

Abstract
Cell spreading and migration associated with the expression of the 92-kD gelatinase (matrix metalloproteinase 9 or MMP-9) are important mechanisms involved in the repair of the respiratory epithelium. We investigated the location of MMP-9 and its potential role in migrating human bronchial epithelial cells (HBEC). In vivo and in vitro, MMP-9 accumulated in migrating HBEC located at the leading edge of a wound and MMP-9 expression paralleled cell migration speed. MMP-9 accumulated through an actin-dependent pathway in the advancing lamellipodia of migrating cells and was subsequently found active in the extracellular matrix (ECM). Lamellipodia became anchored through primordial contacts established with type IV collagen. MMP-9 became amassed behind collagen IV where there were fewer cell–ECM contacts. Both collagen IV and MMP-9 were involved in cell migration because when cell–collagen IV interaction was blocked, cells spread slightly but did not migrate; and when MMP-9 activation was prevented, cells remained fixed on primordial contacts and did not advance at all. These observations suggest that MMP-9 controls the migration of repairing HBEC by remodeling the provisional ECM implicated in primordial contacts.
Keywords: cell migration, gelatinase, matrix metalloproteinase, bronchial epithelium, wound repair
The respiratory epithelium is frequently damaged after exposure to infectious or noninfectious environmental agents. Regardless of the source of injury, lesions can vary from the loss of surface epithelium impermeability, as a result of tight junction impairment, to a more-or-less complete shedding of epithelial cells: the basement membrane may be completely denuded or some clusters of basal cells may remain attached to it (Man and Hulbert 1988). After injury, the respiratory epithelium initiates a wound healing process to restore its barrier integrity. One important aspect is the rapid reepithelialization of the denuded area. Immediately after acute inhalation injury or mechanical deepithelialization of the airway epithelium, the remaining viable epithelial cells at the edge of the wound dedifferentiate, spread, and migrate rapidly over the denuded basement membrane to cover the deepithelialized zone (McDowell et al. 1979; Nikula et al. 1988; Erjefält et al. 1995). Several studies have shown that the most important and first event occurring during the reepithelialization of the denuded airway mucosa is cell migration and not cell proliferation (Wilhelm 1953; Lane and Gordon 1974; Keenan et al. 1982). Cell migration plays an important part in the rapid reconstitution of a cohesive epithelial structure (Hérard et al. 1996b).
Cell locomotion involves a number of interdependent processes including: the formation of cell protrusions or lamellipodia in the direction of movement, retraction of posterior formations, and establishment and rupture of adhesive contacts between the cell and the extracellular matrix (ECM)1 (Zigmond 1989; Small et al. 1996). These interactions between the cell membrane and the ECM are called focal contacts or focal adhesions and link cytoskeletal elements to various ECM molecules. In the lamellipodia, cells adhere to ECM through primordial contacts, which provide actin filament bundles with the first anchorage by which they can exert traction on the ECM, thereby enabling cell migration. Primordial contacts are transient structures that enable cell protrusions to anchor for a short period of time and are rapidly remodeled during migration. Adhesive contacts may be broken by extracellular proteolytic enzymes, such as serine proteinases of the plasmin system or matrix metalloproteinases (MMPs).
Collectively, MMPs are able to degrade all components of the ECM and they play key roles in normal physiologic processes involving ECM remodeling, such as wound healing, angiogenesis, and development. MMPs also participate in inflammation, tumor invasion, and metastasis (Chambers and Matrisian 1997; Woessner 1998). After acute and severe injuries of the airway epithelium, the mucosal damage may result in a total desquamation of the surface epithelial cells leading to a denuded but intact basement membrane (Nordin et al. 1977). Repairing cells spread and migrate onto this continuous thin ECM layer that underlies the respiratory epithelial cell sheet. The basement membrane is mostly synthesized by the cells that lie on it and is essentially composed of type IV collagen, the molecule that gelatinases, an MMP subclass, preferentially degrade. The gelatinases, also called type IV collagenases as a result of their activity, include MMP-2 (72-kD gelatinase) and MMP-9 (92-kD gelatinase). We previously demonstrated that, unlike MMP-2, MMP-9 is strongly expressed by repairing the human bronchial epithelial cells (HBEC) (Buisson et al. 1996a). Those observations supported the idea that MMP-9 is linked to the reepithelialization process and early repair events (Vu and Werb 1998), whereas MMP-2 is important during the prolonged remodeling phase (Ågren 1994; Stricklin et al. 1994).
MMP-9 has been shown to be involved in the migration of several cell types, i.e., macrophages, T lymphocytes, and eosinophils through reconstituted basement membrane (Pluznik et al. 1992; Leppert et al. 1995; Okada et al. 1997). Oligodendrocytes use MMP-9 to put forth processes along astrocytes (Uhm et al. 1998). MMP-9 is also a major factor of human polymorphonuclear cell migration across the basement membrane and elastase contributes to this process by activating pro-MMP-9 (Delclaux et al. 1996). Induction of keratinocyte migration by EGF and hepatocyte growth factor coincides with the induction of MMP-9 activity (McCawley et al. 1998). We previously demonstrated that the epithelial cell–produced MMP-9 actively contributes to the in vitro wound-repair process of the respiratory epithelium (Buisson et al. 1996a); MMP-9 is upregulated during the wound-repair process and is detected in migrating basal epithelial cells. Nevertheless, as Vu and Werb 1998 stated, although much has been learned about MMP-9 involvement in the wound repair of various tissues (Matsubara et al. 1991; Oikarinen et al. 1993; Ågren 1994; Bendeck et al. 1994; Salo et al. 1994), its in vivo substrate and functions remain unclear.
In the present study, we examined how HBEC migrating to repair a wound proceed to anchor to the ECM, how they could pull on these anchorages to advance, and how MMP-9 could break these cell–ECM contacts. To investigate in more detail the precise role of MMP-9 in migrating HBEC, we used new videomicroscopy techniques that enable cell tracking and quantification of cell migration speeds. With these techniques, we were able to study the relationships between migration speeds of individual cells and their cellular MMP-9 content and the speeds of lamellipodia extension and MMP-9 accumulation in these protrusions. We observed that type IV collagen is an essential component that contributes to cell migration by providing the first anchorage (primordial contacts) for HBEC to pull on. Moreover, MMP-9 accumulates just behind collagen IV, at sites of fewer cell–ECM interactions, and its activity is necessary for cell movement. Thus, our working hypothesis is that MMP-9 controls the migration of repairing HBEC by remodeling the provisional ECM involved with primordial contacts.
Materials and Methods
Source of Bronchial Tissue
Human bronchial tissues from patients undergoing surgery for bronchial carcinoma were obtained from microscopically normal areas distant from the tumor. Immediately after excision, the samples were immersed in a Ham's F12/DME (1:3, vol/vol) (GIBCO BRL) supplemented with 80 U/ml penicillin, 80 μg/ml streptomycin (GIBCO BRL), and 50 μg/ml gentamicin (Sigma Aldrich Chimie). Specimens were either processed for cell isolation or for an ex vivo wound repair model. Some tissue samples were also directly embedded in Tissue-Tek OCT compound (Sakura) and frozen in liquid nitrogen.
Cell Culture
HBEC were isolated and cultured according to Buisson et al. 1996b with some modifications. In brief, the bronchial tissues were digested overnight at 4°C with 0.1% Pronase E (Sigma Aldrich Chimie) and dissociated cells were resuspended in Green's culture medium (Green et al. 1979), consisting of Ham's F12/DME (1:3, vol/vol) supplemented with 5 μg/ml insulin, 10 ng/ml EGF, 0.5 μg/ml hydrocortisone, 20 μg/ml adenine, 0.1 nM cholera toxin, 5 μg/ml transferrin, 1.5 ng/ml triiodothyronine, 80 U/ml penicillin, 80 μg/ml streptomycin, 50 μg/ml gentamicin, and 10% FCS (all from Sigma Aldrich Chimie except EGF from Boehringer-Mannheim and FCS from GIBCO BRL). Glass coverslips (20 mm in diameter) were coated with rat type I collagen and prepared as previously described (Buisson et al. 1996a), in the presence of 0.25 μg/ml carbodiimide. The dissociated HBEC were seeded at a density of 1.25 × 106 cells/cm2 on collagen I–coated coverslips and cultured at 37°C in a humidified incubator in the presence of 5% CO2 and 95% air. Confluent primary cultures of HBEC were generally obtained after 2 d.
Wound-Repair Models
In Vitro Wound-Repair Model.
Primary cultures of confluent HBEC were locally injured as previously described (Buisson et al. 1996a) by depositing a 1-μl drop of 1 M sodium hydroxide at the center of the culture. Sodium hydroxide was rapidly neutralized with PBS and a circular wound area of ∼30 mm2 resulted from the sodium hydroxide–induced cellular lysis. Evolution of the remaining surface of the wound area was examined every day with a CCD-WV50 videocamera (Panasonic) connected to a microscope and the corresponding wound areas were calculated. The wound area was expressed as wound relative area, corresponding to the ratio of the denuded surface at a given time to the denuded surface measured immediately after wounding. When the wound had repaired 30–60% of its initial surface (1–2 d), cultures were either processed for the measurement of cell migration or fixed for immunofluorescence labeling studies as follows. Before fixation, HBEC cultures were rapidly washed in PHEM buffer (60 mM Pipes, 23 mM Hepes, 10 mM EGTA, 1 mM MgCl2, adjusted to pH 6.9). Cultures were sequentially fixed for 5 min in 3.7% paraformaldehyde in PBS, washed in PBS, incubated for 5 min in 0.1 M glycine in PBS, washed in PBS, permeabilized for 1 min with 0.5% Triton X-100, rinsed in PBS, and stored at 4°C until used.
Ex Vivo Wound-Repair Model.
Freshly collected human bronchial tissue samples, ∼10 × 10 mm, were locally injured with a metallic probe (2 mm in diameter) frozen with liquid nitrogen and applied for 10 s to the tissue sample with a calibrated pressure of 33 kPa. Under these conditions, only cells of the surface epithelium were damaged and desquamated. After wound induction, tissue samples were maintained in culture for 1 d in Green's culture medium, embedded in Tissue Tek OCT compound, and frozen in liquid nitrogen.
