

Abstract
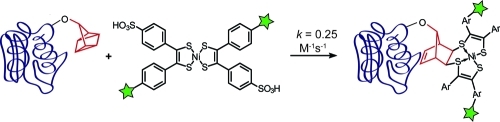
New additions to the bioorthogonal chemistry compendium can advance biological research by enabling multiplexed analysis of biomolecules in complex systems. Here we introduce the quadricyclane ligation, a new bioorthogonal reaction between the highly strained hydrocarbon quadricyclane and Ni bis(dithiolene) reagents. This reaction has a second-order rate constant of 0.25 M–1 s–1, on par with fast bioorthogonal reactions of azides, and proceeds readily in aqueous environments. Ni bis(dithiolene) probes selectively labeled quadricyclane-modified bovine serum albumin, even in the presence of cell lysate. We have demonstrated that the quadricyclane ligation is compatible with, and orthogonal to, strain-promoted azide–alkyne cycloaddition and oxime ligation chemistries by performing all three reactions in one pot on differentially functionalized protein substrates. The quadricyclane ligation joins a small but growing list of tools for the selective covalent modification of biomolecules.
Bioorthogonal chemistry is an enabling tool for biological research as well as a challenging frontier in synthetic methodology.(1) Bioorthogonal reaction development requires attention to functional group reactivity and selectivity, as in any methodology effort, but in addition one must consider issues of water stability, biocompatibility, and reaction kinetics in physiological settings. To invent reactions within the confines of such unusual boundary conditions, chemists have been compelled to explore manifolds of reactivity outside the mainstream of organic synthesis. Some notable success stories have come from adaptations of classic azide chemistry, such as the Staudinger ligation(2) and click chemistry (both Cu-catalyzed(3) and Cu-free versions(4)). These bioorthogonal reactions have made it possible to probe a particular biomolecule labeled with an azide using complementary reagents in vitro, in cells, and in live organisms.(5) However, multiplexed analysis of several biomolecules in a given system requires parallel use of a collection of bioorthogonal reactions. The toolkit has expanded considerably in recent years(1) but is still dwarfed by the compendium of conventional synthetic transformations, underscoring the need for new contributions.
In an effort to develop new bioorthogonal reactions that are themselves orthogonal to the current cohort, we sought to explore unrepresented reactivity space. The published bioorthogonal transformations represent four broad reaction types: 1,3-dipolar cycloadditions,(6) Diels–Alder reactions,(7) metal-catalyzed couplings,3,8 and nucleophilic additions.2,9 Outside of this space lies the [2 + 2 + 2] cycloaddition reaction, a popular choice for the rapid assembly of functionalized ring systems.(10) In practice, such reactions typically are metal-catalyzed as a means to overcome an otherwise significant entropic barrier. However, the highly strained hydrocarbon quadricyclane directly undergoes [2 + 2 + 2] cycloaddition with specific types of π systems (Figure 1A).(11)
Figure 1.

(A) A generic [2 + 2 + 2] reaction between quadricyclane and a π electrophile to yield a norbornene cycloaddition product. (B) 7-Acetoxyquadricyclane (1) reacts with bis(dithiobenzil)nickel(II) (2) to form complex 3. Complex 3 undergoes photodegradation to 2 and 7-acetoxynorbornadiene (4) unless diethyldithiocarbamate (5) is present.
