

Abstract
A series of six novel bis(carboxylato)dichloridobis(ethylamine)platinum(IV) complexes was synthesized and characterized in detail by elemental analysis, FT-IR, ESI-MS, HPLC, multinuclear (1H, 13C, 15N, 195Pt) NMR spectroscopy and in one case by X-ray diffraction. Cytotoxic properties of the complexes were evaluated in four human tumor cell lines originating from ovarian carcinoma (CH1 and SK-OV-3), colon carcinoma (SW480) and non-small cell lung cancer (A549) by means of the MTT colorimetrical assay. In addition, their octanol/water partition coefficients (log P values) were determined. Remarkably the most active (and also most lipophilic) compounds, having 4-propyloxy-4-oxobutanoato and 4-(2-propyloxy)-4-oxobutanoato axial ligands, showed IC50 values down to the low nanomolar range.
Keywords: Platinum complexes, Synthesis, Characterization, Lipophilicity, Cytotoxicity
Graphical abstract
A series of six novel platinum(IV) complexes was synthesized and characterized. Cytotoxicity was evaluated in four human tumor cell lines yielding IC50 values down to the low nanomolar range.
Highlights
▸ Six novel platinum(IV) complexes have been synthesized. ▸ Some complexes have higher cytotoxicity than cisplatin in four human tumor cell lines. ▸ Cytotoxicity is dependent on the lipophilicity.
1. Introduction
Nowadays, 35 years after the serendipitous discovery of the cytotoxic potential of cis-diamminedichloridoplatinum(II) [1], more than 50% of anticancer therapy is platinum based. All platinum containing drugs used in the clinics are platinum(II) compounds (cisplatin, carboplatin, oxaliplatin, nedaplatin, lobaplatin and heptaplatin) [2]. However, it was shown, that Pt(IV) complexes also exhibit strong cytotoxic activity and can have some advantages in comparison to their Pt(II) analogues [3], [4]. Consequences of the higher oxidation state are the introduction of two extra ligands and the change from planar to octahedral geometry. These characteristics, together with their higher kinetic inertness compared to their platinum(II) counterparts, opens up new possibilities in the design of novel platinum-based drugs (easier modulation of the pharmacokinetic properties, more opportunities for targeted therapy, oral administration, etc.) [5]. Nevertheless, no Pt(IV) complex has gained clinical approval up to now [2]. Four Pt(IV) compounds were in clinical trials (Fig. 1): tetraplatin was rejected after phase I because of a high general toxicity [6]; iproplatin was abandoned after phase III clinical trials, because it didn’t show advantages compared with carboplatin [2]; satraplatin was rejected after the SPARC (Satraplatin and Prednisone Against Refractory Cancer) phase III clinical trials [7], because it didn’t show a convincing benefit in terms of overall survival [8]; currently satraplatin is in phase I and II clinical trials in combination regimens [9]; its adamantylamine analogue LA-12 has passed phase I clinical trials.
Fig. 1.

Chemical structures of anticancer Pt(IV) complexes evaluated in clinical trials.
Considering the mechanism of action of Pt(IV) complexes, it is accepted that they act as prodrugs via activation by reduction to their reactive Pt(II) species [10], [11]. Pt(IV) based drugs would have better activity and lower side effects, when they are reduced primarily in the cell; contrary, an extracellular reduction would lead to deactivation and general toxicity. Consequently, the drug’s potential strongly correlates with the rate of reduction which depends on the respective reduction potential [12]. Depending on the nature of the axial ligands, platinum(IV) complexes are reduced more easily in the following order: CF3COO > Cl > CH3COO > OH [13] It was found that complexes with Cl− as axial ligands were reduced very fast and showed a high general toxicity (tetraplatin [14]), on the other hand, complexes with axial hydroxido ligands were not reduced fast enough in the body to express their antitumor activity (iproplatin [15]). In the case of axial carboxylato ligands, an intermediate and optimal redox potential was observed [16], which led to promising results, obtained in preclinical and clinical evaluation of satraplatin and its adamantylamine analogue LA-12.
Recently, a convenient way for obtaining a series of dicarboxylatoplatinum(IV) complexes and modulation of their physicochemical properties such as solubility and lipophilicity was reported by our group [17], [18], [19], [20], [21], [22]. In order to broaden the knowledge with respect to structure–activity relationships for that type of compounds and to find candidates with a promising physicochemical and pharmacological profile, we have synthesized a series of bis(carboxylato)dichloridobis(ethylamine)platinum(IV) complexes. The new compounds were fully characterized by elemental analysis, ATR IR, multinuclear NMR spectroscopy, HPLC and X-ray crystallography in one of the cases. Their cytotoxic properties were evaluated in four human tumor cell lines, originating from ovarian carcinoma (CH1 and SK-OV-3), colon carcinoma (SW480) and non-small cell lung cancer (A549), by means of the MTT colorimetrical assay. In addition, their octanol/water partition coefficients (log P) were determined.
2. Result and discussion
2.1. Synthesis
The new complexes were prepared according to the reaction scheme shown in Fig. 2, starting from K2PtCl4, which was converted to (SP-4-2)-dichloridobis(ethylamine)platinum(II) (complex 1). Oxidation of 1 to the dihydroxido complex 2 was performed in aqueous solution, using 15% hydrogen peroxide as oxidizing agent.
Fig. 2.

Synthesis of novel bis(carboxylato)dichloridobis(ethylamine)platinum(IV) complexes with NMR numbering scheme; i = succinic anhydride/DMF, 70 deg., ii = CDI/DMF, 60 deg., iii = RONa/ROH, RT, iv = cyclopentylamine/DMF, RT.
Subsequent carboxylation of 2 with succinic anhydride was carried out in absolute DMF obtaining the dicarboxylato complex. The latter was used as starting material for the synthesis of complexes 4–8 via activation of its free carboxylic groups with CDI (1,1′-carbonyldiimidazole) in absolute DMF under argon atmosphere. To the imidazolide formed in situ, the respective alcoholate/alcohol mixtures or amine were added to obtain the respective esters (4–6) or amide (8). Purification of the crude products was performed with column chromatography and re-crystallization, when necessary.
When synthesizing complex 7 following the described procedure, a mixture of the desired diisopropylester (7) and asymmetric methylisopropylester (7a) in a ratio of 2:1 (according to 1H NMR) was obtained. Unfortunately separation and isolation of compound 7 was not successful. Formation of the mixed methylisopropylester derivative 7a was also confirmed by ESI-MS. Most likely 7a was formed during separation via column chromatography. Apparently, reaction of 3 after CDI activation with isopropanol is not as fast as in the case of complexes 4–6. As a result, the monoimidazolide platinum complex reacted with methanol, one of the constituents of the mobile phase. By increasing the reaction time from 24 to 72 h and avoiding the use of methanol in the mobile phase for purification, we were able to obtain exclusively pure 7 in satisfactory yield.
2.2. Spectroscopic characterization
Structures of the starting compounds (1, 2) were proven by NMR and ATR IR spectroscopy and the new complexes (3–8) were fully characterized by elemental analysis, one- and two-dimensional multinuclear NMR (1H, 13C, 15N, 195Pt), ESI-MS and ATR IR spectroscopy and in the case of 4 also by X-ray diffraction.
