

Abstract
Ca2+ is an important regulatory ion and alteration of mitochondrial Ca2+ homeostasis can lead to cellular dysfunction and apoptosis. Ca2+ is transported into respiring mitochondria via the Ca2+ uniporter, which is known to be inhibited by Mg2+. This uniporter-mediated mitochondrial Ca2+ transport is also shown to be influenced by inorganic phosphate (Pi). Despite a large number of experimental studies, the kinetic mechanisms associated with the Mg2+ inhibition and Pi regulation of the uniporter function are not well established. To gain a quantitative understanding of the effects of Mg2+ and Pi on the uniporter function, we developed here a mathematical model based on known kinetic properties of the uniporter and presumed Mg2+ inhibition and Pi regulation mechanisms. The model is extended from our previous model of the uniporter that is based on a multistate catalytic binding and interconversion mechanism and Eyring's free energy barrier theory for interconversion. The model satisfactorily describes a wide variety of experimental data sets on the kinetics of mitochondrial Ca2+ uptake. The model also appropriately depicts the inhibitory effect of Mg2+ on the uniporter function, in which Ca2+ uptake is hyperbolic in the absence of Mg2+ and sigmoid in the presence of Mg2+. The model suggests a mixed-type inhibition mechanism for Mg2+ inhibition of the uniporter function. This model is critical for building mechanistic models of mitochondrial bioenergetics and Ca2+ handling to understand the mechanisms by which Ca2+ mediates signaling pathways and modulates energy metabolism.
Introduction
Ca2+ is a key regulatory ion and has multiple roles in mitochondrial function and dysfunction (1–5). Ca2+ is known to mediate signaling pathways between cytosol and mitochondria and modulate mitochondrial energy metabolism. Alteration of mitochondrial Ca2+ homeostasis can lead to mitochondrial dysfunction and mitochondria-mediated cellular injury and apoptosis. Mg2+ is another key regulatory ion and the intracellular concentration of free Mg2+ varies significantly in vivo (0.2–2 mM) under various physiological and pathological conditions (e.g., hormonal stimulation, myocardial ischemia) (6–8). Alteration of cytosolic Mg2+ homoeostasis is known to influence mitochondrial substrate and cation (e.g., Ca2+) transport systems and bioenergetics (e.g., Mg2+ is a cofactor for the operation of F1F0-ATPase and adenine nucleotide translocase; Mg2+ regulates many key enzymes in mitochondria, such as pyruvate dehydrogenase; Mg2+ also inhibits mitochondrial Ca2+ uptake via the Ca2+ uniporter (6–8)). Therefore, it is essential to mechanistically characterize the biophysical and kinetic mechanisms associated with the transport of Ca2+ into mitochondria and how variations in cytosolic Mg2+ influence this Ca2+ transport.
Ca2+ uniporter is the primary influx pathway for Ca2+ in respiring mitochondria, and hence is an important regulator of mitochondrial Ca2+. This uniporter-mediated mitochondrial Ca2+ uptake is known to be influenced by inorganic phosphate (Pi), in addition to the inhibition by Mg2+ (9–13). Thus, the kinetics of Ca2+ uptake not only depends on the catalytic properties of the uniporter and the electrochemical gradient of Ca2+ across the inner mitochondrial membrane (IMM), but also on the mechanisms of Mg2+ inhibition and Pi regulation of the uniporter function. This has been extensively investigated in many experimental (8,9,11–16) and modeling (17–22) studies. Early initial velocity studies of Ca2+ uptake in rat heart and liver mitochondria suggest that Mg2+ is a competitive inhibitor of Ca2+ uptake where it induces a sigmoid kinetics and the degree of sigmoidicity (Km of Ca2+ for the uniporter) increases with increase in [Mg2+], without altering the maximal activity of the uniporter (9,14,15).
However, subsequent study on the kinetics of Ca2+ uptake in rat liver mitochondria reported a noncompetitive type of Mg2+ inhibition of Ca2+ uptake by increasing the Km of Ca2+ and decreasing the maximal activity of the uniporter, with increase in [Mg2+] (11). Furthermore, the Ca2+ uptake is shown to be regulated by Pi (9,10). Specifically, the kinetic study of Crompton et al. (9) on Ca2+ uptake in rat heart mitochondria shows significant differences in the initial Ca2+ uptake, when measured with 0 and 1 mM Pi in the buffer. Though the regulatory roles of Pi on oxidative phosphorylation (23,24) and matrix Ca2+ buffering through Ca2+-Pi complex formation (25) are well recognized, it is not understood how Pi can modulate Ca2+ uptake via the uniporter. Furthermore, though the mechanisms of extramatrix Ca2+ and membrane potential (ΔΨ) dependencies of Ca2+ uptake have been well-described mathematically (17–22), the kinetic mechanism by which Mg2+ inhibits and Pi regulates the uniporter function has not been well characterized in terms of a mechanistic model. The need for such a model is critical for developing mechanistic integrated models of mitochondrial bioenergetics and Ca2+ handling that can be helpful in understanding the mechanisms by which Ca2+ plays a role in mediating signaling pathways and modulating energy metabolism under normal and pathological conditions.
Previously, we introduced a mechanistic mathematical model of Ca2+ uniporter (20) that satisfactorily describes the available data on the kinetics of Ca2+ uptake, measured in respiring mitochondria isolated from rat hearts and rat livers under diverse experimental preparations (14–16). This model was developed based on a multistate catalytic binding and interconversion mechanism and Eyring's free-energy barrier theory for interconversion (26–28). In addition, the model accounts for the allosteric cooperative binding of Ca2+ to the uniporter, observed experimentally (14,15). In a recent work, this model is further improved to accurately characterize the mechanism of ΔΨ-dependency of Ca2+ uptake (22). An interesting prediction of this improved model is that the Ca2+-binding sites on the uniporter are located at equal and negligible distances from the bulk phase on either side of the IMM and the binding affinities of the uniporter to both cytosolic and matrix Ca2+ are the same. This alternate model is shown to be more robust with minimal number of model parameters that accurately describes the available data (14–16). Furthermore, this alternate model is insensitive to physiological variations in matrix [Ca2+]. Though these models are able to characterize the possible mechanisms of both the extramatrix Ca2+ and ΔΨ dependencies of Ca2+ uptake, they do not account for the effects of Mg2+ and Pi on the kinetics of Ca2+ uptake. Furthermore, there exists no mathematical model in the literature that characterizes the inhibitory effect of Mg2+ and regulatory role of Pi on the uniporter-mediated Ca2+ uptake in mitochondria.