Quadricyclane possesses many qualities that render it a promising bioorthogonal reagent. First, it is abiotic, and we envisioned that quadricyclane’s all sp3-hybridized carbon system would be unreactive with native biomolecules.(18) Second, the molecule is relatively small(12) and may therefore be amenable to biosynthetic incorporation into biomolecules. But what intrigued us most was quadricyclane’s reactivity profile. Its ∼80 kcal/mol of strain(13) promotes cycloaddition with diazodicarboxylates and other electron-deficient π systems under mild conditions.(11) By contrast, quadricyclane does not react readily with simple alkenes, alkynes, or even cyclooctynes. As well, previous work has shown that the rates of these cycloaddition reactions are greatly enhanced by the “on water” effect.(14)
To identify a bioorthogonal reaction partner for quadricyclane, we screened a variety of candidates for reactivity at room temperature in aqueous or polar aprotic solvents [Table S1 in the Supporting Information (SI)]. Diazodicarbonyl compounds showed promising kinetics but were not sufficiently stable in water for biological applications.(15) α-Fluoroketones and fluorinated alkenes and alkynes reacted with quadricyclane too slowly for practical utility, and some of these reagents were prone to side reactions. One reagent stood out as a promising lead: bis(dithiobenzil)nickel(II) (2), a Ni bis(dithiolene) complex that reacted cleanly with 7-acetoxyquadricyclane (1) to yield adduct 3 (Figure 1B). Our results were consistent with a 1986 report by Sugimori and co-workers highlighting the rapid reactivity of 2 with quadricyclane.(17) However, transforming this finding into a bioorthogonal reaction—a “quadricyclane ligation”—required that we address the central criteria of water stability, biocompatibility, and reaction kinetics in physiological settings.
First, we assayed the stability of 1 in water and in the presence of biological nucleophiles. We prepared 1 by the photochemical [2 + 2] reaction of 4 as reported previously(19) (Scheme S1 in the SI). Compound 1 was found to be stable in phosphate-buffered saline (PBS, pH 7.4), with no observed degradation after more than 2 months at room temperature (Figure S1 in the SI). We also found 1 to be unreactive with sugars, a variety of oxidants, and free amino acids, most notably cysteine (Figure S2). Furthermore, quadricyclane was stable in the presence of bovine serum albumin (BSA) and cell culture media under conditions emulating those necessary for metabolic labeling experiments. The limited solubility of commercially acquired 2 in polar solvents precluded a thorough stability study, but the compound was found to be unreactive with a variety of nucleophiles in dichloromethane.
We then turned our attention to the stability of norbornene 3, the product formed by ligation of 1 and 2 (Figure 1B). There are reports that quadricyclane’s adduct with 2 undergoes light-induced reversion to norbornadiene (an isomer of quadricyclane) and 2.(21) Accordingly, we exposed 3 to ambient light and monitored the formation of 2 and 4 (Figure 1B). The half-life of the reversion was ∼35 h in CDCl3 (Figures S3 and S4), which would be problematic for many biological labeling applications. We predicted that the photochemical reversion could be inhibited by removal of Ni from the product.(23) After screening a variety of metal chelators (Table S2), we found that diethyldithiocarbamate (5; Figure 1B) entirely prevented the photodegradation of 3 (Figure 1B and Figure S5).
With these promising results, we pursued the synthesis of a Ni bis(dithiolene) reagent that is both water-soluble and functionalized with a probe to detect biomolecule labeling. Compound 15 bearing two sulfonate groups and two biotin moieties was designed for this purpose (Scheme 1). We prepared 15 and a model compound, bis(isopropyl amide) 14, from dithiol-2-one ligand precursors.(24) Briefly, alkyne 6 was combined with xanthogen disulfide 7 in the presence of the radical initiator 1,1′-azobis(cyclohexanecarbonitrile) to yield dithiocarbonate 8.(26) Treatment of 8 with fuming sulfuric acid installed a single sulfonate group and also hydrolyzed the methyl ester, producing 9 in good yield. Standard amide bond-coupling conditions were used to conjugate either isopropylamine or an amine-functionalized biotin derivative to 9, affording 10 and 11, respectively. These intermediates were then converted to anionic Ni bis(dithiolene) species 12 and 13 in situ by treatment with tetramethylammonium hydroxide and NiCl2. Immediate oxidation with 0.5 equiv of iodine afforded the desired neutral complexes 14 and 15.(27) Gratifyingly, we found that 14 and 15 are soluble in PBS at concentrations up to 5 and 10 mM, respectively.
Scheme 1. Water-Soluble Ni Bis(dithiolene) Complexes.

The solubilities of compounds 14 and 15 enabled us to assess the reaction kinetics with quadricyclane in aqueous/organic solvent mixtures as well as to probe in more detail the stability of these Ni bis(dithiolene) complexes in the presence of biomolecules. Complex 14 has a large NIR absorption band at 850 nm that is not present in the adduct with quadricyclane, allowing for pseudo-first-order kinetic measurements by absorption spectroscopy. We found that the second-order rate constant for the reaction of 14 and 1 in a 1:1 PBS/EtOH mixture(28) was 0.25 ± 0.05 M–1 s–1 at room temperature (Figures S6 and S7). This rate constant is comparable to those for cyclooctyne–azide cycloadditions currently used for biological labeling applications(29) and should allow for the use of mild reaction conditions.
