

Abstract
We previously mapped a locus (ahl4) on distal Chromosome 10 that contributes to the age-related hearing loss of A/J strain mice. Here, we report on a refined genetic map position for ahl4 and its association with a mutation in the citrate synthase gene (Cs). We mapped ahl4 to the distal-most 7 Mb of Chromosome 10 by analysis of a new linkage backcross and then further narrowed the interval to 5.5 Mb by analysis of eight C57BL/6J congenic lines with different A/J-derived segments of Chr 10. A nucleotide variant in exon 3 of Cs is the only known DNA difference within the ahl4 candidate gene interval that is unique to the A/J strain and that causes a nonsynonymous codon change. Multiple lines of evidence implicate this missense mutation (H55N) as the underlying cause of ahl4-related hearing loss, likely through its effects on mitochondrial ATP and free radical production in cochlear hair cells. The A/J mouse thus provides a new model system for in vivo studies of mitochondrial function and hearing loss.
Keywords: mouse, genetics, inbred strain, A/J, age-related hearing loss, ahl4, citrate synthase, oxidative stress, mitochondria
1. Introduction
Age-related hearing loss (AHL), or presbycusis, is the most common sensory deficit of elderly individuals and adversely affects their quality of life (Mulrow et al., 1990). Although AHL has a significant genetic component (DeStefano et al., 2003), the identification of susceptibility genes in human populations is extremely difficult because of the late age of manifestation, extensive genetic heterogeneity, and confounding environmental influences such as noise and drug exposures, and only limited success has been achieved (Friedman et al., 2009; Huyghe et al., 2008). Genetically well-defined inbred mouse strains provide a complementary approach that can circumvent many of these difficulties. Inbred strain mice exhibit variable degrees of AHL with characteristics similar to human presbycusis, and they offer the advantages of a short life span and the ability to control both genetic and environmental variation (Henry, 1982; Hunter and Willott, 1987; Noben-Trauth and Johnson, 2009; Ohlemiller, 2006; Zheng et al., 1999). Linkage studies have established a strong genetic basis for AHL in inbred mouse strains and provided chromosomal map positions for multiple underlying genes (Noben-Trauth and Johnson, 2009).
C57BL/6J (B6) and A/J are two popular inbred mouse strains that exhibit AHL. The hearing loss of A/J mice, however, occurs much earlier and progresses more rapidly than that of B6 mice (Zheng et al., 2009). As is typical of AHL in mammals, hearing loss in both A/J and B6 mice occurs earliest and is most severe at high frequencies. The primary cochlear pathology associated with AHL in A/J mice is a rapid loss of sensory hair cells, beginning at the basal turn and progressing toward the apex (Zheng et al., 2009). A similar, but less rapid, pattern of hair cell loss occurs in B6 mice (Spongr et al., 1997). Some evidence indicates that hair cell degeneration may also be a primary underlying cause of hearing loss in elderly humans (Soucek et al., 1986). Because hair cells do not regenerate in mammals, their cumulative loss eventually leads to AHL.
Although B6 and A/J strain mice exhibit dramatically different time courses of AHL, they share the same ahl variant of the cadherin 23 gene (Cdh23) that confers increased AHL susceptibility to many inbred mouse strains (Johnson et al., 2000; Noben-Trauth et al., 2003). Additional genetic factors, therefore, must contribute to the accelerated rate of hearing loss in A/J mice relative to that in B6 mice, and two such factors have been identified: a DNA variant of the mitochondrial tRNA arginine (mt-Tr) gene (Johnson et al., 2001) and a susceptibility allele (ahl4) that recently was mapped to distal Chr 10 (Zheng et al., 2009). The identification and characterization of these contributing factors will provide entry points into molecular pathways and mechanisms that underlie hearing loss in these strains.
Oxidative damage to cochlear cells caused by high levels of reactive oxygen species (ROS) is one mechanism that is known to accelerate AHL in inbred mouse strains, as shown by studies of mice lacking antioxidant enzymes (Keithley et al., 2005; McFadden et al., 1999; Ohlemiller et al., 2000) and supported by age-related changes in oxidative stress markers (Jiang et al., 2007; Staecker et al., 2001). Mitochondrial dysfunction, which decreases cellular energy metabolism and increases ROS production, is thought to play a major role in the aging process of cochlear hair cells (Someya and Prolla, 2010; Yamasoba et al., 2007) and probably underlies the hearing loss attributed to the mt-Tr variant of A/J mice. Although the ahl4 locus contributes more to the hearing loss of A/J mice than mt-Tr, the responsible gene has not been identified, and the pathological mechanisms underlying its effects are unknown (Zheng et al., 2009).
In this report, we provide multiple lines of evidence indicating that a missense mutation of the citrate synthase gene (Cs), unique to the A/J strain, is responsible for the hearing loss attributed to ahl4. Citrate synthase is the pace-making enzyme in the first step of the citric acid cycle, which occurs in the mitochondrial matrix and generates high-energy NADH and FADH2 for oxidative phosphorylation. Impaired citrate synthase activity could thus impact mitochondrial function and exacerbate AHL, similar to the effect caused by the mt-Tr variant of A/J mice. In support of a common mitochondrial pathway of pathology, we show that ahl4 (Cs) and mt-Tr genetically interact to influence AHL severity.
2. Materials and methods
2.1 Mice
All mice used in this study originated from The Jackson Laboratory (http://www.jax.org/), including mice of the C57BL/6J (Stock Number 000664), A/J (Stock Number 000646), C57BL/6J-Chr 10A/J/NaJ (Stock Number 004388), and C57BL/6J-mtA/J/NaJ (Stock Number 005545) inbred strains and genetic admixtures of these strains. Experimental mice were housed in the Research Animal Facility of the Jackson Laboratory, and all procedures involving their use were approved by the Institutional Animal Care and Use Committee. The Jackson Laboratory is accredited by the American Association for the Accreditation of Laboratory Animal Care.
2.2 Hearing assessment of mice by ABR
Hearing in mice was assessed by ABR threshold analysis, as previously described (Zheng et al., 1999). Briefly, the evoked brainstem responses of anesthetized mice were amplified and averaged and their wave patterns displayed on a computer screen. Auditory thresholds were obtained for each specific auditory stimulus by varying the sound pressure level (SPL) to identify the lowest level at which an ABR pattern could be recognized. 100 dB was the maximum SPL presented for all stimuli. With our testing system, average ABR thresholds (in dB SPL) for normal hearing mice are about 30 dB for 8 kHz, 20 dB for 16 kHz, and 45 dB for 32 kHz stimuli. We considered 20 - 40 dB SPL above normal to be a mild impairment, 41 - 60 dB above normal to be intermediate, and greater than 60 dB above normal to be a profound impairment or deafness.
