

Abstract
In experimental autoimmune encephalomyelitis (EAE), an animal model for multiple sclerosis (MS), loss of the blood–brain barrier (BBB) tight junction (TJ) protein claudin-3 correlates with immune cell infiltration into the CNS and BBB leakiness. Here we show that sealing BBB TJs by ectopic tetracycline-regulated expression of the TJ protein claudin-1 in Tie-2 tTA//TRE-claudin-1 double transgenic C57BL/6 mice had no influence on immune cell trafficking across the BBB during EAE and furthermore did not influence the onset and severity of the first clinical disease episode. However, expression of claudin-1 did significantly reduce BBB leakiness for both blood borne tracers and endogenous plasma proteins specifically around vessels expressing claudin-1. In addition, mice expressing claudin-1 exhibited a reduced disease burden during the chronic phase of EAE as compared to control littermates. Our study identifies BBB TJs as the critical structure regulating BBB permeability but not immune cell trafficking into CNS during EAE, and indicates BBB dysfunction is a potential key event contributing to disease burden in the chronic phase of EAE. Our observations suggest that stabilizing BBB barrier function by therapeutic targeting of TJs may be beneficial in treating MS, especially when anti-inflammatory treatments have failed.
Electronic supplementary material
The online version of this article (doi:10.1007/s00401-011-0883-2) contains supplementary material, which is available to authorized users.
Keywords: Blood–brain barrier, Tight junction, Claudin-1, Experimental autoimmune encephalomyelitis, Vascular permeability
Introduction
Under physiological conditions, the blood–brain barrier (BBB) maintains homeostasis of the central nervous system (CNS) by restricting uncontrolled diffusion of water soluble molecules from the blood into the CNS and by limiting immune cell trafficking into the CNS. The BBB is established at the level of CNS microvascular endothelial cells, which are highly specialized to achieve precise control over the substances that leave or enter the brain. Lack of fenestrae and an extremely low pinocytotic activity of BBB endothelial cells inhibit the transcellular passage of molecules across the barrier whereas tight junctions (TJ) between the brain endothelial cells restrict the paracellular diffusion of solutes and ions across the BBB [47]. BBB TJs are unique as in a belt-like region the opposing membranes of adjacent brain endothelial cells form continuous and highly interconnected TJ strands, which appear as a chain of fusion points at the ultrastructural level [10]. In freeze-fracture electron microscopy analysis the fusion points are visualized as strands of TJ particles [46]. At the BBB TJ particles are predominantly associated with the protoplasmic membrane leaflets (P-face) as usually observed in epithelial cells [46], whereas strands of TJs particles of peripheral endothelial cells are rather found associated with the extracellular fracture face (E-face) of the membrane bilayer. TJ strands or particles are composed of integral transmembrane proteins [16], of which occludin and the claudins have been identified to date [40, 41]. Although structurally unrelated, occludin and the claudins are both type II transmembrane proteins bearing 4 transmembrane domains. They are linked to the actin cytoskeleton by TJ associated cytoplasmic peripheral membrane proteins of the MAGUK (membrane associated with a guanylyl kinase-like domain) family, such as zonula occludens (ZO)-1, ZO-2 and ZO-3 [41, 45]. Occludin is highly expressed in TJ of the BBB but not in endothelial TJs of non-neuronal tissues [4, 22], while intact BBB TJs can form in the absence of occludin in vivo [38]. In contrast to occludin, claudins have been shown to be sufficient to induce TJ strands when transfected into fibroblasts [15]. The claudin family is comprised of 24 members, which form the backbone of TJs by establishing homophilic and heterophilic interactions via their extracellular loops [26]. The individual claudins display tissue specific expression patterns and differ in their individual properties in forming dynamic aqueous pores that allow size- and charge-selective paracellular diffusion [17]. Within each tissue, expression of a unique combination of claudins, therefore, determines the respective paracellular tightness of the TJs for different solutes. Claudin-5 has been demonstrated to be the endothelial cell-specific component of TJs strands [34] and is also expressed in BBB TJs [35]. Upon transfection into fibroblasts claudin-5 induces E-face associated TJs [34], whereas claudin-3, which is specifically expressed in TJ of CNS but not other endothelial cells [44, 47], induces P-face associated TJs [16]. This suggests a specific function for claudin-3 in BBB TJs, which is supported further by the recent observation that expression of claudin-3 correlates with BBB maturation in response to Wnt/β-catenin signaling during development [30]. Whereas the exact function of claudin-3 in BBB TJs remains to be investigated, an essential function for claudin-5 in sealing BBB TJs against small molecules up to 800 Da has been demonstrated by genetic deletion of claudin-5 in mice [35]. In addition to claudin-5 and claudin-3, expression of claudin-12 has also been described at the BBB [35]. Previous studies have shown immunostaining for claudin-1 in brain endothelium [31]. Later studies, however, which specifically excluded cross-reactivity of anti-claudin-1 antibodies with claudin-3 failed to detect any immunostaining for claudin-1 in CNS parenchymal microvessels or in primary mouse or human brain endothelial cells [2, 20, 43, 47]. Thus, the combined homophilic and heterophilic interactions between claudin-3, claudin-5, and probably also claudin-12, at the BBB seem to be responsible for the unique barrier function of the BBB TJs [26].
During neurological disorders such as multiple sclerosis (MS) or its animal correlate experimental autoimmune encephalomyelitis (EAE), focal loss of BBB integrity is observed. MS is an inflammatory demyelinating disease of the CNS, in which a large number of inflammatory cells gains access to the CNS parenchyma forming perivascular inflammatory lesions. Lesion formation in MS is known to be associated with focal BBB dysfunction as visualized by gadolinium-enhanced magnetic resonance imaging and occurs early in the pathogenesis of new lesions in MS [24]. Immunohistological analysis of post mortem brain samples of MS patients has identified BBB TJs as the anatomical route for BBB leakiness in MS [29]. In these studies, abnormal distributions of TJ proteins such as occludin and ZO-1 in brain vessels were found to correlate with enhanced BBB leakiness for serum proteins [25, 37]. EAE reliably models the inflammatory phase of MS and BBB alterations observed in EAE resemble those observed in MS [1]. We have previously observed the specific loss of claudin-3 immunostaining from those brain microvessels that were surrounded by inflammatory infiltrates [47], suggesting a direct role for inflammatory cells in disrupting BBB TJs. While much has been learned regarding the sequential steps of immune cell rolling or capture, adhesion and crawling on the BBB, the cellular pathways and the molecular cues mediating immune cell diapedesis across the BBB are only just being unraveled (summarized in [6, 19]). BBB TJs could be disrupted by the immune cells penetrating the BBB on a paracellular route through the endothelial cell–cell contacts or alternatively, immune cells might traverse the BBB on a transcellular route through the brain endothelial cell itself and thus indirectly alter BBB TJ architecture (summarized in [9]).
In the present study, we therefore aimed to delineate the role of BBB TJs in immune cell infiltration and focal BBB leakiness during EAE. Based on our previous observation of the specific loss of claudin-3 from BBB TJs, we hypothesized that in brain endothelial cells ectopic expression of claudin-1, which like claudin-3 induces P-face associated TJs upon transfection into fibroblasts [15], might seal BBB TJs and therefore reduce the paracellular component of immune cell diapedesis across the BBB and/or inflammation-induced BBB leakiness. This notion is supported by previous findings demonstrating that claudin-1 seals TJs in skin epithelial and lung endothelial cells [13, 14]. Furthermore, TJ strands induced by claudin-3 associate with those induced by claudin-1 suggesting that ectopic expression of claudin-1 in BBB TJs would productively integrate into the BBB TJ strands [16]. We, therefore, established transgenic mouse lines with tetracycline (TET)-regulated endothelial cell-specific expression of claudin-1. In two independent transgenic mouse lines, we observed TET-induced expression of claudin-1 in 30–50% of PECAM-1+ CNS microvessels. This partial expression of claudin-1 in BBB TJs sufficed to lead to a significant amelioration of chronic but not acute EAE in double transgenic Tie-2 tTA//TRE–claudin-1 C57BL/6 mice compared to single transgenic littermates.
Materials and methods
Transgene construction
Construction of the Tie-2 tTA vector and production of transgenic mice was described elsewhere [5]. The murine claudin-1 cDNA clone (genbank entry AI663222) was requested from the German Resource Center for Genome Research (RZPD) and sequenced to verify integrity of the open reading frame. The claudin-1 inducible construct was then created by ligating an XhoI/SacII fragment carrying the tet-response element (TRE from pUHD10-3; H. Bujard, Heidelberg), a SacII/BglI claudin-1 fragment, a BamHI/XbaI fragment containing an IRES and the EGFP open reading frame (from pIRES2-EGFP; Clontech, Palo Alto), and an XbaI/XhoI fragment containing the bovine growth hormone poly(A) signal into the XhoI site of pBluescript II. TRE-claudin-1 transgenic mice were then produced by pronuclear injection into fertilized mouse oocytes as described previously [5]. Six TRE-Claudin-1 transgenic founder animals were created from which five independent transgenic lines could be raised.