In this article, we develop a mechanistic mathematical model of Ca2+ uniporter that improves the existing models (17–22) and provides a biophysical basis for the inhibitory effect of Mg2+ and regulatory role of Pi on the uniporter-mediated Ca2+ uptake. The model, extended from our previous models (20,22), is thermodynamically balanced and adequately describes a large number of independent data sets on both Mg2+-dependent and Mg2+-independent Ca2+ uptake via the uniporter (8,9,11,12,14–16). The model accurately depicts the inhibitory effect of Mg2+ on Ca2+ uptake in which the kinetics of Ca2+ uptake is hyperbolic in the absence of Mg2+ and sigmoid in the presence of Mg2+. A mixed type Mg2+ inhibition mechanism is shown to be consistent with the available data.
Mathematical Formulation
Experimental data for model formulation
The kinetic mechanisms associated with the Mg2+ inhibition and Pi regulation of mitochondrial Ca2+ transport is not well established. However, experimental studies in respiring isolated mitochondria (9,11,14–16) and intact permeabilized myocytes (8) provide adequate information about the influences of Mg2+ and Pi on the initial rates of Ca2+ transport into mitochondria. The initial rate data from isolated mitochondria describe the kinetics of extramatrix Ca2+- and ΔΨ-dependent mitochondrial Ca2+ uptake at various extramatrix [Mg2+] and [Pi]. The initial rate data from permeabilized cells demonstrate the nature of mitochondrial Ca2+ uptake in response to physiological variations of cytosolic [Mg2+] and [Ca2+]. Therefore, these kinetic data are appropriate for building kinetic models of the Ca2+ uniporter to characterize the mechanisms by which Mg2+ and Pi regulate the uniporter-mediated mitochondrial Ca2+ transport.
The kinetic studies of Scarpa and colleagues (14,15) report the initial rates of Ca2+ uptake with variations in extramatrix [Ca2+] (0–200 μM), measured in isolated mitochondria from rat livers and rat hearts, in the presence of 2 and 5 mM Mg2+ and 5 and 2 mM Pi in the buffer. These studies reveal a sigmoid kinetics of the extramatrix Ca2+-dependent Ca2+ uptake in the presence of Mg2+ and Pi. The kinetic study of Crompton et al. (9) reports the initial rates of Ca2+ uptake with variations in extramatrix [Ca2+] (0–35 μM), measured in isolated mitochondria from rat hearts, both in the absence and presence of 1 mM Mg2+ and Pi in the buffer. Although this study reveals a similar sigmoid kinetics of the extramatrix Ca2+-dependent Ca2+ uptake in the presence of 1 mM Mg2+ and 1 mM Pi, the kinetics was shown to become hyperbolic in the absence of Mg2+ with 0 and 1 mM Pi present in the buffer. These studies suggest that Mg2+ is a competitive inhibitor of Ca2+ uptake that alters only the Km of Ca2+ for the uniporter. The kinetic study of Bragadin et al. (11) reports the initial rates of Ca2+ uptake with variations in extramatrix [Ca2+] (0–300 μM), measured in isolated mitochondria from rat livers, both in the absence and presence of 2 and 5 mM Mg2+ with 2 mM Pi in the buffer. This study reveals a similar sigmoid kinetics of the extramatrix Ca2+-dependent Ca2+ uptake in the presence of 2 and 5 mM Mg2+, and the kinetic was shown to become hyperbolic in the absence of Mg2+.
However, this study indicates a noncompetitive type of Mg2+ inhibition of Ca2+ uptake that significantly alters both the Km of Ca2+ and the maximal activity of the uniporter. The kinetic study of Wingrove et al. (16) reports the initial rates of Ca2+ uptake with variations in ΔΨ (90–200 mV) for different levels of extramatrix Ca2+ ([Ca2+]e = 0.5, 1.0, and 1.5 μM), measured in isolated mitochondria from rat livers, in the absence of Mg2+ with 0.2 mM Pi present in the buffer. This study reveals a nonlinear, Goldman-Hodgkin-Katz-type ΔΨ-dependency of Ca2+ uptake. The regulatory effect of Pi on Ca2+ uptake seems to be prominent only at low Pi levels (0–1 mM), which is evident from the study of Crompton et al. (9): Pi was shown to increase both the Km of Ca2+ and the maximal activity of the uniporter; Pi did not appreciably affect the kinetics of Ca2+ uptake at higher levels of Pi ([Pi] > 1 mM). This kinetic model of Ca2+ uniporter is developed based on basic physical-chemical principles of ion transport across biomembranes and is parameterized to adequately reproduce these diverse experimental data sets in isolated mitochondria as well as the data sets of Szanda et al. (8) in permeabilized cells.
Proposed mechanism of Mg2+ inhibition of Ca2+ uniporter
Fig. 1 schematizes the proposed kinetic mechanism of Mg2+ inhibition of the uniporter-mediated Ca2+ transport into mitochondria. The uniporter functional unit is assumed to have two binding sites for Ca2+ and two distinct allosteric sites for Mg2+, facing either the external (cytosolic) side or the internal (matrix) side depending on the state of the uniporter (Fig. 1 A). In the forward process, an ionized Ca2+ from the cytosolic side binds to the uniporter T to form the complex T2Ca2+e, which further favors the binding of second ionized Ca2+ (cooperative binding) to form the complex T2Ca2+e. The complex T2Ca2+e then undergoes conformational changes forming the complex 2Ca2+xT in the matrix side. In the reverse process, the complex 2Ca2+xT dissociates in two steps to form two free Ca2+ ions and the unbound uniporter T in the matrix side (Fig. 1 B).
Figure 1.
