

Abstract
The pathogenesis of allergic asthma in childhood remains poorly understood. Environmental factors which appear to contribute to allergic sensitisation, with development of a Th2-biased immunological response in genetically predisposed individuals, include wheezing lower respiratory viral infections in early life and exposure to airborne environmental pollutants. These may activate pattern recognition receptors and/or cause oxidant injury to airway epithelial cells (AECs). In turn, this may promote Th2 polarisation via a “final common pathway” involving interaction between AEC, dendritic cells, and CD4+ T lymphocytes. Potentially important cytokines produced by AEC include thymic stromal lymphopoietin and interleukin-25. Their role is supported by in vitro studies using human AEC, as well as by experiments in animal models. To date, however, few investigations have employed models of the induction phase of childhood asthma. Further research may help to identify interventions that could reduce the risk of allergic asthma.
1. Introduction
Asthma is one of the most common chronic diseases affecting children, especially in economically developed nations. For example, in Australia the prevalence of current asthma in children aged 0–15 years is approximately 11% [1]. Childhood asthma is strongly linked to atopy, which in turn is characteristically associated with a Th2-biased immunological response [2–4]. While this relationship is well documented, the pathogenesis of childhood asthma remains largely unexplained.
However, it is clear that both genetic predisposition and a variety of environmental factors contribute to the development of allergic asthma [4]. Notable among the environmental factors that appear to be crucial in the induction of disease is respiratory viral infection, in particular with rhinovirus (RV) or respiratory syncytial virus (RSV). The association between childhood infections and asthma is complex, because at least in some settings, repeated early-life exposure to infectious agents may reduce the likelihood of developing allergic diseases [5]. Despite this, epidemiological studies strongly suggest that lower respiratory viral infections associated with wheezing, occurring within a critical period of development in early childhood, play an important role in the subsequent development of asthma in children who are repeatedly exposed to inhaled allergens [6–10].
Another clearly defined risk factor for childhood allergic asthma is early-life exposure to airborne environmental irritants. The importance of exposure to environmental tobacco smoke is well established [11, 12]. More recently, a number of large population-based studies, including prospective cohort studies, have clearly defined the increased risk of development of asthma in children exposed to traffic-related particulate pollutants [13–15]. The adverse respiratory effects of such pollutants, especially diesel exhaust particulates (DEPs), are now recognised as a significant public health problem [16].
Somewhat more contentious is the association between the use of paracetamol (acetaminophen) in infancy or childhood and the subsequent development of asthma [17, 18]. The evidence for an increased risk of asthma following exposures to other environmental chemicals is much less convincing [19].
Fundamental questions remain unanswered about the underlying mechanisms by which environmental factors promote the development of childhood asthma. In particular, if atopy and a Th2-biased immunological response are indeed precursors to the development of childhood asthma, a key issue is how does injury by environmental factors drive an allergic response?
A possible “final common pathway,” for which there is now growing support, is based on the interaction between airway epithelial cells, dendritic cells, and CD4+ T-lymphocytes.
2. Driving a Th2-Biased Immunological Response
Dendritic cells (DCs) have long been recognised as playing a crucial role in the induction of Th2 polarisation during an immunological response [20]. As is increasingly being understood, the development of allergic immunological responses may be determined by innate host defence responses after initial exposure to pathogens, allergens, or other irritants [21–23]. These lead to local generation of cytokines that stimulate DC, with effects including upregulation of the expression of costimulatory molecules such as CD40, CD80, CD86, Jagged-1, and OX40L, as well as the production of various chemokines (Figure 1) [24–27]. Such molecules collectively function as “instructive” signals that drive initial Th2 polarisation. The subsequent maintenance of the Th2 bias of the CD4+ T cells may be dependent on epigenetic changes [28, 29], which is now a focus of considerable interest in the study of the pathogenesis of asthma [30, 31].
Figure 1.
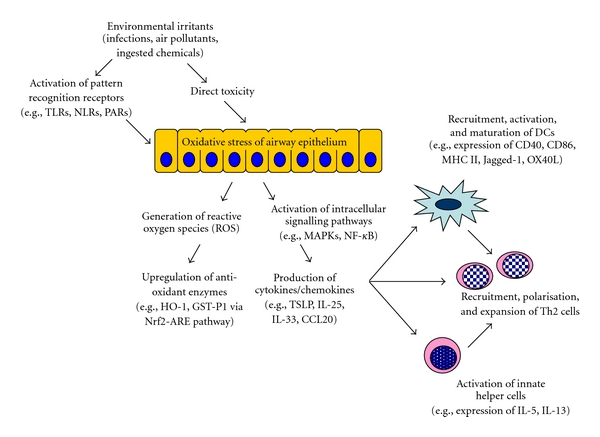
Environmental irritants may activate various pattern recognition receptors on AEC or may be directly toxic to the cells. Exposure to such irritants frequently causes oxidant injury to AEC, leading to generation of reactive oxygen species and upregulation of antioxidant enzyme systems. In parallel, intracellular signalling pathways are activated, triggering production of cytokines that can recruit and stimulate DC, upregulate their expression of costimulatory molecules and promote Th2 polarisation of the CD4+ T-cell response. These cytokines may also activate various populations of innate helper cells, leading to expansion of the Th2 cell population and helping to drive the allergic inflammatory response.
