

Abstract
Aldo-keto reductases (AKRs) are phase I metabolising enzymes that catalyse the reduced nicotinamide adenine dinucleotide (phosphate) (NAD(P)H)-dependent reduction of carbonyl groups to yield primary and secondary alcohols on a wide range of substrates, including aliphatic and aromatic aldehydes and ketones, ketoprostaglan-dins, ketosteroids and xenobiotics. In so doing they functionalise the carbonyl group for conjugation (phase II enzyme reactions). Although functionally diverse, AKRs form a protein superfamily based on their high sequence identity and common protein fold, the (α/(β)8-barrel structure. Well over 150 AKR enzymes, from diverse organisms, have been annotated so far and given systematic names according to a nomenclature that is based on multiple protein sequence alignment and degree of identity. Annotation of non-vertebrate AKRs at the National Center for Biotechnology Information or Vertebrate Genome Annotation (vega) database does not often include the systematic nomenclature name, so the most comprehensive overview of all annotated AKRs is found on the AKR website (http://www.med.upenn.edu/akr/). This site also hosts links to more detailed and specialised information (eg on crystal structures, gene expression and single nucleotide polymorphisms [SNPs]). The protein-based AKR nomenclature allows unambiguous identification of a given enzyme but does not reflect the wealth of genomic and transcriptomic variation that exists in the various databases. In this context, identification of putative new AKRs and their distinction from pseudogenes are challenging. This review provides a short summary of the characteristic features of AKR biochemistry and structure that have been reviewed in great detail elsewhere, and focuses mainly on nomenclature and database entries of human AKRs that so far have not been subject to systematic annotation. Recent developments in the annotation of SNP and transcript variance in AKRs are also summarised.
Keywords: carbonyl reduction, nomenclature, pseudogene, SNP, splice variant
Introduction
Aldo-keto reductases (AKRs) form a superfamily of proteins characterised by their common three-dimensional structure and reaction mechanism in catalysing the reduced nicotinamide adenine dinucleotide (phosphate) (NAD(P)H)-dependent oxido-reduction of carbonyl groups.[1,2] More than 150 enzymes have been identified in a wide range of organisms, including prokaryotes and eukaryotes such as plants, fungi and vertebrates. Based on sequence identity, the proteins fall into 15 different families, termed AKR1-AKR15, and some families (eg AKR1) contain multiple subfamilies.[1,3,4] Fourteen human AKRs (Table 1), which fall into families 1, 6 and 7, have been annotated and their specific structural and kinetic features, as well as substrate and genomic characteristics, have been recently and comprehensively reviewed.[5,6]
Table 1.
Systematic and trivial names of human Aldo-keto reductases (AKRs)
| Systematic name | Trivial name/often-used abbreviations |
|---|---|
| AKR1A1 | Aldehyde reductase; ALR; ALDR1 |
| AKR1B1 | Aldose reductase; AR, ALR2 |
| AKR1B10 | Small intestine-like aldose reductase; ARL-1 |
| AKR1C1 | Dihydrodiol dehydrogenase I; 20α,(3α)-Hydroxysteroid dehydrogenase |
| AKR1C2 | Dihydrodiol dehydrogenase 2; 3α-Hydroxysteroid dehydrogenase, type III |
| AKR1C3 | 3α-Hydroxysteroid dehydrogenase, type II |
| AKR1C4 | Dihydrodiol dehydrogenase 4; chlordecone reductase; 3α-hydroxysteroid dehydrogenase, type I |
| AKR1D1 | Δ4-3-Ketosteroid-5β-reductase |
| AKR1E2 | Aldo-keto reductase family I, member C-like 2; AKRICL2 |
| AKR6A3 | KCNAB I; hKvbeta3; potassium voltage-gated channel, shaker-related subfamily, β-member 1 |
| AKR6A5 | KCNAB 2; hKvbeta2; potassium voltage-gated channel, shaker-related subfamily, β-member 2 |
| AKR6A9 | KCNAB 3; potassium voltage-gated channel, shaker-related subfamily, β-member 3 |
| AKR7A2 | Aflatoxin aldehyde reductase; AFAR1 |
| AKR7A3 | Aflatoxin aldehyde reductase; AFAR2 |
AKRs are, for the most part, cytosolic and are active as monomers. So far, only AKR2, AKR6 and AKR7 family members show evidence of forming multimers, although it is unclear whether quaternary structure is essential for enzyme activity.[7-9] The single amino acid chain of, on average, 320 residues folds into a (α/β)8-barrel, an evolutionarily highly conserved structure originally observed in triose-phosphate isomerase (TIM) and therefore also known as a TIM barrel structure. In contrast to another 32 superfamilies adopting this motif, AKRs do not require metal ions for functionality, and bind NAD(P)H cofactor in an extended anti-conformation so that they can catalyse 4-pro-R hydride transfer and thus belong to the large group of A-face oxidoreductases.