Cell Migration Assay
The migration speeds of cells in injured HBEC cultures were determined as previously described (Zahm et al. 1997). After wound induction, cell nuclei were stained with a fluorescent dye (Hoechst 33258; Molecular Probes, Inc.) and the wounded culture was placed in a small transparent culture chamber of an IM35 inverted microscope (Zeiss). The microscope was equipped with epifluorescence illumination through an excitation filter at 360 nm and an emission filter at 510 nm and with a low level silicon intensified target camera (model 4036; Lhesa). A shutter (Lambda 10–2; Sutter Instrument) was placed in the excitation light path to illuminate the culture for short periods of time (1 s) and to simultaneously digitize the fluorescent images. The images were digitized as 512 × 512 pixels and 8-bit array, using a Sparc-Classic (Sun Microsystems) workstation equipped with an XVideo card (Parallax Graphics). Cell migration was quantified using a previously described software (Zahm et al. 1997) with three main functions: the detection of cell nuclei, the computation of the trajectories of these nuclei, and the analysis of these trajectories. From each nucleus trajectory, the computer calculated the cell migration speed.
When analyzing the effect of the cell's MMP-9 content or the effect of a reagent (cytochalasin D, cycloheximide, monensin, batimastat [British Biotech Pharmaceuticals, Ltd.], anti–MMP-9 antibody [6-6B mAb, a gift from Dr. French, State University of New York at Stony Brook], or anti–collagen IV antibody [20411; Institut Pasteur]) on HBEC migration, we always restricted migration assessment to a population of cells located close to the edge of the wound, i.e., within a distance corresponding to ∼1–2 cells starting from the edge of the wound. Indeed, we previously observed that cell migration speed progressively decreases as the distance from the edge of the wound increases (Zahm et al. 1997).
Determination of Extracellular Gelatinase Activity during Cell Migration Using Immobilized 3H-Gelatin
To assay extracellular gelatinase activity associated to cell migration, we induced the cells to migrate onto 3H-gelatin–containing type I collagen and we measured the accumulation of radioactivity in the culture medium. In short, rat type I collagen was denatured for 1 h at 60°C and radiolabeled with sodium [3H]borohydride (Amersham) to a specific activity of 8 × 106 cpm/mg as previously described (Starkey et al. 1984). Glass coverslips were coated with rat type I collagen containing 10% 3H-gelatin, and then extensively washed. The radioactivity remaining on glass coverslips (6,400 ± 900 cpm/cm2) was measured after an overnight hydrolysis with 1 M sodium hydroxide. During cell migration, the radioactivity released in the culture medium was counted in a 1900 CA liquid scintillation analyzer (Packard).
Preparation of Cell Protein Extracts
Two protein extracts were prepared from migrating HBEC: frozen cells were first extensively washed in water to prepare a cytosolic fraction, and then cells and ECM were extracted with detergent (0.1% SDS) as follows. When injured HBEC cultures had repaired 30–60% of the initial surface of the wound (1–2 d), cell cultures were washed several times with PBS and frozen. A cloning cylinder (6 mm inner diameter, corresponding to the initial wound area) (Bellco Glass, Inc.) was placed on migrating cells, and cells located inside the cylinder were extensively washed with water at 4°C. The aqueous extract was centrifuged (10,000 g for 10 min at 4°C) and lyophilized. The cylinder was filled with 200 μl of 0.1% SDS in water and the liquid was mixed by pipetting several times at 4°C. Cell extraction with SDS was repeated 10 times. The combined detergent extracts were centrifuged (10,000 g for 10 min at 4°C) and lyophilized. Aqueous and detergent lyophilized extracts were resolubilized in Laemmli's sample buffer to achieve a 20-fold sample concentration and used immediately for gelatin zymography, performed as previously described (Buisson et al. 1996a).
Immunocytochemistry
Sections of frozen bronchial tissues were cut (5-μm thick) at−20°C in a 2800 Frigocut cryostat (Cambridge Instruments) and transferred to gelatin-coated slides. Tissue sections and HBEC cultures undergoing repair were immunoreacted with specific antibodies (Ab) using an indirect immunofluorescence labeling technique. All incubations were conducted at room temperature.
A single immunolabeling technique was performed to localize MMP-9 in bronchial tissues and in repairing HBEC cultures. First, nonspecific binding was blocked for 30 min with 3% BSA in PBS. The samples were incubated for 60 min with 3 μg/ml mouse anti–human MMP-9 mAb (GE 209; Oncologix) in 1% BSA in PBS (PBS-BSA). After two washes in PBS for 5 min, and one wash in PBS–BSA for 5 min, the samples were incubated with biotinylated anti–mouse IgG (Amersham) diluted 1:40 in PBS-BSA, for 60 min, and then incubated with FITC-streptavidin (Amersham) diluted 1:50 in PBS for 30 min.
Double immunolabelings with the following Abs were performed to simultaneously localize: MMP-9 (10 μg/ml rabbit polyclonal Ab; Biogenesis) and vinculin (1:200, mouse hVIN-1 mAb, Sigma Aldrich Chimie); type IV collagen (1:1000, rabbit 20411 polyclonal Ab) and MMP-9 (3 μg/ml mouse GE 209 mAb) or vinculin; and cellular fibronectin (1.25 μg/ml rabbit polyclonal Ab; Sigma Aldrich Chimie) and vinculin. In these experiments, two successive immunofluorescence labeling incubations were performed with intervening washes. Mouse mAbs were detected with 2 μg/ml digoxigenin-conjugated sheep anti–mouse F(ab′)2 fragments (Boehringer-Mannheim) in PBS-BSA, and then with 1.3 μg/ml FITC-conjugated sheep antidigoxigenin Fab fragments (Boehringer-Mannheim) in PBS-BSA. Rabbit polyclonal Abs were detected with biotinylated anti–rabbit IgG (Amersham) diluted 1:40 in PBS-BSA and then with Texas red–streptavidin conjugate (Amersham) diluted 1:50 in PBS-BSA. As for fibronectin–vinculin double immunolabeling, rabbit antifibronectin Ab was detected with digoxigenin-conjugated sheep anti–rabbit F(ab′)2 fragments and FITC-conjugated sheep antidigoxigenin Fab fragments, whereas mouse antivinculin mAb was detected with biotinylated anti–mouse IgG and Texas red–conjugated streptavidin. We verified the absence of cross-reactivity by incubating control cultures with nonimmune IgG instead of the primary Ab.
After immunolabeling, cultures were counterstained with Harris hematoxylin (Diagnostica Merck) and mounted in Citifluor antifading solution (Agar Scientific). Actin microfilaments were specifically labeled after incubation with 10 μg/ml FITC–phalloidin (Sigma Aldrich Chimie) and mounted in Citifluor. All fluorescence-labeled preparations were examined with an Axiophot microscope (Zeiss) using successive epifluorescence and Nomarski differential interference illumination. Immunolabelings were also observed with an MRC 600 confocal laser scanning microscope (Bio-Rad Laboratories).
Results
MMP-9 Is Present In Vivo and In Vitro in Migrating Bronchial Epithelial Cells
Immunofluorescent labeling of normal bronchial epithelium detected MMP-9 only in isolated inflammatory cells present in the bronchial submucosa, crossing the basement membrane or infiltrating the pseudostratified surface epithelium (Fig. 1A and Fig. B). Most of these cells had the morphology of polymorphonuclear leukocytes. In remodeled epithelia, MMP-9 was only detected in clusters of flat basal cells overlying the basement membrane, in areas where differentiated cells (goblet and ciliated cells) had been exfoliated (Fig. 1C and Fig. D). These basal cells were previously characterized by the presence of cytokeratins 13 and 14 (Brezillon et al. 1995). We also studied the distribution of MMP-9 in ex vivo epithelia undergoing repair. After inducing a local wound in fresh human bronchial tissues (ex vivo wound-repair model), cells were removed only from the surface epithelium, leaving the basement membrane intact, as demonstrated by the presence of immunoreactive laminin and type IV collagen as a continuous thin layer in the damaged area (data not shown). After 1 d in culture, the epithelial cells had migrated to the edge of the wound to repair it and they appeared as flat cells (Buisson et al. 1996b; Roger et al. 1999). MMP-9 was detected in elongated migrating repairing cells, located close to the edge of the wound (Fig. 1E and Fig. F). When primary and confluent cultures of HBEC were locally wounded, cells at the edge of the wound rapidly spread and migrated to cover the denuded area (Zahm et al. 1991; Buisson et al. 1996a; Zahm et al. 1997). During the wound-repair process, MMP-9 was predominantly found in the forefront of cells migrating into the damaged area (Fig. 1G and Fig. H).
Figure 1.