2.3 Genetic mapping
F1 hybrids of matings between C57BL/6J-Chr 10A/J/NaJ and C57BL/6J strain mice were backcrossed to C57BL/6J-Chr 10A/J/NaJ strain mice. This backcross is herein designated (B6-Chr10A/J × B6) F1 × B6-Chr10A/J. DNA samples from tail tips of backcross progeny (N2 generation) mice were genotyped for multiple polymorphic markers located on Chromosome (Chr) 10. PCR primer pairs designed to amplify specific markers were purchased from Integrated DNA Technologies (Coralville, IA, USA). Most genetic markers were DNA microsatellites genotyped by PCR product size differences, as previously described (Gagnon et al., 2006). Some markers were identified as single nucleotide polymorphisms (SNPs) and genotyped by KBioscience (Hoddesdon, Hertfordshire, UK) facilitated through The Jackson Laboratory’s SNP Genotyping Service. All chromosomal positions (in Mb) given for genomic DNA sequences correspond to NCBI Build m37. ABR thresholds of individual mice were evaluated as quantitative traits, and linkage analysis was performed using the computer program Map Manager QTX (Manly et al., 2001). This program uses a fast regression method to detect and localize quantitative trait loci (QTLs) within intervals defined by genetic markers and can also perform pair-wise locus analysis to search for QTL interactive effects.
2.4 Congenic line development
The C57BL/6J-Chr 10A/J/NaJ chromosome substitution (CS) strain was used to generate eight congenic lines of B6 mice, each with a different sub-region of Chr 10 derived from the A/J strain by backcross introgression. By using a CS strain rather than a standard inbred strain to generate congenic lines, only markers on a single chromosome need to be genotyped and fewer backcrosses are needed for introgression (Nadeau et al., 2000). F1 hybrids of matings between C57BL/6J-Chr 10A/J/NaJ and C57BL/6J strain mice were backcrossed to C57BL/6J mice. N2 and subsequent backcross generation mice were genotyped for markers along the length of Chr 10, loci on all other chromosomes being homozygous for B6 alleles. Mice with selected genotypes were then interbred and their genetically characterized, homozygous progeny used as progenitors to establish eight separate congenic lines. Congenic Line 8, which has the smallest A/J-derived Chr 10 segment containing ahl4, is designated B6.A-(rs3676616-D10Utsw1)/Kjn and is publicly available from The Jackson Laboratory as Stock #14192.
To create a B6.A-ahl4 congenic line of mice with A/J (rather than B6) mitochondria, female mice of the C57BL/6J-mtA/J/NaJ mitochondrial substitution strain were mated with male mice of B6.A-ahl4 Congenic Line 7. Female progeny from this cross, which are heterozygous for ahl4 and have A/J mitochondria, were then backcrossed to Congenic Line 7 male mice. Male and female N2 progeny with ahl4/ahl4 genotypes were selected from this backcross and interbred to establish a new B6.A-ahl4 congenic line with A/J mitochondria.
2.5 Genotyping mice for the citrate synthase SNP by DNA sequence analysis
Genotyping was done by DNA sequence analysis. A 250 bp region around exon 3 of the citrate synthase (Cs) gene, which contains the rs29358506 SNP, was amplified from genomic DNA by PCR with forward primer GGCAGACCAAGAAACGCTAA and reverse primer CCTTCGTGGCACACATCTAA. Synthesized primers were purchased from Integrated DNA Technologies (Coralville, IA, USA). PCR products were purified with the QIAquick PCR Purification Kit (Qiagen Inc., Valencia, CA) in preparation for DNA sequencing. The same primers used for DNA amplification were also used for DNA sequencing on an Applied Biosystems 3700 DNA Sequencer with an optimized Big Dye Terminator Cycle Sequencing method.
3. Results
3.1 Fine mapping of ahl4 by analysis of a new linkage backcross
To reduce genetic variation, we used the C57BL/6J-Chr 10A/J/NaJ chromosome substitution (CS) strain (Nadeau et al., 2000), abbreviated B6-Chr10A/J, instead of the A/J strain for backcross linkage analysis. F1 hybrids between B6-Chr10A/J and B6 mice are heterozygous only for loci on Chr 10. The F1 hybrids were backcrossed to B6-Chr10A/J mice rather than B6 mice because homozygosity for A/J alleles is required for ahl4 manifestation (Zheng et al., 2009). We analyzed 245 N2 mice from this (B6-Chr10A/J × B6) F1 × B6-Chr10A/J backcross for linkage associations of Chr 10 markers with ABR thresholds, analyzed as quantitative traits. Genotypes for segregating loci on Chr 10 were either homozygous for alleles derived from A/J (AA) or heterozygous for alleles derived from A/J and B6 (AB).
The highest linkage association of ABR thresholds was with the distal-most marker evaluated, D10Mit297 (Fig. 1A). LOD scores for D10Mit297 were statistically highly significant (greater than 14) for 16 kHz and 32 kHz thresholds at both 6 and 9 months of age (Supplementary Table 1). The highest LOD score (16.51) was for 16 kHz thresholds at 9 months of age, which could explain 27% of the total (genetic plus environmental) variation exhibited by the backcross mice. In contrast, the highest LOD score (4.53) for linkage with 8 kHz thresholds could explain only 8% of the total variation, demonstrating that ahl4-related hearing loss in the backcross mice was mostly restricted to high frequencies. A conservative 2-LOD interval surrounding the peak LOD score was used to estimate a 7 Mb candidate gene region for ahl4 (Fig. 1A), extending from the 123 Mb position to the distal end of Chr 10 (~130 Mb).
Figure 1. Genetic map position and effect of ahl4 on hearing thresholds of backcross mice.

(A) Linkage analysis of 245 mice from the (B6-Chr10A/J × B6) F1 × B6-Chr10A/J backcross established a 7 Mb candidate gene interval for ahl4, between the 123 Mb position and the distal-most end of Chr 10 (130 Mb). LOD scores are shown for the linkage associations of each marker locus with the 16 kHz ABR thresholds of 6-month-old backcross mice. The peak linkage association (LOD 15.8) was with the distal-most marker evaluated, D10Mit297 at 124.5 Mb. (B) The average16 kHz ABR threshold (in dB SPL, with standard error bars) is shown for backcross mice of each ahl4 genotype tested at 6 and 9 months of age. D10Mit297 was used as a marker for ahl4: mice homozygous for A/J-derived alleles are designated AA (N = 110), and mice heterozygous for A/J and B6 alleles are designated AB (N = 135). The mean threshold differences between the two genotypes at each age were statistically highly significant (2-tailed t-test: P < 9 E-14 for the 6-month test age and P < 1 E-16 for the 9-month test age). The ahl4 locus can explain about 26% of the total 16 kHz ABR threshold variation observed among the backcross mice at both ages.