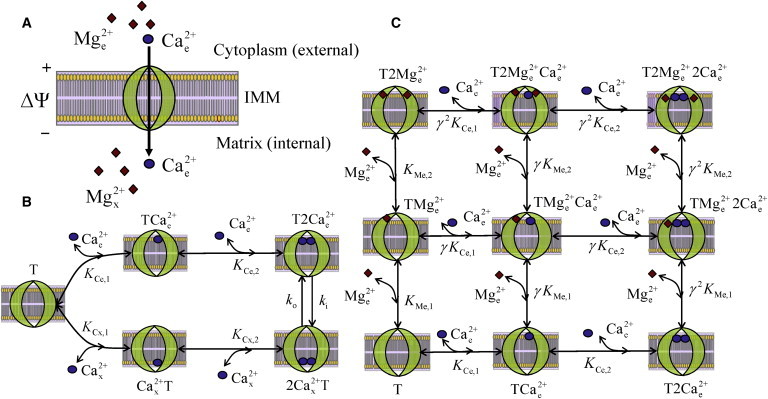
Proposed kinetic mechanism of Mg2+ inhibition of mitochondrial Ca2+ uptake via the Ca2+ uniporter. (A) Schematics of the Ca2+ uniporter, driven by the electrochemical gradient of Ca2+ (circle) across the IMM, exposed to Mg2+ ions (diamond). (B) Schematics of different Ca2+-bound uniporter states during translocation of Ca2+ across the IMM. (C) Schematics of different Ca2+- and Mg2+-bound uniporter states in the cytoplasmic side in a linear mixed-type inhibition mechanism for Mg2+ inhibition of mitochondrial Ca2+ uptake. In the matrix side, the Ca2+-Mg2+ interaction is symmetrical to that in the cytoplasmic side. KCe,1, KCe,2, KMe,1, and KMe,2 are the dissociation constants associated with the binding of first and second cytoplasmic Ca2+ and Mg2+ to the uniporter; 1 ≤ γ ≤ ∞ is a parameter that controls the binding affinity of Ca2+ and Mg2+ to the uniporter. The translocation of Ca2+ across the IMM (T2Cae2+ ↔ 2Cax2+2) is limited by two rate constants ki and ko, which are dependent on ΔΨ.
We consider a general, linear mixed-type inhibition mechanism for Mg2+ inhibition of the uniporter function. In this scheme, the interactions of Mg2+ and Ca2+ with the uniporter in the cytosolic side can be described by Fig. 1 C. Here, in one process, two Mg2+ ions cooperatively bind to the uniporter states E1 (T, TCae2+, T2Cae2+) in two different steps to form the complexes
and
In another process, two ions cooperatively bind to the uniporter states E2 (T, TMge2+, T2Mge2+) in two different steps to form the complexes
and
In the matrix side, the Ca2+-Mg2+ interaction is symmetrical to that in the cytoplasmic side. The complete kinetic scheme for the Mg2+ inhibition of the uniporter function is shown in Fig. S1 of Appendix SA in the Supporting Material. Except for the 2Ca2+-bound uniporter complexes T2Ca2+e and 2Ca2+xT, the other uniporter complexes are assumed not to undergo any conformational changes that give rise to Ca2+ translocation across the IMM.
The translocation of Ca2+ via the uniporter is limited by the interconversion rate constants ki and ko, which are dependent on ΔΨ. KCe and KCx (KMe and KMx) are the dissociation constants for the binding of cytosolic and matrix Ca2+ (Mg2+) to the uniporter. It is assumed that the binding of Mg2+ affects the binding of Ca2+ and vice versa. In this regard, the binding affinities of Ca2+ to the Mg2+-bound uniporter states and Mg2+ to the Ca2+-bound uniporter states decrease with increase in the number of Ca2+ and Mg2+ binding, which is mainly determined by the control parameter γ (1 ≤ γ ≤ ∞). Note that the parameter γ defines the type of Mg2+ inhibition of the uniporter function. For example, γ = 1 represents a noncompetitive inhibition, γ = ∞ represents a competitive inhibition, and 1 < γ < ∞ represents a mixed-type inhibition mechanism.
Ca2+ uniporter flux expression
Based on the proposed mechanism of Mg2+ inhibition of mitochondrial Ca2+ transport (Fig. 1) and the assumption of quasi-steady-state flux and rapid equilibrium binding of Ca2+ and Mg2+, the net Ca2+ transport flux via the uniporter (JCU) can be expressed as (see Appendix SA in the Supporting Material):
| (1) |
where [T]tot represents the total uniporter concentration, and
Model 1
| (2a) |
Model 2
| (2b) |
Model 3
| (2c) |
Note that for γ > 1, the higher-order terms in the uniporter flux expression becomes smaller and smaller compared to the leading order terms, because the concentrations of Ca2+ and Mg2+ bound uniporter states become smaller and smaller. Model 1, Model 2, and Model 3 represent, respectively, the full cooperative, partial cooperative, and no-cooperative binding of Ca2+ and Mg2+ to the uniporter (see Appendix SA in the Supporting Material). The kinetic parameters KCe, KCx, ki, and ko are constrained by the equilibrium relationship
| (3) |
where Keq is the equilibrium constant for transport of 2Ca2+ via the uniporter.
ΔΨ-dependency of the kinetic parameters
We follow a similar approach to that of our previous cation transporter models (20,22,29) to describe the ΔΨ-dependency of the kinetic parameters. Because Mg2+ and Ca2+ have identical valence, similar assumptions and biophysical parameters are used to describe the ΔΨ-dependency of Mg2+ and Ca2+ binding constants for the uniporter. We only summarize below the final equations that are used to express the kinetic parameters Keq, KCe, KCx, KMe, KMx, ki, and ko in terms of ΔΨ:
| (4) |
| (5) |
| (6) |
| (7) |
Here Z = 2 is the valence of Ca2+ or Mg2+; F, R, and T denote the Faraday's constant, ideal gas constant, and absolute temperature, respectively (F = 96484.6 J/mol/mV and R = 8.3145 J/mol/K); αe (αx) is the ratio of the potential difference between Ca2+ or Mg2+ bound to the site of the uniporter facing the external (internal) side of the IMM and Ca2+ or Mg2+ in the bulk phase to the total membrane potential; and βe (βx) is the displacement of external (internal) Ca2+ from the coordinate of maximum potential barrier. The ΔΨ-dependency of ki and ko are derived based on a nonlinear Goldman-Hodgkin-Katz type formalism by expressing βe and βx as functions of ΔΨ (22); nH is an arbitrary number that determines the degree of deviation from the linear Goldman-Hodgkin-Katz formalism.