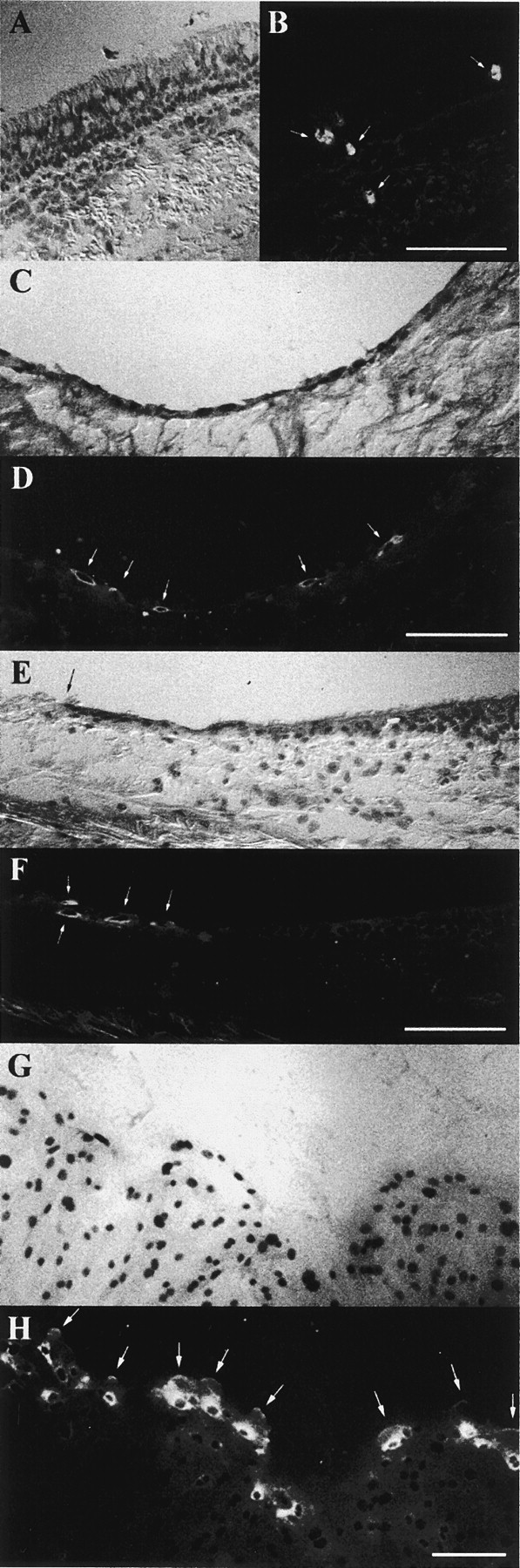
Immunolocalization of MMP-9 in cryosections of human bronchial tissue samples and cultures of repairing HBEC. The localization of MMP-9 was studied by immunofluorescence, using the GE 209 mAb. (A and B) A normal bronchial epithelium in which MMP-9 was seen in sporadic inflammatory cells (B, arrows). (C and D) In a remodeled human bronchial epithelium, MMP-9 was recognized in elongated cells (D, arrows) spread on the basement membrane. (E and F) In a normal human epithelium, which had been locally wounded and maintained in culture for 1 d, MMP-9 was observed only in flat epithelial cells (F, arrows) near the edge of the wound (E, arrow). (G and H) In a confluent primary culture of HBEC, which had been locally injured, MMP-9 was identified 1 d after injury only in cells located at the edge of the wound. In these cells, MMP-9 was predominantly located around the nucleus but had also accumulated at the extremity of cellular protrusions in the direction of cell migration (H, arrows). Bars, 100 μm.
Bronchial Epithelial Cells Expressing MMP-9 Migrate More Rapidly to Repair a Wound
Because we observed that MMP-9 was present in HBEC only when they migrated to repair a wound (Fig. 1), we hypothesized that MMP-9 expression in migrating HBEC may influence HBEC migration. We quantified and studied cell migration speeds as a function of the presence or absence of MMP-9 in these cells. Labeling the nuclei of cells primed to migrate by wound induction enables cell tracking and cell migration speeds to be measured using a previously described method (Zahm et al. 1997). We reported earlier that cell migration speed continuously and significantly decreased with the increasing distance from the edge of the wound (Zahm et al. 1997). Consequently, we analyzed the relationship between the cellular MMP-9 content and the cell migration speed only for cells located near the edge of the wound, i.e., in cells located above the line in Fig. 2 (A–C). Distinguishing between the cells that were strongly immunoreactive for MMP-9 (MMP-9 positive cells) and cells that were weakly labeled or had nonspecific labeling (MMP-9 negative cells), we observed that MMP-9 positive cells migrated 33.7 ± 7.7% (n = 6) more rapidly than MMP-9 negative cells. In five out of six experiments, the migration speed of MMP-9 positive cells was significantly higher (P < 0.05; Fig. 2 D) than that of MMP-9 negative cells.
Figure 2.
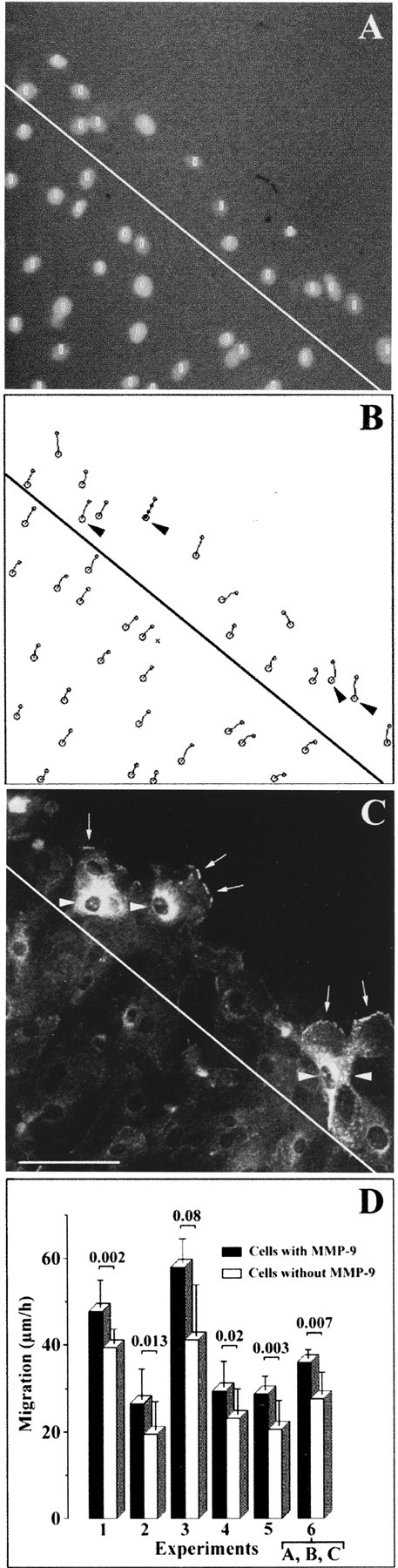
HBEC containing MMP-9 migrate more rapidly to repair a wound. Primary cultures of confluent HBEC were locally injured with sodium hydroxide. (A) Labeling of cell nuclei with a DNA fluorescent dye (Hoechst 33258) at the edge of the wound on day 1 of the wound-repair process. Positions of cell nuclei are identified by white circles. (B) Trajectories of cell nuclei recorded every 10 min and over a 30-min period. Four cells strongly positive for MMP-9 are indicated (arrowheads) and the four successive positions of the nucleus of one of them is shown. (C) Immunofluorescent labeling of MMP-9 after fixation of the culture at the end of the 30-min recording period. Arrowheads indicate the same intensely labeled cells as in B. MMP-9 also accumulated at the forefront of the cellular protrusions (arrows). (D) Mean cell migration speeds ± SD for six different experiments conducted on primary cultures of HBEC derived from six different bronchial samples. Experiment 6 reports the mean migration speeds for the cells located above the line in A–C. For all six experiments, the black bars in D report the mean migration speeds ± SD of cells strongly positive for MMP-9, whereas white bars correspond to the values of the other cells. The significance of the differences between the two groups was determined using t test. Bar, 100 μm.
MMP-9 in Migrating HBEC Is Addressed through an Actin-dependent Pathway to Cellular Extensions Involved in Cell Migration
Because we observed that migrating HBEC contain more MMP-9 than stationary cells, and, moreover, that MMP-9 seems to accumulate at the advancing edge of migrating cells (Fig. 1 H and Fig. 2 C, arrows), we hypothesized that MMP-9 in migrating HBEC may be addressed to cellular extensions involved in cell migration. To investigate this possibility, we studied the dynamics of HBEC spreading and migration together with cellular MMP-9 distribution. During a 20-min period, we observed that cells migrating at the leading edge of a wound did not move forward at a uniform speed. Indeed, some lamellipodia advanced rapidly, whereas other parts of migratory cells did not move (Fig. 3, A–F). As seen in Fig. 1 H and Fig. 2 C, a strong immunoreactivity for MMP-9 was observed in the cytoplasm of migrating cells, with a predominant intense perinuclear distribution, suggesting that MMP-9 is located in the ER and/or the Golgi apparatus (Fig. 3G and Fig. H). It should also be noted that a thin band of MMP-9 lined the tip of those lamellipodia that had moved forward the most during the 20-min period preceding cell fixation. The lamellipodia that had migrated the most also contained more MMP-9 (Fig. 3G and Fig. H, arrowheads) than those that had moved more slowly (arrows).
Figure 3.
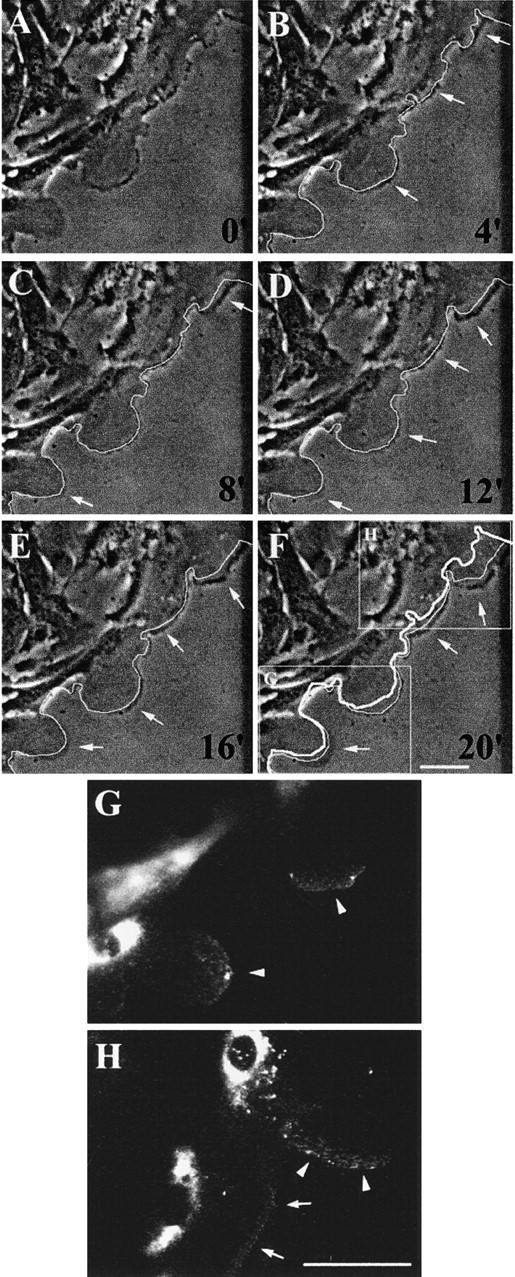
MMP-9 within migrating HBEC accumulates at the tip of extending lamellipodia. 1 d after injuring primary and confluent cultures of HBEC, a series of phase-contrast images of cells migrating and extending lamellipodia at the edge of the wound were taken at 4-min intervals (A–F). In images B–F, the leading front of the cells from the previous image (4 min earlier) has been highlighted with a thin white line. Comparison of this line to the present position of the cells enables the parts of the cells that have advanced during the 4-min period to be visualized (B–F, arrows). In F, the thick white line indicates the initial position of the cells shown in A. At the end of the experiment, the cells were fixed and MMP-9 distribution was assessed by immunofluorescence. (G and H) High magnification of MMP-9 distribution in the areas analyzed for cell movement (F, insets). MMP-9 is detected at the forefront of migrating cells with more intense fluorescence in more rapidly moving lamellipodia (G and H, arrowheads) than in those moving more slowly (H, arrows). Bars, 50 μm.