Configuration of the novel platinum agents can best be established with the help of one- and two-dimensional multinuclear NMR spectroscopy. 1H and 13C chemical shifts of compounds 1–8 were found in the expected range, proving the supposed structure of the complexes. Correct assignment of the signals was based on 1H1H COSY, 1H13C HSQC and 1H13C HMBC spectra of the complexes. In 1H NMR spectra, oxidation and subsequent derivatization can best be judged according to the shift of the NH2 signal from 5.09 ppm in the Pt(II) complex 1 to 5.96 ppm in the dihydroxido complex 2, and to 7.85–7.91 ppm in the dicarboxylato complexes 3–8, respectively. Also indicative are the 15N resonances, where the signal for NH2 shifts from −40.9 ppm in complex 1 to around −21.3 ppm in compounds 3–8. Additionally, successful derivatizations of complex 3 were observed in 13C NMR spectra. The resonance of C-6 (uncoordinated COOH) was shifted upfield upon formation of esters 4–7 or amide 8 by ca. 2 ppm. 195Pt NMR is very powerful technique for investigating the oxidation state and the coordination sphere of platinum complexes. 195Pt signals for complexes 3–8 were detected in the region between 2849 and 2853 ppm, typical for compounds with cis,cis,trans-PtIVCl2N2O2 coordination [23] In comparison, the platinum(II) complex 1 resonates at −601 ppm, more than 3000 ppm upfield to the platinum(IV) analogues. As expected, derivatization of 3 had no influence on the 195Pt signal, because the changes in the molecule are far away from the 195Pt nucleus.
Oxidation of 1 and derivatization of 2 can also be followed in the IR spectra of the complexes. A new and intense signal at 3490 cm−1, corresponding to νPtO–H can be observed in the spectrum of complex 2, in comparison with that of 1. After esterification with succinic anhydride this signal disappeared and in complexes 3–8 new strong bands in the region 1730–1630 cm−1 (νC O) were detected, proving the successful carboxylation. In esters 4–7, bands with 10–25 higher reciprocal wavelengths were detected in comparison with the complex featuring free carboxylic groups (3). However, the signal around 1710 cm−1 is missing in compound 8, because of amide formation and a strong band with a shoulder around 1639 cm−1could be observed.
ESI-MS spectra have also confirmed the identity of the complexes. All new compounds (3–8) were measured in the positive as well as in the negative ion mode. In the positive ion mode, the peak assigned to [M + Na+]+ displayed the highest intensity, whereas in the negative ion mode the highest intensity was detected for [M − H+]−. However, a peak corresponding to [M + Cl−]−, could also be observed in the spectra of esters (complexes 4–7). The detected m/z values as well as the isotopic distribution were in accordance with the expected chemical structures 3–8.
2.3. Crystal structure of complex 4
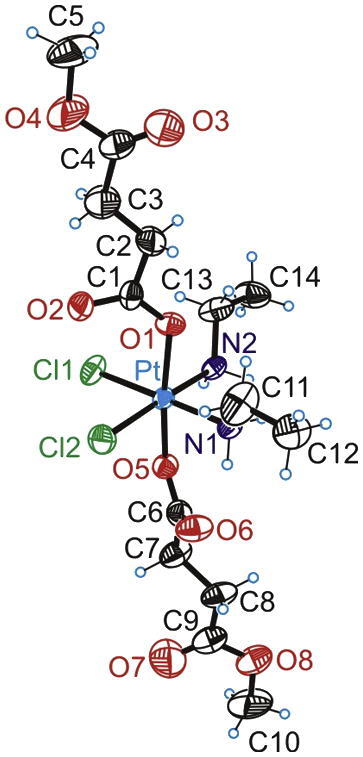
The result from the X-ray diffraction analysis of 4 is shown in Fig. 3. Crystal data, data collection parameters and structure-refinement details are given in the Experimental section. Selected bond lengths and angles are listed in Table 1. The compound crystallized in the triclinic centrosymmetric space group P−1. The Pt(IV) atom has an octahedral coordination geometry with two ethylamine and two chlorido ligands in the equatorial plane and two 4-methoxysuccinates coordinated in axial positions. The Pt–N, Pt–Cl, and Pt–O bond lengths (Table 1) are well comparable with those, observed in structurally similar complexes [17], [18]. Angles in the PtCl2N2O2 octahedron were found between 86.26 and 92.69°, and between 172.70 and 178.18°, respectively. The torsion angles (Pt–O1–C1–O2, and Pt–O5–C6–O6) were found to be close to zero.
Fig. 3.
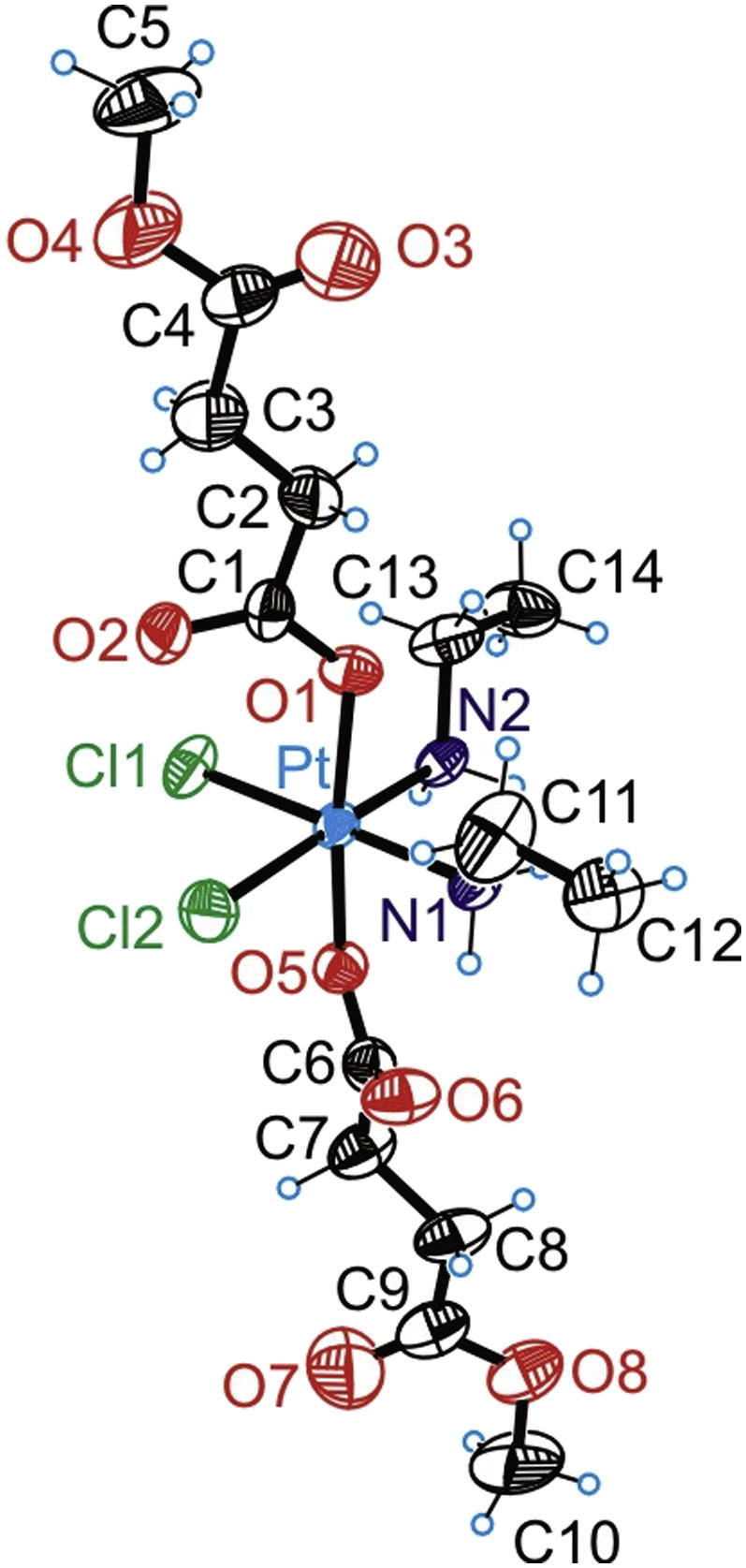
ORTEP diagram of 4 displaying thermal ellipsoids at 50% probability.