The spectrum of AKR substrates is wide and comprises aldehydes, ketones, monosaccharides, ketosteroids, ketoprostaglandins, bile acid precursors, chemical carcinogens and their metabolites -- for example, aflatoxin dialdehydes, nicotine-derived 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone and trans-dihydrodiols of polycyclic aromatic hydrocarbons. Substrate recognition and binding takes place at the C-terminal end of the ellipsoid (α/β)8-barrel body through interaction with amino acids within the three highly variable loops pro-2 10 11 truding out of the barrel structure. [2,10,11] Indeed, sequence conservation among AKRs is lowest in these loops, compared with the α-helices and β-strands forming the barrel structure itself, and suggests that AKRs evolved from a common ancestor by duplication and subsequent divergence to accommodate the binding of the structurally diverse substrates.
Duplication and diversification indeed seem to have been the driving force in the formation of vertebrate AKRs. AKR1B, AKR1C and AKR7A family members appear to be vertebrate specific and in the human genome their genes cluster on chromosomes 7, 10 and 1, respectively.[5] Furthermore, the potential of these three subfamilies to give rise to new functional AKRs is reflected in the existence of several pseudogenes and putative new isoforms in the human genome. By contrast, the genes of all three human AKR6 family members reside in different chromosomal locations, show a high degree of conservation, even to plant AKR6 isoforms, and do not give rise to functional or non-functional (pseudogene) gene duplications.
Biochemistry of AKRs
Almost all AKRs catalyse the reduction of aldehydes and ketones to primary and secondary alcohols, respectively, by use of a reduced nicotinamide cofactor. The reactions involve an ordered bi-bi kinetic mechanism and general acid-base catalysis.[12-14] The cofactor binds first, followed by tethering of the substrate. In the reduction direction, the 4-pro-R hydride from NAD(P)H is transferred to the substrate carbonyl group, followed by protonation of the carbonyl oxygen by a conserved tyrosine acting as a general acid.[15-17] Oxidation proceeds in the reverse sequence with the tyrosine acting as a general base.
Asp 50, Tyr 55, Lys 84 and His 117 form the catalytic tetrad (based on residue numbering in AKR1C9),[2,18,19] where the Tyr is the general acid-base.[17,20] Conservation of these residues is strong and underlines their role in catalysis. Asp 50 is present in 99 per cent, Tyr 55 in 97 per cent, Lys 84 in 97 per cent and His 117 in 88 per cent of all annotated AKRs on the AKR website. Consistent substitution of these key amino acids, however, especially of the histidine, is a characteristic for some AKR subfamilies. In AKR1D isoforms, which catalyse the 5β-reduction of Δ4-3-ketosteroids, glutamate replaces His 117.[2,21] Interestingly, muta-tional analysis in AKR1C9 demonstrates that this single amino acid substitution is sufficient to convert this 3-ketosteroid reductase to a steroid 5β double-bond reductase.[22,23] The reverse change of function has been hypothesised for the respective (E120H) mutation in AKR1D1.[24] In all AKR6A isoforms, the conserved histidine is changed to asparagine. Based on crystal structure comparisons, Di Costanzo et al. hypothesised that in AKR6A isoforms the amino acid in this position is involved in substrate discrimination.[25] AKR6A isoforms, better known as voltage-gated potassium channel (β-subunits (Kvβ or KCNAB), accept a wide range of substrates, but their catalytic efficiency is significantly lower than other AKRs.[26,27] It has been suggested that they may primarily function as redox state sensors in the regulation of potassium channel activity.[27-29] The replacement ofhistidine with asparagine may provide more space to allow for the optimal positioning of bulky lipid and phospholipid aldehydes, formed as products oflipid peroxidation and oxidative stress.
Although these four conserved amino acids seem to play a crucial role in the catalytic mechanism, targeted mutation studies (eg in AKR1B1[15] and AKR1C9[30]) did not indicate a complete loss in enzymatic activity and suggested that, in some instances, proximity of the hydride donor to the acceptor is sufficient to catalyse a reaction. Furthermore, some AKRs, such as ρ-crystallins in amphibian lenses (AKR1C10), appear to have been employed in this tissue basically for structural reasons, having lost almost all enzymatic activity despite unaltered characteristics of the cofactor and substrate binding site.[31]
Nomenclature
The nomenclature for the AKR superfamily, introduced and accepted by the Hugo Genome Nomenclature Committee (HGNC) in 1997, builds solely on sequence and not on functional similarity. Furthermore, systematic annotation through the AKR website requires proof of functionality of the protein in question. This evidence may be RNA or enzyme based, and aims to avoid the annotation of non-functional pseudogenes or predicted genes of unknown physiological function. Sequence submission for the assignment of a systematic AKR superfamily name should be sent directly to the AKR website (http://www.med.upenn.edu/akr/).[3,4] The basic principle in naming a new AKR follows that for the cytochrome P (CYP) superfamily [32,33]. All members of the super-family are annotated with the suffix AKR. The 15 families (Figure 1) into which annotated AKRs fall have less than 40 per cent amino acid identity with each other. Subfamilies, denoted by a capital roman letter, share up to 60 per cent sequence identity. Individual members in the subfamily are numbered according to the chronology of sequence submission to the AKR website. Thus, AKR1A1 (human aldehyde reductase) belongs to family 1, subfamily A and is the first unique protein in the subfamily. If two sequences share 97 per cent or more sequence identity, they are considered as alleles and marked by an additional small roman letter (eg AKR1C10a and AKR1C10b). Two sequences sharing this high degree of identity are not regarded as alleles if they reside in different genomic locations, have distinct 3' untranslated regions or differ in their enzymatic activities. It was also recommended that the corresponding gene be given the same name as the protein, but written in italics.