We previously observed that actin, but not tubulin, polymerization is essential for the migration of nasal epithelial cells (Zahm et al. 1991). The first event occurring during cell migration is the protrusion of lamellipodia resulting from a dynamic and directional polymerization of actin filaments (Small et al. 1996). Since we observed that MMP-9 is present in most migrating cells (Fig. 2) and accumulates at the front line of the most rapidly advancing lamellipodia (Fig. 3), we hypothesized that changes in the cytoskeletal organization and, more specifically, disorganization of the actin microfilament network may alter the trafficking of MMP-9 from its perinuclear pool to the front line of extending lamellipodia. Indeed, treatment of repairing HBEC cultures with 2 μM cytochalasin D, an inhibitor of actin polymerization, rapidly inhibited cell migration (Fig. 4 A) and prevented the accumulation of MMP-9 at the forefront of extending lamellipodia (Fig. 4, B–E).
Figure 4.
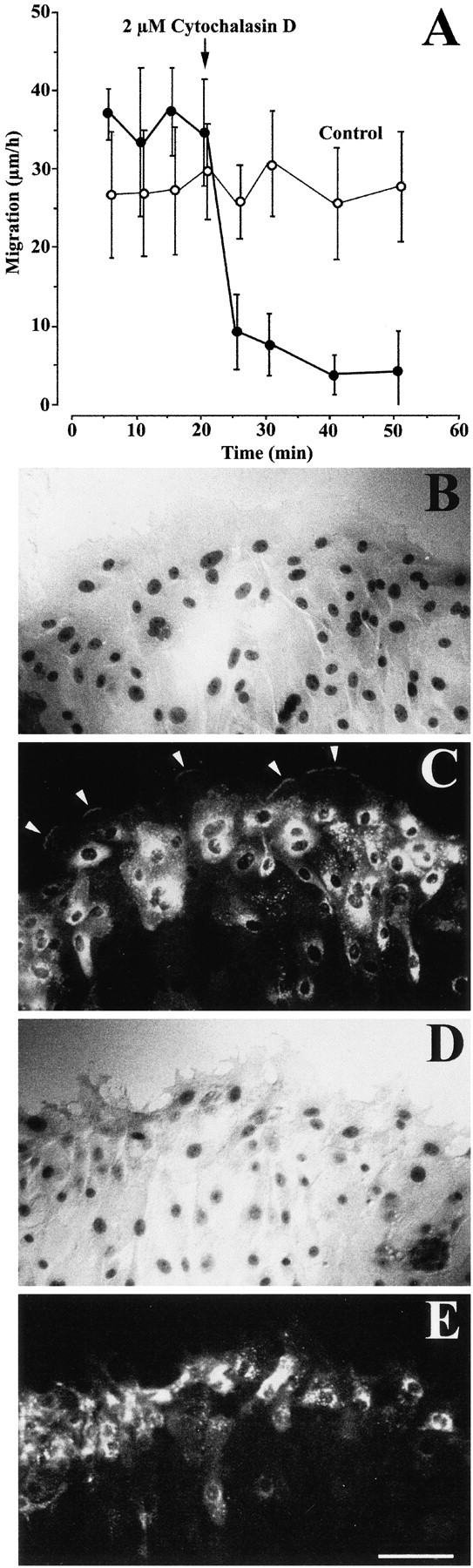
HBEC migration and MMP-9 trafficking to advancing lamellipodia were rapidly inhibited by treatment with cytochalasin D. (A) 1 d after injuring primary and confluent cultures of HBEC, cell migration was measured after fluorescent labeling of their nuclei, as described in Materials and Methods. Migration speeds of cells (n = 20) located at the edge of the wound were measured every 5 min over a 20-min control period. After 20 min, 2 μM cytochalasin D (closed circles) or its vehicle (0.05% DMSO, open circles) was added to the culture medium and the migration speeds of the same cells were monitored again over a 30-min period. Each bar represents the mean migration speed ± SD of 20 cells. (B and D) Phase-contrast micrographs and (C and E) MMP-9 fluorescence immunolabeling of a control culture fixed 3 min after the addition of 0.05% DMSO (B and C) and of another culture fixed 3 min after the addition of 2 μM cytochalasin D (D and E). MMP-9 can be seen at the tip of advancing lamellipodia only in the vehicle control experiment (C, arrowheads). Bar, 100 μm.
MMP-9 Is Identified in Lamellipodia of Migrating HBEC Close to Cell–ECM Interactions and Is Found under Advancing Lamellipodia
Cell migration involves interactions between the cell and the ECM both through weak primordial contacts at the leading edge of the cell and larger spear tiplike focal contacts distributed in the more central part of the cell (Lee et al. 1993). These structures can be visualized by reflection interference contrast microscopy or by the identification of one of the many cytoplasmic proteins associated with focal adhesions (Jockusch et al. 1995). We analyzed the distribution of vinculin, a protein highly concentrated in focal adhesions and located close to the plasma membrane, as a marker of migrating HBEC–ECM interactions. Vinculin accumulated at the leading edge of advancing lamellipodia and was colocalized with type IV collagen (Fig. 5, A–C), suggesting that the latter is actively brought to this site and serves as the first anchorage of migrating cells. When studying the distribution of type IV collagen along with that of vinculin in a series of confocal optical sections through migrating HBEC, we observed that type IV collagen was still present outside the cell in the ECM, suggesting that type IV collagen is actively secreted onto the ECM by migrating HBEC (data not shown). Surprisingly, the accumulation of type IV collagen at the forefront of cell protrusions coincided with MMP-9 accumulation just behind it (Fig. 5, D–F). When the leading edge of a migrating cell was a succession of small protrusions, each protrusion contained MMP-9 in its central part with primordial/focal contacts located slightly ahead or more intracellularly on each side of it (Fig. 5, G–I, arrowheads). In a more central part of a migrating cell, where there was no type IV collagen (Fig. 5, A–C), vinculin was present in focal contacts (Fig. 5B, Fig. H, and Fig. K) and was closely associated with fibronectin (Fig. 5, J–L, arrowheads). In contrast, at the forefront of the cell, where there was no fibronectin (Fig. 5J and Fig. L, arrows), vinculin accumulated with type IV collagen (Fig. 5 C, arrowheads).
Figure 5.
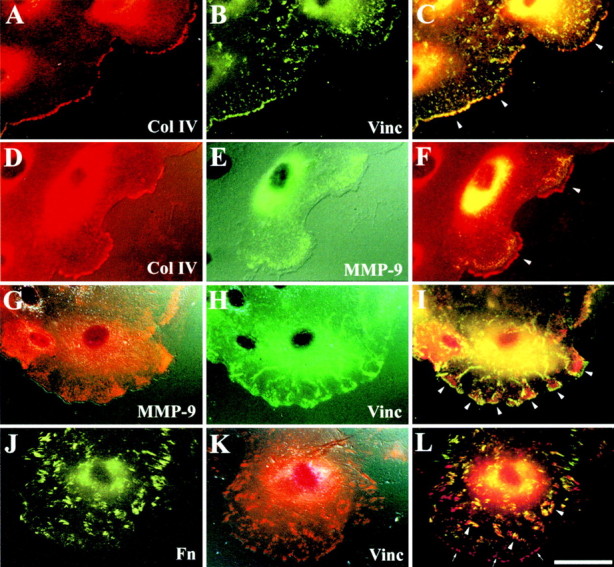
Immunofluorescence analysis of the cellular distribution of MMP-9 in migrating HBEC. Using immunofluorescent double labeling techniques to study repairing HBEC cultures, we evaluated the distribution of the following proteins: (A–C) type IV collagen (Col IV, polyclonal Ab) and vinculin (Vinc, hVIN-1 mAb); (D–F) type IV collagen and MMP-9 (GE 209 mAb); (G–I) MMP-9 (polyclonal Ab) and vinculin; and (J–L) cellular fibronectin (Fn, polyclonal Ab) and vinculin. C, F, I, and L are double exposures taken at excitation frequencies of double-labeled cultures; in these frames, a yellow color indicates the colocalization of red and green fluorescence. (A–C) Type IV collagen and vinculin accumulated at the forefront of a migrating cell (C, arrowheads). (D–F) In two advancing lamellipodia, MMP-9 was amassed behind type IV collagen (F, arrowheads). (G–I) Each cytoplasmic protrusion of a migrating cell is characterized by central accumulation of MMP-9 surrounded by vinculin at the tip of the protrusions and within the cell (I, arrowheads). (J–L) At the basal side and in the central part of a migrating cell, fibronectin accumulates close to vinculin (L, arrowheads) but, at the forefront of the cell, only vinculin accumulates (L, arrows). Bar, 40 μm.
We more precisely investigated MMP-9 distribution in migrating HBEC by using the confocal optical mode for light microscopic immunodetection of MMP-9. At the apical side of migrating cells, MMP-9 was predominantly localized around the nucleus (Fig. 6 A). In the protrusions of migrating cells, vinculin accumulated at the advancing edge (Fig. 6 D), whereas MMP-9 was mainly detected as patches just behind it. Some MMP-9 was also but infrequently seen to be colocalized with vinculin, i.e., in association with primordial/focal contacts (Fig. 6 C). The MMP-9 accumulation was intracellular but close to the basal cell membrane since MMP-9 was observed along with vinculin in the same optical section of the cell (Fig. 6C and Fig. D). When looking at a more basal cellular section, we still observed MMP-9 as patches (Fig. 6 E); then, 0.4-μm lower, MMP-9 took on a diffuse labeling pattern (Fig. 6 G), whereas vinculin could no longer be detected (Fig. 6F and Fig. H). Considering that a lamellipodium is ∼0.2–0.3 μm thick (Abercombie et al. 1970), it can be concluded from these observations that MMP-9, after reaching the extending lamellipodia, was secreted onto the ECM.