Table 1.
Selected bond lengths (Å) and bond angles (°) in complex 4.
| Bond lengths (Å) | |||
| Pt–O1 | 2.039 | Pt–N2 | 2.068 |
| Pt–O5 | 2.039 | Pt–Cl1 | 2.324 |
| Pt–N1 | 2.063 | Pt–Cl2 | 2.309 |
| Bond angles (deg) | |||
| N1–Pt1–O1 | 86.26 | O1–Pt–O5 | 172.70 |
| N1–Pt–N2 | 92.69 | N1–Pt–Cl1 | 178.18 |
| N2–Pt–Cl1 | 86.26 | N2–Pt–Cl2 | 175.68 |
| N1–Pt–Cl2 | 88.72 | Pt–O1–C1–O2 | −1.87 |
| Cl1–Pt–Cl2 | 92.22 | Pt–O5–C6–O6 | −0.45 |
2.4. Cytotoxicity in cancer cell lines
The new compounds were tested in comparison to cisplatin in four human tumor cell lines, originating from ovarian carcinoma (CH1, SK-OV-3), colon carcinoma (SW480) and non-small cell lung cancer (A549) with the help of the colorimetric microculture MTT assay. Except for CH1 cells, these cell lines are resistant to cisplatin, showing IC50 values about one order of magnitude higher than that in CH1 cells. The concentration-effect curves of the tested complexes are shown in Fig. 4 and the obtained IC50 values are summarized in Table 2.
Fig. 4.

Concentration-effect curves of complexes (3–8) in CH1 (A), SK-OV-3 (B), SW480 (C) and A549 (D) obtained by the MTT assay (96 h exposure).
Table 2.
Cytotoxicity of novel complexes (3–8) in comparison to cisplatin in four human cancer cell lines.
| Compound | IC50 (μM)a |
|||
|---|---|---|---|---|
| CH1 | A549 | SW480 | SK-OV-3 | |
| 3/R–COOH | 5.6 ± 1.6 | 50 ± 8 | 40 ± 12 | 120 ± 17 |
| 4/R–COOMe | 0.16 ± 0.05 | 2.5 ± 0.9 | 1.0 ± 0.3 | 2.4 ± 0.1 |
| 5/R–COOEt | 0.061 ± 0.015 | 1.0 ± 0.4 | 0.30 ± 0.05 | 1.2 ± 0.3 |
| 6/R–COOPr | 0.014 ± 0.002 | 0.20 ± 0.03 | 0.11 ± 0.01 | 0.19 ± 0.03 |
| 7/R–COOiPr | 0.0094 ± 0.0012 | 0.78 ± 0.09 | 0.39 ± 0.07 | 0.49 ± 0.11 |
| 8/R–CONHR | 0.75 ± 0.10 | 19 ± 3 | 6.1 ± 0.6 | 13 ± 1 |
| Cisplatin | 0.16 ± 0.03 | 1.3 ± 0.3 | 3.5 ± 0.3 | 1.9 ± 0.3 |
50% Inhibitory concentrations in CH1, A549, SW480 and SK-OV-3 cells in the MTT assay, 96 h exposure. Values are the means ± standard deviations obtained from three independent experiments.
Expectedly, the platinum(IV) precursor 3 featuring two COOH moieties showed the lowest cytotoxicity in all cell lines. In accordance with previously published data [18], [19], [22], lipophilicity as well as cellular accumulation are low for such types of complexes. Conversion of 3 to the corresponding ester derivatives 4–7 has a significant influence on antiproliferative potency. Whereas complex 4 is as cytotoxic as cisplatin in the cisplatin-sensitive CH1 cell line, complexes 5–7 are 3 to 17 times more cytotoxic than cisplatin in the same cell line. An analogous trend was also observed in cisplatin-resistant A549, SW480 and SK-OV-3 cells. Parallel to an increasing lipophilicity of the ester residue (Me, Et, Pr), the IC50 values are decreasing in all cell lines, yielding clear structure–activity relationships. The iPr analogue 7 is similar in cytotoxic potency to the Pr analogue 6 in CH1 cells, but somewhat less potent in the other cell lines. In analogy to previous observations [22], the cyclopentylamide derivative is equipped with a very low antiproliferative potency, despite its high lipophilicity (see below).
2.5. Lipophilicity vs. cytotoxicity
As an important factor for the pharmacokinetics and the cellular accumulation of the new complexes, the lipophilicity was determined by measuring octanol/water partition coefficients (log P) by two different methods; log P values, obtained by RP-HPLC and by the shake-flask method are shown in Table 3.
Table 3.
Log P values for complexes 3–8, R1 and R2, estimated by RP-HPLC and the shake-flask method.
| Compound | Log P values, determined by RP-HPLC in different MeOH concentrations |
Log P, determined by the shake-flask method | |||||
|---|---|---|---|---|---|---|---|
| 0% | 10% | 20% | 30% | 40% | 50% | ||
| R1 | −1.04 | −1.03 | −1.00 | −0.95 | −0.99 | −0.88 | −0.81 |
| 3 | −0.70 | −0.84 | −0.79 | −0.64 | −1.14 | −0.97 | |
| 4 | 0.30 | 0.25 | 0.17 | 0.08 | 0.14 | −0.10 | −0.12 |
| 5 | 0.90 | 0.87 | 0.83 | 0.73 | 0.78 | 0.85 | 0.64 |
| 6 | 1.38 | 1.41 | 1.44 | 1.49 | 1.47 | 1.48 | 1.44 |
| 7 | 1.30 | 1.33 | 1.36 | 1.41 | 1.37 | 1.39 | |
| 8 | 1.11 | 1.14 | 1.17 | 1.23 | 1.20 | 1.25 | 1.21 |
| R2 | 1.39 | 1.41 | 1.44 | 1.48 | 1.44 | 1.46 | 1.69 |
Complexes 4–8 have significantly higher log P values than that reported for cisplatin (−2.59) and platinum(II) complex 1 (−1.47) [24]. However, complex 3 is also more lipophilic than its Pt(II) congener (1), but its lipophilicity is pH-dependent, due to the presence of two underivatized carboxylic groups (lower lipophilicity under physiological conditions is expected). In Fig. 5, a semi-logarithmic graph plotting the cytotoxicity versus log P of the new complexes is shown. In case of compounds 3–7, a linear dependency between lipophilicity and cytotoxicity could be observed – the more lipophilic a complex, the higher its cytotoxicity. The amide complex 8 does not match this trend – according to its lipophilicity, a higher cytotoxicity would be expected. The reasons for the latter finding are yet unclear.
Fig. 5.