Genotyping
Transgenic mice were genotyped by PCR. The primers tTA-FW2 (GACGCCTTAGCCATTGAGATG) and tTA-REV2 (CAGTAGTAGGTGTTTCCCTTTCTTC) amplify a product of 550 bp in size from the DNA of Tie-2 tTA transgenic mice.
PCR using the primers CMV-IE FW (CCATAGAAGACACCGGGACC) and IRES REV2 (AAGCGGCTTCGGCCAGTAAC) amplifies a product of 700 bp in size from the DNA of TRE-Claudin-1 transgenic mice.
Mice
C57BL/6 mice were obtained from Harlan (Horst, The Netherlands). The Tie-2-tTA activator line #7770 was described before [5]. Double transgenic mice capable of TET-inducible expression of claudin-1 were obtained by crossing Tie2 tTA activator transgenic C57BL/6 mice with responder mice carrying a unidirectional construct for claudin–1. The experiments shown in the present study were performed with the TRE-claudin-1 mouse lines #23949 and #23974 backcrossed to the C57BL/6 background for at least 7 generations before being used in the experiments. Double transgenic (Tie-2 tTA//TRE–claudin-1) mice were obtained by breeding Tie-2-tTA activator with TRE-claudin-1 responder mice. Mice in matings were fed with Doxycycline supplemented chow (100 mg/kg; Provimi Kliba, Kaiseraugst, Switzerland) to suppress expression of claudin-1 in double transgenic pups during embryonic development and until weaning at postnatal day 21. Mice were housed in the animal facility of the University of Bern under specific pathogen free conditions in individually ventilated cages. All animal procedures were performed in accordance with the Swiss legislation on the protection of animals and approved by the respective government authorities.
Antibodies and reagents
The following primary antibodies were used at 10–20 μg/ml or as supernatants from hybridomas: rat anti-mouse JAM-A (BV11 and BV12, from E. Dejana, Milan, Italy), rat anti-mouse PECAM-1 (Mec13.3, BD Pharmingen, Heidelberg, Germany); rat anti-mouse CD45 (M1/9) and rat anti-human CD44 (Hermes-1 = 9B5, used as isotype control) (both from ATCC, Rockville, MA, USA), rabbit anti-mouse Claudin-5 (from H. Wolburg/H. Kalbacher, Tübingen, Germany). The following antibodies were obtained from Zymed, San Francisco, USA via LuBioScience GmbH, Lucerne, Switzerland: mouse anti-mouse Claudin-5 (35-2500), rabbit anti-mouse Claudin-1 (51-9000), rabbit anti-mouse Claudin-3 (34-1700), rabbit anti-mouse Claudin-5 (34-1600), rabbit anit-mouse Occludin (71-1500), rabbit anti-mouse ZO-1 (61-7300), rabbit anti-mouse ZO-2 (71-1400). Rabbit anti-human fibronectin (A0245, cross-reacts with mouse fibronectin) and rabbit anti-von Willebrand Factor were from DAKO Cytomation, Glostrup, Denmark.
The following secondary antibodies were used at 3–10 μg/ml: Alexa Fluor 488 goat anti-rat, Alexa Fluor 488 goat anti-rabbit (both from Molecular Probes via Invitrogen via LuBioScience GmbH, Lucerne, Switzerland), Cyanine Cy3 goat anti-rat, Cyanine Cy3 goat anti-rabbit, Cyanine Cy3 donkey anti-mouse (all from Jackson Immunoresearch via Milan Analytica AG, La Roche, Switzerland).
RNA isolation, cDNA synthesis, Gene chip array and qPCR of purified parenchymal mouse brain capillaries
Isolation of PECAM-1+ parenchymal mouse brain capillaries from 4-week-old female C57BL/6 mice (Harlan, Horst, The Netherlands) and RNA isolation from these capillary fragments, cDNA synthesis and gene chip microarray analysis (Mouse Genome 430 2.0 Array, Affymetrix UK, High Wycombe, UK) was performed exactly as described before [32]. qPCR was based on SYBR green (MasterMix Plus for SYBR Assay I Low ROX, Eurogentec, Seraing, Belgium) and carried out on an Applied Biosystems 7500 Real-Time PCR system (Applied Biosystems, Warrington, UK). To compare expression levels of claudin-1 with claudin-5, detection of the endogenous reference probe targeting the cDNA for s16 ribosomal protein was included in each experiment (5 experiments total) and set to 1.0 according to the comparative comparative ΔC
T method (relative expression =  , ΔC
T value = average C
T value of target−average C
T value of endogenous reference). The following primer pairs were used: s16 ribosomal protein (Rps16, NM_013647): forward primer GATATTCGGGTCCGTGTGA, reverse primer TTGAGATGGACTGTCGGATG, length of PCR product 69 bp; claudin-5 (NM_013805): forward primer ACGGGAGGAGCGCTTTAC, reverse primer GTTGGCGAACCAGCAGAG, length of PCR product: 65 bp; claudin-1 (NM_016674): forward primer ACTCCTTGCTGAATCTGAACAGT, reverse primer GGACACAAAGATTGCGATCAG, length of PCR product: 91 bp.
, ΔC
T value = average C
T value of target−average C
T value of endogenous reference). The following primer pairs were used: s16 ribosomal protein (Rps16, NM_013647): forward primer GATATTCGGGTCCGTGTGA, reverse primer TTGAGATGGACTGTCGGATG, length of PCR product 69 bp; claudin-5 (NM_013805): forward primer ACGGGAGGAGCGCTTTAC, reverse primer GTTGGCGAACCAGCAGAG, length of PCR product: 65 bp; claudin-1 (NM_016674): forward primer ACTCCTTGCTGAATCTGAACAGT, reverse primer GGACACAAAGATTGCGATCAG, length of PCR product: 91 bp.
Immunofluorescence stainings
Mice were anesthetized with Isofluorane (Baxter, Arovet AG, Zollikon, Switzerland) and perfused with cold phosphate buffered saline (PBS) through the left ventricle of the heart. Brains were removed, coronally cut into three pieces, embedded in Tissue-Tek (OCT compound, Sysmex Digitana AG, Horgen Switzerland) and snap frozen in an isopentane bath (2-Methylbutane, Grogg Chemie AG, Stettlen/Deisswil, Switzerland) at −80°C. Cryostat sections (6 μm) were air dried overnight before staining. Sections were fixed for 10 min with ethanol at 4°C, followed by 1 min in acetone at room temperature and rehydrated with Tris buffered saline (TBS). Sections were blocked with blocking solution containing 5% (w/v) skimmed milk, 0.3% (v/v) Triton X-100 (Fluka via Grogg Chemie AG, Stettlen/Deisswil, Switzerland) and 0.04% (w/v) NaN3 in TBS for 20 min. Primary antibodies were incubated in the same buffer, unless they were used as supernatants from hybridomas, for 1 h at room temperature. Following the washes with TBS, sections were incubated with secondary antibodies diluted in blocking solution for 1 h at room temperature. Sections were mounted in Mowiol (Calbiochem via Grogg Chemie AG, Stettlen/Deisswil, Switzerland). Sections were analyzed using a Nikon Eclipse E600 microscope connected to a Nikon Digital Camera DXM1200F with the Nikon ACT-1 Version 2.63 software (Nikon, Egg/ZH, Switzerland).
Induction of EAE in C57BL/6 mice
Active EAE was induced exactly as described before [8] by subcutaneous injection of 200 μg of myelin oligodendrocyte glycoprotein peptide (MOGaa35–55) emulsified in complete Freund’s adjuvant (incomplete Freund’s adjuvant (Santa Cruz, Switzerland) supplemented with 4 mg/ml attenuated Mycobacterium tuberculosis (H37 RA, DIFCO Laboratories, Detroit, MI) into 8-to 12-week-old female C57BL/6 wild-type, single, and double transgenic Tie-2 tTA//TRE–claudin-1 mice. 300 ng pertussis toxin from Bordetella pertussis (LuBioScience GmbH, Switzerland) per mouse was administered intraperitoneally at days 1 and 3 post-immunization (p.i.). Assessment of clinical disease activity was performed as described with the following disease scores: 0 = healthy, 0.5 = limp tail, 1 = hind leg paraparesis, 2 = hind leg paraplegia, and 3 = hind leg paraplegia with incontinence [8]. A total of 6 EAE experiments were performed comparing double and single transgenic mice of the Tie-2 tTA//TRE–claudin-1 lines 23949 and 23974. Graph Pad Prism 4.0 software was used to calculate area under the curve (AUC) values of EAE clinical scores and for subsequent statistical analysis utilizing Mann–Whitney test (non-parametric, unpaired).