Figure 6.
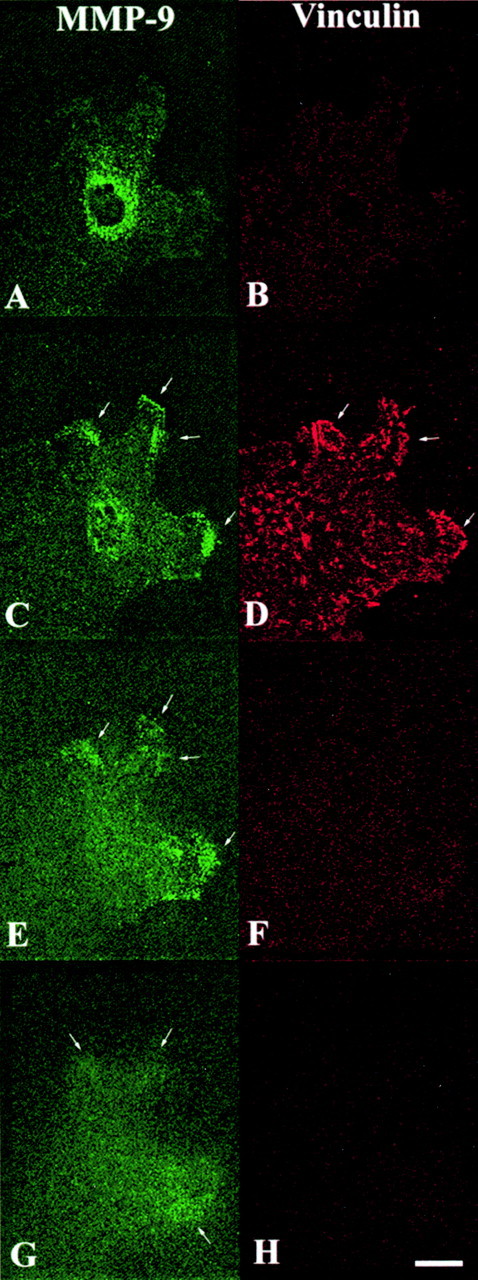
Confocal images of MMP-9 and vinculin distributions in one migratory cell at the edge of the wound in a repairing HBEC culture. After fixation (day 1 postinjury), the culture was processed for double immunofluorescence labeling of MMP-9 (polyclonal Ab) and vinculin. Four serial confocal optical sections (A and B, apical side of the cell; C–H, progressively more basal views) through the migrating cell were taken at 0.4-μm intervals using a Bio-Rad MRC 600 confocal laser scanning microscope and MMP-9 (pseudogreen; A, C, E, and G) and vinculin (pseudored; B, D, F, and H) contents were assessed. MMP-9 was located slightly behind the leading front of advancing lamellipodia (C, E, and G, arrows), whereas vinculin was more intensely identified at the forefront of protrusion (D, arrows). Bar, 20 μm.
Bronchial Epithelial Cell Migration Requires Both the Activity of MMP-9 and the Anchorage of Lamellipodia onto Type IV Collagen
In the advancing lamellipodia of migrating HBEC, type IV collagen accumulated at the extremity of the lamellipodia, whereas MMP-9 was mainly located within the lamellipodia and behind type IV collagen. This observation suggests that both molecules may be actively involved in the spreading and/or migration processes. To investigate this possibility in more detail, we studied the migration of HBEC in the presence of either the 6-6B mAb, known to block MMP-9 activation (Ramos-DeSimone et al. 1993) or a high affinity polyclonal Ab specific to type IV collagen. After exposure to either Ab, the migration speed of the cells at the edge of the wound progressively declined (Fig. 7A and Fig. B). After exposure to a control Ab or the anti–MMP-9 mAb, stress fibers and spotty actin deposits were seen at the migrating front of the cells (Fig. 7C and Fig. D). In contrast, in the presence of the anti–collagen IV Ab, cells spread without notable actin accumulation at the leading edge of the cell, even though microfilament bundles were still present in the central part of the cell (Fig. 7 E).
Figure 7.
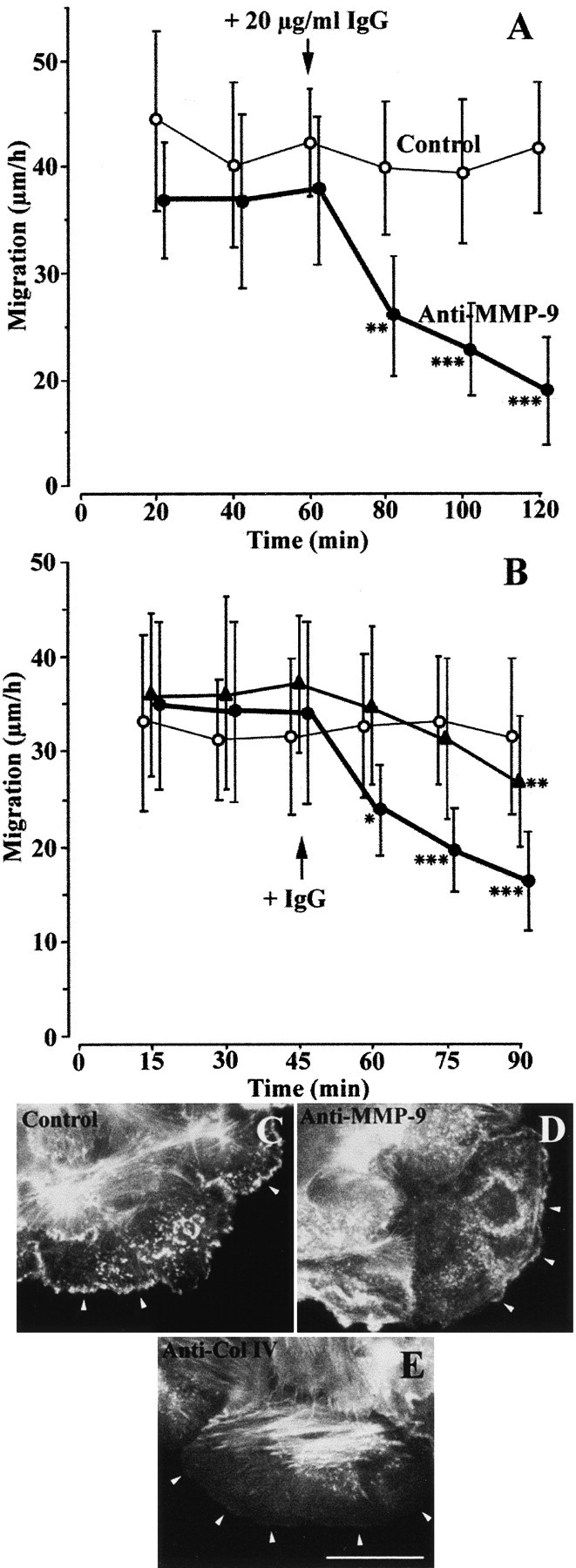
Anti–MMP-9 or anti–collagen IV Ab inhibits HBEC migration. (A and B) 1 d after injuring primary cultures of HBEC, cell migration was measured after fluorescent labeling of cell nuclei, as described in Materials and Methods. Migration speeds of the cells (n = 20) located at the edge of the wound were determined every 20 min (A) or 15 min (B) over a 60-min (A) or 45-min (B) control period. (A) At 60 min, 20 μg/ml of 6-6B anti–MMP-9 mAb (closed circles) or 20 μg/ml of control mouse IgG (open circles) was added to the culture medium and the migration speeds of the same cells were monitored over an additional 60-min period. (B) At 45 min, 30 μg/ml (closed circles) or 15 μg/ml (closed triangles) of anti–collagen IV Ab or 30 μg/ml of control IgG (open circles) was added to the culture medium, and the migration speeds of the same cells were monitored for an additional 45-min period. Each bar represents the mean ± SD of the migration speed of 20 cells. Cell migration at each time and in the presence of Ab was compared (paired t test) to cell migration during the control period: *P < 0.05, **P < 0.01, ***P < 0.001. (C–E) FITC–phalloidin labeling of actin in a culture exposed to a control Ab (C), the anti–MMP-9 6-6B mAb (D), or the anti–collagen IV Ab (E). In C and D, numerous sites of intense actin labeling are seen at the tip of cell protrusions (arrowheads). After exposure to the anti–collagen IV Ab, actin labeling at the forefront of the cell became very faint (E, arrowheads), whereas strongly labeled microfilament bundles radiating from focal contacts remained in the central part of the cell. Bar, 40 μm.
After sustained exposure of repairing HBEC to the anti–MMP-9 mAb, only tiny movements of cells at the edge of the wound could be detected (Fig. 8 D). When observed by phase-contrast microscopy, the leading edge of these cells appears as a dark and thick border, suggesting the presence of denser cytoplasmic constituents and/or stronger interactions with the ECM (Fig. 8 A). On the other hand, after exposure to the anti–collagen IV Ab, the cell border was more faint (Fig. 8 E). In the latter situation, small forward and backward movements of the cell border were observed (Fig. 8, F–H), suggesting that the cells made small movements ahead but were not able to go any further, probably as a consequence of defective cell anchorage depriving them of their traction system. We can conclude the following from these experiments: in the presence of 6-6B anti–MMP-9 mAb, HBEC attach firmly to type IV collagen but are unable to modify this anchorage and, thus, remain stuck to collagen IV; and in the presence of the anti–collagen IV Ab, HBEC initiate small spreading movements but are unable to go any further because no definite contact can be established with the ECM to form the traction mechanism.
Figure 8.