Semi-logarithmic plot of lipophilicity (log P determined with RP-HPLC) of complexes 3–8 vs. cytotoxicity (IC50) in CH1, A549, SW480 and SK-OV-3 cells.
2.6. Induction of apoptosis and necrosis
In order to compare the capacities of inducing apoptosis and necrosis of one representative, namely compound 7, with those of cisplatin, growing SW480 cultures were treated with these compounds in various concentrations for 48 h, then double-stained with annexin V-FITC and propidium iodide and analyzed by fluorescence-activated cell sorting (FACS). This method allows to discriminate necrotic (stained by propidium iodide only), early apoptotic (stained by annexin V-FITC) and late apoptotic (stained by both) from viable (unstained) cells. From the dot plots (Fig. 6), it becomes obvious that the apoptosis-inducing potency of compound 7 is much higher than that of cisplatin. 50 μM of compound 7 reduce the amount of viable cells to 50% by induction of both apoptosis (29%) and necrosis (21%), while the same concentration of cisplatin has very little effect within the chosen exposure time in the intrinsically cisplatin-resistant SW480 (colon cancer) cells.
Fig. 6.

FACS analysis of annexin V- and PI-stained SW480 colon cancer cells. Left column: untreated control; right column: after treatment with 50 μM cisplatin/compound 7 for 48 h. The upper left quadrant contains the necrotic (stained by PI only), the lower right early apoptotic (stained by annexin V-FITC only), the upper right late apoptotic (stained by both) and the lower left quadrant the viable (unstained) fraction of cell populations.
A synopsis of all data (Fig. 7) further illustrates that compound 7 is a powerful apoptotic agent, which effectively causes cell death in a dose-dependent way.
Fig. 7.

Concentration-effect curves for compound 7 (A) and cisplatin (B) with regard to apoptosis and necrosis induction in SW480 cells after 48 h exposure, measured by FACS using annexin V-FITC/propidium iodide staining.
3. Conclusions
Six novel bis(carboxylato)platinum(IV) complexes have been synthesized and fully characterized. The new compounds were investigated for their lipophilic properties and their in vitro cytotoxicity in four human tumor cell lines. Remarkably, IC50 values down to the nanomolar range, up to 32 times lower compared to cisplatin, were found. Whether the very high cytotoxicity is also accompanied by manageable systemic toxicity in vivo will be evaluated in future work.
4. Experimental protocols
4.1. Materials and methods
All reagents and solvents were obtained from commercial suppliers, and were used without further purification. Methanol and ethanol were dried, according to standard procedures. For column chromatography, silica gel 60 (Fluka) was used. (OC-6-33)-Dichloridobis(ethylamine)dihydroxidoplatinum(IV) (complex 2, Fig. 2) was synthesized starting from K2PtCl4 and ethylamine, using Dhara’s [25] method with some modifications. The resulting dichlorido complex 1 was oxidized with 15% H2O2.
1H, 13C, 15N, 195Pt and two-dimensional 1H1H COSY, 1H13C and 1H15N HSQC, and 1H13C HMBC NMR spectra were recorded with a Bruker Avance III 500 MHz NMR spectrometer at 500.32 (1H), 125.81 (13C), 107.55 (195Pt), and 50.70 MHz (15N) in DMF-d7 at ambient temperature, using the solvent residual peak for 1H and 13C as internal reference. The splitting of proton resonances in the 1H NMR spectra are defined as s = singlet, bs = broad singlet, d = doublet, t = triplet, and m = multiplet. 15N chemical shifts were referenced relative to external NH4Cl; whereas 195Pt chemical shifts were referenced relative to external K2[PtCl4] (see Fig. 2 for NMR numbering scheme).
IR spectra were obtained with a Perkin–Elmer 370 FT-IR 2000 instrument (4000–400 cm−1) by using an ATR unit. Intensities of reported IR bands are defined as br = broad, s = strong, m = medium, and w = weak. Electrospray ionization mass spectrometry was carried out with a Bruker Esquire 3000 instrument using MeOH as solvent. Elemental analyses were performed with a Perkin–Elmer 2400 CHN-Elemental Analyzer by the Microanalytical Laboratory of the University of Vienna. Analyses indicated by the symbols of the elements or functions were within ±0.4% of the theoretical values. Purity of novel compounds was additionally proved by analytical reversed-phase HPLC.
4.2. Synthesis
4.2.1. (OC-6-33)-Bis(3-carboxypropanoato)dichloridobis(ethylamine)platinum(IV) (3)
Succinic anhydride (674.3 mg, 6.738 mmol) and 653 mg (1.675 mmol) of (OC-6-33)-dichloridobis(ethylamine)dihydroxidoplatinum(IV) were suspended in 9 mL of absolute DMF and the reaction mixture was stirred at 65 °C for 40 min and then for 2 more hours at room temperature. During this time, the solid material dissolved to form a yellow-brown solution. DMF was removed under reduced pressure. The residue was dissolved in acetone and filtered to give a clear, yellow solution. This solution was concentrated under reduced pressure, and subsequent addition of diethyl ether led to precipitation of a pale yellow solid. The precipitate was filtrated and dried in vacuo, while heating at 40 °C. Yield: 398 mg (40%). Anal. C12H24Cl2N2O8Pt (C, H, N). ESI-MS: m/z 612.6 [M + Na+]+, 588.7 [M − H+]−. 1H NMR: δ = 12.52 (bs, 2H, COOH), 7.87 (bs, 4H, NH2), 3.10 (m, 4H, H-2), 2.75 (m, 4H, H-4), 2.68 (m, 4H, H-5), 1.44 (t, 3JH,H = 7.2 Hz, 6H, H-1) ppm. 13C NMR: δ = 181.3 (C-3), 174.1 (C-6), 39.9 (C-2), 31.2 (C-4), 29.9 (C-5), 14.3 (C-1) ppm. 15N NMR7: δ = −21.2 ppm. 195Pt NMR: δ = 2851 ppm. IR (ATR): 3241 m, 3197 br; 2977 br; 1710 s (νC O), 1642 m (νC O); 1357 m, 1344 m; 1238 s 1216 m, 1175 m; 1081 w, 1024 w (νC–N) cm−1.
4.2.2. (OC-6-33)-Dichloridobis(ethylamine)bis((4-methoxy)-4-oxobutanoato)platinum(IV) (4)
CDI (241.1 mg, 1.4869 mmol) in absolute DMF (10 mL) was added to a solution of 3 (433.4 mg, 0.7342 mmol) in absolute DMF (6 mL), and the mixture was heated to 60 °C. After 10 min of being stirred, the solution was cooled to room temperature and CO2 was removed by flushing with argon. Sodium methanolate (a piece of Na in 10 mL of absolute MeOH) in absolute MeOH was added and the solution was stirred for 24 h at room temperature. Methanol and DMF were removed under reduced pressure to form a yellow oil. The crude product was purified by column chromatography (EtOAc/MeOH, 7:1) to yield a yellow solid, which was dried in vacuo. Yield: 106 mg (23%). Anal. C14H28Cl2N2O8Pt (C, H, N). ESI-MS: m/z 640.8 [M + Na+]+, 616.5 [M − H+]−, 652.8 [M + Cl−]−. 1H NMR: δ = 7.85 (bs, 4H, NH2), 3.82 (s, 6H, H-7), 3.10 (m, 4H, H-2), 2.78 (m, 4H, H-4), 2.71 (m, 4H, H-5), 1.45 (t, 3JH,H = 7.2 Hz, 6H, H-1) ppm. 13C NMR: δ = 181.0 (C-3), 173.1 (C-6), 51.2 (C-7), 39.9 (C-2), 31.1 (3JC,Pt = 38.2 Hz, C-4), 29.8 (C-5), 14.3 (3JC,Pt = 35.0 Hz, C-1) ppm. 15N NMR: δ = −21.3 ppm. 195Pt NMR: δ = 2853 ppm. IR (ATR): 3221 w, 3188 m; 2894 br; 1729 s (νC O), 1647 s (νC O); 1362 m 1333s; 1259 s, 1198s, 1176 s; 1088 m, 1026 w (νC–N); 683 w cm−1. Crystals, suitable for X-ray data collection, were obtained after vapor diffusion of diethyl ether into a methanol solution of 4.