Figure 1.
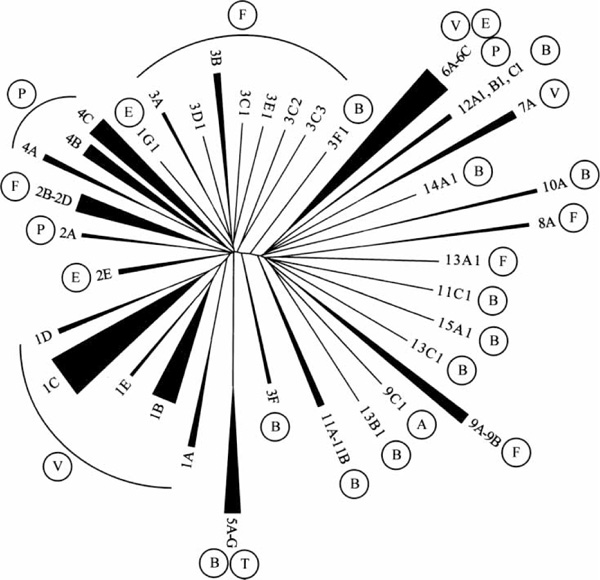
Dendogram of all systemically annotated AKR enzymes. All sub-trees comprising more than a single member have been collapsed and the width of the resulting wedge reflects the number of annotated sequences in this category. Circled capital letters denote the species of origin A, archeae; B, bacteria; E, ecdysozoa; F, fungi; P, plants; T, trypanosomatidae; V, vertebrate.
The nomenclature has the advantage that it assigns a unique gene name to each single AKR through establishing its amino acid sequence similarity to other families and subfamilies. In this way, it is independent of enzyme properties, species of origin and functional homologies. Therefore, it is not surprising that most families at the same time constitute clusters of closely related organisms rather than similar function, and that an AKR of a more distantly related organism will fall into a different subfamily (Figure 1). For example, bacterial AKRs form their own families with multiple subfamilies owing to the high diversification that has occurred in the 3,900 million years since bacteria and archeae split. At the same time, vertebrate, but especially mammalian, AKR sequences (mammalians appeared about 300 million years ago) form sequence clusters with multiple, highly similar members in each subfamily. Inspection of the vertebrate AKR sequences reveals that the AKR1 family evolved separately from families AKR6 and AKR7. Furthermore, only potassium channel β-subunit sequences have been so well conserved during evolution that plant and arthropod sequences both cluster in the AKR6 family.
A search for AKRs on NCBI, HGNC and Ensembl
Although the new nomenclature came into effect nearly 12 years ago--and has been widely accepted by the AKR research community--it has not yet become widely adopted by database annotators. This is especially true for non-mammalian AKRs. Of the 160 proteins annotated on the AKR website (http://www.med.upenn.edu/akr/), 62 give problematic results if searched with their systematic name against the National Center for Biotechnology Information (NCBI) database. For 45 AKRs, not a single database entry can be found. This illustrates that regular updates of protein and nucleotide annotations have failed to notice and acknowledge the proposed systematic nomenclature.
Annotation and nomenclature of human AKRs, however, are far more consistent. A database search for human AKRs at the National Institutes of Health (http://www.ncbi.nlm.nih.gov), the European Molecular Biology Laboratory (http://www.ensembl.org), the Sanger Institute (http://vega.sanger.ac.uk/index.html) and the HGNC (http://www.genenames.org) identified 13 of the 14 proteins listed on the AKR website (Table 1). AKR1E2, which appears in all four databases under its synonym AKR1CL2, was originally annotated as LoopADR in 2000 and later named AKR1C like 2 (AKR1CL2), based on its sequence similarity to AKR1C isoforms. In 2003 and 2004, two publications described the enzymatic activity of this protein towards 9,10-phenanthrenequinone, but not steroid hormones, and termed it human testis-specific AKR (htAKR), based on its restricted expression pattern.[34,35] The protein shares 74 per cent sequence identity with the previously described murine AKR1E1[36] and hence was systematically named AKR1E2.
Unclassified and predicted human AKR database entries
The database search for human AKRs at NCBI also revealed a number of other sequences annotated in context with AKRs. The total of 58 database entries comprised the 14 systematically annotated and well-described human AKRs and 12 separate annotations of pseudogenes. The search also revealed that in 22 cases, the respective protein belonged to a different protein family but appears in a biological context with an AKR. The remaining ten database entries (Table 2) resemble unclassified or predicted human AKR sequences that are related to three different AKR subfamilies: AKR1C, AKR1B and AKR7A.
Table 2.