The stability of 14 was also monitored using the NIR absorption band, which is dependent on both the oxidation state of the complex (i.e., 14 vs 12) and the connectivity of the dithiolene ligands.(30) The absorption at 850 nm remained essentially unaltered when 14 was incubated in PBS for 20 h (Figure S8). As well, exposure to amino acids had either no effect or a minimal effect on the absorption intensity (Figure S9). A marked exception was cysteine, which caused an immediate decrease in the intensity of the 850 nm absorption band and the concomitant appearance of a new band at ∼900 nm (Figure S10). This transformation is consistent with reduction of 14 to 12,(30b) and treatment of 14 with other reducing agents yielded similar results (Figure S11). The neutral complex could be regenerated by addition of K3Fe(CN)6, as judged by the reappearance of the absorption band at 850 nm and restoration of the reactivity with 1 (Figures S12 and S13). Concerned that 14 would undergo redox reactions with free thiol groups of protein-associated cysteine residues, we monitored the integrity of the Ni bis(dithiolene) complex in the presence of BSA, which possesses a solvent-exposed cysteine side chain.(31) Incubation with BSA led to reduction of 14 but at a lower rate than observed with free cysteine (Figure S14). Therefore, protein-mediated reduction should not undermine the much faster quadricyclane ligation. Notably, 14 was stable toward oxidized insulin over a 5 h period (Figure S15).
The quadricyclane ligation was then subjected to the first critical test of bioorthogonality: selective protein labeling. We modified lysine residues on BSA with quadricyclane p-nitrophenyl carbonate 16 (Figure 2A).(32) We then treated either quadricyclane-modified BSA (QC-BSA) or native BSA with 50 μM 15 for various amounts of time and, after quenching with excess 5 and 1, assayed the products by Western blot probing with an α-biotin antibody conjugated to horseradish peroxidase (α-biotin HRP). We were excited to see that 15 selectively labeled QC-BSA in a time-dependent manner with very little background labeling of unmodified BSA, even upon prolonged exposure of the Western blot (Figure 2B). Similarly, when the reaction time was held constant (30 min) and the concentration of 15 was varied, dose-dependent labeling was observed, again with minimal nonspecific reactivity (Figure 2C). As well, pretreatment of QC-BSA with tetrasulfonated Ni bis(dithiolene) 17(33) quenched the protein-bound quadricyclane moiety, as demonstrated by reduced labeling with 15 (Figure 2D). To determine whether the quadricyclane ligation possessed the heightened selectivity required for labeling of target biomolecules within more complex samples, we combined 1.5 μg of QC-BSA (or unmodified BSA) with 25 μg of E. coli lysate. This mixture was treated with 50 μM 15 for 30 min, quenched as described above, and analyzed by Western blot. Selective labeling of QC-BSA was observed, but the signal was weak (Figure S16). We hypothesized that the diminished signal was due to reduction of 15 by a species present in the lysate. Gratifyingly, when we added K3Fe(CN)6 (1 mM) to the reaction mixture, robust and selective labeling of QC-BSA was observed (Figure 2E).
Figure 2.