We used the closely linked marker D10Mit297 to infer ahl4 genotypes of backcross mice. At 6 months of age, the average 16 kHz threshold for mice with AA genotypes (ahl4/ahl4) was 46.4 compared with 26.5 for AB mice (ahl4/+), and at 9 months of age the average 16 kHz threshold was 71.8 for AA mice (ahl4/ahl4) compared with 46.3 for AB mice (ah4/+). Mean differences between genotypes at both ages were statistically highly significant (Fig. 1B), confirming that the ahl4 locus had a major effect on the hearing loss susceptibility of the backcross mice.
3.2 Refinement of the ahl4 candidate gene region by congenic strain analysis
C57BL/6J congenic lines with different A/J-derived segments of Chr 10 were developed to further refine the location of ahl4 and to determine the isolated effect of the ahl4/ahl4 genotype on hearing loss. We used the C57BL/6J-Chr 10A/J/NaJ chromosome substitution strain (Nadeau et al., 2000) and backcross introgression, as described in Section 2.4, to derive the eight congenic lines shown in Fig. 2.
Figure 2. Refined localization of ahl4 by means of congenic line analysis.
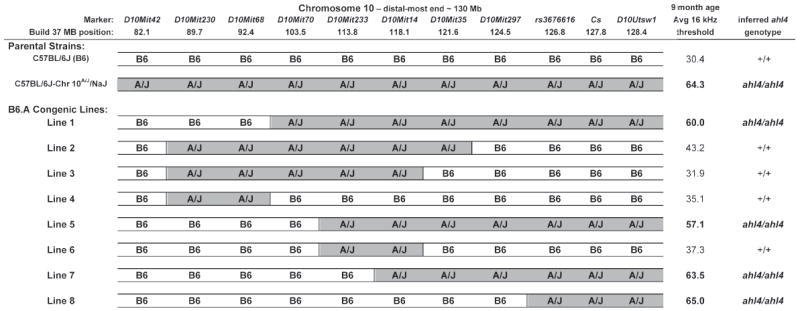
Eight congenic lines, derived from the C57BL/6J-Chr 10A/J/NaJ chromosome substitution strain, were constructed with different A/J-derived segments of Chr 10 introgressed into the C57BL/6J genome. The average 16 kHz ABR thresholds of 9-month-old mice were used to infer ahl4 genotypes for each congenic line. Comparisons of these genotypes with genotypes of Chr 10 marker loci indicate that ahl4 is located distal to D10Mit297, narrowing the candidate region to a 5.5 Mb interval (124.5-130 Mb), which includes the citrate synthase (Cs) candidate gene. The average ABR thresholds of the parental C57BL/6J and C57BL/6J-Chr 10A/J/NaJ strains are shown for comparisons.
ABR threshold averages, standard deviations, and standard errors were calculated for mice from each congenic line, tested at 6 and 9 months of age with 8 kHz, 16 kHz, and 32 kHz auditory stimuli (Supplementary Table 2). Mice of the two parental strains used to create the congenic lines, C57BL/6J and C57BL/6J-Chr 10A/J/NaJ, were also tested for comparisons. Of all auditory stimulus-test age combinations, the 16 kHz ABR thresholds of 9-month-old mice showed the greatest differences between the two parental strains (Supplementary Table 2) and were chosen to infer ahl4 genotypes for each of the derivative congenic lines (Fig. 2). Comparisons of these inferred ahl4 genotypes with the known genotypes of Chr 10 marker loci indicated that ahl4 is distal to D10Mit297 (Fig. 2). Congenic Line 8, which has the most restricted region of A/J-derived Chr 10, was particularly informative and narrowed the ahl4 candidate gene region to the distal-most 5.5 Mb (124.5-130 Mb) of Chr 10, a region that includes the citrate synthase gene (Cs).
The congenic strain results also demonstrate that homozygosity for the A/J allele of ahl4 by itself can accelerate hearing loss in otherwise B6 mice and does not require contributions from other loci for its manifestation on this strain background.
3.3 Evidence that a citrate synthase mutation underlies ahl4-related hearing loss
The 5.5 Mb candidate region for ahl4 includes 104 annotated genes and a cluster of 59 olfactory receptor genes (Mouse Genome Database, http://www.informatics.jax.org). To identify the causative gene mutation for ahl4, we first examined known inbred strain SNPs in the candidate gene region. A/J is the only strain we have studied that shows an ahl4 influence on hearing loss (Zheng et al., 2009). The ahl4 locus was not linked with ABR thresholds in previously analyzed backcrosses involving BUB/BnJ, DBA/2J, NOD/ShiLtJ, BALBcByJ, and C57BL/6J strains, even though these strains have the same predisposing ahl allele as A/J (Johnson et al., 2000). We therefore reasoned that the A/J allele of the gene responsible for ahl4 must differ from the alleles of these other inbred strains. We first compared A/J with two other strains having high SNP densities: C57BL/6J, the mouse genome reference strain, and BALB/cByJ, a strain that is genetically closely related to A/J (Petkov et al., 2004). All three of these strains have been genotyped for more than 8 million SNPs (Frazer et al., 2007). We searched the Mouse Phenome SNP Database (http://phenome.jax.org/SNP/) to identify A/J-specific variants within the ahl4 candidate interval on Chr 10 (124.5-130 Mb). A total of 15,816 SNPs within the 5.5 Mb interval have allele data for all three strains, 699 of these occur within known exons. The A/J allele differs from both BALB/cByJ and C57BL/6J alleles at 116 SNPs in the candidate interval, but only one of these occurs within an exon – SNP rs29358506, which is located in exon 3 of the citrate synthase gene (Cs).