Substituting Eq. 4 for Keq, Eq. 5 for KCe and KCx, and Eq. 6 for ki and ko into Eq. 3, the following kinetic and thermodynamic constraints were obtained:
| (8) |
From our previous analyses of the ΔΨ-dependency of mitochondrial Ca2+ uptake (22), it is apparent that the Ca2+ (Mg2+) binding sites on the uniporter are located at equal and negligible distances from the bulk phase on either side of the IMM (i.e., αe = αe ≈ 0) and the binding affinities of the uniporter to both cytosolic and matrix Ca2+ (Mg2+) are the same (K0Ce = K0Cx and K0Me and K0Mx). The kinetic constraint in Eq. 8, then, provides ki0 = ko0 and the biophysical constraint in Eq. 8 is automatically satisfied. The estimate nH = 2.65 was found to well describe the ΔΨ-dependencies of the biophysical parameters βe and βx. We followed similar assumptions for binding of Ca2+ and Mg2+ to the uniporter and translocation of Ca2+ via the uniporter, so that αe = αe = α ≈ 0, K0Ce = K0Cx = K0C, K0Me = K0Mx = K0M, ki0 = ko0 = k0, and nH = 2.65. With this, the number of unknown parameters for estimation in this model is further reduced from ten to four (k0, K0C, K0M, and γ).
Pi-dependency of the kinetic parameters
Low extramatrix Pi levels have been shown to modulate the kinetics of Ca2+ uniporter in isolated rat heart mitochondria. Specifically, the kinetic study of Crompton et al. (9) shows changes in both the Km of Ca2+ and the maximal activity of the uniporter at 0 and 1 mM Pi, in the presence and absence of extramatrix Mg2+. However, other initial rate studies with higher concentrations of Pi (>1 mM) do not show any significant effect of Pi on the kinetics of Ca2+ uniporter (11,14,15). In view of these experimental observations, we incorporate the Pi effect on the uniporter function by exclusively modifying the apparent Km of Ca2+ and Mg2+ for the uniporter (K0C and K0M) as
and
| (9) |
where KPi is the Pi binding constant for the uniporter; K0C and K0M on the right side of Eq. 9 are the true (intrinsic) dissociation constants of Ca2+ and Mg2+ for the uniporter. Further justification for the proposed mechanism of Pi regulation of the kinetic parameters K0C and K0M is given in Appendix SB in the Supporting Material. With negligible pH gradient across IMM, Pi can quickly equilibrate across the IMM via the Pi-H+ cotransporter (23,24,30). Therefore, we assumed that [Pi]e = [Pi]x = [Pi]. With this modification, the total number of unknown parameters for estimation in this uniporter model becomes five.
Parameter estimation and sensitivity analysis
To estimate the unknown kinetic parameters of the model, a combined least-squares estimation technique was used in multiple steps to fit the model-simulated outputs to the available experimental data,
| (10) |
where Nexp is the number of experiments and Ndata is the number of data points in a particular experiment, are the experimental data and are the corresponding model simulated outputs, and is the maximum value of . The accuracy and robustness of the model fitting to the data for a particular model are assessed based on the value of mean residual error E(ϕ) and its sensitivities to perturbations in the optimal parameter estimates.
The model sensitivity to infinitesimal changes in the parameter values can be computed by
| (11) |
where ϕi is the ith parameter; denotes the normalized sensitivity index for the parameter ϕi, whereas SϕE denote the combined normalized sensitivity index for all parameters together. A higher sensitivity index indicates that a small change in a given parameter (or all parameters) can result in a significant change in the model output. The most accurate model and parameters can have higher and SϕE with lower least-squared error E(ϕ).
Results
In this section, we provide the detailed parameterization and independent validation of the three versions of the kinetic model of mitochondrial Ca2+ uniporter. The three kinetic models (Model 1, full cooperativity; Model 2, partial cooperativity; Model 3, no cooperativity) are simulated to exclusively fit the available data sets (9,11,14–16) on the kinetics of extramatrix Ca2+- and ΔΨ-dependent mitochondrial Ca2+ uptake measured both in the absence and presence of different amounts of Mg2+ and Pi in the buffer. The model fittings of these data sets are demonstrated through Fig. 2. The dashed, solid, and dotted lines represent the fittings for Model 1, Model 2, and Model 3, respectively. The model is further validated by comparing the model predictions to another data set (8) on the kinetics of mitochondrial Ca2+ uptake measured in permeabilized cells with physiological variations in cytosolic [Ca2+] and [Mg2+], which is demonstrated in Fig. 3. The estimated parameter values associated with these three variant models are provided in Table 1. Additional model simulations describing the nature of extramatrix Ca2+ and ΔΨ dependencies and the nature of Mg2+ inhibition of mitochondrial Ca2+ uptake are demonstrated through Fig. S3, Fig. S4, and Fig. S5 of Appendix SC in the Supporting Material.
Figure 2.
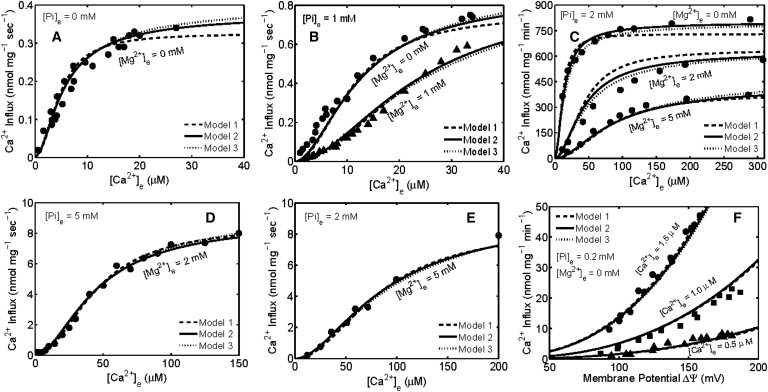
Comparison of the Ca2+ uniporter models (lines) to the experimental data (points) on the extramatrix Ca2+ and ΔΨ-dependencies of mitochondrial Ca2+ uptake measured both in the presence and absence of extramatrix Mg2+. Shown are the fittings of three different versions of the kinetic model (Model 1, Model 2, and Model 3) to the kinetic data of (A and B) Crompton et al. (9) in which the initial rates of Ca2+ influx in response to variations in extramatrix [Ca2+] were measured in energized mitochondria of rat hearts suspended (A) in a medium containing 0 mM Mg2+ and 0 mM Pi and (B) in a medium containing 0 and 1 mM Mg2+ and 1 mM Pi; (C) Bragadin et al. (11) in which the initial rates of Ca2+ influx in response to variations in extramatrix [Ca2+] were measured in energized mitochondria of rat livers suspended in a medium containing different levels of Mg2+ ([Mg2+]e = 0, 2, and 5 mM) and 2 mM Pi; (D) Vinogradov and Scarpa (14) in which the initial rates of Ca2+ influx in response to variations in extramatrix [Ca2+] were measured in energized mitochondria of rat livers suspended in a medium containing 2 mM Mg2+ and 5 mM Pi; (E) Scarpa and Graziotti (15) in which the initial rates of Ca2+ influx in response to variations in extramatrix [Ca2+] were measured in energized mitochondria of rat hearts suspended in a medium containing 5 mM Mg2+ and 2 mM Pi; and (F) Wingrove et al. (16) in which the initial rates of Ca2+ influx as a function of ΔΨ were measured in energized mitochondria of rat livers suspended in a medium containing three different levels of Ca2+ ([Ca2+]e = 0.5, 1.0, and 1.5 μM), with 0 mM Mg2+ and 0.2 mM Pi present in the medium. The models were fitted to these data sets by setting ΔΨ at 140 mV for the data corresponding to 0 mM Pi (A) and 190 mV for the data corresponding to ≥ 1 mM Pi (B–E). Matrix [Ca2+] was fixed at 250 nM and matrix [Mg2+] was fixed at 0.4 mM.