In vivo BBB permeability assays
BBB leakiness for the small molecular tracert Hoechst nuclear stain (HOE 33258, 534D, Calbiochem-Novabiochem, Laufelfingen, Switzerland) and for endogenous fibronectin was assessed in single and double transgenic Tie-2 tTA//TRE–claudin-1 mice of lines 23949 and 23974 at day 50 post EAE induction. 100 μl of 2% Hoechst nuclear stain in PBS was injected intravenously into each mouse. 30 min after dye circulation, mice were decapitated, brains were removed, coronally cut into three pieces, which were embedded side by side in Tissue-Tek (OCT compound, Sysmex Digitana AG, Horgen Switzerland) and snap frozen in an isopentane bath (2-Methylbutane, Grogg Chemie AG, Stettlen/Deisswil, Switzerland) at −80°C. Brain cryosections of 4 double and 6 single transgenic mice were immunofluorescently labeled for CD45 and claudin-1. From each brain at least 10 cryosections (each showing 3 brain sections) were analyzed, with at least 70 μm between individual sections analyzed. Inflammatory cuffs in the brain parenchyma were identified by perivascular accumulation of CD45pos cells and subdivided into 4 leakiness groups according to claudin-1 expression and Hoechst leakage: claudin-1pos/Hoechstpos, claudin-1pos/Hoechstneg, claudin-1neg/Hoechstpos and claudin-1neg/Hoechstneg. The number of inflammatory cuffs was expressed either in percentage per brain or as a ratio of Hoechstpos to Hoechstneg cuffs and statistically analyzed with unpaired t test (Graph Pad Prism 4.0).
For quantification of endogenous fibronectin (FN) leakage across the BBB brain cryosections were performed as described above on day 50 p.i. of 6 double and 9 single transgenic Tie-2 tTA//TRE–claudin-1 mice (lines 23949 and 23974). Pairs of serial sections were immunofluorescently double-stained for CD45/claudin-1 and CD45/fibronectin, respectively. Per individual brain section-pairs were analyzed with at least 70 μm between them. Inflammatory cuffs in the brain parenchyma were identified by perivascular accumulation of CD45pos cells and subdivided into 4 fibronectin leakiness groups according to claudin-1 expression and fibronectin leakage: claudin-1pos/fibronectinpos, claudin-1pos/fibronectinneg, claudin-1neg/fibronectinpos or claudin-1neg/fibronectinneg. The number of inflammatory cuffs was expressed either in percentage per brain or as a ratio of fibronectinpos to fibronectinneg cuffs and statistically analyzed with unpaired t test (Graph Pad Prism 4.0).
Quantification of inflammatory cuffs
The number of CD45pos inflammatory cellular infiltrates surrounding CNS blood vessels (“inflammatory cuffs”) was counted in a total of 10 double transgenic and 15 single transgenic Tie-2 tTA//TRE–claudin-1 mice from lines 23949 and 23974 at day 50 post-immunization and were classified based on their localization as leptomeningeal cuffs surrounding meningeal vessels, as periventricular when localized in direct vicinity to the CNS ventricles and as parenchymal cuffs when surrounding parenchymal blood vessels. The analysis was restricted to the brain as merging of parenchymal and meningeal inflammatory infiltrates in the spinal cord prohibited quantitation of individual inflammatory cuffs in spatial relation to individual blood vessels. The number of inflammatory cuffs per mouse was calculated as mean number of inflammatory cuffs per brain section and median number of inflammatory cuffs for all mice was statistically analyzed using the Mann–Whitney U test (Graph Pad Prism 4.0).
MOG specific T cell priming
In vitro antigen-recall assays were carried out with MOGaa35–55-immunized single, and double transgenic Tie-2 tTA//TRE–claudin-1 mice, as described before [42]. At day 10 post-immunization, mice were killed and draining lymph nodes were dissected. Single-cell suspensions were produced and cells were seeded in 96-well plates at 2 × 105 cells/well in RPMI 1640 supplemented with 10% FBS (GIBCO, LuBiosience, Lucerne, Switzerland), 10 U/mL penicillin/streptomycin, 2 mM l-glutamine, 1% nonessential amino acids, 1 mM sodium pyruvate and 0.05 mM β-mercaptoethanol (Grogg Chemie AG). Increasing concentrations of MOGaa35–55 were used to test for antigen-specific T cell proliferation. T cell proliferation induced by cross-linking of CD3 and CD28 with 0.1 μg/ml of the respective antibodies (clones 145-2C11 and 37.51, Pharmingen BD Biosciences, Switzerland) was used as a positive control. T cell proliferation in medium without antigen or with 10 μg/ml of purified protein derivate of Mycobacterium tuberculosis (PPD, CFA-component) was used as negative and adjuvants control, respectively. All samples were plated in triplicates. [3H]Thymidine (1 μCi/mL) was added 16 h prior to harvesting the cultures on glass fiber filters using a cell harvester (Inotech, Dottikon, Switzerland) and incorporation of [3H]thymidine was measured by liquid scintillation counting.
Statistics
Statistical analysis was performed with Graph Pad Prism 4.0, as described in the corresponding paragraphs.
Results
Expression of tight junction molecules in CNS microvessels in C57BL/6 mice
Descriptions on the presence or absence of claudin-1 in tight junctions (TJs) of blood vessels of the brain have been controversial. To clarify the molecular architecture of brain endothelial TJs and claudin-1 expression in the brain we have, therefore, performed a thorough immunofluorescence analysis for TJ and TJ associated proteins in frozen brain sections of wild-type C57BL/6 mice differentiating between their expression in brain parenchymal blood vessels, meningeal blood vessels, the fenestrated choroid plexus endothelium, the blood-cerebrospinal fluid barrier forming choroid plexus epithelium and ependymal cells lining the ventricles (Table 1). As previously shown [47] we detected occludin, ZO-1 and ZO-2, claudin-3 and claudin-5 but not claudin-1 localized to TJs of parenchymal and meningeal blood vessel endothelial cells. Interestingly, diffuse claudin-1 immunostaining was observed in meningeal blood vessel endothelial cells (Table 1; Fig. 2). Endothelial cells from choroid plexus fenestrated microvessels lacked occludin, claudin-1 and claudin-3, but stained positive for claudin-5. Confirming our previous observations [48] TJs of the choroid plexus epithelium expressed all molecules analyzed (including claudin-1 and claudin-2) except claudin-5. Similarly, although ependymal cells lining the ventricles do not form a tight barrier, they stained positive for all TJ molecules analyzed with the exception of claudin-5.
Table 1.
Detection of tight junction proteins by immunofluorescence staining of the healthy brain
| Parenchymal blood vessel endothelium | Meningeal blood vessel endothelium | Choroid plexus endothelium | Choroid plexus epithelium | Ependymal cells | |
|---|---|---|---|---|---|
| Claudin-1 | – | +a | – | + | + |
| Claudin-2 | – | – | – | + | + |
| Claudin-3 | + | + | – | + | ± |
| Claudin-5 | + | + | + | – | – |
| Occludin | + | + | – | + | + |
| ZO-1 | + | + | + | + | + |
| ZO-2 | + | + | + | + | + |
aDiffuse rather than junctional staining
Fig. 2.
Expression of claudin-1 in the brains of Tie-2 tTA//TRE-claudin-1 double and single transgenic mice. Immunofluorescence staining for claudin-1+ (red) and PECAM-1+ (green) in frozen brain sections of double transgenic Tie-2 tTA//TRE–claudin-1 mice and single transgenic littermates from line 23949 is shown. Expression of claudin-1 was detected in PECAM-1 + parenchymal blood vessels in the brain of Tie-2 tTA//TRE-claudin-1 double transgenic mice but not in single transgenic littermates. In addition, diffuse claudin-1 immunostaining was detected in PECAM-1+ vascular endothelial cells within the leptomeninges and in cell-junctions of choroid plexus epithelial cells in Tie-2 tTA//TRE-claudin-1 double transgenic and single transgenic mice. Scale bar 20 μm
To confirm the absence of claudin-1 expression in mouse brain endothelial cells, we analyzed claudin-1 mRNA expression from freshly isolated PECAM-1pos brain parenchymal microvascular fragments by microarray and quantitative RT-PCR. Both analyses confirmed claudin-5 and the absence of claudin-1 mRNA expression in brain endothelial cells (Fig. 1; [32]. Microarray data (CEL, CHP, and RMA files) are available at GEO (accession number GSE14375).
Fig. 1.