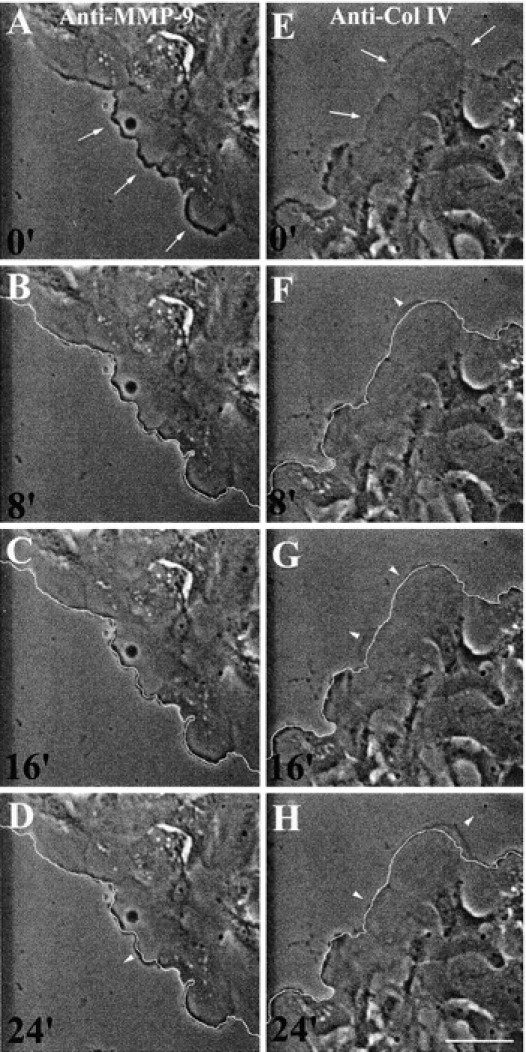
After exposure to the 6-6B anti–MMP-9 mAb, HBEC become immobilized, whereas in the presence of an anti–collagen IV Ab, they spread but do not migrate. 1 d after injuring primary cultures of HBEC, 20 μg/ml of 6-6B anti–MMP-9 mAb (A–D) or 30 μg/ml of polyclonal anti–collagen IV Ab (E–H) was added for 90 min. A series of phase-contrast images of cells at the edge of the wound were taken at 8-min intervals. In all images, the leading edge of the cells at the beginning of the recording (A and E) is indicated by a thin white line in B–D and F–H, respectively. Comparison of this line with the subsequent positions of the cells enables parts of the cells that had spread or retracted during the 8-min period to be localized (arrowheads). In the presence of the anti–MMP-9 mAb and under the phase-contrast microscope, the cell border appears dark and thick, whereas in the presence of the anti–collagen IV Ab, the cell border is more difficult to see (A and E, arrows). Bar, 70 μm.
HBEC Migration Coincides with an Increased Extracellular Gelatinase Activity and an Accumulation of Active MMP-9 in the ECM
We previously observed that MMP-9 was activated during the in vitro wound-repair process of the respiratory epithelium (Buisson et al. 1996a). The ability of anti–MMP-9 mAb to block HBEC migration suggests that MMP-9 must be activated for migration to occur. Moreover, we have observed that MMP-9 is present in the ECM underlying advancing lamellipodia of migrating HBEC (Fig. 6). Therefore, we hypothesized that the MMP-9 that is deposited on the ECM is active and able to degrade the surrounding ECM. When 3H-gelatin, a substrate for MMP-9, was incorporated into type I collagen on which repairing HBEC migrate, we observed a progressive release of radioactivity in the culture medium (Fig. 9A and Fig. B, control), suggesting a degradation of the ECM by gelatinase. Interestingly, every experimental condition that decreased migration of repairing HBEC (cycloheximide, batimastat, and an absence of growth factors) also decreased the degradation of radioactive ECM (Fig. 9A and Fig. B).
Figure 9.
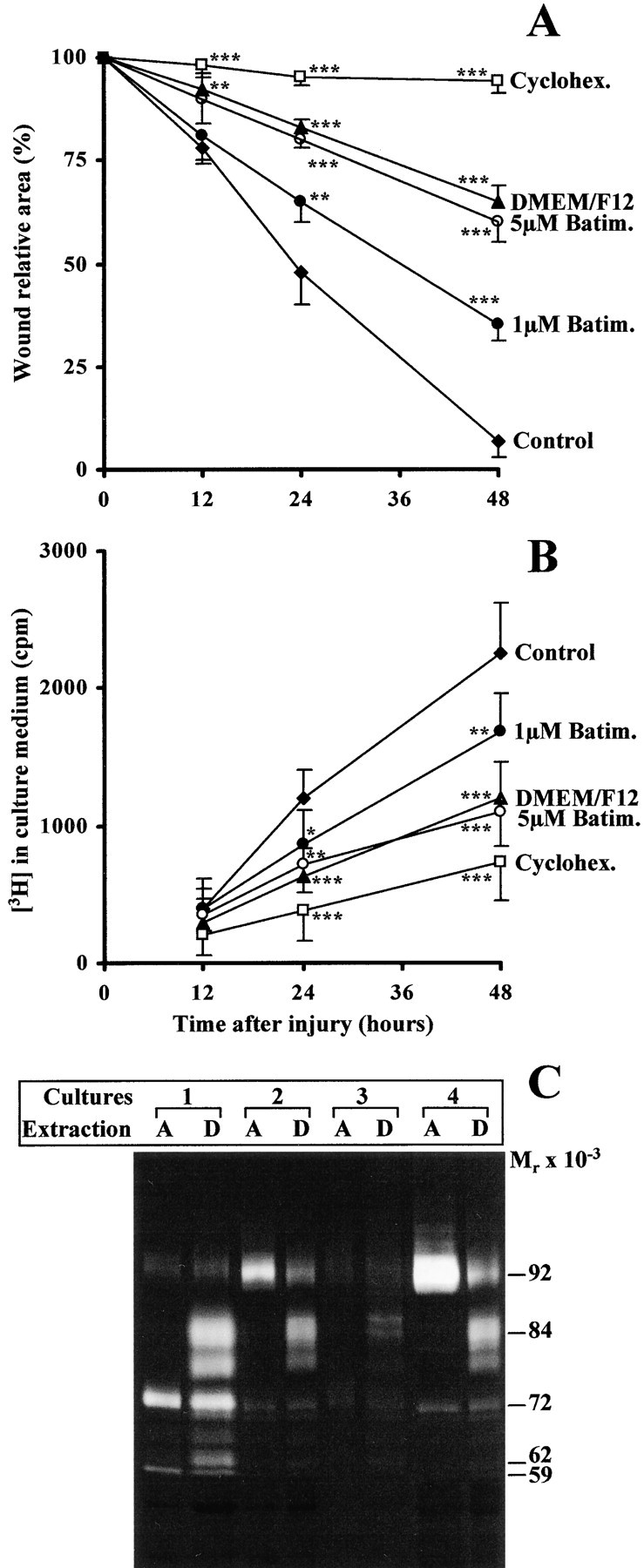
Extracellular gelatinase activity during HBEC migration. (A and B) HBEC were cultured on 3H-gelatin–containing type I collagen and locally injured at confluency with sodium hydroxide. The wound-repair process was followed for 48 h in the presence of Green's culture medium either complete (Control) or without any supplements (DMEM/F12) or in the presence of 35 μM cycloheximide (Cyclohex), of 1 or 5 μM batimastat (Batim). During the wound-repair process, the wound relative area (A) and the [3H]radioactivity released in the culture medium (B) were evaluated in each condition and compared (t test) at each time to the same parameter in the control experiment: *P < 0.05, ** P< 0.01, *** P< 0.001. Each condition (mean ± SD) was run in quintuplicates. (C) 1 d after injuring primary and confluent cultures of HBEC, derived from four different bronchial samples (1–4), an aqueous (A) or detergent (D) protein extract from migrating HBEC repairing a wound was prepared and subjected to gelatin zymography as described in Materials and Methods. The location of gelatinolytic bands with the appropriate molecular mass in kilodaltons is reported on the right.
As we already observed with puromycin (Zahm et al. 1991), no repair occurred in the presence of cycloheximide, which is another inhibitor of protein synthesis. When cycloheximide was tested in a migration assay, we observed that cycloheximide significantly inhibited cell migration after an incubation period of 1–2 h, suggesting that this time is needed to clear the intracellular pool of proteins, including MMP-9 and type IV collagen, involved in HBEC migration (data not shown). Monensin, added to migrating HBEC at a 5-μM concentration, inhibited cell migration within 10 min (data not shown). These results suggest that cell migration is closely dependent upon the production and secretion of proteins. Batimastat, a broad spectrum MMP inhibitor, known to inhibit invadopodial degradation of fibronectin by invasive MDA-MB-231 breast cancer cells (Kelly et al. 1998), inhibited in a dose-dependent way HBEC migration and 3H-gelatin degradation (Fig. 9A and Fig. B). The inhibition of HBEC migration that we observed in the different experimental conditions paralleled the inhibition of 3H-gelatin degradation. These results suggest that cell migration involves the gelatinase-dependent degradation of the ECM on which cells migrate. In an aqueous protein extract of migrating HBEC, MMP-9 is present only in its 92-kD inactive precursor form (Fig. 9 C, lanes A). After the removal of the cytosol, a detergent extraction of both membranes of migrating cells and their associated ECM revealed that MMP-9 is mainly present in these compartments in its 84-kD active form (Fig. 9 C, lanes D). This observation suggests that MMP-9, present at the cell membrane or deposited on the ECM during cell migration, is active.
Discussion
Our results clearly demonstrate that MMP-9 is actively involved in the migration of repairing HBEC: MMP-9 is only detected in migrating HBEC and its expression coincides with the cell migration speed; MMP-9 in migrating HBEC is addressed to lamellipodia advancing in the direction of migration and is found active on the ECM; HBEC migration coincides with an increased extracellular gelatinase activity; blocking MMP-9 activation or MMP-9 activity results in less cell migration; the MMP-9 substrate, type IV collagen, is present in primordial contacts of migrating HBEC; and MMP-9 accumulates close behind this rapidly remodeled collagen IV at sites with fewer cell–ECM interactions.
We observed that MMP-9 was upregulated only in migrating HBEC and more specifically in those cells lining the edge of a wound. MMP-9 has been shown to be involved in the migration of many cells (Pluznik et al. 1992; Leppert et al. 1995; Buisson et al. 1996a; Delclaux et al. 1996; Okada et al. 1997; McCawley et al. 1998; Uhm et al. 1998). In most of those studies, MMP-9 involvement in cell migration was demonstrated by the following observations: MMP-9 was upregulated and activated during migration; migration was partially inhibited in the presence of MMPs, or more specifically, MMP-9 inhibitors (tissue inhibitor of MMP-1, specific blocking Ab); induction or inhibition of cell migration coincided with induction or repression of MMP-9; and cell migration across an ECM barrier was concomitant with collagen degradation. The in vitro experimental approaches that we have developed enabled us to investigate more extensively the details of MMP-9 involvement in cell migration. Indeed, the quantification of cell migration coupled with the MMP-9 localization in migrating cells gives a new insight into the study of MMP-9 during the migration process. We demonstrated that, within the cells located close to the edge of the wound, only the cells that migrated rapidly expressed large amounts of MMP-9. The MMP-9 location at the edge of the wound could be explained by the faster migration of cells at the edge of the wound than those located further away (Zahm et al. 1997) and the expression of an active migratory phenotype only by cells at the edge of the wound, whereas cells further from this front can migrate as sheets and their migration may be directed by the cells at the forefront (Zahm et al. 1991).