Non-classified human AKRs in the NCBI database
| Name | Alternative names | Gene IDa | Chromosomal location | Related to | Probable identity |
|---|---|---|---|---|---|
| AKR1B1I | ARL, HSI | 9405 | 7 | AKR1B | Variance of AKR1B10 |
| AKR1B10L | tcag7.1260 | 441282 | 7q33 | AKR1B | New AKR or pseudogene of AKR1B10 |
| LOC340888 | 340888 | 10q2l.3 | AKR1B | Retro-transposed AKR1B10 pseudogene | |
| LOC643582 | 643582 | I8q22.l | AKR1B | Retro-transposed AKR1B10 pseudogene | |
| AKR1CL1 | RAKc | 340811 | 10pI5.I | AKR1C | New AKR1C or pseudogene |
| AKR1C-pseudo | 266745 | 10pI5-10pI4 | AKR1C | Variance of AKR1C2 | |
| LOC6485I7 | 648517 | Unclear | AKR1C | AKR1C pseudogene or transcript variant | |
| LOC100I34257 | 100134257 | Unclear | AKR1C | AKR1C pseudogene or transcript variant | |
| LOC643789 | 643789 | 10pI5.I | AKR1C | Pseudogene | |
| LOC648947 | 648947 | 10pI5.I | AKR1C | AKR1CL1 pseudogene | |
| tAKR | 389932 | 10pI5.I | AKR1C | AKR1C pseudogene | |
| AKR7L | AKR7A4, AFAR3 | 246181 | Ip35-Ip36.I | AKR7A | New AKR7A or pseudogene |
| AFARPI | 246182 | IpI2 | AKR7A | Retro-transposed AKR7 pseudogene |
a At NCBI (http://www.ncbi.nlm.nih.gov).
AKR1CL1 is located between AKR1C3 and AKR1C4 on chromosome 10. Ambiguous reports at NCBI describe two different forms of the gene. In one case, exons 1-4 of eight exons encode a 129-amino acid peptide (Accession No. NP001007537.1). This protein includes the complete catalytic tetrad but appears to be too short to be a functional AKR. In the second case, AKR1CL1, similarly to the other four AKR1C members, consists ofnine exons and encodes a complete 326-amino acid protein (Accession No. EAW86451) with 68 per cent sequence identity to AKR1C1-4. The shorter version exactly matches residues 1-129 of the longer form and resembles a differently transcribed and processed version of the same gene. A few messenger RNA (mRNA) sequences are annotated that match AKR1CL1 with deletions and frameshifts, but a Blast search with the longer version against the expressed sequence tag (EST) database does not yield any AKR1CL1 transcripts. Therefore, it remains unclear whether AKR1CL1 is a processed pseudogene or a new functional AKR1C member.
AKRlC-pseudo is an old, discontinued entry that resembles AKR1C2. AKR1C2 shares 97 per cent sequence identity with AKR1C1 but exhibits a unique expression pattern and substrate specificity.[37-41]LOC648517 and LOC100134257 contain sequence elements that completely match exons of AKR1C1 and AKR1C2 . Their chromosomal location could not be determined, however, and therefore it is not clear whether they might be transcript variants of either AKR1C1 or AKR1C2, or are separate pseudogenes.
Four entries are related to AKR1B10. While AKR1B11 is a discontinued record replaced by AKR1B10, LOC340888 and LOC643582 are Gnomon predictions and appear to be retrotransposed pseudogenes of AKR1B10. AKR1B10L resides next to AKR1B10 on chromosome 7 and also consists of ten exons. The two proteins share 91 per cent amino acid identity. The presence of a single EST from placenta (BX350113.2) that completely matches residues 23-209 of the predicted 316-amino acid AKR1B10L transcript suggests that AKR1B10L could indeed resemble a new and thus far uncharacterised human AKR family member. Alternatively, the presence of the mRNA could result from a processed pseudogene.
Two additional database entries relate to the AKR7 family. AKR7L clusters together with AKR7A2 and AKR7A3 on chromosome 1p35-1p36.1.[3,42] Alternative transcription leads to two different transcript variants. Variant 1 is the longer and comprises seven exons that encode a 331-amino acid protein. This form may indeed resemble a new functional AKR that has 92 per cent and 88 per cent identity with AKR7A3 and AKR7A2, respectively. Variant 2 lacks exons 4 and 5 and codes for a probably non-functional alternative peptide. This deletion results in a frameshift-eliminating sequence similarity to AKRs in the last 80 amino acids of the 250-amino acid peptide. The AKR7 family pseudogene (AFARP1)isaretro-transposed AKR7, where the complete processed mRNA has reintegrated into the genome.
In addition, Barski et al.[5] report on three putative AKRs not identified in the above-described database search. The database entry LOC643789 was originally designated as the AKR1C pseudo-gene but was discontinued in March 2008. LOC648947 is a predicted gene with nine exons, encodes a 316-amino acid protein and shares 82 per cent sequence identity with the longer version of AKR1CL1; however, this protein sequence harbours a stop codon following amino acid 199. At least three mRNA sequences for this gene are present in the EST database. They do not completely match the predicted protein sequence of LOC648947, and a region corresponding to amino acids 192-227 is deleted. As this deletion covers a region conserved in all AKR1C isoforms, it is very likely that LOC648947 represents a processed pseudogene of AKR1CL1. Finally, tAKR shows similarity to other AKR1C isoforms and the gene is located between AKR1CL2 and AKR1C1 on chromosome 10. Original annotation predicts a gene of 11 exons, with exons 3-11 encoding a putative protein with more than 300 amino acids. A transcript exists only for the first 125 amino acids, however, and hence tAKR is thus far classified as a processed pseudogene.