4.2.3. (OC-6-33)-Dichloridobis((4-ethoxy)-4-oxobutanoato)bis(ethylamine)platinum(IV) (5)
The synthesis was carried out as described for 4. The crude product was purified by column chromatography (EtOAc/MeOH, 9:1), then recrystallized from ethyl acetate and diethyl ether to yield a pale yellow solid. The final product was dried in vacuo. Yield: 24 mg (16%). Anal. C16H32Cl2N2O8Pt (C, H, N). ESI-MS: m/z 669.1 [M + Na+]+, 644.3 [M − H+]−, 681.0 [M + Cl−]−. 1H NMR: δ = 7.86 (bs, 4H, NH2), 4.26 (m, 3JH,H = 7.1 Hz, 4H, H-7), 3.10 (m, 4H, H-2), 2.77 (t, 4H, H-4), 2.69 (t, 4H, H-5), 1.45 (t, 3JH,H = 7.2 Hz, 6H, H-1), 1.39 (t, 3JH,H = 7.1 Hz, 6H, H-8) ppm. 13C NMR: δ = 181.0 (C-3), 172.7 (C-6), 60.2 (C-7), 39.9 (C-2), 31.1 (3JC,Pt = 37.3 Hz, C-4), 30.0 (C-5), 14.4 (3JC,Pt = 34.5 Hz, C-1), 14.0 (C-8) ppm. 15N NMR: δ = −21.3 ppm. 195Pt NMR: δ = 2850 ppm. IR (ATR): 3180 w, 3153 w; 2987 w; 1727 s (νC O); 1658 m, 1631 m (νC O); 1369 m; 1260 s, 1231 s; 1163 s; 1085 w; 1039 m, 1023 s (νC–N); 857 w, 681 m cm−1.
4.2.4. (OC-6-33)-Dichloridobis(ethylamine)bis((4-propyloxy)-4-oxobutanoato)platinum(IV) (6)
The synthesis was carried out as described for 4. The crude product was purified by column chromatography (EtOAc/MeOH, 11:1), then recrystallized from ethyl acetate and diethyl ether to yield a pale yellow solid. The final product was dried in vacuo. Yield: 33 mg (11%). Anal. C18H36Cl2N2O8Pt (C, H, N). ESI-MS: m/z 697.1 [M + Na+]+, 672.9 [M − H+]−, 708.9 [M + Cl−]−. 1H NMR: δ = 7.86 (bs, 4H, NH2), 4.18 (t, 3JH,H = 6.7 Hz, 4H, H-7), 3.10 (m, 4H, H-2), 2.78 (m, 4H, H-4), 2.71 (m, 4H, H-5), 1.80 (m, 3JH,H = 7.3 Hz, 4H, H-8), 1.44 (t, 3JH,H = 7.2 Hz, 6H, H-1), 1.09 (t, 3JH,H = 7.4 Hz, 6H, H-9) ppm. 13C NMR: δ = 181.0 (C-3), 172.7 (C-6), 65.8 (C-7), 39.9 (C-2), 31.1 (3JC,Pt = 31.1 Hz, C-4), 29.9 (C-5), 22.0 (C-8), 14.4 (3JC,Pt = 34.7 Hz, C-1), 10.1 (C-9) ppm. 15N NMR: δ = −21.4 ppm. 195Pt NMR: δ = 2850 ppm. IR (ATR): 3222 m, 3192 m; 2970 w; 1731 s (νC O); 1671 m, 1654 s (νC O); 1370 m, 1326 m; 1164 s, 1088 w; 684 w cm−1.
4.2.5. (OC-6-33)-Dichloridobis(ethylamine)bis((4-(2-propyloxy))-4-oxobutanoato)platinum(IV) (7)
CDI (184 mg, 1.135 mmol) in absolute DMF (6 mL) was added to a solution of 3 (325 mg, 0.551 mmol) in absolute DMF (5 mL), and the mixture was heated to 70 °C. After 10 min of being stirred, the solution was cooled to room temperature and CO2 was removed by flushing with argon. 12 mL of sodium 2-propanolate (a piece of Na dissolved in 2-propanol, HPLC grade) was added to the solution and heated to 40 °C. The mixture was then stirred for 72 h at room temperature. 2-Propanol and DMF were removed under reduced pressure to form a yellow oil. The crude product was purified by column chromatography (EtOAc/2-propanol, 10:1) and then precipitated with Et2O and cooled to 0 °C to give an almost white solid, which was dried in vacuo. Yield: 110 mg (30%). Anal. C18H36Cl2N2O8Pt (C, H, N). ESI-MS: m/z 696.8 [M + Na+]+, 673.4 [M − H+]−, 708.8 [M + Cl−]−. 1H NMR: δ = 7.70 (bs, 4H, NH2), 4.93 (m, 3JH,H = 6.3 Hz, 2H, H-7), 2.94 (m, 4H, H-2), 2.59 (m, 4H, H-4), 2.49 (m, 4H, H-5), 1.27 (t, 3JH,H = 7.2 Hz, 6H, H-1), 1.21 (d, 3JH,H = 6.3 Hz, 12H, H-8) ppm. 13C NMR: δ = 180.9 (C-3), 172.0 (C-6), 67.4 (C-7), 39.8 (C-2), 30.9 (3JC,Pt = 37.5 Hz, C-4), 30.1(C-5), 21.3 (C-8), 14.2 (3JC,Pt = 32.9 Hz, C-1) ppm. 15N NMR: δ = −21.3 ppm. 195Pt NMR: δ = 2850 ppm. IR (ATR): 3282 m, 3206 m; 2981 w, 2937 w; 1722 s, 1702 s (νC O); 1668 s, 1637 s (νC O); 1375 m, 1362 m; 1304 s, 1265 s, 1239s; 1106 m, 1087 m cm−1.