In agreement with our postulate that the MMP-9 distribution specifically reflects the migration ability of epithelial cells, we also observed that, over a 30-min period, cell migration at the edge of a wound was heterogeneous but paralleled the MMP-9 content of migrating cells, whereas over a period of several hours, cell migration was uniform. These observations suggest that HBEC migration is a step-by-step process in which MMP-9 expression is highly and rapidly regulated. Induction of MMP-9 expression in migrating HBEC might reflect a modification of the ECM onto which the cells are progressing. For example, collagenase 1 (MMP-1) is expressed by migrating keratinocytes that have moved off an intact basement membrane and are in contact with dermal and provisional matrices (Saarialho-Kere et al. 1993). Recently, Yao et al. 1998 observed a lower constitutive production of MMP-9 by HBEC cultured on type IV collagen than on type I and III collagens, suggesting that type IV collagen is associated with a homeostatic phenotype and type I and III collagens with a matrix resorption phenotype. In contrast, our results indicate that type IV collagen is actively involved in HBEC migration because collagen IV was produced by migrating HBEC; collagen IV was specifically amassed at primordial contacts, the first cell–ECM contacts involved in cell migration; and the anchorage of cells onto collagen IV was essential for subsequent cell migration. Another possible mechanism of regulation of MMP-9 expression in migrating HBEC is via cell–cell adhesion molecules and changes in cell shape (Vu and Werb 1998). We observed that the disorganization of the actin cytoskeleton altered MMP-9 distribution in migrating HBEC. Recently, Chintala et al. 1998 reported that actin polymerization was required for the induction of MMP-9 and subsequent invasive properties of human glioma cells. Thus, actin is probably a key molecule controlling cell migration, MMP-9 expression, and trafficking in HBEC repairing a wound.
The active involvement of MMP-9 in HBEC migration led us to examine the question of the sequence of events occurring during cell migration and how MMP-9 could participate in it. Cell locomotion is a dynamic interplay of various processes, such as cell–ECM adhesion, extension of the leading edge of the cell, and retraction of the trailing edge. To move along the ECM, cells must first adhere to it, through cell–ECM contacts, with sufficiently strong links to allow subsequent spreading of the cell margin. In rapidly moving cells, adequate cell–ECM contacts are provided by highly labile regions of close contact, called primordial contacts, distributed at the cell front heading in the direction of migration (Lee et al. 1993; Small et al. 1996). Primordial contacts are present at the leading edge of migrating HBEC and they are characterized by a dense accumulation of actin filaments and the presence of vinculin and type IV collagen. These primordial contacts are very short-lived structures. Assuming that lamellipodia in migrating HBEC move at 0.5–1.5 μm/min (Fig. 3) and the collagen–vinculin band is ∼2.5-μm wide (Fig. 6), the life span of primordial contacts should be 1–5 min. The rapid remodeling of primordial contacts may be attributed to MMP-9 because of the following: MMP-9 is detected within and around cell–ECM contacts, most MMP-9 being identified just behind primordial contacts at sites with fewer cell–ECM interactions; behind the MMP-9 band in extending lamellipodia, a migrating cell establishes new contacts with the ECM through fibronectin-containing focal contacts; when MMP-9 activation is blocked, cells remain fixed on the previously established primordial contacts and no longer move; and confocal microscopy and zymography analysis of ECM-associated MMP-9 showed that MMP-9 was subsequently found active on the ECM, which is consistent with an extracellular action of MMP-9 on the preceding cell–ECM interactions. When studying the location and activation of gelatinases in endothelial cells, Partridge et al. 1997 noted that gelatinases colocalized with β1-integrin, thereby indicating the incorporation of gelatinases into focal contacts.
Those authors suggested that endothelial cells release MMPs into the extracellular milieu, and then direct and activate gelatinases at the focal contacts. More recently, MMP-9 was shown to be accumulated and released at the tip of pseudopods of endothelial cells invading a collagen gel (Nguyen et al. 1998). In our study, MMP-9 was not observed at the leading edge of advancing lamellipodia. Taken together, these observations suggest that MMP-9 distribution in invasive cells during angiogenesis (three-dimensional migration), i.e., association of MMP-9 with cell–ECM contacts, differs from that in repairing migratory cells (two-dimensional migration), i.e., location of MMP-9 close to cell–ECM contacts. An invasive cell would first have to digest to some extent its extracellular microenvironment before moving, whereas a cell migrating along the ECM would first have to establish contacts with this substrate, and then remodel these contacts to advance. Whereas the extending lamellipodia of repairing HBEC formed primordial contacts via type IV collagen, we observed that in the more central part of a migrating cell, type IV collagen disappeared from the cell–ECM contacts and cells are anchored to fibronectin at sites of focal contacts. We previously reported that fibronectin and the corresponding α5β1 integrin play an important role in the process of airway epithelium wound repair (Hérard et al. 1996a). These findings suggest that migrating HBEC produce different ECM molecules to be used for anchorage and that these distinct molecules play specific roles in the different cell–ECM contacts.
In conclusion, we hypothesize that wound repair by HBEC involves the following sequence of migrating events: anchorage onto type IV collagen via primordial contacts; MMP-9 trafficking just behind type IV collagen, to rapidly remodel these primordial contacts through the specific degradation of type IV collagen; and establishment of more stable fibronectin-containing contacts in the central part of the cell, while new primordial contacts with type IV collagen are made at the forefront.
Acknowledgments
The authors thank Drs. Edith Puchelle (INSERM Unité 514), Hervé Emonard, William Hornebeck, and Jean-Claude Monboisse (all from UPRESA CNRS 6021) for helpful comments and discussions, Dr. Deborah L. French for the gift of the 6-6B mAb, Dr. Shi-Ping Jiang (Oncologix, Inc., Gaithersburg, MD) for the gift of the GE 209 mAb, and Dr. Dominique Ploton (UPRES EA2063) and his associates for their help in the confocal microscopy study.
Claire Legrand is the recipient of a fellowship from the Ministère de l'Enseignement Supérieur et de la Recherche, France.
Footnotes
1.used in this paper: Ab, antibody; ECM, extracellular matrix; HBEC, human bronchial epithelial cells; MMP, matrix metalloproteinase
References
- Abercombie M., Heaysman J.E.M., Pegrum S.M. The locomotion of fibroblasts in culture. Exp. Cell Res. 1970;67:359–367. doi: 10.1016/0014-4827(71)90420-4. [DOI] [PubMed] [Google Scholar]
- Ågren M.S. Gelatinase activity during wound healing. Br. J. Dermatol. 1994;131:634–640. doi: 10.1111/j.1365-2133.1994.tb04974.x. [DOI] [PubMed] [Google Scholar]
- Bendeck M.P., Zempo N., Clowes A.W., Galardy R.E., Reidy M.A. Smooth muscle cell migration and matrix metalloproteinase expression after arterial injury in the rat. Circ. Res. 1994;75:539–545. doi: 10.1161/01.res.75.3.539. [DOI] [PubMed] [Google Scholar]
- Brezillon S., Dupuit F., Hinnrasky J., Marchand V., Kälin N., Tümmler B., Puchelle E. Decreased expression of the CFTR protein in remodeled human nasal epithelium from non-cystic fibrosis patients. Lab. Invest. 1995;72:191–200. [PubMed] [Google Scholar]
- Buisson A.C., Zahm J.M., Polette M., Pierrot D., Bellon G., Puchelle E., Birembaut P., Tournier J.M. Gelatinase B is involved in the in vitro wound repair of human respiratory epithelium J. Cell. Physiol. 166 1996. 413 426a [DOI] [PubMed] [Google Scholar]
- Buisson A.C., Gilles C., Polette M., Zahm J.M., Birembaut P., Tournier J.M. Wound repair-induced expression of stromelysins is associated with the acquisition of a mesenchymal phenotype in human respiratory epithelial cells Lab. Invest. 74 1996. 658 669b [PubMed] [Google Scholar]
- Chambers A.F., Matrisian L.M. Changing views of the role of matrix metalloproteinases in metastasis. J. Natl. Cancer Inst. 1997;89:1260–1269. doi: 10.1093/jnci/89.17.1260. [DOI] [PubMed] [Google Scholar]
- Chintala S.K., Sawaya R., Aggarwal B.B., Majumder S., Giri D.K., Kyritsis A.P., Gokaslan Z.L., Rao J.S. Induction of matrix metalloproteinase-9 requires a polymerized actin cytoskeleton in human malignant glioma cells. J. Biol. Chem. 1998;273:13545–13551. doi: 10.1074/jbc.273.22.13545. [DOI] [PubMed] [Google Scholar]
- Delclaux C., Delacourt C., d'Ortho M.P., Boyer V., Lafuma C., Harf A. Role of gelatinase B and elastase in human polymorphonuclear neutrophil migration across basement membrane. Am. J. Respir. Cell Mol. Biol. 1996;14:288–295. doi: 10.1165/ajrcmb.14.3.8845180. [DOI] [PubMed] [Google Scholar]
- Erjefält J.S., Erjefält I., Sundler F., Persson C.G.A. In vivo restitution of airway epithelium. Cell Tissue Res. 1995;281:305–316. doi: 10.1007/BF00583399. [DOI] [PubMed] [Google Scholar]
- Green H., Kehinde O., Thomas J. Growth of cultured human epidermal cells into multiple epithelia suitable for grafting. Proc. Natl. Acad. Sci. USA. 1979;76:5665–5668. doi: 10.1073/pnas.76.11.5665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hérard A.L., Pierrot D., Hinnrasky J., Kaplan H., Sheppard D., Puchelle E., Zahm J.M. Fibronectin and its α5β1-integrin receptor are involved in the wound-repair process of airway epithelium Am. J. Physiol. 271 1996. L726 L733a [DOI] [PubMed] [Google Scholar]
- Hérard A.L., Zahm J.M., Pierrot D., Hinnrasky J., Fuchey C., Puchelle E. Epithelial barrier integrity during in vitro wound repair of the airway epithelium Am. J. Respir. Cell Mol. Biol. 15 1996. 624 632b [DOI] [PubMed] [Google Scholar]
- Jockusch B.M., Bubeck P., Giehl K., Kroemker M., Moshner J., Rothkegel M., Rüdiger M., Schlüter K., Stanke G., Winkler J. The molecular architecture of focal adhesions. Annu. Rev. Cell Dev. Biol. 1995;11:379–416. doi: 10.1146/annurev.cb.11.110195.002115. [DOI] [PubMed] [Google Scholar]
- Keenan K.P., Combs J.W., McDowell E.M. Regeneration of hamster tracheal epithelium after mechanical injury. I. Focal lesionsquantitative morphologic study of cell proliferation. Virchows Arch. B. 1982;41:193–214. doi: 10.1007/BF02890281. [DOI] [PubMed] [Google Scholar]
- Kelly T., Yan Y., Osborne R.L., Athota A.B., Rozypal T.L., Colclasure J.C., Chu W.S. Proteolysis of extracellular matrix by invadopodia facilitates human breast cancer cell invasion and is mediated by matrix metalloproteinases. Clin. Exp. Metastasis. 1998;16:501–512. doi: 10.1023/a:1006538200886. [DOI] [PubMed] [Google Scholar]
- Lane B.P., Gordon R. Regeneration of rat tracheal epithelium after mechanical injury. The relationship between mitotic activity and cellular differentiation. Proc. Soc. Exp. Biol. Med. 1974;145:1139–1144. doi: 10.3181/00379727-145-37968. [DOI] [PubMed] [Google Scholar]
- Lee J., Ishihara A., Jacobson K. How do cells move along surfaces? Trends Cell Biol. 1993;3:366–370. doi: 10.1016/0962-8924(93)90084-e. [DOI] [PubMed] [Google Scholar]
- Leppert D., Waubant E., Galardy R., Bunnett N.W., Hauser S.L. T cell gelatinases mediate basement membrane transmigration in vitro. J. Immunol. 1995;154:4379–4389. [PubMed] [Google Scholar]
- Man, S.F.P., and W.C. Hulbert. 1988. Airway repair and adaptation to inhalation injury. In Pathophysiology and Treatment of Inhalation Injuries. Lung Biology in Health and Disease. J. Loke, editor. Marcel Dekker, New York. 1–47.