Figure 3.
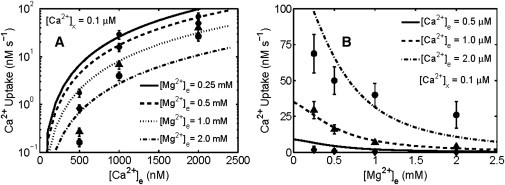
Comparison of the Ca2+ uniporter model (lines) to the experimental data (points) on the cytosolic Ca2+ and Mg2+ dependencies of mitochondrial Ca2+ uptake measured in permeabilized cells by Szanda et al. (8). Shown are the simulations and comparisons of Model 2 of the uniporter to the kinetic data in which the initial rates of Ca2+ uptake were measured (A) as a function of cytosolic [Ca2+] (logarithmic y axis) with four different levels of cytosolic Mg2+ ([Mg2+]e = 0.25, 0.5, 1, and 2 mM), and (B) as a function of cytosolic [Mg2+] (linear y axis) with three different levels of cytosolic Ca2+ ([Ca2+]e = 0.5, 1.0, and 2.0 μM). Model simulations were done using ΔΨ = 190 mV, matrix [Ca2+] = 0.1 μM, and matrix [Mg2+] = 0.4 mM. The rate constant k0 was found to be 5.96 nM/s (0.006 × 10−3 nmol/mg/s), as 1 mg mitochondrial protein = 1 μL mitochondrial volume.
Table 1.
Estimated parameter values and the corresponding normalized sensitivity coefficients for the kinetic model of mitochondrial Ca2+ uniporter
| Parameters(ϕi) | Model 1 |
Model 2 |
Model 3 |
References | ||||
|---|---|---|---|---|---|---|---|---|
| Value | Value | Value | ||||||
| k0 (nmol/mg/s) | 1.27 × 10−3 | 0.0671 | 1.49 × 10−3 | 0.2692 | 1.58 × 10−3 | 0.3302 | (9) | |
| 20.32 × 10−3 | 0.0671 | 22.35 × 10−3 | 0.2692 | 22.12 × 10−3 | 0.3302 | (11) | ||
| 16.51 × 10−3 | 0.0671 | 19.37 × 10−3 | 0.2692 | 20.54 × 10−3 | 0.3302 | (14) | ||
| 27.94 × 10−3 | 0.0671 | 30.55 × 10−3 | 0.2692 | 29.23 × 10−3 | 0.3302 | (15) | ||
| 63.5 × 10−3 | 0.0671 | 62.58 × 10−3 | 0.2692 | 60.04 × 10−3 | 0.3302 | (16) | ||
| K0C (M) | 3.965 × 10−6 | 0.1131 | 2.786 × 10−6 | 0.4946 | 1.955 × 10−6 | 0.7319 | All | |
| K0M (M) | 0.655 × 10−3 | 0.0564 | 0.542 × 10−3 | 0.1725 | 0.403 × 10−3 | 0.4218 | All | |
| γ (Unitless) | 3.9 | 0.1784 | 5.7 | 0.1739 | 9.2 | 0.1412 | All | |
| KPi (M) | 0.2 × 10−3 | 0.0319 | 0.2 × 10−3 | 0.1678 | 0.2 × 10−3 | 0.2696 | All | |
| Eϕ; SEϕ | 0.077; 0.447 | 0.058; 1.278 | 0.053; 1.895 | Eqs. 10 and 11 | ||||
Rate constant k0 is redefined as k0 = [T]tot · k0. Note that the apparent values of K0C and K0M calculated using Eq. 9 for different [Pi] matches well with the estimates in Table S1 of Appendix SB in the Supporting Material, obtained without considering the Pi regulation mechanism of K0C and K0M.
We followed a multistep modular approach for parameterization of the model:
-
Step 1.
The kinetic parameters k0 and K0C were estimated based on the Pi- and Mg2+-independent data ([Pi]e = 0 mM; [Mg2+]e = 0 mM) of Crompton et al. (9) (Fig. 2 A). The values of the kinetic parameters KPi, K0M, and γ were reasonably chosen (fixed) to fit this data set, because these parameters are insensitive to this data set. The ΔΨ was set at 140 mV, corresponding to 0 mM Pi, based on a 2003 study by Bose et al. (23).
-
Step 2.
The Pi-dependent data of Crompton et al. (9) (Fig. 2 B) both with and without Mg2+ ([Pi]e = 1 mM; [Mg2+]e = 0 and 1 mM) were used to estimate KPi, K0M, and γ, as these data sets are suitable to estimate the Pi and Mg2+ binding constants. In this step, the values of k0 and K0C remained the same, as estimated in the first step. The ΔΨ was set at 190 mV, corresponding to a typical state-2 respiration with Pi ≥ 0.2 mM (23,24,30).
-
Step 3.
