astrogliosis. Brain Pathol. 1994;4:229–237. doi: 10.1111/j.1750-3639.1994.tb00838.x. [DOI] [PubMed] [Google Scholar]
- 42.Saha RN, Pahan K. Signals for the induction of nitric oxide synthase in astrocytes. Neurochem Int. 2006;49:154–163. doi: 10.1016/j.neuint.2006.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Saha RN, Jana M, Pahan K. MAPK p38 regulates transcriptional activity of NF-kappaB in primary human astrocytes via acetylation of p65. J Immunol. 2007;179:7101–7109. doi: 10.4049/jimmunol.179.10.7101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nagele RG, Wegiel J, Venkataraman V, Imaki H, Wang KC, Wegiel J. Contribution of glial cells to the development of amyloid plaques in Alzheimer’s disease. Neurobiol Aging. 2004;25:663–674. doi: 10.1016/j.neurobiolaging.2004.01.007. [DOI] [PubMed] [Google Scholar]
- 45.Brahmachari S, Fung YK, Pahan K. Induction of glial fibrillary acidic protein expression in astrocytes by nitric oxide. J Neurosci. 2006;26:4930–4939. doi: 10.1523/JNEUROSCI.5480-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jana A, Hogan EL, Pahan K. Ceramide and neurodegeneration: susceptibility of neurons and oligodendrocytes to cell damage and death. J Neurol Sci. 2009;278:5–15. doi: 10.1016/j.jns.2008.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Miller A. Current and investigational therapies used to alter the course of disease in multiple sclerosis. South Med J. 1997;90:367–375. doi: 10.1097/00007611-199704000-00001. [DOI] [PubMed] [Google Scholar]
- 48.Barkhatova VP, Zavalishin IA, Askarova LSh, Shavratskii VKh, Demina EG. Changes in neurotransmitters in multiple sclerosis. Neurosci Behav Physiol. 1998;28:341–344. doi: 10.1007/BF02464784. [DOI] [PubMed] [Google Scholar]
- 49.Nair B. Final report on the safety assessment of benzyl alcohol, benzoic acid, and sodium benzoate. Int J Toxicol. 2001;20 (Suppl 3):23–50. doi: 10.1080/10915810152630729. [DOI] [PubMed] [Google Scholar]
- 50.Leonard JV, Morris AA. Urea cycle disorders. Semin Neonatol. 2002;7:27–35. doi: 10.1053/siny.2001.0085. [DOI] [PubMed] [Google Scholar]
- 51.Scaglia F, Carter S, O’Brien WE, Lee B. Effect of alternative pathway therapy on branched chain amino acid metabolism in urea cycle disorder patients. Mol Genet Metab. 2004;81 (Suppl 1):S79–S85. doi: 10.1016/j.ymgme.2003.11.017. [DOI] [PubMed] [Google Scholar]