- Matsubara M., Girard M.T., Kublin C.L., Cintron C., Fini M.E. Differential roles for two gelatinolytic enzymes of the matrix metalloproteinase family in the remodeling cornea. Dev. Biol. 1991;147:425–439. doi: 10.1016/0012-1606(91)90300-r. [DOI] [PubMed] [Google Scholar]
- McCawley L.J., O'Brien P., Hudson L.G. Epidermal growth factor (EGF)- and scatter factor/hepatocyte growth factor (SF/HGF)-mediated keratinocyte migration is coincident with induction of matrix metalloproteinase (MMP-9) J. Cell. Physiol. 1998;176:255–265. doi: 10.1002/(SICI)1097-4652(199808)176:2<255::AID-JCP4>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- McDowell E.M., Becci P.J., Schurch W., Trump B.F. The respiratory epithelium. VII. Epidermoid metaplasia of hamster tracheal epithelium during regeneration following mechanical injury. J. Natl. Cancer Inst. 1979;62:995–1008. [PubMed] [Google Scholar]
- Nguyen M., Arkell J., Jackson C.J. Active and tissue inhibitor of matrix metalloproteinase-free gelatinase B accumulates within human microvascular endothelial vesicles. J. Biol. Chem. 1998;273:5400–5404. doi: 10.1074/jbc.273.9.5400. [DOI] [PubMed] [Google Scholar]
- Nikula K.J., Wilson D.W., Giri S.N., Plopper C.G., Dungworth D.L. The response of the rat tracheal epithelium to ozone exposureinjury, adaptation and repair. Am. J. Pathol. 1988;131:373–384. [PMC free article] [PubMed] [Google Scholar]
- Nordin U., Engström B., Jansson B., Lindholm C.E. Surface structure and vascular anatomy of the tracheal wall under normal conditions and after intubation Acta Anaesthesiol. Scand. 345Suppl.1977. 35 56 [Google Scholar]
- Oikarinen A., Kylmäniemi M., Autio-Harmainen H., Autio P., Salo T. Demonstration of 72-kDa and 92-kDa forms of type IV collagenase in human skinvariable expression in various blistering diseases, induction during re-epithelialization, and decrease by topical glucocorticoids. J. Invest. Dermatol. 1993;101:205–210. doi: 10.1111/1523-1747.ep12363823. [DOI] [PubMed] [Google Scholar]
- Okada S., Kita H., George J., Gleich G.J., Leiferman K. Migration of eosinophils through basement membrane components in vitrorole of matrix metalloproteinase-9. Am. J. Respir. Cell Mol. Biol. 1997;17:519–528. doi: 10.1165/ajrcmb.17.4.2877. [DOI] [PubMed] [Google Scholar]
- Partridge C.A., Phillips P.G., Niedbala M.J., Jeffrey J.J. Localization and activation of type IV collagenase/gelatinase at endothelial focal contacts. Am. J. Physiol. 1997;272:L813–L822. doi: 10.1152/ajplung.1997.272.5.L813. [DOI] [PubMed] [Google Scholar]
- Pluznik D.H., Fridman R., Reich R. Correlation in the expression of type IV collagenase and the invasive and chemotactic abilities of myelomonocytic cells differentiation into macrophages. Exp. Hematol. 1992;20:57–63. [PubMed] [Google Scholar]
- Ramos-DeSimone N., Moll U.M., Quigley J.P., French D.L. Inhibition of matrix metalloproteinase 9 activation by a specific monoclonal antibody. Hybridoma. 1993;12:349–363. doi: 10.1089/hyb.1993.12.349. [DOI] [PubMed] [Google Scholar]
- Roger P., Puchelle E., Bajolet-Laudinat O., Tournier J.-M., Debordeaux C., Plotkowski M.-C., Cohen J.H.M., Sheppard D., de Bentzmann S. Fibronectin and the α5β1 integrin mediate binding of Pseudomonas aeruginosa to repairing airway epithelium. Eur. Respir. J. 1999;In press [PubMed] [Google Scholar]
- Saarialho-Kere U.K., Kovacs S.O., Pentland A.P., Olerud J.E., Welgus H.G., Parks W.C. Cell–matrix interactions modulate interstitial collagenase expression by human keratinocytes actively involved in wound healing. J. Clin. Invest. 1993;92:2858–2866. doi: 10.1172/JCI116906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salo T., Mäkelä M., Kylmäniemi M., Autio-Harmainen H., Larjava H. Expression of matrix metalloproteinase-2 and -9 during early human wound healing. Lab. Invest. 1994;70:176–182. [PubMed] [Google Scholar]
- Small J.V., Anderson K., Rottner K. Actin and the coordination of protrusion, attachment and retraction in cell crawling. Biosci. Rep. 1996;16:351–368. doi: 10.1007/BF01207261. [DOI] [PubMed] [Google Scholar]
- Starkey J.R., Hosick H.L., Stanford D.R., Liggitt H.D. Interaction of metastatic tumor cells with bovine lens capsule basement membrane. Cancer Res. 1984;44:1585–1594. [PubMed] [Google Scholar]
- Stricklin G.P., Li L., Nanney L.B. Localization of mRNAs representing interstitial collagenase, 72-kDa gelatinase, and TIMP in healing porcine burn wounds. J. Invest. Dermatol. 1994;103:352–358. doi: 10.1111/1523-1747.ep12394926. [DOI] [PubMed] [Google Scholar]
- Uhm J.H., Dooley N.P., Oh L.Y.S., Yong V.W. Oligodendrocytes utilize a matrix metalloproteinase, MMP-9, to extend processes along an astrocyte extracellular matrix. Glia. 1998;22:53–63. doi: 10.1002/(sici)1098-1136(199801)22:1<53::aid-glia5>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- Vu T.H., Werb Z. Gelatinase Bstructure, regulation and function. In: Parks W.C., Mecham R.P., editors. Matrix Metalloproteinases (Biology of Extracellular Matrix Series) Academic Press; New York: 1998. pp. 115–148. [Google Scholar]
- Wilhelm D.L. Regeneration of tracheal epithelium. J. Pathol. Bacteriol. 1953;5025:543–550. doi: 10.1002/path.1700650226. [DOI] [PubMed] [Google Scholar]
- Woessner J.F. The matrix metalloproteinase family. In: Parks W.C., Mecham R.P., editors. Matrix Metalloproteinases (Biology of Extracellular Matrix Series) Academic Press; New York: 1998. pp. 1–14. [Google Scholar]
- Yao P.M., Delclaux C., d'Ortho M.P., Maitre B., Harf A., Lafuma C. Cell–matrix interactions modulate 92-kD gelatinase expression by human bronchial epithelial cells. Am. J. Respir. Cell Mol. Biol. 1998;18:813–822. doi: 10.1165/ajrcmb.18.6.2984. [DOI] [PubMed] [Google Scholar]
- Zahm J.M., Chevillard M., Puchelle E. Wound repair of human surface respiratory epithelium. Am. J. Respir. Cell Mol. Biol. 1991;5:242–248. doi: 10.1165/ajrcmb/5.3.242. [DOI] [PubMed] [Google Scholar]
- Zahm J.M., Kaplan H., Hérard A.L., Doriot F., Pierrot D., Somelette P., Puchelle E. Cell migration and proliferation during the in vitro wound repair of the respiratory epithelium. Cell Motil. Cytoskelet. 1997;37:33–43. doi: 10.1002/(SICI)1097-0169(1997)37:1<33::AID-CM4>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- Zigmond S.H. Cell locomotion and chemotaxis. Curr. Opin. Cell Biol. 1989;1:80–86. doi: 10.1016/s0955-0674(89)80041-9. [DOI] [PubMed] [Google Scholar]