4.2.6. (OC-6-33)-Dichloridobis((4-cyclopentylamino)-4-oxobuta-noato)bis(ethylamine)platinum(IV) (8)
CDI (106.5 mg, 0.6568 mmol) in absolute DMF (7 mL) was added to a solution of 3 (184.5 mg, 0.3126 mmol) in absolute DMF (5 mL), and the mixture was heated to 60 °C. After 10 min of being stirred, the solution was cooled to room temperature and CO2 was removed by flushing with argon. Cyclopentylamine (75 μL, 0.7516 mmol) in 4 mL of absolute DMF was added to the solution and stirred for 30 h in the dark at room temperature. DMF was removed under reduced pressure to form a brown oil. The crude product was purified by column chromatography (EtOAc/MeOH, 4:1) and then precipitated with Et2O (ultrasonic) and cooled to 0 °C to give an almost white solid, which was filtered off, washed with Et2O and EtOAc and dried in vacuo. Yield: 64 mg (28%). Anal. C22H42N4O6PtCl2 (C, H, N). ESI-MS: m/z 747.3 [M + Na+]+, 723.1 [M − H+]−. 1H NMR: δ = 7.95 (d, 3JH,H = 6.9 Hz, 2H, NH-amide), 7.91 (bs, 4H, NH2), 4.26 (m, 3JH,H = 6.8 Hz, 2H, H-7), 3.13 (m, 4H, H-2), 2.68 (m, 4H, C-4), 2.56 (m, 4H, H-5), 2.00 (m, 4H, H-8), 1.84 (m, 4H, H-9), 1.70 (m, 4H, H-9), 1.62 (m, 4H, H-8), 1.45 (t, 3JH,H = 7.1 Hz, 6H, H-1) ppm. 13C NMR: δ = 181.8 (C-3), 171.3 (C-6), 51.0 (C-7), 40.0 (C-2), 32.7 (C-8), 32.1 (C-4), 31.8 (C-5), 23.8 (C-9), 14.4 (C-1) ppm. 15N NMR: δ = 107.7 (CONH-amide), −20.3 (NH2) ppm. 195Pt NMR: δ = 2489 ppm. IR (ATR): 3352 m, 3210 br, 3073 br; 2871 w; 1639 s (νC O); 1535 s, 1356 w, 1258 m, 1246 w cm−1.
4.3. Crystallographic structure determination
X-ray diffraction measurement was performed on a Bruker X8 APEXII CCD diffractometer. Single crystal of 4 was positioned at 40 mm from the detector, and 1839 frames were measured, each for 20 s over 1° scan width. The data were processed using SAINT software [26]. Crystal data, data collection parameters, and structure-refinement details are given in Table 4. The structures were solved by direct methods and refined by full-matrix least-squares techniques. Non-H atoms were refined with anisotropic displacement parameters. H atoms were inserted in calculated positions and refined with a riding model. The isotropic thermal parameters were estimated to be 1.2 times the values of the equivalent isotropic thermal parameters of the atoms to which hydrogens were bonded. Structure solution was achieved with SHELXS-97 and refinement with SHELXL-97 [27], and graphics were produced with ORTEP-3 [28].
Table 4.
Crystallographic data for complex 4.
| 4 | |
|---|---|
| Empirical formula | C14H28Cl2N4O8Pt |
| Fw | 618.37 |
| Space group | P−1 |
| a [Å] | 6.0519(3) |
| b [Å] | 13.1190(6) |
| c [Å] | 14.0840(5) |
| α [°] | 79.521(2) |
| β [°] | 83.694(2) |
| γ [°] | 85.073(3) |
| V [Å3] | 1090.41(8) |
| Z | 2 |
| λ [Å] | 0.71073 |
| ρcalcd [g cm−3] | 1.883 |
| Crystal size [mm3] | 0.20 × 0.13 × 0.12 |
| T [K] | 100(2) |
| μ [mm−1] | 6.719 |
| R1a | 0.0357 |
| wR2b | 0.0858 |
| GOFc | 0.989 |
R1 = Σ||Fo| − |Fc||/Σ|Fo|.
wR2 = {Σ[w(Fo2 − Fc2)2]/Σ[w(Fo2)2]}1/2.
GOF = {Σ[w(Fo2 − Fc2)2]/(n − p)}1/2, where n is the number of reflections and p is the total number of parameters refined.
4.4. Determination of lipophilicity
Lipophilicity of new complexes was determined by the shake-flask method and by reversed-phased HPLC.
4.4.1. Shake-flask method
The log P determination of compounds (4, 5, 6 and 8) was carried out, according to the guidelines for the shake-flask method [29] with slight modifications [20]. Weighted amounts of platinum complexes were dissolved in HPLC-grade water, which was pre-saturated with n-octanol, and mixed by sonication for 5 min. Afterward, the solutions were centrifuged for 5 min and the concentration of Pt was determined by ICP-MS. Weighted amounts of that solutions were mixed with the same volume of n-octanol (pre-saturated with water) and shaken for 15. After phase separation, the Pt concentration in the aqueous phase was again determined by ICP-MS and the partition coefficients were calculated.
The platinum content in the aqueous phase was determined by ICP-MS (Agilent 7500ce, Waldbronn, Germany), equipped with a CETAC ASX-520 autosampler (Neuss, Germany), a Scott double pass spray chamber, and a MicroMist nebulizer. For the analysis, the samples were diluted 1:1000 with 2.5% HCl. Every sample contained 0.5 ppb In as internal standard (CPI International, Santa Rosa, CA, USA).
4.4.2. Reversed-phase HPLC method
HPLC analysis was performed on a Dionex Summit system controlled by the Dionex Chromeleon 6.60 software. The experimental conditions were as follows: Agilent ZORBAX Bonus-RP column (4.6 mm × 250 mm); 0.1% TFA water/MeOH based mobile phases; UV–vis detection set up at 210 nm; temperature of the column: 25 °C; flow rate: 1 mL min−1; concentration of the investigated complexes: 2.5 mM, (1 mM KI as internal standard was added); 25 μL injection volume. The capacity factors k′ = (tR − t0)/t0 (tR is the retention time of the species analyzed and k0 is the retention time of the unretained substance, used as a standard (here KI)) of the investigated compounds were determined at different MeOH/water ratios (from 60:40 for the most lipophilic to 10:90 for the most hydrophilic compounds). Using the linear relationship between log k’ and the percentage of MeOH in the mobile phase: log k′ = log k′w – %MeOH. log k′ values for all complexes were calculated for 0, 10, 20, 30, 40 and 50% of MeOH in the mobile phase. Complexes 3–8 and for comparison (OC-6-33)-dichlorido(ethane-1,2-diamine)bis{(4-methoxy)-4-oxobutanoato}platinum(IV) (R1) and (OC-6-33)-bis{(5-butyloxy)-5-oxo-3-methylpentanoato}dichlorido(ethane-1,2-diamine)platinum(IV) (R2) described in Ref. [22] were investigated. Calibration curves for different MeOH concentrations were created on the basis of determined log P values of 4-6, 8, R1, and R2. The equations derived from the calibration curves for different percentage of MeOH together with their R2 values, are shown in Table 5. From these equations log P values for all investigated complexes were calculated.
Table 5.
Results from the calibration curves log P = f (log k′) under different experimental conditions.
| %MeOH | Equation | R2 |
|---|---|---|
| 0 | 0.8842× −2.1484 | 0.92 |
| 10 | 0.9126× −1.6351 | 0.93 |
| 20 | 0.9586× −1.1251 | 0.95 |
| 30 | 0.9736× −0.4783 | 0.98 |
| 40 | 1.0174× +0.081 | 0.96 |
| 50 | 1.0063× +0.7582 | 0.98 |
4.5. Cell lines and culture conditions
CH1 (ovarian carcinoma, human) cells were donated by Lloyd R. Kelland (CRC Center for Cancer Therapeutics, Institute of Cancer Research, Sutton, U.K.). A549 (non-small cell lung cancer, human) and SW480 (colon carcinoma, human) cells were kindly provided by Brigitte Marian (Institute of Cancer Research, Department of Medicine I, Medical University of Vienna, Austria), and SK-OV-3 (ovarian carcinoma, human) cells by Evelyn Dittrich (General Hospital, Medical University of Vienna, Austria). Cells were grown in 75 cm2 culture flasks (Iwaki/Asahi Technoglass) as adherent monolayer cultures in Minimal Essential Medium (MEM) supplemented with 10% heat-inactivated fetal bovine serum, 1 mM sodium pyruvate, and 2 mM l-glutamine (all purchased from Sigma–Aldrich) without antibiotics. Cultures were maintained at 37 °C in a humidified atmosphere containing 5% CO2 and 95% air.