Ni bis(dithiolene) reagents selectively label quadricyclane-modified BSA. (A) Modification of BSA’s solvent-exposed lysine residues with compound 16 and subsequent labeling with biotinylated Ni bis(dithiolene) reagent 15. (B, C) Ni bis(dithiolene) 15 displays (B) time- and (C) dose-dependent labeling of QC-BSA. BSA (−) or QC-BSA (+) (5 μg) was treated with (B) 50 μM 15 for various amounts of time or (C) various concentrations of 15 for 30 min. The reactions were quenched with 1 and 5, and the presence of product was detected by Western blot using α-biotin-HRP. (D) Reaction of 15 with QC-BSA can be prevented by pretreatment with tetrasulfonated Ni bis(dithiolene) 17. BSA (−) or QC-BSA (+) (5 μg) was treated with various amounts of 17 for 30 min followed by 50 μM 15 for 30 min. The reactions were quenched with 1 and 5, and the samples were analyzed by Western blot probing with α-biotin-HRP. (E) QC-BSA can be selectively labeled in a mixture of proteins. To 25 μg of lysate in the presence of 1 mM K3Fe(CN)6 was added no BSA or reagent (lane 1), BSA (1.5 μg) and 50 μM 15 (lane 2), and QC-BSA (1.5 μg) and 50 μM 15 (lane 3). After 30 min, the reactions were quenched with 1 and 5 and analyzed by Western blot. The BSA monomer and dimer bands are visible in the QC-BSA-treated sample. The bands denoted with asterisks represent a biotinylated E. coli protein. In (B–E), equal protein loading was verified by Ponceau stain.
New additions to the bioorthogonal reaction compendium are most powerful when they can be used in conjunction with other bioorthogonal chemistries. We therefore aimed to determine whether the quadricyclane ligation can be performed simultaneously with two established bioorthogonal reactions, Cu-free click chemistry and oxime formation, which are already known to be mutually compatible.(34) A mixture containing equal protein loads of QC-BSA (MW ∼66 kDa), azidohomoalanine-containing dihydrofolate reductase (Az-DHFR, MW ∼23 kDa),(35) and aldehyde-tagged maltose binding protein (CHO-MBP, MW ∼42 kDa)(36) was treated with nickel complex 15, an azacyclooctyne conjugated to fluorescein (DIMAC-fluor),(37) and an aminooxy-functionalized FLAG peptide (H2NO-FLAG)(36) (Figure 3A). After incubation for 3 h, the mixture was separated into three portions, each of which was analyzed by Western blot probing with one of the following antibodies: α-biotin-HRP, α-fluorescein-HRP, or α-FLAG-HRP (Figure 3B). As shown in Figure 3B, each labeling reagent, including 15, reacted only with its complementary bioorthogonal partner.(38) Like the cyclooctyne and aminooxy probes, compound 15 showed no significant labeling of proteins lacking its partner (quadricyclane), nor did it interfere with the other bioorthogonal reactions.
Figure 3.

The quadricyclane ligation is orthogonal to Cu-free click chemistry and the oxime ligation. (A) A mixture of QC-BSA, Az-DHFR, and CHO-MBP (8 μg each) was treated with 15 (150 μM), DIMAC-fluor (250 μM), and H2NO-FLAG (1 mM) for 3 h at 37 °C, pH 4.5. This mixture was basified with 850 mM tris buffer and quenched with excess 1, 5, and 2-azidoethanol. (B) The mixture was separated into three portions, and each portion was analyzed by Western blot probing with a different antibody: α-biotin-HRP (quadricyclane ligation), α-fluorescein-HRP (Cu-free click chemistry), or α-FLAG-HRP (oxime ligation). The Ponceau stain indicates that all three proteins were present. Oligomer bands were observed for BSA and DHFR.(38)
In summary, the quadricyclane ligation is a promising bioorthogonal reaction that can be used for selective protein labeling alongside other popular bioorthogonal chemistries. The two reaction partners reliably form their covalent adduct in environs as complex as cell lysates. The next challenge for this chemistry will be applications to cell labeling.(39) However, the quadricyclane ligation in its present form is best suited for in vitro labeling experiments; improvements will be important for its use in living systems, where stabilizing additives such as K3Fe(CN)6 and diethyldithiocarbamate are not ideal.(40) More detailed studies into the balance of Ni bis(dithiolene) reactivity and redox stability are warranted, as well as investigations of the adduct’s photochemistry. Nonetheless, our results herein suggest that reactions of quadricyclane constitute a fertile sector of reactivity space for bioorthogonal chemistry.
Acknowledgments
We thank N. Agard and J. Hudak for Az-DHFR and CHO-MBP samples and R. Bergman, C. Gordon, J. Jewett, and K. Palaniappan for helpful discussions. This work was funded by a grant to C.R.B. from the NIH (GM058867). E.M.S. was supported by a predoctoral fellowship from the Organic Division of the ACS.