The Sanger - Wellcome Trust Mouse Genome Project has sequenced the entire genomes of 14 inbred strains at over 20x coverage with reference to the C57BL/6J genome, including A/J, BALB/cJ, DBA/2J, and NOD/ShiLtJ, (http://www.sanger.ac.uk/resources/mouse/genomes/). A search of this database for SNPs and short insertion/deletions that differ between A/J and these other strains identified the Cs SNP variant but failed to find any other verifiable A/J-specific exonic DNA sequence differences in the 5.5 Mb ahl4 candidate region. Thus, both the inbred strain SNP data and the DNA sequence data strongly support the Cs variant (rather than an undiscovered variant in a closely linked gene) as the mutation responsible for the ahl4 hearing loss phenotype of A/J mice. In the following sections we provide additional lines of evidence supporting this conclusion.
Further analysis of SNP databases showed that A/J is the only one of 110 strains and substrains surveyed that has an adenine nucleotide at the Cs SNP; all other strains have cytosine (Supplementary Fig. 1). The unique A/J variant is predicted to cause an amino acid change of histidine to asparagine at position 55 (H55N) of the mouse CS protein (NP_080720). The histidine at this position is evolutionarily conserved in the orthologous CS proteins of all mammalian species examined and in many other vertebrate and invertebrate species (Supplementary Table 3), indicating a constrained functional importance for this residue.
We examined the genealogy of A strain lineages to determine the origin of the A/J-specific Cs mutation (Fig. 3A). The original A strain was developed by L.C. Strong in 1921, and from this lineage the A/WySn strain was separated in 1927, the A/J strain in 1928, and the A/He strain in 1938 (Festing; Ho et al., 2004). The derivative AXB and BXA recombinant inbred (RI) strains were separated from the A/J production colony around 1975 (Nesbitt and Skamene, 1984), and the B6.A chromosome substitution (CS) strains were separated from A/J in the mid 1990s (Nadeau et al., 2000). Genotyping of the rs29358506 SNP in these strains revealed that the A/WySnJ and A/HeJ inbred strains have the common C nucleotide, whereas the more recently derived AXB and BXA RI strains and B6.A CS strains have the mutated A nucleotide (Fig. 3A, B). An archived DNA sample from A/J mice sampled in 1983 also had the A nucleotide. We conclude that the C to A mutation must have occurred in the A/J lineage between 1928 (when the A/J line was separated from the other A strains) and 1975 (when the derivative AXB and BXA RI strains were developed). Consistent with their Cs genotypes (Fig. 3B), the hearing loss of A/J mice occurs earlier and progresses more rapidly than in mice of the genetically closely related A/WySnJ and A/HeJ strains (Fig. 3C).
Figure 3. Genealogy and hearing loss phenotypes of A-strain mice and their associations with a Cs DNA variant.
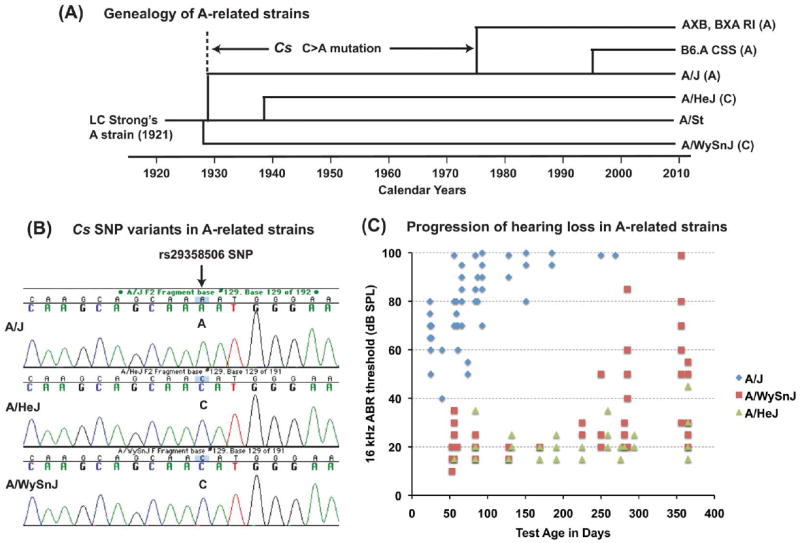
(A) Genealogy of inbred strains that have been derived from the original A strain developed by L.C. Strong in 1921 (A/St). Shown are the approximate dates when these A-related inbred strains were separated from one another. The nucleotide (C or A) corresponding to the rs29358506 SNP, which is located in exon 3 of Cs, is shown for each strain and indicates that the C to A mutation must have occurred in the A/J strain lineage sometime between 1928 and 1975. (B) DNA sequence analysis of the rs29358506 SNP in A/HeJ and A/WySnJ strain mice. Both of these closely related strains have the conserved C nucleotide at this position, in contrast to the A nucleotide of the A/J strain. (C) Hearing loss occurs earlier and progresses more rapidly in A/J mice than in A/HeJ or A/WySnJ mice. A scatter plot shows the 16 kHz ABR thresholds of individual mice from each strain tested at multiple ages (number of data points: A/J 100, A/WySnJ 86, A/HeJ 67).
Evidence for a genetic interaction of ahl4 (Cs) with mt-Tr
We previously reported that a variant of the mitochondrial arginine tRNA gene (mt-Tr) contributes to the hearing loss of A/J mice (Johnson et al., 2001). Our evidence that a Cs mutation underlies ahl4 led us to investigate whether a genetic interaction exists between ahl4 and mt-Tr, hypothesizing that both mutations impair mitochondrial function. Because mitochondria are strictly maternally inherited, we first tested for a maternal influence on hearing loss. B.A-ahl4 congenic line mice are homozygous for the A/J-derived ahl4 variant but otherwise have a B6 genome and B6 mitochondria. Individual male and female mice of B6.A-ahl4 Congenic Line 5 (Fig. 2) were mated with A/J mice to produce reciprocal F1 hybrids. Because they have identical nuclear genomes, any differences in the phenotypes of the reciprocal hybrids are limited to sex-related influences of the parental strains, including possible mitochondrial effects. We assessed hearing in the reciprocal F1 hybrids at 6 and 9 months of age. The average 16 kHz ABR threshold of (A/J female × B6.A-ahl4 male) F1 hybrids was 9 dB higher than that of reciprocal (B6.A-ahl4 female × A/J male) F1 hybrids at 6 months of age and about 30 dB higher at 9 months (Fig. 4A). Threshold differences at both test ages were statistically significant. These results demonstrate a maternal effect of the A/J strain that worsens the hearing loss in genetically identical F1 hybrids with ahl4/ahl4 genotypes.
Figure 4. A/J-derived mitochondria worsen the hearing loss of B6.A-ahl4 congenic mice.