In summary, systematic integration of these AKR database entries into the AKR nomenclature is still pending owing to a lack of sufficient experimental data to support an assignment.
Annotated variants of AKRs
Efforts have been made systematically to describe genetic variants and splice isoforms in annotated human AKRs and to identify the location and classification of putative pseudogenes (Table 3). Data on single nucleotide polymorphisms (SNPs) are accumulating rapidly, and the SNP database build 130 (NCBI), as of 30th April 2009, contains over 17.8 million SNP clusters, with over 7.3 million SNPs located in genes. Nomenclature for SNPs in AKRs is based on that for sulfotransferase (SULT) genes[43] and takes only coding SNPs into account.[4] Systematic annotation of the SNP is marked by the gene name followed by an asterisk, an Arabic number indicating synonymous ('1') or non-synonymous ('2'-'n') character and a letter distinguishing between suballeles; the nomenclature is chronologically based. While this seems to be a feasible approach for synonymous and non-synonymous SNPs, it neglects SNPs leading to fra-meshifts and those in the non-coding gene region that have the potential to alter the transcription efficiency, splicing and stability of mRNA. For all AKRs SNP frequency is significantly higher than the estimated 1 SNP/kilobase.[44,45] Although the total number of SNPs per gene is only a snapshot in this time of extremely rapid data growth, it appears that some AKRs (eg AKR1D1, AKR6A3 and AKR6A9) have accumulated significantly fewer single nucleotide variations in their coding region than others.
Table 3.
Genomic and transcriptomic variations in annotated human AKRs
| AKR | Gene IDa | Chromosomal location | No. of SNPsb (in gene/coding region/non-synonymous/frameshift) | SNP frequency (per 1 kilobase: in gene/in coding region) | Peptide variants | Additional processed transcripts | Pseudogenes on vega (HGNC) |
|---|---|---|---|---|---|---|---|
| 1A1 | 10327 | 1p33-p32 | 154/9/5/1 | 8.01/9.2 | 3 | 10 | 0 |
| 1B1 | 231 | 7q35 | 160/13/9/0 | 9.53/13.67 | 3 | 12 | 5(8) |
| 1B10 | 57016 | 7q33 | 127/16/5/0 | 9.65/16.82 | 1 | 3 | 4 |
| 1C1 | 1645 | 10p15-p14 | 160/16/6/0 | 10.88/16.46 | 3 | 2 | 0 |
| 1C2 | 1646 | 10p15-p14 | 353/16/8/0 | 12.5/16.46 | 1 | 1 | 0 |
| 1C3 | 8644 | 10p15-p14 | 261/23/13/2 | 19.61/23.66 | 1 | 3 | 0 |
| 1C4 | 1109 | 10p15 -p14 | 198/7/6/0 | 8.95/7.2 | 1 | 1 | 0 |
| 1D1 | 6718 | 7q32-q33 | 275/1/0/0 | 6.57/1.02 | 4 | 2 | 2(1) |
| 1E2 | 83592 | 10p15.1 | 211/12/7/0 | 9.66/12.46 | 2 | 4 | 0 |
| 6A3 | 7881 | 3q26.1 | 1888/1/0/0 | 4.51/0.81 | 10 | 0 | 0 |
| 6A5 | 8514 | 1p36.3 | 414/8/2/1 | 7.15/7.25 | 8 | 6 | 0 |
| 6A9 | 9196 | 17p13.1 | 42/3/1/2 | 6.24/2.47 | 1 | 2 | 0 |
| 7A2 | 8574 | 1p36.13 | 71/10/8/0 | 8.68/9.26 | 1 | 0 | 4 |
| 7A3 | 22977 | 1p36.13 | 67/12/5/0 | 10.76/12.05 | 1 | 0 | 0 |
aAt NCBI (http://www.ncbi.nlm.nih.gov).
bDeduced from the vega database (http://vega.sanger.ac.uk/index.html).
The table does not include the putative transcript variants and pseudogenes listed in Table 2.
In addition to SNPs, other variations have been annotated over the years. The most well known are pseudogenes, and one or several have been identified for a few AKRs. For example, the pseudogene forms of AKR1B1 are well documented and systematically annotated as AKR1B1P1-8, where P stands for pseudogene. For example, AKR1B1P4 denotes the fourth pseudogene annotated for AKR1B1 . Analysis of the non-systematically named AKRs (Table 2) suggests that many more pseudogenes, in addition to the few already annotated, might exist. This may also bethecaseforalternativespliceforms, wherethe vega database (http://vega.sanger.ac.uk/index.html) from the Sanger Institute offers the most systematic annotation approach. Gene transcripts are numbered in the order of their annotation and designated with the suffix '-n' added to the specific gene name. For example, AKR1C1 has five different transcripts, annotated as AKR1C1-001 to AKR1C1-005. For half of the human AKRs, transcript processing leads to one or more alternative peptides. Furthermore, non-coding RNAs have been identified for 11 of the 14 genes. While, in the past, these fragments have been ignored, being perceived as resulting from errors in transcription or splicing, increasing evidence suggests that they play important regulatory functions (eg in gene expression, imprinting, RNA stability, splicing and translation).[46] Their role in AKR expression and function remains to be elucidated.