Supporting Information Available
Experimental procedures and supporting figures, schemes, and tables. This material is available free of charge via the Internet at http://pubs.acs.org.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Sletten E. M.; Bertozzi C. R. Angew. Chem., Int. Ed. 2009, 48, 6974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxon E.; Bertozzi C. R. Science 2000, 287, 2007. [DOI] [PubMed] [Google Scholar]
- Rostovtsev V. V.; Green L. G.; Fokin V. V.; Sharpless K. B. Angew. Chem., Int. Ed. 2002, 41, 2596. [DOI] [PubMed] [Google Scholar]
- Jewett J. C.; Bertozzi C. R. Chem. Soc. Rev. 2010, 39, 1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sletten E. M.; Bertozzi C. R. Acc. Chem. Res. 2011, 44, 666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Agard N. J.; Prescher J. A.; Bertozzi C. R. J. Am. Chem. Soc. 2004, 126, 15046. [DOI] [PubMed] [Google Scholar]; b Song W.; Wang Y.; Qu J.; Madden M. M.; Lin Q. Angew. Chem., Int. Ed. 2008, 47, 2832. [DOI] [PubMed] [Google Scholar]; c Sanders B. C.; Friscourt F.; Ledin P. A.; Mbua N. E.; Arumugam S.; Guo J.; Boltje T. J.; Popik V. V.; Boons G.-J. J. Am. Chem. Soc. 2011, 133, 949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Blackman M. L.; Royzen M.; Fox J. M. J. Am. Chem. Soc. 2008, 130, 13518. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Devaraj N. K.; Weissleder R.; Hilderbrand S. A. Bioconjugate Chem. 2008, 19, 2297. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Pipkorn R.; Waldeck W.; Didinger B.; Koch M.; Mueller G.; Wiessler M.; Braun K. J. Pept. Sci. 2009, 15, 235. [DOI] [PubMed] [Google Scholar]
- a Lin Y. A.; Chalker J. M.; Floyd N.; Bernardes G. J. L.; Davis B. G. J. Am. Chem. Soc. 2008, 130, 9642. [DOI] [PubMed] [Google Scholar]; b Kodama K.; Fukuzawa S.; Nakayama H.; Sakamoto K.; Kigawa T.; Yabuki T.; Matsuda N.; Shirouzu M.; Takio K.; Yokoyama S.; Tachibana K. ChemBioChem 2007, 8, 232. [DOI] [PubMed] [Google Scholar]
- a Zeng Y.; Ramya T. N. C.; Dirksen A.; Dawson P. E.; Paulson J. C. Nat. Methods 2009, 6, 207. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Ren H.; Xiao F.; Zhan K.; Kim Y.-P.; Xie H.; Xia Z.; Rao J. Angew. Chem., Int. Ed. 2009, 48, 9658. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Ou W.; Uno T.; Chiu H.-P.; Grunewald J.; Cellitti S. E.; Crossgrove T.; Hao X.; Fan Q.; Quinn L. L.; Patterson P.; Okach L.; Jones D. H.; Lesley S. A.; Brock A.; Geierstanger B. H. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 10437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chopade P. R.; Louie J. Adv. Synth. Catal. 2006, 348, 2307. [Google Scholar]
- Petrov V. A.; Vasil’ev N. V. Curr. Org. Synth. 2006, 3, 215. [Google Scholar]
- Hill W. E.; Szechi J.; Hofstee C.; Dane J. H. Environ. Sci. Technol. 1997, 31, 651. [Google Scholar]
- Watson W. H.; Tavanaiepour I.; Marchand A. P.; Dave P. R. Acta Crystallogr. 1987, C43, 1356. [Google Scholar]
- Kabakoff D. S.; Buenzil J.-C. G.; Oth J. F. M.; Hammond W. B.; Berson J. A. J. Am. Chem. Soc. 1975, 97, 1510. [Google Scholar]
- a Domingo L. R.; Saez J. A.; Zaragoza R. J.; Arno M. J. Org. Chem. 2008, 73, 8791. [DOI] [PubMed] [Google Scholar]; b Narayan S.; Muldoon J.; Finn M. G.; Fokin V. V.; Kolb H. C.; Sharpless K. B. Angew. Chem., Int. Ed. 2005, 44, 3275. [DOI] [PubMed] [Google Scholar]
- a LeBlanc B. F.; Sheridan R. S. J. Am. Chem. Soc. 1985, 107, 4554. [Google Scholar]; b Keana J. F. W.; Guzikowski A. P.; Ward D. D.; Morat C.; Van Nice F. L. J. Org. Chem. 1983, 48, 2654. [Google Scholar]
- Kajitani M.; Kohara M.; Kitayama T.; Asano Y.; Sugimori A. Chem. Lett. 1986, 2109. [Google Scholar]
- Gassman P. G.; Patton D. S. J. Am. Chem. Soc. 1968, 90, 7276. [Google Scholar]
- a Kajitani M.; Kohara M.; Kitayama T.; Akiyama T.; Sugimori A. J. Phys. Org. Chem. 1989, 2, 131–145. [Google Scholar]; b Norbornadiene also reacts with 2 to form an identical adduct, but at a rate that is 1000-fold lower than observed with quadricyclane. See ref (21a).