Claudin-1 is not expressed in parenchymal CNS microvessels. mRNA expression level of Claudin-1 and Claudin-5 in parenchymal CNS microvascular endothelial cells was tested by quantitative RT-PCR with the s16 ribosomal protein (Rps16) set to 1.0. As a source for RNA mouse brain microvessels were used immediately after their isolation. Data express mean values of 5 qPCRs carried out in triplicates ± standard error. b.d. below limit of detection
Establishment and characterization of Tie2-tTA//TRE-claudin-1 double transgenic mice
To determine the influence of TJ integrity on immune cell recruitment across the BBB and BBB leakiness during EAE we established transgenic mice with endothelial cell-specific and TET-inducible expression of claudin-1 using the TET-OFF system [5]. Inducibility of claudin-1 expression in responder lines was tested by mating TRE-claudin-1 mice with Tie-2 tTA activator mice expressing tTA in an endothelial cell-specific manner to generate double transgenic mice [5]. Five TRE-claudin-1 responder mice lines were characterized for expression of claudin-1 and GFP (11 mice for line 23949, 12 mice for line 23954, 10 mice for line 23970, 19 mice for line 23971 and 8 mice for line 23974). Whereas expression of GFP was only detected in line 23974, TET-induced expression of claudin-1 was observed in all five lines. However, only ~30% of PECAM-1+ parenchymal vessels displayed positive staining for TET-regulated claudin-1 correctly localized to the cell-to-cell junctions (e.g. 61 claudin-1+PECAM-1+ from 182 PECAM-1+ brain microvessels in line 23949 and 70 claudin-1+PECAM-1+ from 211 PECAM-1+ brain microvessles in line 23974; Fig. 2). TRE-claudin-1 responder lines #23949 and #23974 revealed the highest endothelial expression of claudin-1 and were chosen for further analysis. Wild-type C57BL/6 mice (Table 1) and single transgenic mice (Fig. 2) did not show positive staining for claudin-1 in brain parenchymal vessels, although we detected endogenous junctional claudin-1 staining in the epithelial cells of the choroid plexus and diffuse rather than junctional claudin-1 staining in endothelial cells of meningeal vessels.
Double transgenic Tie-2 tTA//TRE-claudin-1 C57BL/6 mice did not show any apparent phenotypic differences in the overall anatomy of their organs when compared to single transgenic and wild-type littermates. Histological analysis of PECAM-1+ blood vessels in tissue sections of the brain, spinal cord and the secondary lymphoid organs of double transgenic Tie-2 tTA//TRE-claudin-1 C57BL/6 mice revealed no obvious differences in the vascular architecture of these organs compared to the organs in single transgenic or wild-type littermates. Moreover, size and cellular composition of the secondary lymphoid organs was not altered in double transgenic Tie-2 tTA//TRE-claudin-1 C57BL/6 mice compared to single transgenic and wild-type littermates as determined by FACS analysis (data not shown). Interestingly, TET-induced claudin-1 was not expressed in high endothelial venules of double transgenic Tie-2 tTA//TRE-claudin-1 C57BL/6 mice and would, therefore, not interfere with lymphocyte recirculation to lymph nodes.
Ectopic TET-regulated claudin-1 ameliorates chronic but not acute experimental autoimmune encephalomyelitis (EAE)
We next asked, if TET-regulated claudin-1, despite limited expression levels in PECAM-1+ brain microvessels, seals BBB TJs or impacts pathogenesis of MOGaa35–55-induced EAE. We, therefore, compared EAE development in double transgenic Tie-2 tTA//TRE-claudin-1 C57BL/6 mice versus in single transgenic Tie-1tTA or TRE-claudin-1 littermates (lines #23949 and #23974, respectively). In addition, in the first 3 experiments wild-type C57BL/6 littermates were included to test for changes in EAE pathogenesis in single transgenic C57BL/6 mice (data not shown). No difference in the day of onset of clinical EAE nor the maximum clinical score was observed between Tie-2 tTA//TRE-claudin-1 C57BL/6 mice, and single transgenic or wild-type littermates (Fig. 3 and data not shown). In contrast, starting from day 30 p.i., we observed a significantly milder disease score for Tie-2 tTA//TRE-claudin-1 C57BL/6 mice as compared to single transgenic or wild-type littermates (Fig. 3a and data not shown), resulting in overall milder chronic EAE in Tie-2 tTA//TRE-claudin-1 C57BL/6 mice (Fig. 3b). Combined with our observation that MOG-specific T cell priming was unaltered in Tie-2 tTA//TRE-claudin-1 C57BL/6 mice compared to single transgenic littermates (data not shown), the data suggest that TET-regulated claudin-1 expression ameliorates chronic but not acute EAE.
Fig. 3.
TET-regulated claudin-1 ameliorates chronic EAE Active EAE was induced in Tie2-tTA//TRE-claudin-1 double transgenic mice and single transgenic littermates from lines 23949 and 23974, respectively, by subcutaneous immunization with MOGaa35–55 in CFA. a Data show mean disease scores of 4 animals per group ± SEM, evaluated daily following induction of EAE. Black line with open squares indicate single transgenic mice line 23974; dark line with filled squares indicate double transgenic mice line 23974; gray line with open circles indicate single transgenic mice line 23949; light gray line with filled circles indicate double transgenic mice line 23949. One representative experiment out of six comparing a total of 20 Tie2-tTA//TRE-claudin-1 double transgenic mice with 19 single transgenic mice from line 23974 and 31 double transgenic mice with 33 single transgenic mice from line 23949 is shown. b The area under the curve (AUC) was calculated from the EAE clinical course of each mouse between day 30 and day 50 post-immunization. Shown are the mean AUC values for each group ± SEM. Statistical analysis of AUC values with the Mann–Whitney test revealed significantly reduced EAE severity in double transgenic (filled bars) compared to single transgenic (open bars) mice in both TRE-claudin-1 lines. *P < 0.05; **P < 0.005
TET-regulated claudin-1 does not influence immune cell infiltration into the CNS during EAE
To determine if TET-regulated claudin-1 reduces immune cell infiltration across the BBB, we quantified CD45+ inflammatory infiltrates surrounding PECAM-1+ microvessels (inflammatory cuffs) in brain sections at day 50 after induction of EAE. Based on brain localization we categorized inflammatory cuffs in 3 groups: (1) meningeal cuffs localized around leptomeningeal vessels, (2) periventricular cuffs localized directly adjacent to a ventricle, and (3) parenchymal cuffs localized around parenchymal brain microvessels. We detected no difference in the overall number of inflammatory cuffs present in the brains of Tie-2//TRE-claudin-1 C57BL/6 double transgenic mice versus single transgenic littermates nor in the number or ratio of inflammatory cuffs in meningeal, periventricular and parenchymal compartments (Fig. 4). Thus, TET-regulated claudin-1 did not influence immune cell recruitment across the BBB during EAE. An additional analysis counting 549 and 353 parenchymal cuffs in the brains of two Tie-2//TRE-claudin-1 C57BL/6 double transgenic mice and 2 single transgenic littermates, respectively, during the first clinical episode of EAE (day 18 p.i., clinical score 1) further supports the notion that TET-regulated claudin-1 did not interfere with the migration of immune cells across the BBB during EAE.
Fig. 4.
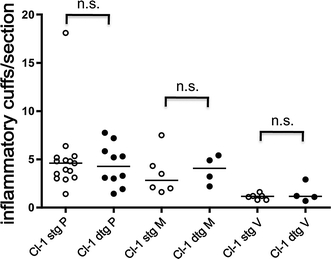
TET-regulated claudin-1 does not influence the number of inflammatory cuffs in the brain. Inflammatory cuffs were counted in a total of 10 double transgenic (dtg) and 15 single transgenic (stg) Tie2-tTA//TRE-claudin-1 mice of lines 23974 and 23949 in 168 versus 147 brain sections, respectively. A total of 1020 versus 1338 parenchymal cuffs (P), 474 versus 510 leptomeningeal cuffs (M) and 197 versus 170 periventricular cuffs (V) in double transgenic versus single transgenic mice, respectively, are included into the analysis. Each dot represents the mean number of inflammatory cuffs/per brain section of one mouse. Horizontal lines depict median for each group. Mann–Whitney U analysis revealed no significant differences comparing the number of cuffs in stg versus dtg mice
TET-regulated claudin-1 reduces BBB permeability in chronic EAE
To investigate, if amelioration of chronic EAE in Tie-2 tTA//TRE-claudin-1 C57BL/6 mice correlated with reduced BBB leakiness, in vivo BBB permeability was examined using intravenous injection of Hoechst nuclear dye, which measures BBB permeability at a particular time point and with high sensitivity due to its small size of 540 Da. Further, staining for endogenous plasma fibronectin, which accumulates in the brain parenchyma over time during EAE, allows a retrospective view of BBB leakage.