(A) Maternal effects on hearing loss in reciprocal (B6.A-ahl4 × A/J) F1 hybrids. In the figure (A/J female × B6.A-ahl4 male) F1 hybrids are denoted as A/J×B6, and (B6.A-ahl4 female × A/J male) F1 hybrids as B6×A/J. Both have identical nuclear genomes and are homozygous for ahl4. Average 16 kHz ABR thresholds (with standard error bars) are shown for mice tested at 6 and 9-months of age. Mean thresholds of A/J×B6 hybrids are statistically significantly higher than those of the reciprocal B6×A/J hybrids (2-tailed t-test: * P < 0.05 for the 6-month test age, and ** P < 0.01 for the 9-month test age). (B) Genetic interaction of ahl4 with A/J-derived mitochondria. B6.A-ahl4 congenic mice with B6 mitochondria (ahl4/ahl4, B6 mtDNA), B6.A-ahl4 congenic mice with A/J mitochondria (ahl4/ahl4, A/J mtDNA), B6 mice with A/J mitochondria (+/+, A/J mtDNA), and B6 mice with B6 mitochondria (+/+, B6 mtDNA) were assessed for hearing at 9 months of age, and their average 16 kHz ABR thresholds (with standard error bars) compared. Mean thresholds of B6.A-ahl4/ahl4 mice with A/J mtDNA were statistically significantly higher than those with B6 mtDNA (* 2 tailed t-test: P < 0.05). Although higher, the average thresholds of B6 mice with A/J mtDNA were not statistically different from those of B6 mice with B6 mtDNA.
It is possible that a maternal influence other than mitochondria was responsible for the differences we observed in the hearing loss phenotypes of the reciprocal F1 hybrids. To directly test for mitochondrial effects, we constructed a B6.A-ahl4 congenic line with A/J-derived mitochondria to compare with the previously described B6.A-ahl4 congenic lines with B6 mitochondria. Mice of B6.A-ahl4 congenic Line 7 (Fig. 2), which are homozygous for ahl4 and have B6-derived mitochondria (ahl4/ahl4, B6 mtDNA) and mice of the C57BL/6J-mtA/J/NaJ mitochondrial substitution strain (+/+, A/J mtDNA) were used to create a new B6.A-ahl4 congenic line with A/J-derived mitochondria (ahl4/ahl4, A/J mtDNA), as described in Section 2.4. Except for A/J-derived ahl4 and mtDNA differences, all three strains are genetically identical to B6 mice (+/+, B6 mtDNA). The average 16 kHz ABR thresholds of 9-month-old mice from each of these four strains are shown in Fig. 4B. Thresholds of B6.A-ahl4 congenic mice with A/J mtDNA (ahl4/ahl4, A/J mtDNA) were on average 10.4 dB higher than those of B6.A-ahl4 congenic mice with B6 mtDNA (ahl4/ahl4, B6 mtDNA), a statistically significant difference. Although 5 dB higher, the average thresholds of C57BL/6J-mtA/J/NaJ mice (+/+, A/J mtDNA) were not statistically different from those of B6 mice (+/+, B6 mtDNA). These results confirm that A/J mitochondria exacerbate hearing loss in mice with ahl4/ahl4 genotypes, providing genetic evidence that ahl4 is likely to be involved in a mitochondrial-related mechanism of pathology and further supporting Cs as the gene responsible for ahl4.
4. Discussion
A splice variant of the gene encoding CDH23, a component of the tip link in hair cell stereocilia (Siemens et al., 2004), is thought to underlie the progressive hearing loss attributed to the ahl locus (Noben-Trauth et al., 2003). This variant (Cdh23ahl) encodes a defective CDH23 isoform that may weaken tip links or impede their repair. A consequent increase in the frequency of broken tip links could stress cochlear hair cells and lead to their degeneration over time (Noben-Trauth and Johnson, 2009). The Cdh23ahl/ahl genotype appears to sensitize hair cells to the detrimental effects of other gene mutations, which otherwise may not be apparent until much older ages (Johnson et al., 2006). All of the mice examined in the present study have the predisposing Cdh23ahl/ahl genotype, so phenotypic differences cannot be ascribed to this gene.
We provide several lines of evidence that a missense mutation of Cs is responsible for the ahl4-related hearing loss of A/J strain mice. Our backcross linkage analysis placed ahl4 within the distal-most 7 Mb of Chr 10, and this result was confirmed by congenic strain analysis, which further narrowed the candidate region to 5.5 Mb. The entire genomes of A/J and four strains known to lack an ahl4 hearing loss effect (C57BL/6J, BALB/cJ, DBA/2J, and NOD/ShiLtJ) have been sequenced at 20x coverage (http://www.sanger.ac.uk/resources/mouse/genomes/). The rs29358506 SNP, which is located in exon 3 of Cs, is the only exon or splice site variant within the 5.5 Mb candidate region that is unique to the A/J strain. Mice of the genetically closely related strains A/HeJ and A/WySnJ do not have this mutation and do not exhibit the early onset and rapid progression of hearing loss characteristic of A/J mice. Examination of A-related mouse strains for the presence of this mutation combined with knowledge of their genealogy enabled us to deduce that the Cs mutation must have occurred in the A/J strain lineage at The Jackson Laboratory sometime between 1928 and 1975 and then became fixed in the breeding stocks.
The C to A nucleotide change in exon 3 of Cs in A/J mice causes a non-conservative amino acid change, from histidine (H) to asparagine (N) at position 55 of the CS protein (H55N). This histidine is highly conserved in the orthologous proteins of all mammals and in many other species (Supplementary Table 3), indicating that it is likely to be functionally important. The PolyPhen computer tool (http://coot.embl.de/PolyPhen/), which uses both phylogenetic and structural information to assess the impact of amino acid substitutions on protein function, predicts that the H55N mutation is probably damaging. But the strongest evidence for the deleterious nature of the H55N mutation on protein function comes from a recent investigation into the cause of low CS activity (50-65% reduction) in skeletal muscle of A/J mice compared with other strains (Ratkevicius et al., 2010). This study showed that A/J mice have the same CS and mitochondrial protein content as mice of other strains, but differ in CS enzyme kinetics and catalytic properties, thereby implicating the H55N mutation as the most likely cause for the low CS activity of this strain.
Citrate synthase in mammals is encoded by a single nuclear gene. After translation in the cytosol, CS is transported into the mitochondrial matrix, where it functions as the first and rate-limiting enzyme of the citric acid cycle (Cheng et al., 2009) and thus plays a decisive role in regulating energy generation and ROS production of mitochondrial respiration. A missense mutation of CS might therefore be expected to have deleterious effects on mitochondrial function. Mitochondrial dysfunction can lead to progressively impaired energy metabolism, continuous ROS production, oxidative damage, and eventual cell death (Balaban et al., 2005; Dirks et al., 2006).