Key factors that may activate and/or drive the maturation of conventional or myeloid DC, to promote Th2-biased differentiation of CD4+ T-lymphocytes, include the cytokines granulocyte-macrophage colony-stimulating factor (GM-CSF), thymic stromal lymphopoietin (TSLP), interleukin (IL)-25 and IL-33 [32]. Because airway epithelial cells (AEC) can secrete GM-CSF, TSLP, IL-25, and IL-33 in response to injury, as well as chemoattractants for DC such as CCL20, the airway epithelium appears likely to play a critical role in promoting recruitment/survival of DC and Th2 polarisation of the immune response (Figure 1) [33–35].
2.1. Role of TSLP
Accumulating evidence indicates that TSLP activates DC to prime CD4+ T cells for inflammatory Th2 differentiation [36]. In the context of the induction of childhood asthma, a role for AEC-derived TSLP in the induction phase is strongly supported by in vitro studies. Following exposure to DEP in vitro, human AEC generated reactive oxygen species (ROS) and secreted TSLP, which caused DC precursors to enhance expression of OX40L and Jagged-1, which in turn promoted upregulation of Th2 responses [37, 38]. Similarly, AEC exposed to double-stranded RNA or infected with RV or RSV in vitro exhibited marked upregulation of expression of TSLP [39, 40]. In vivo, transgenic overexpression of TSLP in the lungs promoted an antigen-driven allergic inflammatory response [41]. Injury by proteases has also been shown to elicit generation of ROS by epithelial cells, leading to oxidation of lipids, signalling through Toll-like receptor (TLR) 4, and the production of TSLP that drives Th2 responses following subcutaneous immunisation [42]. Because many allergens exhibit endogenous protease activity, it is therefore also possible that inhaled allergens might themselves contribute to enhanced expression of TSLP by AEC.
To date, however, there are no reported in vitro studies using AEC isolated from children nor have there been studies on the role of TSLP in an animal model of childhood asthma.
Further support for a role for TSLP in the development of asthma comes from studies associating single nucleotide polymorphisms in the TSLP gene or its promoter region with an increased risk of developing asthma in childhood [43, 44].
2.2. Role of IL-25
Epithelial cell-derived IL-25 is increasingly recognised as being important in the induction of allergic inflammation [45]. TSLP-activated DC induce strong upregulation of the receptor for IL-25 on Th2 cells, thus linking these two cytokine pathways [46]. Recent studies have identified novel populations of cells involved in the innate host response, which contribute to maintaining and enhancing the Th2-biased response by secreting cytokines such as IL-5 and IL-13 in response to IL-25 [47].
We have shown that IL-25 produced by AEC plays a key role in the induction of a Th2-biased inflammatory response following respiratory viral infection [35]. Furthermore, we have recently provided convincing evidence that IL-25 is of crucial importance in the induction phase of childhood asthma. For the latter studies, we used a novel animal model of neonatal infection with pneumonia virus of mice (PVM), a species-specific paramyxovirus which simulates RSV infection in human infants [48]. Following subsequent intranasal sensitisation with ovalbumin and long-term low-level challenge, these mice developed inflammation and remodelling typical of chronic asthma, together with a Th2-biased immunological response [49]. By themselves, neither infection nor allergen exposure led to the development of an asthmatic phenotype. This model therefore simulates the interaction between early childhood infection with RSV and sensitisation to inhaled allergens during the development of childhood asthma.
In this model, we found that there was significant upregulation of expression of IL-25 following neonatal infection with PVM. Therefore, we tested the effects of administration of a neutralising antibody to IL-25 on the development of the asthmatic phenotype, in comparison to administration of an antibody to IL-4, which has long been recognised as having a crucial role in the induction of Th2 responses [50]. Anti-IL-25, administered either during chronic challenge or in early life alone, prevented key changes of airway remodelling such as subepithelial fibrosis and epithelial hypertrophy, and suppressed development of a Th2 response. Anti-IL-4 was more effective in inhibiting allergic inflammation, prevented goblet cell change but not other features of remodelling, and also suppressed development of a Th2 response [51]. These novel findings suggest that blocking induction of a Th2 response during the neonatal period or later in childhood could be effective for primary prevention of asthma, and that IL-25 might play a crucial role in this process.
2.3. Role of Other Factors
GM-CSF clearly plays a key role in the development of DC [52] and differentiated human respiratory epithelial cells release significant amounts of GM-CSF when exposed to DEP in vitro [53]. However, there is currently no direct evidence of a role for GM-CSF produced by AEC during the induction phase of childhood asthma.