Summary
Proteins of the AKR superfamily are present in all phyla and are characterised by their common three-dimensional structure and reaction mechanism. Fourteen human AKRs have so far been annotated with systematic gene names in accordance with a systematic nomenclature provided through the AKR website that is based on amino acid sequence similarity and proof of protein functionality. Additional non-systematic annotations of human AKRs are present in the NCBI database that may resemble new, uncharacterised proteins or pseudogenes of existing AKRs. Furthermore, the rapid and continuous increase in data on SNPs and transcript variants challenges a systematic annotation of these phenomena and adds to the complexity of defining the functionality of a given gene product. Despite these complexities, the AKR website provides the most comprehensive and up-to-date portal for tracking the development of AKR annotation and provides links to in-depth information on each single AKR superfamily member in other databases.
Acknowledgements
The writing of this paper was funded in part by NIH grants P30-ES013508, R01-DK47015, R01-CA90744 and R01-CA39504 awarded to T.M.P
References
- Jez JM, Flynn TG, Penning TM. A nomenclature system for the aldo-keto reductase superfamily. Adv Exp Med Biol. 1997;414:579–600. doi: 10.1007/978-1-4615-5871-2_66. [DOI] [PubMed] [Google Scholar]
- Jez JM, Bennett MJ, Schlegel BP, Lewis M, Penning TM. Comparative anatomy of the aldo-keto reductase superfamily. Biochem J. 1997;326:625–636. doi: 10.1042/bj3260625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyndman D, Bauman DR, Heredia VV, Penning TM. The aldo-keto reductase superfamily homepage. Chem Biol Interact. 2003;143-144:621–631. doi: 10.1016/s0009-2797(02)00193-x. [DOI] [PubMed] [Google Scholar]
- Drury JE, Hyndman D, Jin Y, In: Enzymology and Molecular Biology of Carbonyl Metabolism. Weiner H, Maser E, Lindhal R, editor. Purdue Press, West Lafayette, IN; 2007. The aldo-keto reductase superfamily homepage: 2006 update; pp. 184–197. [Google Scholar]
- Barski OA, Tipparaju SM, Bhatnagar A. The aldo-keto reductase superfamily and its role in drug metabolism and detoxification. Drug Metab Rev. 2008;40:553–624. doi: 10.1080/03602530802431439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Y, Penning TM. Aldo-keto reductases and bioactivation/detoxication. Annu Rev Pharmacol Toxicol. 2007;47:263–292. doi: 10.1146/annurev.pharmtox.47.120505.105337. [DOI] [PubMed] [Google Scholar]
- Kozma E, Brown E, Ellis EM, Lapthorn AJ. The crystal structure of rat liver AKR7A1. A dimeric member of the aldo-keto reductase superfamily. J Biol Chem. 2002;277:16285–16293. doi: 10.1074/jbc.M110808200. [DOI] [PubMed] [Google Scholar]
- Kavanagh KL, Klimacek M, Nidetzky B, Wilson DK. The structure of apo and holo forms of xylose reductase, a dimeric aldo-keto reductase from Candida tenuis. Biochemistry. 2002;41:8785–8795. doi: 10.1021/bi025786n. [DOI] [PubMed] [Google Scholar]
- Gulbis JM, Mann S, Mackinnon R. Structure of a voltage-dependent K+ channel β subunit. Cell. 1999;97:943–952. doi: 10.1016/S0092-8674(00)80805-3. [DOI] [PubMed] [Google Scholar]
- Bennett MJ, Albert RH, Jez JM, Ma H. et al. Steroid recognition and regulation of hormone action: Crystal structure of testosterone and NADP+ bound to 3α-hydroxysteroid/dihydrodiol dehydrogenase. Structure. 1997;5:799–812. doi: 10.1016/S0969-2126(97)00234-7. [DOI] [PubMed] [Google Scholar]
- Couture JF, De Jesus-Tran KP, Roy AM, Cantin L. et al. Comparison of crystal structures of human type 3 3α-hydroxysteroid dehydrogenase reveals an "induced-fit" mechanism and a conserved basic motif involved in the binding of androgen. Protein Sci. 2005;14:1485–1497. doi: 10.1110/ps.051353205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Askonas LJ, Ricigliano JW, Penning TM. The kinetic mechanism catalysed by homogeneous rat liver 3α-hydroxysteroid dehydrogenase. Evidence for binary and ternary dead-end complexes containing non-steroidal anti-inflammatory drugs. Biochem J. 1991;278:835–841. doi: 10.1042/bj2780835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuhauser W, Haltrich D, Kulbe KD, Nidetzky B. NAD(P)H-dependent aldose reductase from the xylose-assimilating yeast Candida tenuis. Isolation, characterization and biochemical properties of the enzyme. Biochem J. 1997;326:683–692. doi: 10.1042/bj3260683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trauger JW, Jiang A, Stearns BA, Lograsso PV. Kinetics of allopregnanolone formation catalyzed by human 3α-hydroxysteroid dehydrogenase type III (AKR1C2) Biochemistry. 2002;41:13451–13459. doi: 10.1021/bi026109w. [DOI] [PubMed] [Google Scholar]