Poor performance in endurance exercises suggests that A/J mice may have reduced energy metabolism, consistent with the possibility that the H55N mutation negatively affects mitochondrial ATP production. In a study of genetic variation in aerobic capacity, A/J mice exhibited the shortest treadmill duration time among the 10 inbred mouse strains tested (Lightfoot et al., 2001). A more recent study also showed poor treadmill performance in A/J mice, estimating the endurance capacity of BALB/c mice to be 52% greater than that of C57BL/6J mice and 217% greater than that of A/J mice (Haramizu et al., 2009). However, without support from genetic linkage studies or other independent evidence, a causal relationship between the CS mutation and exercise endurance in A/J mice remains speculative.
Mitochondrial dysfunction associated with excess ROS generation and decreased energy metabolism is thought to play a central role in many age-associated neurodegenerative diseases (Balaban et al., 2005; Lin and Beal, 2006), including AHL (Jiang et al., 2007; Someya and Prolla, 2010; Yamasoba et al., 2007). The sensory hair cells of the inner ear are metabolically active and require high levels of ATP for such activities as actin treadmilling, phosphoinositide turnover, myosin-based motility, and calcium pumping (Shin et al., 2007). Because of their high energy requirement, hair cells may be especially sensitive to impaired mitochondrial function such as may be caused by the CS mutation in A/J mice. Diminished CS activity may decrease the production of ATP by slowing the rate of oxidative phosphorylation or decreasing the rate of glycolysis. This disruption in energy metabolism could increase ROS production and oxidative stress in high-energy demanding cells and trigger stress-activated or other signal transduction pathways resulting in cell death. A study of yeast cells that lack the mitochondrial citrate synthase gene provides some support for a relationship between CS deficiency and stress-induced cell death. These cells were shown to be hyper susceptible to heat- or aging-induced apoptosis, which was preceded by a depletion of glutathione and an accumulation of ROS in the mitochondria (Lee et al., 2007). A similar mechanism involving CS deficiency, impaired mitochondrial function, and oxidative stress may contribute to the progressive hair cell death that is a characteristic feature of cochlear pathology in A/J mice (Zheng et al., 2009).
In addition to ahl and ahl4 alleles, which are encoded by nuclear genes, A/J mice have a variant of the mitochondrial arginine tRNA gene (mt-Tr) that also contributes to AHL susceptibility (Johnson et al., 2001). Cell lines carrying mitochondrial DNA (mt DNA) with the A/J strain variant of mt-Tr were shown to have a lower per-mitochondrion respiration capacity and an overall increase in ROS production than lines carrying wild-type mtDNA (Moreno-Loshuertos et al., 2006), suggesting that negative effects of excess ROS production may underlie the hearing loss associated with the mt-Tr variant. Because deleterious variants of mt-Tr and Cs (the putative ahl4 gene) are both expected to impair mitochondrial function, we undertook genetic experiments to examine possible interactive effects of these genes. We found that hearing loss in ahl4/ahl4 mice is indeed exacerbated by the presence of A/J-derived mitochondria (Fig. 4), suggesting a common mitochondrial pathway of pathology and indirectly supporting Cs as the gene underlying ahl4. These results identify a new nuclear-mitochondrial DNA interaction and underscore the magnitude of effects that nuclear genes can have on modifying the penetrance and severity of hearing loss associated with mtDNA mutations (Carelli et al., 2003; Kokotas et al., 2007).
Although there appears to be a strong association between mitochondrial dysfunction and progressive hearing loss, little is known about the underlying genetic factors and molecular mechanisms that may be involved. Most of what we know about mitochondrial functions relating to hearing loss in mice has been gleaned from phenotypes associated with acquired mtDNA mutations in somatic tissues. Mice engineered to carry a mutator allele of mitochondrial polymerase gamma (POLG) display a large increase in accumulated mtDNA mutations and display features of accelerated aging (Kujoth et al., 2005), including AHL (Niu et al., 2007; Someya et al., 2008; Yamasoba et al., 2007). The A/J mouse model described here provides a complementary in vivo system to study mitochondrial function and hearing loss, but with respect to specific heritable factors rather than acquired somatic mtDNA mutations.
Supplementary Material
Acknowledgments
We thank Sandra Gray for her skilled management of the inbred strains, linkage crosses, and congenic lines of mice analyzed in this study. We also thank The Jackson Laboratory’s Genome Sciences service for DNA sequencing and SNP genotyping assays. The research reported here was supported by grant DC005827 (KRJ) from the National Institutes of Health (NIH), National Institute on Deafness and Other Communication Disorders (NIDCD). The Jackson Laboratory institutional shared services are supported by NIH National Cancer Institute (NCI) support grant CA34196.