There has been much speculation about the possible contribution of IL-33 to the development of asthma, given the capacity of this cytokine to promote DC maturation towards a Th2-inducing phenotype in vitro [54]. While IL-33 is expressed by airway epithelium in asthmatics [55], there is once again a paucity of direct evidence for a role in the induction phase of allergic asthma.
3. Triggering Cytokine Release by Airway Epithelial Cells
There is now considerable evidence that an important mechanism by which environmental irritants cause injury to AEC is via inducing the production of reactive oxygen species (ROS). This has been well studied in vitro in response to injury by RSV [56, 57]. Furthermore, activation of antioxidant defence mechanisms, through binding of the transcription factor Nrf2 to antioxidant response elements (AREs), has been shown to be an important part of the host response to RSV infection in vivo. RSV infection caused induction of various ARE-driven enzymes, and gene-targeted mice deficient in Nrf2 had significantly lower levels of reduced glutathione in the lungs, higher levels of oxidative modification of lung proteins and lipids, and developed significantly more severe RSV disease, both in terms of inflammation and epithelial injury [58]. Similarly, in vitro studies have demonstrated that DEP induce oxidative stress, with greater effects on AEC than on other target cells such as pulmonary macrophages [59]. There is clear evidence that paracetamol (acetaminophen) decreases intracellular levels of reduced glutathione and thus predisposes to injury by ROS [60].
Oxidative stress leads to activation of key intracellular signalling mechanisms, for example, the mitogen-activated protein kinase (MAPK) and NF-κB pathways, and these can be inhibited by administration of antioxidants [61, 62]. Activation in turn leads to the generation of chemoattractants and proinflammatory cytokines. Whether similar events occur in vivo during the induction phase of paediatric asthma has not been formally demonstrated. However, the concept that oxidant injury may be a key early event is supported by evidence that functional polymorphisms in oxidant defence genes increase the risk of developing asthma in childhood [63, 64].
Environmental irritants may also trigger AEC via pattern recognition receptors, including the predominantly cell surface or endosomal TLRs and the cytoplasmic Nod-like receptors (NLRs). These receptors are key components of host innate defences, capable of recognising conserved molecular patterns associated with pathogens or with cellular damage [65]. TLRs signal via adaptor proteins, notably MyD88, leading to activation of MAPK and NF-κB signalling pathways [66]. Whether TLR-dependent signalling promotes the development of a Th2-biased response following exposure to viral infection or environmental irritants is not altogether clear. However, the potential importance of TLR-mediated responses is supported by evidence that polymorphisms in genes for TLR2 and TLR4 are related to increased prevalence of asthma from birth up to the age of 8 years [67]. Members of the NLR family assemble into large multiprotein complexes, termed inflammasomes, which activate caspase-1, a proteolytic enzyme that cleaves and thus activates cytokines such as IL-1β and IL-18 for secretion [65]. Expression of NLRs has been demonstrated in the airway epithelium [68], but their role in the response to injury by environmental irritants, or in driving allergic inflammation, has not hitherto been investigated.
Another potentially important mechanism by which cytokine release from AEC might be triggered is via the enzymatic activity of some allergens on protease-activated receptors. For example, fungal antigens appear to be able to elicit secretion of TSLP via PAR-2 activation [69].
4. A Proasthmatic Epithelial Phenotype
An intriguing question, especially in relation to early-life viral infection, is whether environmental injury might lead to induction of a relatively stable epithelial phenotype that characterises and/or promotes the development of asthma [70]. There is no doubt that the airway epithelium of asthmatics is different to that of nonasthmatics, with evidence of abnormal proliferation/repair and enhanced production of proinflammatory cytokines [71–73]. Notably, this includes enhanced expression of TSLP and GM-CSF by asthmatic AEC [74, 75]. Furthermore, impaired production of interferons has been related to the increased susceptibility of asthmatics to viral infections [76, 77]. However, what is not clear is whether this is a reason for development of asthma, or an effect of a predisposing factor or of asthma itself. The issue is relevant to the ongoing debate about the relationship between viral infection and asthma [78]. Interestingly, recent evidence suggests that in a Th2-biased environment, which induces IL-13-driven mucous cell change in the airway epithelium, the altered epithelial lining may be more susceptible to infection by rhinovirus [79]. Thus, the mucous cell hyperplasia and other remodelling of the airway epithelium associated with the development of childhood asthma [80] may predispose to progression of the asthmatic phenotype, by promoting a vicious cycle of viral and allergic inflammation.
5. Conclusion
Injury to AEC by environmental factors may have an important role in initiating a cascade of responses that can lead to the development of asthma in childhood. Oxidant stress by environmental injurious agents may activate a variety of intracellular signalling pathways, that in turn drive the synthesis and secretion of multiple cytokines able to promote the recruitment of T cells and their polarisation towards a Th2 cytokine-secreting phenotype. However, at present there are many gaps in our understanding of the pathogenetic sequence of events. Further studies in animal models of childhood asthma may help to identify interventions that could reduce the risk of allergic asthma. Caution is warranted, however, because the mechanisms are likely to be complex and targeting single cytokines or regulatory pathways is rarely successful in asthma [81, 82].