Hoechst dye was injected intravenously into Tie-2 tTA//TRE-claudin-1 C57BL/6 mice and single transgenic littermates at day 50 after induction of EAE, after 30 min of dye circulation, brains were removed, and immunofluorescently stained for claudin-1 and for infiltrating leukocytes with the pan-leukocyte marker CD45. In both Tie-2 tTA//TRE-claudin-1 C57BL/6 mice and single transgenic littermates Hoechst dye leakage was restricted to vessels with concurrent perivascular accumulation of CD45+ leukocytes.
Since Hoechst dye leakage was observed in all inflammatory cuffs surrounding meningeal brain and periventricular microvessels, further quantitative analysis was restricted to parenchymal inflammatory cuffs. In Tie-2 tTA//TRE-claudin-1 C57BL/6 mice as compared to single transgenic littermates, a significantly increased percentage of parenchymal inflammatory cuffs around brain microvessels were impermeable to Hoechst dye (Fig. 5a). These data suggest amelioration of chronic EAE as observed in Tie-2 tTA//TRE-claudin-1 C57BL/6 mice may be a consequence of claudin-1 mediated tightening of BBB TJs leading to reduced BBB permeability during chronic EAE. Therefore, for each microvessel TET-regulated claudin-1 expression was correlated with BBB leakiness to Hoechst dye. Parenchymal inflammatory cuffs were categorized in 4 groups by junctional claudin-1 expression and Hoechst leakage as follows: (1) claudin-1pos/Hoechstpos, (2) claudin-1pos/Hoechstneg, (3) claudin-1neg/Hoechstpos, and (4) claudin-1neg/Hoechstneg. All 4 categories of inflammatory cuffs were observed in Tie-2 tTA//TRE-claudin-1 C57BL/6 mice, whereas only claudin-1neg/Hoechstpos and claudin-1neg/Hoechstneg inflammatory cuffs were present in single transgenic littermates (Fig. 5b). As expected, no significant difference in Hoechstneg/Hoechstpos ratios in claudin-1neg inflammatory cuffs in Tie-2 tTA//TRE-claudin-1 C57BL/6 mice versus single transgenic littermates was found (Fig. 5c). In contrast, Hoechstneg/Hoechstpos ratios of inflammatory cuffs around claudin-1pos versus claudin-1neg microvessels revealed a significant increase supporting the conclusion that TET-regulated claudin-1 increases TJ integrity and reduces BBB leakiness for Hoechst dye in chronic EAE.
Fig. 5.
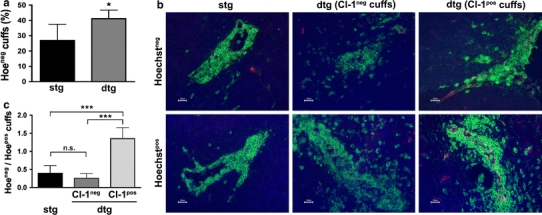
TET-regulated claudin-1 induces BBB tightening for Hoechst dye in chronic EAE. At the end of an EAE experiment (day 50 p.i.), Hoechst dye was injected intravenously into 6 single and 4 double transgenic Tie2-tTA//TRE-claudin-1 mice. Brain cryosections of these mice were stained for Claudin-1 (red) and CD45 (green). Leakage of the nuclear dye Hoechst can be observed in blue. a The mean percentage (±SD) of inflammatory cuffs per brain localized around Hoechst-impermeable BBB vessels is significantly increased in double transgenic compared to single transgenic mice. b The following parenchymal cuffs are shown: inflammatory cuffs in single and double transgenic mice that localized around BBB vessels that do not stain positive for TET-regulated laudin-1 and are either permeable for Hoechst dye or not; and inflammatory cuffs in double transgenic mice localized around claudin-1-expressing BBB vessels which are either permeable for Hoechst dye or not. Scale bar 20 μm. c The mean ratio (±SD) of Hoechst-impermeable to Hoechst-permeable inflammatory cuffs localized around claudin-1 negative vessels (in single and double transgenic mice) or claudin-1 positive vessels (in double transgenic mice) demonstrates highly significant tightening of the BBB for Hoechst dye in the presence of TET-regulated claudin-1. *P < 0.05; ***P < 0.001
To extend these observations, we additionally assessed BBB permeability by staining for the endogenous plasma marker fibronectin, which is found to accumulate around leaky brain microvessels during EAE. Consistent with Hoechst dye data Tie-2 tTA//TRE-claudin-1 C57BL/6 mice displayed a significantly increased number of fibronectin-impermeable inflammatory cuffs versus single transgenic littermates (Fig. 6a). Parenchymal inflammatory cuffs were again subdivided into 4 groups based on expression of endothelial expression of claudin-1 and presence of perivascular fibronectin, with claudin-1neg/fibronectinneg and claudin-1neg/fibronectinpos inflammatory cuffs present in both double and single transgenic mice and claudin-1pos/fibronectinneg and claudin-1pos/fibronectinpos inflammatory cuffs found only in double transgenic mice (Fig. 6b). Based on these groups, we quantified the inflammatory cuffs and calculated the ratio of fibronectinneg/fibronectinpos parenchymal cuffs versus claudin-1 expression as a parameter of BBB tightness (Fig. 6c). Again as expected, no significant difference in fibronectinneg/fibronectinpos ratios of claudin-1neg inflammatory cuffs was observed in Tie-2 tTA//TRE-claudin-1 C57BL/6 mice versus single transgenic littermates (Fig. 6c). In contrast, fibronectinneg/fibronectinpos ratios for claudin-1pos inflammatory cuffs versus claudin-1neg inflammatory cuffs revealed a significant increase supporting the conclusion that during EAE, TET-regulated claudin-1 expression increases TJ integrity thereby reducing BBB leakiness to fibronectin. Determining the ratio of fibronectinneg/fibronectinpos parenchymal cuffs versus claudin-1 expression in mice during the first clinical episode of EAE (day 18 p.i.) confirmed this finding. Taken together, our data demonstrate that TET-regulated claudin-1 expression reduces leakiness of brain microvessels surrounded by inflammatory cells to exogenous tracers and endogenous plasma proteins during EAE.
Fig. 6.
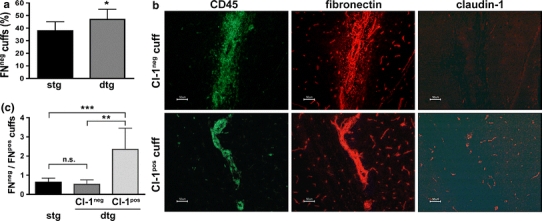
TET-regulated claudin-1 induces BBB tightening for endogenous fibronectin in EAE. Brains of 9 single and 6 double transgenic TET-claudin-1 mice at day 50 post induction of aEAE were prepared, cryo-sectioned and stained for CD45 (green) and claudin-1 (red) as well as for CD45 (green) and fibronectin (red) on consecutive sections. a The mean percentage (±SD) of inflammatory cuffs per brain that have formed around fibronectin-impermeable BBB vessels is significantly increased in double transgenic compared to single transgenic mice. b Shown are representative parenchymal cuffs in a double transgenic mouse. One has formed around a claudin-1neg BBB vessel and is leaky for plasma fibronectin (upper row), one stains positive for claudin-1 and is impermeable for plasma fibronectin (lower row). Note that fibronectin-labeling also stains the basement membranes of BBB vessels. Scale bar 50 μm. c The mean ratio (±SD) of fibronectin-impermeable to fibronectin-permeable inflammatory cuffs that formed around claudin-1neg vessels (in single and double transgenic mice) or claudin-1pos vessels (in double transgenic mice) demonstrates highly significant tightening of the BBB for plasma fibronectin in the presence of transgenic claudin-1. *P < 0.05; **P < 0.005; ***P < 0.001
Discussion
Increased BBB permeability is a characteristic hallmark of the pathological alterations observed in the CNS in MS and in its animal model EAE [27]. In fact, breakdown of the BBB identified by gadolinium-enhanced magnetic resonance imaging is the earliest detectable event in the development of most new CNS lesions in MS and is known to be associated with inflammation [24, 28]. Amelioration of clinical symptoms is observed, when CNS inflammation resolves and BBB function is restored, supporting the notion that lesion formation in MS is driven by inflammatory processes [28] and suggesting a causal relationship between inflammatory cell recruitment into the CNS and BBB dysfunction. Similarly, in EAE, loss of BBB integrity has been recognized as a fundamental event in pathogenesis and further the magnitude of BBB dysfunction has been correlated with disease severity [11]. The close association of inflammation-induced BBB dysfunction with immune cell entry into the CNS has, however, hampered attempts to elucidate the specific contribution of BBB dysfunction to MS pathogenesis.