Footnotes
Disclosure statement There are no actual or potential conflicts of interest regarding the authors of this manuscript and the work described herein.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Balaban RS, Nemoto S, Finkel T. Mitochondria, oxidants, and aging. Cell. 2005;120:483–95. doi: 10.1016/j.cell.2005.02.001. [DOI] [PubMed] [Google Scholar]
- Carelli V, Giordano C, d’Amati G. Pathogenic expression of homoplasmic mtDNA mutations needs a complex nuclear-mitochondrial interaction. Trends Genet. 2003;19:257–62. doi: 10.1016/S0168-9525(03)00072-6. [DOI] [PubMed] [Google Scholar]
- Cheng TL, Liao CC, Tsai WH, Lin CC, Yeh CW, Teng CF, Chang WT. Identification and characterization of the mitochondrial targeting sequence and mechanism in human citrate synthase. Journal of Cellular Biochemistry. 2009;107:1002–15. doi: 10.1002/jcb.22200. [DOI] [PubMed] [Google Scholar]
- DeStefano AL, Gates GA, Heard-Costa N, Myers RH, Baldwin CT. Genomewide linkage analysis to presbycusis in the Framingham Heart Study. Arch Otolaryngol Head Neck Surg. 2003;129:285–9. doi: 10.1001/archotol.129.3.285. [DOI] [PubMed] [Google Scholar]
- Dirks AJ, Hofer T, Marzetti E, Pahor M, Leeuwenburgh C. Mitochondrial DNA mutations, energy metabolism and apoptosis in aging muscle. Ageing Res Rev. 2006;5:179–95. doi: 10.1016/j.arr.2006.03.002. [DOI] [PubMed] [Google Scholar]
- Festing MFW. Mouse Genome Database (MGD) Mouse Genome Informatics, The Jackson Laboratory; Bar Harbor4, Maine: 2008. Inbred strains of mice. [Google Scholar]
- Frazer KA, Eskin E, Kang HM, Bogue MA, Hinds DA, Beilharz EJ, Gupta RV, Montgomery J, Morenzoni MM, Nilsen GB, Pethiyagoda CL, Stuve LL, Johnson FM, Daly MJ, Wade CM, Cox DR. A sequence-based variation map of 8.27 million SNPs in inbred mouse strains. Nature. 2007;448:1050–3. doi: 10.1038/nature06067. [DOI] [PubMed] [Google Scholar]
- Friedman RA, Van Laer L, Huentelman MJ, Sheth SS, Van Eyken E, Corneveaux JJ, Tembe WD, Halperin RF, Thorburn AQ, Thys S, Bonneux S, Fransen E, Huyghe J, Pyykko I, Cremers CW, Kremer H, Dhooge I, Stephens D, Orzan E, Pfister M, Bille M, Parving A, Sorri M, Van de Heyning PH, Makmura L, Ohmen JD, Linthicum FH, Jr, Fayad JN, Pearson JV, Craig DW, Stephan DA, Van Camp G. GRM7 variants confer susceptibility to age-related hearing impairment. Hum Mol Genet. 2009;18:785–796. doi: 10.1093/hmg/ddn402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gagnon LH, Longo-Guess CM, Berryman M, Shin JB, Saylor KW, Yu H, Gillespie PG, Johnson KR. The chloride intracellular channel protein CLIC5 is expressed at high levels in hair cell stereocilia and is essential for normal inner ear function. J Neurosci. 2006;26:10188–98. doi: 10.1523/JNEUROSCI.2166-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haramizu S, Nagasawa A, Ota N, Hase T, Tokimitsu I, Murase T. Different contribution of muscle and liver lipid metabolism to endurance capacity and obesity susceptibility of mice. Journal of Applied Physiology. 2009;106:871–9. doi: 10.1152/japplphysiol.90804.2008. [DOI] [PubMed] [Google Scholar]
- Henry KR. Age-related auditory loss and genetics: an electrocochleographic comparison of six inbred strains of mice. J Gerontol. 1982;37:275–82. doi: 10.1093/geronj/37.3.275. [DOI] [PubMed] [Google Scholar]
- Ho M, Post CM, Donahue LR, Lidov HG, Bronson RT, Goolsby H, Watkins SC, Cox GA, Brown RH., Jr Disruption of muscle membrane and phenotype divergence in two novel mouse models of dysferlin deficiency. Hum Mol Genet. 2004;13:1999–2010. doi: 10.1093/hmg/ddh212. [DOI] [PubMed] [Google Scholar]
- Hunter KP, Willott JF. Aging and the auditory brainstem response in mice with severe or minimal presbycusis. Hear Res. 1987;30:207–218. doi: 10.1016/0378-5955(87)90137-7. [DOI] [PubMed] [Google Scholar]
- Huyghe JR, Van Laer L, Hendrickx JJ, Fransen E, Demeester K, Topsakal V, Kunst S, Manninen M, Jensen M, Bonaconsa A, Mazzoli M, Baur M, Hannula S, Maki-Torkko E, Espeso A, Van Eyken E, Flaquer A, Becker C, Stephens D, Sorri M, Orzan E, Bille M, Parving A, Pyykko I, Cremers CW, Kremer H, Van de Heyning PH, Wienker TF, Nurnberg P, Pfister M, Van Camp G. Genome-wide SNP-based linkage scan identifies a locus on 8q24 for an age-related hearing impairment trait. Am J Hum Genet. 2008;83:401–7. doi: 10.1016/j.ajhg.2008.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H, Talaska AE, Schacht J, Sha SH. Oxidative imbalance in the aging inner ear. Neurobiol Aging. 2007;28:1605–12. doi: 10.1016/j.neurobiolaging.2006.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson KR, Zheng QY, Erway LC. A major gene affecting age-related hearing loss is common to at least ten inbred strains of mice. Genomics. 2000;70:171–180. doi: 10.1006/geno.2000.6377. [DOI] [PubMed] [Google Scholar]
- Johnson KR, Zheng QY, Bykhovskaya Y, Spirina O, Fischel-Ghodsian N. A nuclear-mitochondrial DNA interaction affecting hearing impairment in mice. Nat Genet. 2001;27:191–4. doi: 10.1038/84831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson KR, Zheng QY, Noben-Trauth K. Strain background effects and genetic modifiers of hearing in mice. Brain Res. 2006;1091:79–88. doi: 10.1016/j.brainres.2006.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keithley EM, Canto C, Zheng QY, Wang X, Fischel-Ghodsian N, Johnson KR. Cu/Zn superoxide dismutase and age-related hearing loss. Hear Res. 2005;209:76–85. doi: 10.1016/j.heares.2005.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kokotas H, Petersen MB, Willems PJ. Mitochondrial deafness. Clinical Genetics. 2007;71:379–91. doi: 10.1111/j.1399-0004.2007.00800.x. [DOI] [PubMed] [Google Scholar]
- Kujoth GC, Hiona A, Pugh TD, Someya S, Panzer K, Wohlgemuth SE, Hofer T, Seo AY, Sullivan R, Jobling WA, Morrow JD, Van Remmen H, Sedivy JM, Yamasoba T, Tanokura M, Weindruch R, Leeuwenburgh C, Prolla TA. Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science. 2005;309:481–4. doi: 10.1126/science.1112125. [DOI] [PubMed] [Google Scholar]