Acknowledgment
Work in the authors' laboratories is supported by grants from the National Health & Medical Research Council of Australia.
References
- 1.Marks GB, Poulos L, Ampon RD, Waters AM. Prevalence asthma in Australia 2008. Canberra. 2008:26–48. [Google Scholar]
- 2.Bacharier LB, Boner A, Carlsen KH, et al. Diagnosis and treatment of asthma in childhood: a PRACTALL consensus report. Allergy. 2008;63(1):5–34. doi: 10.1111/j.1398-9995.2007.01586.x. [DOI] [PubMed] [Google Scholar]
- 3.Anderson GP. Endotyping asthma: new insights into key pathogenic mechanisms in a complex, heterogeneous disease. The Lancet. 2008;372(9643):1107–1119. doi: 10.1016/S0140-6736(08)61452-X. [DOI] [PubMed] [Google Scholar]
- 4.Sly PD, Boner AL, Björksten B, et al. Early identification of atopy in the prediction of persistent asthma in children. The Lancet. 2008;372(9643):1100–1106. doi: 10.1016/S0140-6736(08)61451-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Garn H, Renz H. Epidemiological and immunological evidence for the hygiene hypothesis. Immunobiology. 2007;212(6):441–452. doi: 10.1016/j.imbio.2007.03.006. [DOI] [PubMed] [Google Scholar]
- 6.Sigurs N, Gustafsson PM, Bjarnason R, et al. Severe respiratory syncytial virus bronchiolitis in infancy and asthma and allergy at age 13. American Journal of Respiratory and Critical Care Medicine. 2005;171(2):137–141. doi: 10.1164/rccm.200406-730OC. [DOI] [PubMed] [Google Scholar]
- 7.Kristjansson S, Bjarnarson SP, Wennergren G, et al. Respiratory syncytial virus and other respiratory viruses during the first 3 months of life promote a local TH2-like response. Journal of Allergy and Clinical Immunology. 2005;116(4):805–811. doi: 10.1016/j.jaci.2005.07.012. [DOI] [PubMed] [Google Scholar]
- 8.Kusel MMH, de Klerk NH, Kebadze T, et al. Early-life respiratory viral infections, atopic sensitization, and risk of subsequent development of persistent asthma. Journal of Allergy and Clinical Immunology. 2007;119(5):1105–1110. doi: 10.1016/j.jaci.2006.12.669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jackson DJ, Gangnon RE, Evans MD, et al. Wheezing rhinovirus illnesses in early life predict asthma development in high-risk children. American Journal of Respiratory and Critical Care Medicine. 2008;178(7):667–672. doi: 10.1164/rccm.200802-309OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wu P, Dupont WD, Griffin MR, et al. Evidence of a causal role of winter virus infection during infancy in early childhood asthma. American Journal of Respiratory and Critical Care Medicine. 2008;178(11):1123–1129. doi: 10.1164/rccm.200804-579OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gilmour MI, Jaakkola MS, London SJ, Nel AE, Rogers CA. How exposure to environmental tobacco smoke, outdoor air pollutants, and increased pollen burdens influences the incidence of asthma. Environmental Health Perspectives. 2006;114(4):627–633. doi: 10.1289/ehp.8380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wigle DT, Arbuckle TE, Turner MC, et al. Epidemiologic evidence of relationships between reproductive and child health outcomes and environmental chemical contaminants. Journal of Toxicology and Environmental Health—Part B. 2008;11(5-6):373–517. doi: 10.1080/10937400801921320. [DOI] [PubMed] [Google Scholar]
- 13.Gehring U, Wijga AH, Brauer M, et al. Traffic-related air pollution and the development of asthma and allergies during the first 8 years of life. American Journal of Respiratory and Critical Care Medicine. 2010;181(6):596–603. doi: 10.1164/rccm.200906-0858OC. [DOI] [PubMed] [Google Scholar]
- 14.McConnell R, Islam T, Shankardass K, et al. Childhood incident asthma and traffic-related air pollution at home and school. Environmental Health Perspectives. 2010;118(7):1021–1026. doi: 10.1289/ehp.0901232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pénard-Morand C, Raherison C, Charpin D, et al. Long-term exposure to close-proximity air pollution and asthma and allergies in urban children. European Respiratory Journal. 2010;36(1):33–40. doi: 10.1183/09031936.00116109. [DOI] [PubMed] [Google Scholar]
- 16.Balmes JR. How does diesel exhaust impact asthma? Thorax. 2011;66(1):4–6. doi: 10.1136/thx.2010.145391. [DOI] [PubMed] [Google Scholar]