The five parameters k0, K0C, K0M, KPi and γ were further optimized and fine-tuned to jointly fit the model to the three data sets of Crompton et al. (9), along with the kinetic data of Bragadin et al. (11) ([Pi]e = 2 mM; [Mg2+]e = 0, 2, and 5 mM, see our Fig. 2 C); Vinogradov and Scarpa (14) ([Pi]e = 5 mM; [Mg2+]e = 2 mM, see our Fig. 2 D); Scarpa and Graziotti (15) ([Pi]e = 2 mM; [Mg2+]e = 5 mM, see our Fig. 2 E); and Wingrove et al. (16) ([Pi]e = 0.2 mM; [Mg2+]e = 0 mM, see our Fig. 2 F).
The parameter k0 was allowed to vary among data sets from different groups to account for different tissue types (i.e., mitochondria from rat livers versus rat hearts) and diverse experimental preparations that affect the overall uniporter activity (e.g., heterogeneity of mitochondrial population and differential energization or ΔΨ). The biophysical parameter αe = αe = α was set at zero and nH was set at 2.65, based on our previous uniporter model (22). Matrix [Ca2+] was set at 250 nM; matrix [Mg2+] was set at 0.4 mM, a value within the physiological range, based on the extensive work of Jung et al. (31); and ΔΨ was set at 190 mV for Pi ≥ 0.2 mM (9,23). We note here that, this extended uniporter has all the characteristics of our previous uniporter model (20,22). The model is insensitive to physiological variations in matrix [Ca2+], which is demonstrated in Fig. S3, A and B, in Appendix SC in the Supporting Material. Hence, the parameter estimates would depend neither on matrix [Ca2+], nor on Ca2+ buffering and Ca2+-Pi complex formation in the matrix.
One of the key observations from the model analyses of the data (fitting) is that, the fitting is able to provide unique and accurate estimates of the kinetic parameters K0C, K0M, KPi, and γ for all the available data sets (Table 1), irrespective of the amounts of Mg2+ and Pi present in the buffer. Furthermore, the fitting gives a unique and accurate estimate of k0 for all the data sets obtained from the same source (e.g., k0 is same for all the data curves of Crompton et al.). The fitting is able to provide a unique and accurate estimate of the parameter KPi (KPi ≈ 0.2 mM) that governs the Pi-dependencies of the kinetic parameters (K0C and K0M). Most importantly, Pi only seems to modulate the K0C and K0M in low Pi range (0–1 mM); for [Pi]e > 1 mM; it does not alter K0C and K0M significantly, which is consistent with the available data.
Another important observation from the fitting is that this estimated value of K0C is found to be much lower (e.g., K0C ≈ 4.14 μM using Model 1 for all data sets) compared to our previously estimated values (20,22) (e.g., K0C = 46 μM and 88 μM using Model 1 from the data sets of Scarpa and colleagues (14,15)). This is due to the presence of 2 and 5 mM Mg2+ and 5 and 2 mM Pi in their buffer, which significantly modifies the apparent Km of Ca2+ for the uniporter; this was not accounted for in our previous uniporter models (20,22). Even in the presence of 2 or 5 mM Pi, the apparent Km of Ca2+ becomes ∼9 μM, which is still much lower than that of the previous estimates. A small Km value indicates that Ca2+ uptake attains saturation for a lower level of extramatrix Ca2+, compared to our previous uniporter models (20,22), which is demonstrated in Fig. S4, A and B, of Appendix SC in the Supporting Material. Overall, this uniporter model accurately depicts the kinetics of mitochondrial Ca2+ uptake over a wide range of experimental conditions, in which the kinetics of Ca2+ uptake is hyperbolic in the absence of Mg2+, sigmoid in presence of Mg2+, and the degree of sigmoidicity increases with increase in [Mg2+], which is depicted in Fig. S4, C and D, of Appendix SC in the Supporting Material.
From the fittings of the models to the data (Fig. 2), it is apparent that both Model 2 and Model 3 adequately explain the available data compared to Model 1. This is also evident from the combined least-square error (Eϕ) and normalized sensitivity coefficients for individual parameters and all parameters together ( and SEϕ) (Table 1). Specifically, Eϕ is higher and SEϕ is lower for Model 1, whereas Eϕ is lower and SEϕ is higher for Model 2 and Model 3. Additional statistical test based on the Akaike's Information Criterion (AIC) is also conducted to verify the goodness of the fits. The computed value of AIC (AIC = Ndataln(Eϕ) + 2Nϕ) was found to be higher for Model 1 compared to Model 2 and Model 3. This is obvious because the number of unknown parameters (Nϕ) is the same in all three models. Therefore, AIC is strictly dependent on Eϕ for a given model. From model parameterization and sensitivity analyses (Table 1) it is made clear that the kinetic parameters K0C, K0M, and γ are very accurate and consistent over a wide variety of experimental preparations at physiological levels of Pi. The estimated values of these parameters varied between different versions of the model due to the nature of binding assumptions. Therefore, either of the models (Model 2 or Model 3) can be considered as the most accurate model of the uniporter that best describes the available data compared to Model 1.
Based on these estimated values of the kinetic parameters (K0C, K0M, KPi, γ) and the previously estimated values of the biophysical parameters (α, nH), we further validated the uniporter model by simulating the extramatrix Ca2+ and Mg2+ dependencies of mitochondrial Ca2+ uptake in permeabilized cells (8). Fig. 3 A shows the model comparisons (logarithmic y axis) to the data as a function of cytosolic [Ca2+] for four different cytosolic [Mg2+] values ([Mg2+]e = 0.25, 0.5, 1, and 2 mM; [Pi]e = 1 mM), whereas Fig. 3 B shows the model comparisons (linear y axis) to the data as a function of cytosolic [Mg2+] for three different cytosolic [Ca2+] values ([Ca2+]e = 0.5, 1, and 2 μM; [Pi]e = 1 mM). We used one of the most accurate models of the uniporter (Model 2) for these model simulations. The value of the rate constant k0 was first converted from the units of nmol/mg/s to the units of nM/s by using the conversion factor 1 mg protein = 3.7 μL, then modified until the model predictions closely match the data. Matrix [Ca2+] was set at 250 nM, matrix [Mg2+] was set at 0.4 mM, and ΔΨ was set at 190 mV. Considering that no other kinetic parameter was adjusted, the model is able to adequately predict the data and the nature of Mg2+ inhibition of mitochondrial Ca2+ uptake in intact permeabilized cells (8), under physiological variations of cytosolic [Ca2+] and [Mg2+], supporting the accuracy of the model. Similar model predictions on % Mg2+ inhibition of Ca2+ uptake as a function of extramatrix [Ca2+] and [Mg2+] are demonstrated through Fig. S5 of Appendix SC in the Supporting Material.