- Kunkely H.; Volger A. Inorg. Chim. Acta 2001, 319, 183. [Google Scholar]
- a Madhu V.; Das S. K. Inorg. Chem. 2008, 47, 5055. [DOI] [PubMed] [Google Scholar]; b Dalgleish S.; Robertson N. Chem. Commun. 2009, 46, 5826. [DOI] [PubMed] [Google Scholar]
- Gareau Y.; Beauchemin A. Heterocycles 1998, 48, 2003. [Google Scholar]
- No oxidation of biotin was observed.
- Compound 1’s hydrophobicity mandated the use of ethanol.
- a Baskin J. M.; Prescher J. A.; Laughlin S. T.; Agard N. J.; Chang P. V.; Miller I. A.; Lo A.; Codelli J. A.; Bertozzi C. R. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 16793. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Ning X.; Guo J.; Wolfert M. A.; Boons G.-J. Angew. Chem., Int. Ed. 2008, 47, 2253. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Debets M. F.; van Berkel S. S.; Schoffelen S.; Rutjes F. P. J. T.; van Hest J. C. M.; van Delft F. L. Chem. Commun. 2010, 46, 97. [DOI] [PubMed] [Google Scholar]
- a Miller T. R.; Dance I. G. J. Am. Chem. Soc. 1973, 95, 6970. [Google Scholar]; b Nakazumi H.; Takamura R.; Kitao T. J. Soc. Dyers Colour. 1991, 107, 459. [Google Scholar]; c Chen C.-T.; Liao S.-Y.; Lin K.-J.; Chen C.-H.; Lin T-Y.J. Inorg. Chem. 1999, 38, 2734. [Google Scholar]
- a Brown J. R.Albumin Structure, Function and Uses; Pergamon Press: New York, 1977; pp 27–51. [Google Scholar]; b An Ellman’s test for sulfhydryl groups indicated that 30–40% of the predicted free cysteine residues on commercial BSA were available for disulfide exchange.
- For the synthesis of 16, see Scheme S1.
- For the synthesis of 17, see Scheme S2.
- Chang P. V.; Prescher J. A.; Hangauer M. J.; Bertozzi C. R. J. Am. Chem. Soc. 2007, 129, 8400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiick K. L.; Saxon E.; Tirrell D. A.; Bertozzi C. R. Proc. Natl. Acad. Sci. U.S.A. 2002, 99, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrico I. S.; Carlson B. L.; Bertozzi C. R. Nat. Chem. Biol. 2007, 3, 321. [DOI] [PubMed] [Google Scholar]
- For the synthesis of DIMAC-fluorescein, see Scheme S3.
- Controls for Figure 3B can be found in Figure S17.
- Notably, compound 17 showed no significant toxicity to cultured mammalian cells at concentrations up to 500 μM after 1 h of treatment. By contrast, Cu(I) displays significant toxicity under these conditions. See Figures S18 and S19.
- Cultured mammalian cells tolerate diethyldithiocarbamate at millimolar concentrations without apparent toxicity (Figures S20 and S21).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.