- 17.Beasley R, Clayton T, Crane J, et al. Association between paracetamol use in infancy and childhood, and risk of asthma, rhinoconjunctivitis, and eczema in children aged 6-7 years: analysis from Phase Three of the ISAAC programme. The Lancet. 2008;372(9643):1039–1048. doi: 10.1016/S0140-6736(08)61445-2. [DOI] [PubMed] [Google Scholar]
- 18.Amberbir A, Medhin G, Alem A, Britton J, Davey G, Venn A. The role of acetaminophen and geohelminth infection on the incidence of wheeze and eczema: a longitudinal birth-cohort study. American Journal of Respiratory and Critical Care Medicine. 2011;183(2):165–170. doi: 10.1164/rccm.201006-0989OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dietert RR, Zelikoff JT. Early-life environment, developmental immunotoxicology, and the risk of pediatric allergic disease including asthma. Birth Defects Research Part B—Developmental and Reproductive Toxicology. 2008;83(6):547–560. doi: 10.1002/bdrb.20170. [DOI] [PubMed] [Google Scholar]
- 20.Vermaelen K, Pauwels R. Pulmonary dendritic cells. American Journal of Respiratory and Critical Care Medicine. 2005;172(5):530–551. doi: 10.1164/rccm.200410-1384SO. [DOI] [PubMed] [Google Scholar]
- 21.Sabroe I, Parker LC, Dockrell DH, Davies DE, Dower SK, Whyte MKB. Targeting the networks that underpin contiguous immunity in asthma and chronic obstructive pulmonary disease. American Journal of Respiratory and Critical Care Medicine. 2007;175(4):306–311. doi: 10.1164/rccm.200606-777PP. [DOI] [PubMed] [Google Scholar]
- 22.Holt PG, Strickland DH. Interactions between innate and adaptive immunity inasthma pathogenesis: new perspectives from studies onacute exacerbations. Journal of Allergy and Clinical Immunology. 2010;125(5):963–972. doi: 10.1016/j.jaci.2010.02.011. [DOI] [PubMed] [Google Scholar]
- 23.Kaiko GE, Foster PS. New insights into the generation of Th2 immunity and potential therapeutic targets for the treatment of asthma. Current Opinion in Allergy and Clinical Immunology. 2011;11(1):39–45. doi: 10.1097/ACI.0b013e328342322f. [DOI] [PubMed] [Google Scholar]
- 24.Amsen D, Blander JM, Lee GR, Tanigaki K, Honjo T, Flavell RA. Instruction of distinct CD4 T helper cell fates by different notch ligands on antigen-presenting cells. Cell. 2004;117(4):515–526. doi: 10.1016/s0092-8674(04)00451-9. [DOI] [PubMed] [Google Scholar]
- 25.von Garnier C, Wikstrom ME, Zosky G, et al. Allergic airways disease develops after an increase in allergen capture and processing in the airway mucosa. The Journal of Immunology. 2007;179(9):5748–5759. doi: 10.4049/jimmunol.179.9.5748. [DOI] [PubMed] [Google Scholar]
- 26.Lloyd CM. Dust mites’ dirty dealings in the lung. Nature Medicine. 2009;15(4):366–367. doi: 10.1038/nm0409-366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lambrecht BN, Hammad H. Biology of lung dendritic cells at the origin of asthma. Immunity. 2009;31(3):412–424. doi: 10.1016/j.immuni.2009.08.008. [DOI] [PubMed] [Google Scholar]
- 28.Janson PCJ, Winerdal ME, Winqvist O. At the crossroads of T helper lineage commitment-Epigenetics points the way. Biochimica et Biophysica Acta. 2009;1790(9):906–919. doi: 10.1016/j.bbagen.2008.12.003. [DOI] [PubMed] [Google Scholar]
- 29.van Panhuys N, Le Gros G, McConnell MJ. Epigenetic regulation of Th2 cytokine expression in atopic diseases. Tissue Antigens. 2008;72(2):91–97. doi: 10.1111/j.1399-0039.2008.01068.x. [DOI] [PubMed] [Google Scholar]
- 30.Kumar RK, Hitchins MP, Foster PS. Epigenetic changes in childhood asthma. DMM Disease Models and Mechanisms. 2009;2(11-12):549–553. doi: 10.1242/dmm.001719. [DOI] [PubMed] [Google Scholar]
- 31.Martino D, Prescott S. Epigenetics and prenatal influences on asthma and allergic airways disease. Chest. 2011;139(3):640–647. doi: 10.1378/chest.10-1800. [DOI] [PubMed] [Google Scholar]
- 32.Holt PG, Strickland DH, Bosco A, Jahnsen FL. Pathogenic mechanisms of allergic inflammation: atopic asthma as a paradigm. Advances in immunology. 2009;104:51–113. doi: 10.1016/S0065-2776(08)04003-0. [DOI] [PubMed] [Google Scholar]
- 33.Weckmann M, Collison A, Simpson JL, et al. Critical link between TRAIL and CCL20 for the activation of TH2 cells and the expression of allergic airway disease. Nature Medicine. 2007;13(11):1308–1315. doi: 10.1038/nm1660. [DOI] [PubMed] [Google Scholar]