Our present study provides evidence for the specific contribution of focal BBB leakiness to disease pathogenesis in EAE and identifies the BBB TJs as potentially critical structures for the observed BBB dysfunction during EAE. Specifically, sealing BBB TJs by ectopic expression of claudin-1 in Tie-2 tTA//TRE-claudin-1 double transgenic C57BL/6 mice had no influence on immune cell trafficking across the BBB during EAE and therefore did not influence the onset nor the severity of the first clinical episode of EAE. However, expression of TET-regulated claudin-1 significantly reduced BBB leakiness for blood borne tracers or endogenous plasma proteins specifically in claudin-1 expressing vessels. Further, a reduced disease burden in the chronic phase of EAE was observed in Tie-2 tTA//TRE-claudin-1 mice compared to control littermates. Our study, which dissociates BBB leakiness from immune cell trafficking into the CNS during EAE, identifies the BBB TJs as critical structures regulating BBB permeability, and defines BBB dysfunction as a potentially crucial underlying event contributing to the disease burden in the chronic phase of MS.
Previous studies on post mortem MS brains have already suggested a relationship of BBB TJ abnormalities with BBB hyperpermeability in MS. In these studies, evaluation of a large number of brain vessels by immunofluorescence staining and semi-quantitative thick-section confocal analysis demonstrated TJ abnormalities manifested by beading, interruption or absence of junctional immunostaining or appearance of diffuse cytoplasmic staining for the TJ proteins ZO-1, occludin and junctional adhesion molecule-A (JAM-A) in up to 40% of the blood vessels in MS brain tissue from primary progressive and secondary progressive disease states [25, 29, 36, 37]. Aberrant TJ morphology observed in MS brains correlated with enhanced BBB leakiness for serum proteins, highlighting that BBB TJ disruption underlies BBB leakiness in MS [25, 37]. Based on these observations, these authors have proposed to consider BBB TJ pathology—besides inflammation and demyelination—a distinct entity of tissue injury in MS [37].
EAE reliably models the inflammatory phase of MS and a number of observations support the view that EAE equally well allows to model the BBB TJ pathology observed in MS. Mirroring the observations made in MS tissue samples, disrupted immunostaining for ZO-1 was observed in brain microvessels within areas of focal inflammatory lesions before the onset of clinical EAE and was maintained throughout the clinical disease course [1]. Similarly, we previously observed a focal loss of claudin-3 immunostaining from brain microvessels surrounded by inflammatory cells during EAE [47]. Alterations of the molecular composition of BBB TJs during EAE were found to be accompanied by an aberrant junctional morphology detected at the ultrastructural level, namely pronounced TJ convolution and thereby an apparent extension of the intercellular cleft revealing many kissing points [47].
Although the pathological alterations of BBB TJs described above are indicative of abnormal BBB TJ function as well, observation of altered distribution or lack of an individual TJ protein at the BBB in MS and EAE tissues may not suffice to define BBB TJ pathology or to conclude on deficient BBB TJ function in vivo. For instance, occludin has been shown to be expressed at high levels in brain endothelial cells [4, 22] yet the complete absence is compatible with the development of intact BBB TJs in vivo [38]. In contrast expression of specific occludin splice variants (summarized in [12] or changes in phosphorylation state may influence BBB TJ function. The latter is supported by the observation of occludin dephosphorylation in spinal cord microvessels in a rat model of EAE corresponding with the onset of CNS inflammation [33].
It has been suggested that during MS and EAE immune cells migrate across the BBB via a paracellular route through endothelial cell–cell junctions, thereby causing BBB leakiness and TJ pathology. This view is supported by the observation that mice deficient for the cell–cell contact molecule PECAM-1 develop aggravated EAE due to impaired BBB cell–cell contact integrity combined with increased inflammatory cell recruitment across the BBB into the CNS [18]. On the other hand, there is mounting evidence that immune cell diapedesis across the BBB during EAE might rather occur through the endothelial cell proper, leaving BBB TJs untouched (summarized in [9]). Therefore, alternative or more complex mechanisms of inflammation-induced BBB TJ pathology must be considered. It is conceivable that in the latter scenario soluble mediators released by immune cells might trigger BBB TJs alterations as implied by observations showing that at least in vitro Th17 cells can impair BBB integrity through the combined production of IL-17A and IL-22 leading to downregulation of occludin and to a lesser extent of ZO-1 in brain endothelial cells [23]. In addition, CD8 T cells were shown to initiate BBB TJ disruption in vivo through a non-apoptotic perforin-dependent mechanism [39].
In the present study, we hypothesized that experimental sealing of BBB TJs during CNS inflammation would allow us to determine the contribution of BBB TJs to immune cell diapedesis across the BBB as compared to inflammation-induced BBB leakiness during EAE. Our present study shows that TET-regulated endothelial cell-specific claudin-1 integrated into BBB TJs and significantly reduced BBB leakiness for both a small molecular tracer and endogenous plasma derived fibronectin. Interestingly, although TET-induced expression of claudin-1 could only be observed in 30–50% of brain vessels, claudin-1 induced sealing of BBB TJs during EAE was found to significantly ameliorate the chronic phase of EAE in two independent transgenic mouse lines. Our results, therefore, provide direct functional evidence identifying BBB TJs as the critical component in regulating barrier function of the BBB during EAE and confirming the notion that continued BBB TJ alterations are the basis of BBB dysfunction, which contributes to disease pathogenesis in this animal model of MS.
At the same time, we failed to find any difference in the overall number or size of inflammatory cuffs detected in the brains of Tie-2//TRE-claudin-1 double transgenic mice versus their single transgenic littermates demonstrating that claudin-1 induced sealing of BBB TJs did not influence immune cell recruitment across the BBB during EAE. Unimpeded extravasation of immune cells across the BBB at sites of claudin-1 expression, therefore, implies a limited contribution of the paracellular pathway in immune cell diapedesis across the BBB during EAE and suggests that transcellular immune cell trafficking across the brain endothelial cells proper suffices to fully establish inflammatory infiltrates in the CNS. Lack of any significant differences observed in the onset and severity of the first clinical episode of EAE between Tie-2//TRE-claudin-1 double transgenic mice and their single transgenic littermates suggests that the early phase of EAE pathogenesis is driven by immune cells infiltrating the CNS and might explain the therapeutic efficacy of drugs such as natalizumab in treating MS because of their inhibiting this process [7]. Although our study does not allow the elucidation of the molecular mechanisms initiating inflammation-induced BBB leakiness in EAE, it provides experimental in vivo evidence demonstrating that BBB dysfunction during EAE is due to BBB TJ disruption and identifies BBB TJ pathology as a specific pathogenetic entity contributing to overall disease pathogenesis in EAE.
As the degree of demyelination and/or axonal damage have been described as morphological correlates to increasing disability in MS and clinical severity of EAE [21], we undertook an effort to determine if ameliorated chronic EAE in Tie-2//TRE-claudin-1 C57BL/6 double transgenic mice was due to reduced CNS pathology. To this end, we performed preliminary double immunofluorescence stainings comparing macrophage infiltration and axonal injury in correlation to demyelination in brain and spinal cord sections of Tie-2//TRE-claudin-1 C57BL/6 double transgenic mice and single transgenic littermates during chronic EAE. Immunostaining for F4/80 showed indistinguishable levels of macrophage accumulation in the CNS of Tie-2//TRE-claudin-1 C57BL/6 double transgenic mice and single transgenic littermates. Similarly, immunostaining for MPB and Luxol Fast Blue staining revealed comparable degrees of demyelination in brain and spinal cord sections of Tie-2//TRE-claudin-1 C57BL/6 double transgenic mice and single transgenic littermates. Furthermore, similar degrees of enhanced neurofilament immunofluorescence in spinal cord tissue sections of Tie-2//TRE-claudin-1 C57BL/6 double transgenic mice and single transgenic littermates were indicative of comparable levels of axonal pathology. These observations suggest that claudin-1 induced sealing of BBB TJs does not readily translate into reduced CNS pathology but might lead to an improvement of the physiological conditions in the CNS of Tie-2//TRE-claudin-1 C57BL/6 double transgenic mice sustaining electrical activity and thus proper function of CNS neurons. Induced sealing of BBB TJs could reduce edema and radical formation, and/or improve ion homeostasis in the CNS, and thus lead to a neuroprotective outcome.