- Lee YJ, Hoe KL, Maeng PJ. Yeast cells lacking the CIT1-encoded mitochondrial citrate synthase are hypersusceptible to heat- or aging-induced apoptosis. Mol Biol Cell. 2007;18:3556–67. doi: 10.1091/mbc.E07-02-0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lightfoot JT, Turner MJ, Debate KA, Kleeberger SR. Interstrain variation in murine aerobic capacity. Medicine and Science in Sports and Exercise. 2001;33:2053–7. doi: 10.1097/00005768-200112000-00012. [DOI] [PubMed] [Google Scholar]
- Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006;443:787–95. doi: 10.1038/nature05292. [DOI] [PubMed] [Google Scholar]
- Manly KF, Cudmore RH, Jr, Meer JM. Map Manager QTX, cross-platform software for genetic mapping. Mamm Genome. 2001;12:930–2. doi: 10.1007/s00335-001-1016-3. [DOI] [PubMed] [Google Scholar]
- McFadden SL, Ding D, Burkard RF, Jiang H, Reaume AG, Flood DG, Salvi RJ. Cu/Zn SOD deficiency potentiates hearing loss and cochlear pathology in aged 129,CD-1 mice. J Comp Neurol. 1999;413:101–12. [PubMed] [Google Scholar]
- Moreno-Loshuertos R, Acin-Perez R, Fernandez-Silva P, Movilla N, Perez-Martos A, Rodriguez de Cordoba S, Gallardo ME, Enriquez JA. Differences in reactive oxygen species production explain the phenotypes associated with common mouse mitochondrial DNA variants. Nat Genet. 2006;38:1261–8. doi: 10.1038/ng1897. [DOI] [PubMed] [Google Scholar]
- Mulrow CD, Aguilar C, Endicott JE, Velez R, Tuley MR, Charlip WS, Hill JA. Association between hearing impairment and the quality of life of elderly individuals. J Am Geriatr Soc. 1990;38:45–50. doi: 10.1111/j.1532-5415.1990.tb01595.x. [DOI] [PubMed] [Google Scholar]
- Nadeau JH, Singer JB, Matin A, Lander ES. Analysing complex genetic traits with chromosome substitution strains. Nat Genet. 2000;24:221–5. doi: 10.1038/73427. [DOI] [PubMed] [Google Scholar]
- Nesbitt MN, Skamene E. Recombinant inbred mouse strains derived from A/J and C57BL/6J: a tool for the study of genetic mechanisms in host resistance to infection and malignancy. Journal of Leukocyte Biology. 1984;36:357–64. doi: 10.1002/jlb.36.3.357. [DOI] [PubMed] [Google Scholar]
- Niu X, Trifunovic A, Larsson NG, Canlon B. Somatic mtDNA mutations cause progressive hearing loss in the mouse. Exp Cell Res. 2007;313:3924–34. doi: 10.1016/j.yexcr.2007.05.029. [DOI] [PubMed] [Google Scholar]
- Noben-Trauth K, Zheng QY, Johnson KR. Association of cadherin 23 with polygenic inheritance and genetic modification of sensorineural hearing loss. Nat Genet. 2003;35:21–23. doi: 10.1038/ng1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noben-Trauth K, Johnson KR. Inheritance patterns of progressive hearing loss in laboratory strains of mice. Brain Res. 2009;1277:42–51. doi: 10.1016/j.brainres.2009.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohlemiller KK, McFadden SL, Ding DL, Lear PM, Ho YS. Targeted mutation of the gene for cellular glutathione peroxidase (Gpx1) increases noise-induced hearing loss in mice. J Assoc Res Otolaryngol. 2000;1:243–54. doi: 10.1007/s101620010043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohlemiller KK. Contributions of mouse models to understanding of age- and noise-related hearing loss. Brain Res. 2006;1091:89–102. doi: 10.1016/j.brainres.2006.03.017. [DOI] [PubMed] [Google Scholar]
- Petkov PM, Ding Y, Cassell MA, Zhang W, Wagner G, Sargent EE, Asquith S, Crew V, Johnson KA, Robinson P, Scott VE, Wiles MV. An efficient SNP system for mouse genome scanning and elucidating strain relationships. Genome Res. 2004;14:1806–11. doi: 10.1101/gr.2825804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratkevicius A, Carroll AM, Kilikevicius A, Venckunas T, McDermott KT, Gray SR, Wackerhage H, Lionikas A. H55N polymorphism as a likely cause of variation in citrate synthase activity of mouse skeletal muscle. Physiol Genomics. 2010;42A:96–102. doi: 10.1152/physiolgenomics.00066.2010. [DOI] [PubMed] [Google Scholar]
- Shin JB, Streijger F, Beynon A, Peters T, Gadzala L, McMillen D, Bystrom C, Van der Zee CE, Wallimann T, Gillespie PG. Hair Bundles Are Specialized for ATP Delivery via Creatine Kinase. Neuron. 2007;53:371–86. doi: 10.1016/j.neuron.2006.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siemens J, Lillo C, Dumont RA, Reynolds A, Williams DS, Gillespie PG, Muller U. Cadherin 23 is a component of the tip link in hair-cell stereocilia. Nature. 2004;428:950–5. doi: 10.1038/nature02483. [DOI] [PubMed] [Google Scholar]
- Someya S, Yamasoba T, Kujoth GC, Pugh TD, Weindruch R, Tanokura M, Prolla TA. The role of mtDNA mutations in the pathogenesis of age-related hearing loss in mice carrying a mutator DNA polymerase gamma. Neurobiology of Aging. 2008;29:1080–92. doi: 10.1016/j.neurobiolaging.2007.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Someya S, Prolla TA. Mitochondrial oxidative damage and apoptosis in age-related hearing loss. Mechanisms of Ageing and Development. 2010;131:480–6. doi: 10.1016/j.mad.2010.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soucek S, Michaels L, Frohlich A. Evidence for hair cell degeneration as the primary lesion in hearing loss of the elderly. J Otolaryngol. 1986;15:175–83. [PubMed] [Google Scholar]
- Spongr VP, Flood DG, Frisina RD, Salvi RJ. Quantitative measures of hair cell loss in CBA and C57BL/6 mice throughout their life spans. J Acoust Soc Am. 1997;101:3546–53. doi: 10.1121/1.418315. [DOI] [PubMed] [Google Scholar]
- Staecker H, Zheng QY, Van De Water TR. Oxidative stress in aging in the C57B16/J mouse cochlea. Acta Otolaryngol. 2001;121:666–72. doi: 10.1080/00016480152583593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamasoba T, Someya S, Yamada C, Weindruch R, Prolla TA, Tanokura M. Role of mitochondrial dysfunction and mitochondrial DNA mutations in age-related hearing loss. Hear Res. 2007;226:185–93. doi: 10.1016/j.heares.2006.06.004. [DOI] [PubMed] [Google Scholar]
- Zheng QY, Johnson KR, Erway LC. Assessment of hearing in 80 inbred strains of mice by ABR threshold analyses. Hear Res. 1999;130:94–107. doi: 10.1016/s0378-5955(99)00003-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng QY, Ding D, Yu H, Salvi RJ, Johnson KR. A locus on distal chromosome 10 (ahl4) affecting age-related hearing loss in A/J mice. Neurobiol Aging. 2009;30:1693–1705. doi: 10.1016/j.neurobiolaging.2007.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.