- 34.Hammad H, Chieppa M, Perros F, Willart MA, Germain RN, Lambrecht BN. House dust mite allergen induces asthma via Toll-like receptor 4 triggering of airway structural cells. Nature Medicine. 2009;15(4):410–416. doi: 10.1038/nm.1946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kaiko GE, Phipps S, Angkasekwinai P, Dong C, Foster PS. NK cell deficiency predisposes to viral-induced Th2-type allergic inflammation via epithelial-derived IL-25. The Journal of Immunology. 2010;185(8):4681–4690. doi: 10.4049/jimmunol.1001758. [DOI] [PubMed] [Google Scholar]
- 36.Ziegler SF, Artis D. Sensing the outside world: TSLP regulates barrier immunity. Nature Immunology. 2010;11(4):289–293. doi: 10.1038/ni.1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bleck B, Tse DB, Curotto De Lafaille MA, Zhang F, Reibman J. Diesel exhaust particle-exposed human bronchial epithelial cells induce dendritic cell maturation and polarization via thymic stromal lymphopoietin. Journal of Clinical Immunology. 2008;28(2):147–156. doi: 10.1007/s10875-007-9149-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bleck B, Tse DB, Gordon T, Ahsan MR, Reibman J. Diesel exhaust particle-treated human bronchial epithelial cells upregulate Jagged-1 and OX40 ligand in myeloid dendritic cells via thymic stromal lymphopoietin. The Journal of Immunology. 2010;185(11):6636–6645. doi: 10.4049/jimmunol.1000719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kato A, Favoreto S, Avila PC, Schleimer RP. TLR3- and Th2 cytokine-dependent production of thymic stromal lymphopoietin in human airway epithelial cells. The Journal of Immunology. 2007;179(2):1080–1087. doi: 10.4049/jimmunol.179.2.1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Qiao J, Li A, Jin X. TSLP from RSV-stimulated rat airway epithelial cells activates myeloid dendritic cells. Immunology and Cell Biology. 2010;89:231–238. doi: 10.1038/icb.2010.85. [DOI] [PubMed] [Google Scholar]
- 41.Headley MB, Zhou B, Shih WX, Aye T, Comeau MR, Ziegler SF. TSLP conditions the lung immune environment for the generation of pathogenic innate and antigen-specific adaptive immune responses. The Journal of Immunology. 2009;182(3):1641–1647. doi: 10.4049/jimmunol.182.3.1641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tang H, Cao W, Kasturi SP, et al. The T helper type 2 response to cysteine proteases requires dendritic cell-basophil cooperation via ROS-mediated signaling. Nature Immunology. 2010;11(7):608–617. doi: 10.1038/ni.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hunninghake GM, Soto-Quirós ME, Avila L, et al. TSLP polymorphisms are associated with asthma in a sex-specific fashion. Allergy. 2010;65(12):1566–1575. doi: 10.1111/j.1398-9995.2010.02415.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Harada M, Hirota T, Jodo AI, et al. Thymic stromal lymphopoietin gene promoter polymorphisms are associated with susceptibility to bronchial asthma. American Journal of Respiratory Cell and Molecular Biology. 2011;44:787–793. doi: 10.1165/rcmb.2009-0418OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bulek K, Swaidani S, Aronica M, Li X. Epithelium: the interplay between innate and Th2 immunity. Immunology and Cell Biology. 2010;88(3):257–268. doi: 10.1038/icb.2009.113. [DOI] [PubMed] [Google Scholar]
- 46.Wang YH, Liu YJ. Thymic stromal lymphopoietin, OX40-ligand, and interleukin-25 in allergic responses. Clinical and Experimental Allergy. 2009;39(6):798–806. doi: 10.1111/j.1365-2222.2009.03241.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Saenz SA, Noti M, Artis D. Innate immune cell populations function as initiators and effectors in Th2 cytokine responses. Trends in Immunology. 2010;31(11):407–413. doi: 10.1016/j.it.2010.09.001. [DOI] [PubMed] [Google Scholar]
- 48.Domachowske JB, Bonville CA, Rosenberg HF. Animal models for studying respiratory syncytial virus infection and its long term effects on lung function. Pediatric Infectious Disease Journal. 2004;23(11):S228–S234. doi: 10.1097/01.inf.0000144672.81955.a4. [DOI] [PubMed] [Google Scholar]