From the model analyses of the available data (Figs. 2 and 3), it is evident that this uniporter model is able to explain adequately the extramatrix Pi-, Mg2+-, Ca2+-, and ΔΨ-dependent kinetics of mitochondrial Ca2+ uptake in a wide variety of experimental preparations. To further validate the proposed mechanisms of Mg2+ inhibition of the uniporter function, we integrated this uniporter model and our recently developed 3Na+-1Ca2+ antiporter model (29) to our previously developed model of mitochondrial Ca2+ handling and bioenergetics (19). The integrated model is used to simulate the experimental data of Cox and Matlib (32) and McCormack et al. (12) on matrix free [Ca2+] as a function of extramatrix free [Ca2+], [Mg2+], and [Na+]. Detailed analyses and fittings of the integrated model to these data sets are described in Appendix SD (Figs. S6 and S7) in the Supporting Material. The model appropriately described these data sets, further signifying the accuracy and robustness of this uniporter model and the proposed Mg2+ and Pi regulation mechanisms.
Discussion
The work reported in this article provides the detailed development of a mechanistic mathematical model of mitochondrial Ca2+ uniporter that characterizes the kinetic mechanisms by which cytosolic Mg2+ inhibits and Pi regulates the uniporter-mediated Ca2+ transport into mitochondria. The model is extended from our previous models of the uniporter (20,22) and developed based on a similar multistate catalytic binding and interconversion mechanism for carrier-mediated facilitated transport and Eyring free energy barrier theory for interconversion and electrodiffusion (26–28). The model is parameterized and validated by comparing the model-simulated outputs to a large number of independent data sets on the kinetics of extramatrix Ca2+- and ΔΨ-dependent Ca2+ uptake via the uniporter, obtained in respiring mitochondria isolated from rat hearts and livers (9,11,14–16) and intact permeabilized myocytes (8) both in the absence and presence of different amounts of extramatrix Mg2+ and Pi. The model precisely describes the inhibitory effect of extramatrix Mg2+ and the regulatory role of Pi on the uniporter-mediated mitochondrial Ca2+ uptake, observed in these experiments.
Although this uniporter model has all the characteristics of our previous uniporter models (20,22), it significantly differs from the earlier models in that it mechanistically characterizes the inhibitory effect of Mg2+ and regulatory role of Pi on the uniporter function. It identifies the most plausible Mg2+ inhibition and Pi regulation mechanisms based on a large number of experimental data sets (8,9,11,12,14–16). This uniporter model clarifies the earlier contradictory results on the type of Mg2+ inhibition of the uniporter function. Particularly, whereas the kinetic studies of Crompton et al. (9) and Scarpa and colleagues (14,15) showed a competitive type of Mg2+ inhibition, the later kinetic study of Bragadin et al. (11) showed a noncompetitive type of Mg2+ inhibition of Ca2+ uptake. Our model suggests a mixed-type mechanism for Mg2+ inhibition of Ca2+ uptake, in which the kinetics of Ca2+ uptake is hyperbolic in the absence of Mg2+ and sigmoid in the presence of Mg2+. This is manifested by a marked increase in the apparent Km of Ca2+ for the uniporter and decrease in the maximal activity of the uniporter with increasing extramatrix [Mg2+]. Furthermore, the degree of sigmoidicity increases with increase in extramatrix [Mg2+], which is consistent with experimental observations.
This uniporter model also accurately explains the regulatory effect of cytosolic Pi on mitochondrial Ca2+ uptake, typically seen at low Pi levels (0–1 mM) in the kinetic study of Crompton et al. (9). Accordingly, the Km of Ca2+ and Mg2+ for the uniporter (K0C and K0M) were expressed in terms of Pi, in which Pi is shown to increase the value of K0C and decrease the value of K0M, and the Pi effect is shown to be negligible at higher Pi concentrations ([Pi] > 1 mM), consistent with the available data. On the other hand, without considering the Pi regulation mechanism, the model did not fit to the available data with unique K0C and K0M values for all data sets. This is apparent from the detailed model analyses presented in Appendix SB in the Supporting Material, which justifies this formulation of Pi regulation of K0C and K0M parameters. However, additional kinetic data at submillimolar Pi concentrations may be necessary to mechanistically evaluate the precise regulatory effect of Pi on the uniporter function.
Despite significant technical advances, the structure and composition of mitochondrial Ca2+ uniporter and the regulation mechanisms of the uniporter function by divalent cations (e.g., Mg2+) and metabolites (e.g., Pi) are still largely unknown. Our present uniporter model is based on the assumption that there exist at least two binding sites for Ca2+ ions and two separate allosteric sites for Mg2+ ions on the uniporter (Fig. 1). We explored three possible Ca2+ and Mg2+ binding schemes (Model 1, full cooperativity; Model 2, partial cooperativity; and Model 3, no cooperativity) to resolve the most likely Ca2+ and Mg2+ binding mechanism that can explain the observed kinetics of mitochondrial Ca2+ uptake under diverse experimental preparations (8,9,11,12,14–16). With our model analyses of the available data, though we are not able to fully distinguish between the three versions of the kinetic model, Model 2 and Model 3 seem to describe the data most accurately compared to Model 1. Alternatively, by assuming 1), two binding sites for Ca2+ ions and one separate allosteric site for Mg2+ ions on the uniporter or 2), two binding sites for Ca2+ ions and Mg2+ ions competing for the Ca2+ binding sites, we were not able to fit the two reduced models to any of the available data sets (see Figs. S8–S10 and Appendix SE in the Supporting Material).
In this mixed-type Mg2+ inhibition scheme for the Ca2+ uniporter, we consider binding of two Ca2+ ions and two Mg2+ ions to the uniporter, and the order of Ca2+ and Mg2+ binding does not affect the uniporter conformational states. In addition, the translocation of Ca2+ across the IMM occurs only via the conformational changes of the 2Ca2+-bound uniporter complexes T2Ca2+e and 2Ca2+xT (Fig. 1); other Mg2+ and Ca2+-bound uniporter states do not undergo any conformational changes. However, alternate mechanisms are possible, in which the Mg2+-bound uniporter states can undergo conformational changes, leading to Mg2+ influx via the Ca2+ uniporter. This is not considered here because it not only introduces additional parameter, but also there is lack of experimental evidence and data available on the kinetics of Mg2+ transport via the Ca2+ uniporter. Another salient feature of this uniporter model is that, using a single control parameter γ, the model can display all possible types of Mg2+ inhibition mechanisms (e.g., γ = 1, noncompetitive inhibition; γ = ∞, competitive inhibition; and 1 < γ < ∞, a mixed-type inhibition). Consequently, by analyzing a large number of experimental data sets (8,9,11,12,14–16), the model confirmed that a mixed-type inhibition mechanism is most viable for the operation of the Ca2+ uniporter.