4.6. Cytotoxicity tests in cancer cell lines
Cytotoxicity in the cell lines mentioned above was determined by the colorimetric MTT assay (MTT = 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide, purchased from Fluka). Cells were harvested from culture flasks by trypsinization and seeded in 100 μL aliquots in MEM supplemented with 10% heat-inactivated fetal bovine serum, 1 mM sodium pyruvate, 2 mM l-glutamine, and 1% non-essential amino acids (100×) into 96-well microculture plates (Iwaki/Asahi Technoglass) in the following densities, to ensure exponential growth of untreated controls throughout the experiment: 1.5 × 103 (CH1), 3.5 × 103 (SK-OV-3), 4.0 × 103 (A549), and 2.5 × 103 (SW480) viable cells per well. Cells were allowed to settle and resume exponential growth in drug-free complete culture medium for 24 h, followed by the addition of dilutions of the test compounds in 100 μL/well of the same medium. After continuous exposure for 96 h, the medium was replaced by a 100 μL/well RPMI 1640 medium (supplemented with 10% heat-inactivated fetal bovine serum and 4 mM l-glutamine) plus 20 μL/well solution of MTT in phosphate-buffered saline (5 mg/mL) (all purchased from Sigma–Aldrich). After incubation for 4 h, medium/MTT mixtures were removed, and the formazan product formed by viable cells was dissolved in DMSO (150 μL/well). Optical densities at 550 nm were measured with a microplate reader (Tecan Spectra Classic), using a reference wavelength of 690 nm to correct for unspecific absorption. The quantity of viable cells was expressed as percentage of untreated controls, and 50% inhibitory concentrations (IC50) were calculated from concentration-effect curves by interpolation. Evaluation is based on means from three independent experiments, each comprising six replicates per concentration level.
4.7. Apoptosis/necrosis assay
Cell death was analyzed by fluorescence-activated cell sorting (FACS) using FITC-conjugated annexin V (BioVision, USA) and propidium iodide (PI; Fluka) staining (Table 6).
Table 6.
Viable, apoptotic and necrotic cell fractions (in %) of SW480 cells upon treatment with cisplatin or compound 7 for 48 h, analyzed by FACS using annexin V and PI staining.
| Concentration, μM | Viable cells | Early apoptosis | Late apoptosis | Necrosis |
|---|---|---|---|---|
| Compound 7 (in %) | ||||
| 0 | 98.3 ± 0.6 | 0.7 ± 0.3 | 0.8 ± 0.5 | 0.2 ± 0.1 |
| 5 | 97.2 ± 1.2 | 1.5 ± 0.8 | 1.0 ± 0.4 | 0.4 ± 0.3 |
| 10 | 87.9 ± 9.8 | 3.3 ± 1.9 | 5.1 ± 5.0 | 3.8 ± 3.7 |
| 15 | 77.7 ± 8.3 | 3.9 ± 2.9 | 10.6 ± 5.9 | 7.9 ± 2.6 |
| 25 | 62.4 ± 4.9 | 5.8 ± 4.9 | 14.7 ± 6.2 | 17.1 ± 7.8 |
| 50 | 47.5 ± 2.6 | 4.8 ± 0.8 | 26.3 ± 8.0 | 21.5 ± 7.0 |
| Cisplatin (in %) | ||||
| 0 | 98.1 ± 0.7 | 0.7 ± 0.6 | 0.5 ± 0.4 | 0.7 ± 0.5 |
| 5 | 96.1 ± 2.1 | 1.0 ± 0.2 | 1.1 ± 0.7 | 1.9 ± 2.2 |
| 10 | 93.5 ± 2.0 | 2.5 ± 0.8 | 1.5 ± 1.2 | 2.5 ± 3.1 |
| 15 | 95.9 ± 1.0 | 2.9 ± 0.3 | 1.0 ± 0.6 | 0.2 ± 0.1 |
| 25 | 94.9 ± 3.5 | 3.3 ± 3.4 | 1.3 ± 0.6 | 0.6 ± 0.5 |
| 50 | 93.8 ± 3.5 | 3.7 ± 2.6 | 1.6 ± 0.8 | 0.9 ± 0.7 |
| 100 | 78.7 ± 11.3 | 8.0 ± 3.5 | 4.6 ± 4.0 | 8.7 ± 4.7 |
SW480 cells were seeded into 6-well plates (Iwaki/Asahi Technoglass, Gyouda, Japan) in amounts of 2 × 105 cells per well in complete medium (as described above) and allowed to settle for 24 h. The cells were exposed to cisplatin and compound 7 for 48 h at 37 °C. After the incubation, cells were gently trypsinized, washed with PBS, and suspended with FITC-conjugated annexin V (0.25 μg/mL) in binding buffer (10 mM HEPES/NaOH pH 7.4, 140 mM NaCl, 2.5 mM CaCl2) at room temperature for 15 min. PI (1 μg/mL) was added shortly before the measurement. Stained cells were analyzed with a FACS Calibur instrument (Becton Dickinson, Franklin Lakes, NJ, USA) using Cell-QuestPro software. At least three independent experiments were conducted, and 10,000 cells were counted per analysis.
Acknowledgments
H. V. is thankful for financial support of the University of Vienna within the doctoral program “Initiativkolleg Functional Molecules” IKI041-N. The authors are indebted to the FFG – Austrian Research Promotion Agency, the Austrian Council for Research and Technology Development, the FWF (Austrian Science Fund) and COST D39. We are thankful to Anna Bytzek for determination of log P values via the shake-flask method. We are indebted to Prof. Verena Dirsch and Daniel Schachner (Institute of Pharmacognosy, University of Vienna, Austria) for providing FACS equipment and technical assistance.
Footnotes
Supplementary data related to this article can be found online at doi:10.1016/j.ejmech.2011.09.006.
Contributor Information
Mathea S. Galanski, Email: mathea.galanski@univie.ac.at.
Bernhard K. Keppler, Email: bernhard.keppler@univie.ac.at.
Appendix. Supplementary material
Crystallographic data have been deposited with the Cambridge Crystallographic Data Center with number CCDC 821855. Copies of data can be obtained, free of charge, on application to CCDC, 12 Union Road, Cambridge CB2 1EZ, UK (deposit@ccdc.com.ac.uk).