- 49.Siegle JS, Hansbro N, Herbert C, et al. Interaction between early-life viral infection and allergen exposure induces an asthmatic phenotype in mice. Respirology. 2009;14, article A16 doi: 10.1186/1465-9921-11-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Paul WE, Zhu J. How are TH2-type immune responses initiated and amplified? Nature Reviews Immunology. 2010;10(4):225–235. doi: 10.1038/nri2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Siegle JS, Hansbro N, Dong C, Angkasekwinai P, Foster PS, Kumar RK. Blocking induction of T helper type 2 responses prevents development of disease in a model of childhood asthma. Clinical and Experimental Immunology. 2011;165(1):19–28. doi: 10.1111/j.1365-2249.2011.04392.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Watowich SS, Liu YJ. Mechanisms regulating dendritic cell specification and development. Immunological Reviews. 2010;238(1):76–92. doi: 10.1111/j.1600-065X.2010.00949.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Auger F, Gendron MC, Chamot C, Marano F, Dazy AC. Responses of well-differentiated nasal epithelial cells exposed to particles: role of the epithelium in airway inflammation. Toxicology and Applied Pharmacology. 2006;215(3):285–294. doi: 10.1016/j.taap.2006.03.002. [DOI] [PubMed] [Google Scholar]
- 54.Borish L, Steinke JW. Interleukin-33 in asthma: how big of a role does it play? Current Allergy and Asthma Reports. 2010;11:7–11. doi: 10.1007/s11882-010-0153-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Préfontaine D, Nadigel J, Chouiali F, et al. Increased IL-33 expression by epithelial cells in bronchial asthma. Journal of Allergy and Clinical Immunology. 2010;125(3):752–754. doi: 10.1016/j.jaci.2009.12.935. [DOI] [PubMed] [Google Scholar]
- 56.Hosakote YM, Liu T, Castro SM, Garofalo RP, Casola A. Respiratory syncytial virus induces oxidative stress by modulating antioxidant enzymes. American Journal of Respiratory Cell and Molecular Biology. 2009;41(3):348–357. doi: 10.1165/rcmb.2008-0330OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mochizuki H, Todokoro M, Arakawa H. RS virus-induced inflammation and the intracellular glutathione redox state in cultured human airway epithelial cells. Inflammation. 2009;32(4):252–264. doi: 10.1007/s10753-009-9128-0. [DOI] [PubMed] [Google Scholar]
- 58.Cho HY, Imani F, Miller-DeGraff L, et al. Antiviral activity of Nrf2 in a murine model of respiratory syncytial virus disease. American Journal of Respiratory and Critical Care Medicine. 2009;179(2):138–150. doi: 10.1164/rccm.200804-535OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Li N, Wang M, Oberley TD, Sempf JM, Nel AE. Comparison of the pro-oxidative and proinflammatory effects of organic diesel exhaust particle chemicals in bronchial epithelial cells and macrophages. The Journal of Immunology. 2002;169(8):4531–4541. doi: 10.4049/jimmunol.169.8.4531. [DOI] [PubMed] [Google Scholar]
- 60.James LP, Mayeux PR, Hinson JA. Acetaminophen-induced hepatotoxicity. Drug Metabolism and Disposition. 2003;31(12):1499–1506. doi: 10.1124/dmd.31.12.1499. [DOI] [PubMed] [Google Scholar]
- 61.Hashimoto S, Gon Y, Takeshita I, et al. Diesel exhaust particles activate p38 MAP kinase to produce interleukin 8 and RANTES by human bronchial epithelial cells and N-acetylcysteine attenuates p38 MAP kinase activation. American Journal of Respiratory and Critical Care Medicine. 2000;161(1):280–285. doi: 10.1164/ajrccm.161.1.9904110. [DOI] [PubMed] [Google Scholar]
- 62.Takizawa H, Abe S, Okazaki H, et al. Diesel exhaust particles upregulate eotaxin gene expression in human bronchial epithelial cells via nuclear factor-κB-dependent pathway. American Journal of Physiology. 2003;284(6):L1055–L1062. doi: 10.1152/ajplung.00358.2002. [DOI] [PubMed] [Google Scholar]
- 63.Salam MT, Lin PC, Avol EL, Gauderman WJ, Gilliland FD. Microsomal epoxide hydrolase, glutathione S-transferase P1, traffic and childhood asthma. Thorax. 2007;62(12):1050–1057. doi: 10.1136/thx.2007.080127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Castro-Giner F, Künzli N, Jacquemin B, et al. Traffic-related air pollution, oxidative stress genes, and asthma (ECHRS) Environmental Health Perspectives. 2009;117(12):1919–1924. doi: 10.1289/ehp.0900589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fukata M, Vamadevan AS, Abreu MT. Toll-like receptors (TLRs) and Nod-like receptors (NLRs) in inflammatory disorders. Seminars in Immunology. 2009;21(4):242–253. doi: 10.1016/j.smim.2009.06.005. [DOI] [PubMed] [Google Scholar]