- Bohren KM, Grimshaw CE, Lai CJ, Harrison DH. et al. Tyrosine-48 is the proton donor and histidine-110 directs substrate stereochemical selectivity in the reduction reaction of human aldose reductase: Enzyme kinetics and crystal structure of the Y48H mutant enzyme. Biochemistry. 1994;33:2021–2032. doi: 10.1021/bi00174a007. [DOI] [PubMed] [Google Scholar]
- Penning TM. Molecular determinants of steroid recognition and catalysis in aldo-keto reductases. Lessons from 3α-hydroxysteroid dehydrogenase. J Steroid Biochem Mol Biol. 1999;69:211–225. doi: 10.1016/S0960-0760(99)00038-2. [DOI] [PubMed] [Google Scholar]
- Schlegel BP, Jez JM, Penning TM. Mutagenesis of 3α-hydroxysteroid dehydrogenase reveals a "push-pull" mechanism for proton transfer in aldo-keto reductases. Biochemistry. 1998;37:3538–3548. doi: 10.1021/bi9723055. [DOI] [PubMed] [Google Scholar]
- Jez JM, Flynn TG, Penning TM. A new nomenclature for the aldo-keto reductase superfamily. Biochem Pharmacol. 1997;54:639–647. doi: 10.1016/S0006-2952(97)84253-0. [DOI] [PubMed] [Google Scholar]
- Bennett MJ, Schlegel BP, Jez JM, Penning TM, Lewis M. Structure of 3α-hydroxysteroid/dihydrodiol dehydrogenase com-plexed with NADP+ Biochemistry. 1996;35:10702–10711. doi: 10.1021/bi9604688. [DOI] [PubMed] [Google Scholar]
- Grimshaw CE, Bohren KM, Lai CJ, Gabbay KH. Human aldose reductase: pK of tyrosine 48 reveals the preferred ionization state for catalysis and inhibition. Biochemistry. 1995;34:14374–14384. doi: 10.1021/bi00044a014. [DOI] [PubMed] [Google Scholar]
- Kondo KH, Kai MH, Setoguchi Y, Eggertsen G. et al. Cloning and expression of cDNA of human Δ4-3-oxosteroid 5β-reductase and substrate specificity of the expressed enzyme. Eur J Biochem. 1994;219:357–363. doi: 10.1111/j.1432-1033.1994.tb19947.x. [DOI] [PubMed] [Google Scholar]
- Penning TM, Ma H, Jez JM. Engineering steroid hormone specificity into aldo-keto reductases. Chem Biol Interact. 2001;130-132:659–671. doi: 10.1016/s0009-2797(00)00257-x. [DOI] [PubMed] [Google Scholar]
- Jez JM, Penning TM. Engineering steroid 5β-reductase activity into rat liver 3α-hydroxysteroid dehydrogenase. Biochemistry. 1998;37:9695–9703. doi: 10.1021/bi980294p. [DOI] [PubMed] [Google Scholar]
- Drury JE, Penning TM. In: Enzymology and Molecular Biology of Carbonyl Metabolism. Weiner H, Maser E, Lindhal R, editor. Purdue Press, West Lafayette, IN; 2007. Δ4-3-ketosteroid 5β-reductase (AKR1D1): Properties and role in bile acid synthesis; pp. 332–340. [Google Scholar]
- Di Costanzo L, Penning TM, Christianson DW. Aldo-keto reductases in which the conserved catalytic histidine is substituted. Chem Biol Interact. 2009;178:127–133. doi: 10.1016/j.cbi.2008.10.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tipparaju SM, Barski OA, Srivastava S, Bhatnagar A. Catalytic mechanism and substrate specificity of the β-subunit of the voltage-gated potassium channel. Biochemistry. 2008;47:8840–8854. doi: 10.1021/bi800301b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weng J, Cao Y, Moss N, Zhou M. Modulation of voltage-dependent Shaker family potassium channels by an aldo-keto reductase. J Biol Chem. 2006;281:15194–15200. doi: 10.1074/jbc.M513809200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tipparaju SM, Saxena N, Liu SQ, Kumar R, Bhatnagar A. Differential regulation of voltage-gated K+ channels by oxidized and reduced pyridine nucleotide coenzymes. Am J Physiol Cell Physiol. 2005;288:C366–C376. doi: 10.1152/ajpcell.00354.2004. [DOI] [PubMed] [Google Scholar]