References
- 1.Rosenberg M., VanCamp L., Krigas T. Inhibition of cell division in Escherichia coli by electrolysis products from a platinum electrode. Nature. 1965;205:698–699. doi: 10.1038/205698a0. [DOI] [PubMed] [Google Scholar]
- 2.Wheate N.J., Walker S., Craig G.E., Oun R. The status of platinum anticancer drugs in the clinic and in clinical trials. Dalton Trans. 2010;39:8113–8127. doi: 10.1039/c0dt00292e. [DOI] [PubMed] [Google Scholar]
- 3.Hall M.D., Mellor H.R., Callaghan R., Hambley T.W. Basis for design and development of platinum(IV) anticancer complexes. J. Med. Chem. 2007;50(15):3403–3411. doi: 10.1021/jm070280u. [DOI] [PubMed] [Google Scholar]
- 4.Galanski M. Recent developments in the field of anticancer platinum complexes. Recent Pat. Anticancer Drug Discov. 2006;1:285–295. doi: 10.2174/157489206777442287. [DOI] [PubMed] [Google Scholar]
- 5.Galanski M., Jakupec M.J., Keppler B.K. Update of the preclinical situation of anticancer platinum complexes: novel design strategies and innovative analytical approaches. Curr. Med. Chem. 2005;12(18):2075–2094. doi: 10.2174/0929867054637626. [DOI] [PubMed] [Google Scholar]
- 6.Schilder R.J., LaCreta F.P., Perez R.P., Johnson S.W., Brennan J.M., Rogatko A., Nash S., McAleer C., Hamilton T.C., Roby D., Young R.C., Ozols R.F., O’Dwyer P.J. Phase I and pharmacokinetic study of ormaplatin (tetraplatin, NSC 363812) administered on a day 1 and day 8 schedule. Cancer Res. 1994;54(3):709–717. [PubMed] [Google Scholar]
- 7.USA Food and Drug Administration; 2007. G.B. Inc, Orplanta. Satraplatin Capsules, in Advisory Committee Briefing Document. [Google Scholar]
- 8.European Medicines Agency . 2008. Withdrawal Assessment Report for Orplanta. 1–37. [Google Scholar]
- 9.http://www.clinicaltrials.gov, U.S National Library of Medicine, Bethesda, 1993.
- 10.Novakova O., Vrana O., Kiseleva V.I., Brabec V. DNA interactions of antitumor platinum(IV) complexes. Eur. J. Biochem. 1995;228(3):616–624. doi: 10.1111/j.1432-1033.1995.tb20301.x. [DOI] [PubMed] [Google Scholar]
- 11.Galanski M., Keppler B.K. Is reduction required for antitumor activity of platinum(IV) compounds? Characterisation of a platinum(IV)-nucleotide adduct [enPt(OCOCH3)3(5′-GMP)] by NMR spectroscopy and ESI-MS. Inorg. Chim. Acta. 2000;300–302:783–789. [Google Scholar]
- 12.Ellis L.T., Er H.M., Hambley T.W. The influence of the axial ligands of a series of platinum(IV) anti-cancer complexes on their reduction to platinum(II) and reaction with DNA. Aust. J. Chem. 1995;48(4):793–806. [Google Scholar]
- 13.Choi S., Filotto C., Bisanzo M., Delaney S., Lagasee D., Whitworth J.L., Jusko A., Li C., Wood N.A., Willingham J., Schwenker A., Spaulding K. Reduction and anticancer activity of platinum(IV) complexes. Inorg. Chem. 1998;37(10):2500–2504. [Google Scholar]
- 14.Chaney S.G., Wyrick S., Till G.K. In vitro biotransformations of tetrachloro(d, l-trans)-1,2-diaminocyclohexaneplatinum(IV) (tetraplatin) in rat plasma. Cancer Res. 1990;50:4539–4545. [PubMed] [Google Scholar]
- 15.Pendyala L., Cowens J.W., Chheda G.B., Dutta S.P., Creaven P.J. Identification of cis-dichloro-bis-isopropylamine platinum(II) as a major metabolite of iproplatin in humans. Cancer Res. 1988;48:3533–3536. [PubMed] [Google Scholar]
- 16.Barefoot R.R. Speciation of platinum compounds: a review of recent applications in studies of platinum anticancer drugs. J. Chromatogr. B. 2001;751:205–211. doi: 10.1016/s0378-4347(00)00498-9. [DOI] [PubMed] [Google Scholar]
- 17.Reithofer M., Galanski M., Roller A., Keppler B.K. An entry to novel platinum complexes: carboxylation of dihydroxoplatinum(IV) complexes with succinic anhydride and subsequent derivatization. Eur. J. Inorg. Chem. 2006;13:2612–2617. [Google Scholar]
- 18.Reithofer M.R., Valiahdi S.M., Jakupec M.A., Arion V.B., Egger A., Galanski M., Keppler B.K. Novel di- and tetracarboxylatoplatinum(IV) complexes. Synthesis, characterization, cytotoxic activity, and DNA platination. J. Med. Chem. 2007;50:6692–6699. doi: 10.1021/jm070897b. [DOI] [PubMed] [Google Scholar]
- 19.Reithofer M.R., Schwarzinger A., Valiahdi S.M., Galanski M., Jakupec M.A., Keppler B.K. Novel bis(carboxylato)dichlorido(ethane-1,2-diamine)platinum(IV) complexes with exceptionally high cytotoxicity. J. Inorg. Biochem. 2008;102:2072–2077. doi: 10.1016/j.jinorgbio.2008.07.006. [DOI] [PubMed] [Google Scholar]
- 20.Reithofer M.R., Valiahdi S.M., Galanski M., Jakupec M.A., Arion V.B., Keppler B.K. Novel endothall containing platinum(IV) complexes – synthesis, characterization, and cytotoxic activity. Chem. Biodiv. 2008;5:2160–2170. doi: 10.1002/cbdv.200890197. [DOI] [PubMed] [Google Scholar]
- 21.Bytzek A., Reithofer M., Galanski M., Groessl M., Keppler B.K., Hartinger C.G. The first example of MEEKC-ICP-MS coupling and its application for the analysis of anticancer platinum complexes. Electrophoresis. 2010;31:1144–1150. doi: 10.1002/elps.200900522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Reithofer M.R., Bytzek A.K., Valiahdi S.M., Kowol C.R., Groessl M., Hartinger C.G., Jakupec M.A., Galanski M., Keppler B.K. Tuning of lipophilicity and cytotoxic potency by structural variation of anticancer platinum(IV) complexes. J. Inorg. Biochem. 2011;105:46–51. doi: 10.1016/j.jinorgbio.2010.09.006. [DOI] [PubMed] [Google Scholar]
- 23.Ang W.H., Pilet S., Scopelliti R., Bussy F., Juillerat-Jeanneret L., Dyson P.J. Synthesis and characterization of platinum(IV) anticancer drugs with functionalized aromatic carboxylate ligands: influence of the ligands on drug efficacies and uptake. J. Med. Chem. 2005;48:8060–8069. doi: 10.1021/jm0506468. [DOI] [PubMed] [Google Scholar]
- 24.Tetko I.V., Jaroszewicz I., Platts J.A., Kuduk-Jaworska J. Calculation of lipophilicity for Pt(II) complexes: experimental comparison of several methods. J. Inorg. Biochem. 2008;102(7):1424–1437. doi: 10.1016/j.jinorgbio.2007.12.029. [DOI] [PubMed] [Google Scholar]
- 25.Dhara S. A rapid method for the synthesis of cis-[Pt(NH3)2Cl2] Indian J. Chem. 1970;8:193–194. [Google Scholar]
- 26.Bruker-Nonius AXS Inc.; Madison, WI: 2004. SAINT-Plus, Version 7.06a and APEX2. [Google Scholar]
- 27.Sheldrick G.M. A short history of SHELX. Acta Cryst. 2008;A64:112–122. doi: 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
- 28.Johnson G.K. OAK Ridge National Laboratory; Oak Ridge, TN: 1976. Report ORNL-5138. [Google Scholar]
- 29.OECD; Paris: 1995. OECD Guidelines for Testing of Chemicals, No. 107. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.