Persistent endothelial abnormalities and BBB leakage have been described in primary and secondary progressive MS [29] indicating the existence of the same pathogenic axis in patients. In light of the fact that progressing disability at later time points of the MS disease course is not prevented by the current anti-inflammatory treatments available [3], our present findings demonstrating a continuous contribution of BBB TJs pathology to EAE pathogenesis point to therapeutic sealing of BBB TJs as an attractive alternative for the restoration of BBB function and eventually CNS homeostasis. The challenges faced for developing such methods are our present lack of in depth knowledge on the precise molecular architecture of BBB TJs as well as our incomplete understanding of the function and regulation of expression of the individual TJ components in BBB TJs. The recent discovery of the Wnt/β-catenin signaling pathway involved in brain angiogenesis and claudin-3 expression may open such a novel avenue for therapeutic stabilization of BBB TJs during CNS inflammation.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgments
Special thanks go to Dr. Silke Tauber for preparing the claudin-constructs and to Therese Périnat and Claudi Blatti for expert technical assistance. Furthermore, we wish to thank Dr. Axinia Döring for her tireless help in the daily clinical scoring of EAE mice, Nannette Rink for microinjection, Sorin Ciocan and Thomas Zeis for help with immunofluorescence stainings and Svetlozar Tsonev and Isabelle Oswald for state-of-the art mouse husbandry and care. This work was funded by the the Swiss National Foundation, the European Stroke Network (ESN, EU FP7 N° 201024 and N° 202213) and Integrating Project JUSTBRAIN (EU, FP7 N° HEALTH-2009-241861).
Conflict of interest
The authors declare that they have no conflict of interest.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Footnotes
F. Pfeiffer and J. Schäfer contributed equally to this work.
References
- 1.Bennett J, Basivireddy J, Kollar A, Biron KE, Reickmann P, Jefferies WA, McQuaid S. Blood-brain barrier disruption and enhanced vascular permeability in the multiple sclerosis model EAE. J Neuroimmunol. 2010;229(1–2):180–191. doi: 10.1016/j.jneuroim.2010.08.011. [DOI] [PubMed] [Google Scholar]
- 2.Coisne C, Dehouck L, Faveeuw C, Delplace Y, Miller F, Landry C, Morissette C, Fenart L, Cecchelli R, Tremblay P, Dehouck B. Mouse syngenic in vitro blood-brain barrier model: a new tool to examine inflammatory events in cerebral endothelium. Lab Invest. 2005;85:734–746. doi: 10.1038/labinvest.3700281. [DOI] [PubMed] [Google Scholar]
- 3.Compston A, Coles A. Multiple sclerosis. Lancet. 2008;372(9648):1502–1517. doi: 10.1016/S0140-6736(08)61620-7. [DOI] [PubMed] [Google Scholar]
- 4.Daneman R, Zhou L, Agalliu D, Cahoy JD, Kaushal A, Barres BA. The mouse blood-brain barrier transcriptome: a new resource for understanding the development and function of brain endothelial cells. PLoS One. 2010;5(10):e13741. doi: 10.1371/journal.pone.0013741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Deutsch U, Schlaeger TM, Dehouck B, Doring A, Tauber S, Risau W, Engelhardt B. Inducible endothelial cell-specific gene expression in transgenic mouse embryos and adult mice. Exp Cell Res. 2008;314(6):1202–1216. doi: 10.1016/j.yexcr.2007.12.026. [DOI] [PubMed] [Google Scholar]
- 6.Engelhardt B. T cell migration into the central nervous system during health and disease: different molecular keys allow access to different central nervous system compartments. Clin Exp Neuroimmunol. 2010;1:1–15. doi: 10.1111/j.1759-1961.2009.00005.x. [DOI] [Google Scholar]
- 7.Engelhardt B, Kappos L. Natalizumab: targeting alpha4-integrins in multiple sclerosis. Neurodegener Dis. 2008;5(1):16–22. doi: 10.1159/000109933. [DOI] [PubMed] [Google Scholar]
- 8.Engelhardt B, Kempe B, Merfeld-Clauss S, Laschinger M, Furie B, Wild MK, Vestweber D. P-selectin glycoprotein ligand 1 is not required for the development of experimental autoimmune encephalomyelitis in SJL and C57BL/6 mice. J Immunol. 2005;175(2):1267–1275. doi: 10.4049/jimmunol.175.2.1267. [DOI] [PubMed] [Google Scholar]
- 9.Engelhardt B, Wolburg H. Mini-review: transendothelial migration of leukocytes: through the front door or around the side of the house? Eur J Immunol. 2004;34(11):2955–2963. doi: 10.1002/eji.200425327. [DOI] [PubMed] [Google Scholar]
- 10.Engelhardt B, Wolburg H. The blood-brain barrier in EAE. In: ELaCS Constantinescu., editor. Experimental models of multiple sclerosis. New York: Springer Science + Business Media. Inc; 2005. pp. 415–449. [Google Scholar]
- 11.Fabis MJ, Scott GS, Kean RB, Koprowski H, Hooper DC. Loss of blood-brain barrier integrity in the spinal cord is common to experimental allergic encephalomyelitis in knockout mouse models. Proc Natl Acad Sci USA. 2007;104(13):5656–5661. doi: 10.1073/pnas.0701252104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Feldman GJ, Mullin JM, Ryan MP. Occludin: structure, function and regulation. Adv Drug Deliv Rev. 2005;57(6):883–917. doi: 10.1016/j.addr.2005.01.009. [DOI] [PubMed] [Google Scholar]
- 13.Fujibe M, Chiba H, Kojima T, Soma T, Wada T, Yamashita T, Sawada N. Thr203 of claudin-1, a putative phosphorylation site for MAP kinase, is required to promote the barrier function of tight junctions. Exp Cell Res. 2004;295(1):36–47. doi: 10.1016/j.yexcr.2003.12.014. [DOI] [PubMed] [Google Scholar]
- 14.Furuse M, Hata M, Furuse K, Yoshida Y, Haratake A, Sugitani Y, Noda T, Kubo A, Tsukita S. Claudin-based tight junctions are crucial for the mammalian epidermal barrier: a lesson from claudin-1-deficient mice. J Cell Biol. 2002;156(6):1099–1111. doi: 10.1083/jcb.200110122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Furuse M, Sasaki H, Fujimoto K, Tsukita S. A single gene product, claudin-1 or -2, reconstitutes tight junction strands and recruits occludin in fibroblasts. J Cell Biol. 1998;143(2):391–401. doi: 10.1083/jcb.143.2.391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Furuse M, Sasaki H, Tsukita S. Manner of interaction of heterogeneous claudin species within and between tight junction strands. J Cell Biol. 1999;147(4):891–903. doi: 10.1083/jcb.147.4.891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Furuse M, Tsukita S. Claudins in occluding junctions of humans and flies. Trends Cell Biol. 2006;16(4):181–188. doi: 10.1016/j.tcb.2006.02.006. [DOI] [PubMed] [Google Scholar]
- 18.Graesser D, Solowiej A, Bruckner M, Osterweil E, Juedes A, Davis S, Ruddle NH, Engelhardt B, Madri JA. Altered vascular permeability and early onset of experimental autoimmune encephalomyelitis in PECAM-1-deficient mice. J Clin Invest. 2002;109(3):383–392. doi: 10.1172/JCI13595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Greenwood J, Heasman SJ, Alvarez JI, Prat A, Lyck R, Engelhardt B. Leukocyte-endothelial cell crosstalk at the blood-brain barrier: a prerequisite for successful immune cell entry to the brain. Neuropathol Appl Neurobiol. 2011;37(1):24–39. doi: 10.1111/j.1365-2990.2010.01140.x. [DOI] [PubMed] [Google Scholar]
- 20.Hamm S, Dehouck B, Kraus J, Wolburg-Buchholz K, Wolburg H, Risau W, Cecchelli R, Engelhardt B, Dehouck MP. Astrocyte mediated modulation of blood-brain barrier permeability does not correlate with a loss of tight junction proteins from the cellular contacts. Cell Tissue Res. 2004;315(2):157–166. doi: 10.1007/s00441-003-0825-y. [DOI] [PubMed] [Google Scholar]
- 21.Herrero-Herranz E, Pardo LA, Gold R, Linker RA. Pattern of axonal injury in murine myelin oligodendrocyte glycoprotein induced experimental autoimmune encephalomyelitis: implications for multiple sclerosis. Neurobiol Dis. 2008;30(2):162–173. doi: 10.1016/j.nbd.2008.01.001. [DOI] [PubMed] [Google Scholar]