- Pan Y, Weng J, Cao Y, Bhosle RC, Zhou M. Functional coupling between the Kv1.1 channel and aldoketoreductase Kvβ1. J Biol Chem. 2008;283:8634–8642. doi: 10.1074/jbc.M709304200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlegel BP, Ratnam K, Penning TM. Retention of NADPH-linked quinone reductase activity in an aldo-keto reductase following mutation of the catalytic tyrosine. Biochemistry. 1998;37:11003–11011. doi: 10.1021/bi980475r. [DOI] [PubMed] [Google Scholar]
- Fujii Y, Watanabe K, Hayashi H, Urade Y. et al. Purification and characterization of rho-crystallin from Japanese common bullfrog lens. J Biol Chem. 1990;265:9914–9923. [PubMed] [Google Scholar]
- Dayhoff MO, Barker WC, Hunt LT. Establishing homologies in protein sequences. Methods Enzymol. 1983;91:524–545. doi: 10.1016/s0076-6879(83)91049-2. [DOI] [PubMed] [Google Scholar]
- Nebert DW, Adesnik M, Coon MJ. et al. The P450 gene superfamily: recommended nomenclature. DNA. 1987;6(1):1–11. doi: 10.1089/dna.1987.6.1. [DOI] [PubMed] [Google Scholar]
- Nishinaka T, Azuma Y, Ushijima S, Miki S. et al. Human testis specific protein: A new member of aldo-keto reductase superfamily. Chem Biol Interact. 2003;143-144:299–305. doi: 10.1016/s0009-2797(02)00187-4. [DOI] [PubMed] [Google Scholar]
- Azuma Y, Nishinaka T, Ushijima S, Soh J. et al. Characterization of htAKR, a novel gene product in the aldo-keto reductase family specifically expressed in human testis. Mol Hum Reprod. 2004;10:527–533. doi: 10.1093/molehr/gah062. [DOI] [PubMed] [Google Scholar]
- Bohren KM, Barski OA, Gabbay KH. Characterization of a novel murine aldo-keto reductase. Adv Exp Med Biol. 1997;414:455–464. doi: 10.1007/978-1-4615-5871-2_52. [DOI] [PubMed] [Google Scholar]
- Ciaccio PJ, Tew KD. cDNA and deduced amino acid sequences of a human colon dihydrodiol dehydrogenase. Biochim Biophys Acta. 1994;1186:129–132. doi: 10.1016/0005-2728(94)90144-9. [DOI] [PubMed] [Google Scholar]
- Qin KN, New MI, Cheng KC. Molecular cloning of multiple cDNAs encoding human enzymes structurally related to 3α-hydroxysteroid dehydrogenase. J Steroid Biochem Mol Biol. 1993;46:673–679. doi: 10.1016/0960-0760(93)90308-J. [DOI] [PubMed] [Google Scholar]
- Stolz A, Hammond L, Lou H, Takikawa H. et al. cDNA cloning and expression of the human hepatic bile acid-binding protein. A member of the monomeric reductase gene family. J Biol Chem. 1993;268:10448–10457. [PubMed] [Google Scholar]
- Penning TM, Jin Y, Heredia VV, Lewis M. Structure-function relationships in 3α-hydroxysteroid dehydrogenases: A comparison of the rat and human isoforms. J Steroid Biochem Mol Biol. 2003;85:247–255. doi: 10.1016/S0960-0760(03)00236-X. [DOI] [PubMed] [Google Scholar]
- Penning TM, Jin Y, Steckelbroeck S, Lanisnik-Rizner T, Lewis M. Structure-function of human 3α-hydroxysteroid dehydrogenases: Genes and proteins. Mol Cell Endocrinol. 2004;215:63–72. doi: 10.1016/j.mce.2003.11.006. [DOI] [PubMed] [Google Scholar]
- Praml C, Savelyeva L, Schwab M. Aflatoxin B1 aldehyde reductase (AFAR) genes cluster at 1p35-1p36.1 in a region frequently altered in human tumour cells. Oncogene. 2003;22:4765–4773. doi: 10.1038/sj.onc.1206684. [DOI] [PubMed] [Google Scholar]
- Blanchard RL, Freimuth RR, Buck J, Weinshilbourn RM, Coughtrie MW. A proposed nomenclature system for the cytosolic sulfotransferase (SULT) superfamily. Pharmacogenetics. 2004;14:199–211. doi: 10.1097/00008571-200403000-00009. [DOI] [PubMed] [Google Scholar]
- Kwok PY, Deng Q, Zakeri H, Taylor SL, Nickerson DA. Increasing the information content of STS-based genome maps: Identifying polymorphisms in mapped STSs. Genomics. 1996;31:123–126. doi: 10.1006/geno.1996.0019. [DOI] [PubMed] [Google Scholar]
- Wang DG, Fan JB, Siao CJ, Berno A. et al. Large-scale identification, mapping, and genotyping of single-nucleotide polymorphisms in the human genome. Science. 1998;280:1077–1082. doi: 10.1126/science.280.5366.1077. [DOI] [PubMed] [Google Scholar]
- Mattick JS, Makunin IV. Non-coding RNA. Hum Mol Genet. 2006;15:R17–R29. doi: 10.1093/hmg/ddl046. [DOI] [PubMed] [Google Scholar]