- 66.Li X, Jiang S, Tapping RI. Toll-like receptor signaling in cell proliferation and survival. Cytokine. 2010;49(1):1–9. doi: 10.1016/j.cyto.2009.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kerkhof M, Postma DS, Brunekreef B, et al. Toll-like receptor 2 and 4 genes influence susceptibility to adverse effects of traffic-related air pollution on childhood asthma. Thorax. 2010;65(8):690–697. doi: 10.1136/thx.2009.119636. [DOI] [PubMed] [Google Scholar]
- 68.Bogefors J, Rydberg C, Uddman R, et al. Nod1, Nod2 and Nalp3 receptors, new potential targets in treatment of allergic rhinitis? Allergy. 2010;65(10):1222–1226. doi: 10.1111/j.1398-9995.2009.02315.x. [DOI] [PubMed] [Google Scholar]
- 69.Kouzaki H, O’Grady SM, Lawrence CB, Kita H. Proteases induce production of thymic stromal lymphopoietin by airway epithelial cells through protease-activated receptor-2. The Journal of Immunology. 2009;183(2):1427–1434. doi: 10.4049/jimmunol.0900904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Holtzman MJ, Tyner JW, Kim EY, et al. Acute and chronic airway responses to viral infection: implications for asthma and chronic obstructive pulmonary disease. Proceedings of the American Thoracic Society. 2005;2(2):132–140. doi: 10.1513/pats.200502-015AW. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lordan JL, Bucchieri F, Richter A, et al. Cooperative effects of Th2 cytokines and allergen on normal and asthmatic bronchial epithelial cells. The Journal of Immunology. 2002;169(1):407–414. doi: 10.4049/jimmunol.169.1.407. [DOI] [PubMed] [Google Scholar]
- 72.Bosson J, Stenfors N, Bucht A, et al. Ozone-induced bronchial epithelial cytokine expression differs between healthy and asthmatic subjects. Clinical and Experimental Allergy. 2003;33(6):777–782. doi: 10.1046/j.1365-2222.2003.01662.x. [DOI] [PubMed] [Google Scholar]
- 73.Stevens PT, Kicic A, Sutanto EN, Knight DA, Stick SM. Dysregulated repair in asthmatic paediatric airway epithelial cells: the role of plasminogen activator inhibitor-1. Clinical and Experimental Allergy. 2008;38(12):1901–1910. doi: 10.1111/j.1365-2222.2008.03093.x. [DOI] [PubMed] [Google Scholar]
- 74.Corrigan CJ, Wang W, Meng Q, et al. Allergen-induced expression of IL-25 and IL-25 receptor in atopic asthmatic airways and late-phase cutaneous responses. Journal of Allergy and Clinical Immunology. 2011;128(1):119–124. doi: 10.1016/j.jaci.2011.03.043. [DOI] [PubMed] [Google Scholar]
- 75.Hackett TL, Singhera GK, Shaheen F, et al. Intrinsic phenotypic differences of asthmatic epithelium and its inflammatory responses to RSV and air pollution. doi: 10.1165/rcmb.2011-0031OC. American Journal of Respiratory Cell and Molecular Biology. In press. [DOI] [PubMed] [Google Scholar]
- 76.Wark PAB, Johnston SL, Bucchieri F, et al. Asthmatic bronchial epithelial cells have a deficient innate immune response to infection with rhinovirus. Journal of Experimental Medicine. 2005;201(6):937–947. doi: 10.1084/jem.20041901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Contoli M, Message SD, Laza-Stanca V, et al. Role of deficient type III interferon-λ production in asthma exacerbations. Nature Medicine. 2006;12(9):1023–1026. doi: 10.1038/nm1462. [DOI] [PubMed] [Google Scholar]
- 78.Sly PD, Kusel M, Holt PG. Do early-life viral infections cause asthma? Journal of Allergy and Clinical Immunology. 2010;125(6):1202–1205. doi: 10.1016/j.jaci.2010.01.024. [DOI] [PubMed] [Google Scholar]
- 79.Lachowicz-Scroggins ME, Boushey HA, Finkbeiner WE, Widdicombe JH. Interleukin-13-induced mucous metaplasia increases susceptibility of human airway epithelium to rhinovirus infection. American Journal of Respiratory Cell and Molecular Biology. 2010;43(6):652–661. doi: 10.1165/rcmb.2009-0244OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Rogers DF. Pulmonary mucus: pediatric perspective. Pediatric Pulmonology. 2003;36(3):178–188. doi: 10.1002/ppul.10322. [DOI] [PubMed] [Google Scholar]
- 81.Kumar RK, Herbert C, Webb DC, Li L, Foster PS. Effects of anticytokine therapy in a mouse model of chronic asthma. American Journal of Respiratory and Critical Care Medicine. 2004;170(10):1043–1048. doi: 10.1164/rccm.200405-681OC. [DOI] [PubMed] [Google Scholar]
- 82.Collison A, Herbert C, Siegle JS, Mattes J, Foster PS, Kumar RK. Altered expression of microRNA in the airway wall in chronic asthma: miR-126 as a potential therapeutic target. BMC Pulmonary Medicine. 2011;11, article 29 doi: 10.1186/1471-2466-11-29. [DOI] [PMC free article] [PubMed] [Google Scholar]