- 22.Hirase T, Staddon JM, Saitou M, Ando-Akatsuka Y, Itoh M, Furuse M, Fujimoto K, Tsukita S, Rubin LL. Occludin as a possible determinant of tight junction permeability in endothelial cells. J Cell Sci. 1997;110(Pt 14):1603–1613. doi: 10.1242/jcs.110.14.1603. [DOI] [PubMed] [Google Scholar]
- 23.Kebir H, Kreymborg K, Ifergan I, Dodelet-Devillers A, Cayrol R, Bernard M, Giuliani F, Arbour N, Becher B, Prat A. Human TH17 lymphocytes promote blood-brain barrier disruption and central nervous system inflammation. Nat Med. 2007;13(10):1173–1175. doi: 10.1038/nm1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kermode AG, Thompson AJ, Tofts P, MacManus DG, Kendall BE, Kingsley DP, Moseley IF, Rudge P, McDonald WI. Breakdown of the blood-brain barrier precedes symptoms and other MRI signs of new lesions in multiple sclerosis. Pathogenetic and clinical implications. Brain. 1990;113(Pt 5):1477–1489. doi: 10.1093/brain/113.5.1477. [DOI] [PubMed] [Google Scholar]
- 25.Kirk J, Plumb J, Mirakhur M, McQuaid S. Tight junctional abnormality in multiple sclerosis white matter affects all calibres of vessel and is associated with blood-brain barrier leakage and active demyelination. J Pathol. 2003;201(2):319–327. doi: 10.1002/path.1434. [DOI] [PubMed] [Google Scholar]
- 26.Krause G, Winkler L, Mueller SL, Haseloff RF, Piontek J, Blasig IE. Structure and function of claudins. Biochim Biophys Acta. 2008;1778(3):631–645. doi: 10.1016/j.bbamem.2007.10.018. [DOI] [PubMed] [Google Scholar]
- 27.Lassmann H. Comparative neuropathology of chronic experimental allergic encephalomyelitis and multiple sclerosis. Berlin: Springer Verlag; 1983. [PubMed] [Google Scholar]
- 28.Lassmann H, Wekerle H. The pathology of multiple sclerosis. In: Compston A, editor. Mcalpine’s multiple sclerosis. Edinburgh: Elsevier Chruchill Livingstone; 2005. pp. 557–599. [Google Scholar]
- 29.Leech S, Kirk J, Plumb J, McQuaid S. Persistent endothelial abnormalities and blood-brain barrier leak in primary and secondary progressive multiple sclerosis. Neuropathol Appl Neurobiol. 2007;33(1):86–98. doi: 10.1111/j.1365-2990.2006.00781.x. [DOI] [PubMed] [Google Scholar]
- 30.Liebner S, Corada M, Bangsow T, Babbage J, Taddei A, Czupalla CJ, Reis M, Felici A, Wolburg H, Fruttiger M, Taketo MM, von Melchner H, Plate KH, Gerhardt H, Dejana E. Wnt/beta-catenin signaling controls development of the blood-brain barrier. J Cell Biol. 2008;183(3):409–417. doi: 10.1083/jcb.200806024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liebner S, Fischmann A, Rascher G, Duffner F, Grote E-H, Kalbacher H, Wolburg H. Claudin-1 and claudin-5 expression and tight junction morphology are altered in blood vessels of human glioblastoma multiforme. Acta Neuropathol. 2000;100:323–331. doi: 10.1007/s004010000180. [DOI] [PubMed] [Google Scholar]
- 32.Lyck R, Ruderisch N, Moll AG, Steiner O, Cohen CD, Engelhardt B, Makrides V, Verrey F. Culture-induced changes in blood-brain barrier transcriptome: implications for amino-acid transporters in vivo. J Cereb Blood Flow Metab. 2009;29(9):1491–1502. doi: 10.1038/jcbfm.2009.72. [DOI] [PubMed] [Google Scholar]
- 33.Morgan L, Shah B, Rivers LE, Barden L, Groom AJ, Chung R, Higazi D, Desmond H, Smith T, Staddon JM. Inflammation and dephosphorylation of the tight junction protein occludin in an experimental model of multiple sclerosis. Neuroscience. 2007;147(3):664–673. doi: 10.1016/j.neuroscience.2007.04.051. [DOI] [PubMed] [Google Scholar]
- 34.Morita K, Sasaki H, Furuse M, Tsukita S. Endothelial claudin: claudin-5/TMVCF constitutes tight junction strands in endothelial cells. J Cell Biol. 1999;147(1):185–194. doi: 10.1083/jcb.147.1.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nitta T, Hata M, Gotoh S, Seo Y, Sasaki H, Hashimoto N, Furuse M, Tsukita S. Size-selective loosening of the blood-brain barrier in claudin-5-deficient mice. J Cell Biol. 2003;161:653–660. doi: 10.1083/jcb.200302070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Padden M, Leech S, Craig B, Kirk J, Brankin B, McQuaid S. Differences in expression of junctional adhesion molecule-A and beta-catenin in multiple sclerosis brain tissue: increasing evidence for the role of tight junction pathology. Acta Neuropathol. 2007;113(2):177–186. doi: 10.1007/s00401-006-0145-x. [DOI] [PubMed] [Google Scholar]
- 37.Plumb J, McQuaid S, Mirakhur M, Kirk J. Abnormal endothelial tight junctions in active lesions and normal-appearing white matter in multiple sclerosis. Brain Pathol. 2002;12(2):154–169. doi: 10.1111/j.1750-3639.2002.tb00430.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Saitou M, Furuse M, Sasaki H, Schulzke JD, Fromm M, Takano H, Noda T, Tsukita S. Complex phenotype of mice lacking occludin, a component of tight junction strands. Mol Biol Cell. 2000;11(12):4131–4142. doi: 10.1091/mbc.11.12.4131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Suidan GL, McDole JR, Chen Y, Pirko I, Johnson AJ. Induction of blood brain barrier tight junction protein alterations by CD8 T cells. PLoS One. 2008;3(8):e3037. doi: 10.1371/journal.pone.0003037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tsukita S, Furuse M. Claudin-based barrier in simple and stratified cellular sheets. Curr Opin Cell Biol. 2002;14(5):531–536. doi: 10.1016/S0955-0674(02)00362-9. [DOI] [PubMed] [Google Scholar]
- 41.Tsukita S, Furuse M, Itoh M. Structural and signalling molecules come together at tight junctions. Curr Opin Cell Biol. 1999;11(5):628–633. doi: 10.1016/S0955-0674(99)00016-2. [DOI] [PubMed] [Google Scholar]
- 42.Uboldi C, Doring A, Alt C, Estess P, Siegelman M, Engelhardt B. l-Selectin-deficient SJL and C57BL/6 mice are not resistant to experimental autoimmune encephalomyelitis. Eur J Immunol. 2008;38(8):2156–2167. doi: 10.1002/eji.200838209. [DOI] [PubMed] [Google Scholar]
- 43.Weksler BB, Subileau EA, Perriere N, Charneau P, Holloway K, Leveque M, Tricoire-Leignel H, Nicotra A, Bourdoulous S, Turowski P, Male DK, Roux F, Greenwood J, Romero IA, Couraud PO. Blood-brain barrier-specific properties of a human adult brain endothelial cell line. Faseb J. 2005;19(13):1872–1874. doi: 10.1096/fj.04-3458fje. [DOI] [PubMed] [Google Scholar]
- 44.Witt KA, Mark KS, Hom S, Davis TP. Effects of hypoxia-reoxygenation on rat blood-brain barrier permeability and tight junctional protein expression. Am J Physiol Heart Circ Physiol. 2003;285(6):H2820–H2831. doi: 10.1152/ajpheart.00589.2003. [DOI] [PubMed] [Google Scholar]
- 45.Wolburg H, Lippoldt A. Tight Junctions of the blood-brain barrier. development, composition and regulation. Vasc Pharmacol. 2002;28:323–337. doi: 10.1016/S1537-1891(02)00200-8. [DOI] [PubMed] [Google Scholar]
- 46.Wolburg H, Neuhaus J, Kniesel U, Krauss B, Schmid EM, Ocalan M, Farrell C, Risau W. Modulation of tight junction structure in blood-brain barrier endothelial cells. Effects of tissue culture, second messengers and cocultured astrocytes. J Cell Sci. 1994;107(Pt 5):1347–1357. doi: 10.1242/jcs.107.5.1347. [DOI] [PubMed] [Google Scholar]
- 47.Wolburg H, Wolburg-Buchholz K, Kraus J, Rascher-Eggstein G, Liebner S, Hamm S, Duffner F, Grote EH, Risau W, Engelhardt B. Localization of claudin-3 in tight junctions of the blood-brain barrier is selectively lost during experimental autoimmune encephalomyelitis and human glioblastoma multiforme. Acta Neuropathol (Berl) 2003;105(6):586–592. doi: 10.1007/s00401-003-0688-z. [DOI] [PubMed] [Google Scholar]
- 48.Wolburg H, Wolburg-Buchholz K, Liebner S, Engelhardt B. Claudin-1, claudin-2 and claudin-11 are present in tight junctions of choroid plexus epithelium of the mouse. Neurosci Lett. 2001;307(2):77–80. doi: 10.1016/S0304-3940(01)01927-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.