Another important feature of this uniporter model is that it provides a unique and consistent estimates of the kinetic parameters associated with the binding of Ca2+, Mg2+, and Pi to the uniporter (K0C, K0M, KPi, and γ) over a large number of experimental data sets (9,11,14–16). Most importantly, the estimate of Km of Ca2+ for the uniporter (K0C) is found to be much lower compared to that reported in our earlier studies (20,22), and is unique over all the available data sets, irrespective of the amounts of extramatrix Mg2+ and Pi and the tissue types (hearts versus livers). For example, in this uniporter model, K0C is found to be ∼4.14 μM using Model 1 for all the available data sets, whereas Model 1 of our previous uniporter models (20,22) estimated a much higher value of K0C, at ∼44 μM for rat liver mitochondria (14) and 88 μM for rat heart mitochondria (15), due to the presence of different amount of Mg2+ and Pi in the buffer. In this model, this effect is attributed to changes in the binding affinities of Ca2+ for the uniporter with different amount of extramatrix Mg2+ and Pi. Accordingly, Ca2+ uptake reaches saturation at a much lower level of extramatrix Ca2+ (≈30 μM) in the absence of extramatrix Mg2+ compared to our previous uniporter models (20,22), when simulated over the entire ΔΨ-range; in the presence of 2 mM Mg2+, a higher level of extramatrix Ca2+ (≈100 μM) is required for the Ca2+ uptake to attain saturation (see Fig. S4, A and B, of Appendix SC in the Supporting Material).
Though the pathways of mitochondrial Ca2+ transport have been known for last three decades, their functional consequences are becoming increasingly clear in recent years in relation to the cell physiology and pathophysiology. Most importantly, mitochondrial Ca2+ uptake plays a substantial role in shaping cytosolic Ca2+ signals in many cell types and regulating local Ca2+ concentration in cellular microdomains (3,33,34). It is also apparent that mitochondria can considerably alter the kinetics of intracellular Ca2+ oscillations and waves generated by Ca2+ release from the endoplasmic reticulum (35). A decline in mitochondrial Ca2+ uptake can have significant effect on the spatiotemporal characteristics of cellular Ca2+ signaling and mitochondrial energy metabolism.
Considering its significance in tuning Ca2+-mediated cellular responses, it is important to understand each factor that modulates mitochondrial Ca2+ transport. In this regard, Mg2+ is known to be a key regulator of mitochondrial cation and substrate transport systems, and inhibition of mitochondrial Ca2+ uptake by physiological elevations of cytosolic Mg2+ has been well recognized in many experimental studies (8,9,11). However, the consequences of this inhibition are poorly understood at cellular levels and it is largely unknown whether Mg2+ has any potential role in modifying cellular Ca2+ signals. Therefore, this model of Ca2+ uniporter is very useful for building biophysically based integrated models of mitochondrial and intracellular Ca2+ handling, to understand the mechanisms by which Ca2+ plays a critical role in the signal transduction processes and modulates mitochondrial and cellular energy metabolisms.
Acknowledgments
The authors are thankful to the anonymous reviewers for their useful comments that helped in improving the manuscript significantly.
This work was supported by National Institutes of Health grants No. R01-HL095122 (to R.K.D.) and No. R01-HL072011 (to D.A.B.), and American Heart Association grant No. SDG-0735093N (to R.K.D.).
Footnotes
Fenq Qi's present address is Sanford-Burnham Medical Research Institute at Lake Nona, Orlando, FL.
Supporting Material
References
- 1.Gunter T.E., Gunter K.K., Gavin C.E. Mitochondrial calcium transport: physiological and pathological relevance. Am. J. Physiol. 1994;267:C313–C339. doi: 10.1152/ajpcell.1994.267.2.C313. [DOI] [PubMed] [Google Scholar]
- 2.Bernardi P. Mitochondrial transport of cations: channels, exchangers, and permeability transition. Physiol. Rev. 1999;79:1127–1155. doi: 10.1152/physrev.1999.79.4.1127. [DOI] [PubMed] [Google Scholar]
- 3.Duchen M.R. Mitochondria and Ca2+ in cell physiology and pathophysiology. Cell Calcium. 2000;28:339–348. doi: 10.1054/ceca.2000.0170. [DOI] [PubMed] [Google Scholar]
- 4.O'Rourke B., Cortassa S., Aon M.A. Mitochondrial ion channels: gatekeepers of life and death. Physiology (Bethesda) 2005;20:303–315. doi: 10.1152/physiol.00020.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gunter T.E., Sheu S.S. Characteristics and possible functions of mitochondrial Ca2+ transport mechanisms. Biochim. Biophys. Acta. 2009;1787:1291–1308. doi: 10.1016/j.bbabio.2008.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Romani A., Scarpa A. Hormonal control of Mg2+ transport in the heart. Nature. 1990;346:841–844. doi: 10.1038/346841a0. [DOI] [PubMed] [Google Scholar]
- 7.Murphy E., Steenbergen C., London R.E. Cytosolic free magnesium levels in ischemic rat heart. J. Biol. Chem. 1989;264:5622–5627. [PubMed] [Google Scholar]
- 8.Szanda G., Rajki A., Spät A. Effect of cytosolic Mg2+ on mitochondrial Ca2+ signaling. Pflugers Arch. 2009;457:941–954. doi: 10.1007/s00424-008-0551-0. [DOI] [PubMed] [Google Scholar]
- 9.Crompton M., Sigel E., Carafoli E. A kinetic study of the energy-linked influx of Ca2+ into heart mitochondria. Eur. J. Biochem. 1976;69:429–434. [Google Scholar]
- 10.Crompton M., Hediger M., Carafoli E. The effect of inorganic phosphate on calcium influx into rat heart mitochondria. Biochem. Biophys. Res. Commun. 1978;80:540–546. doi: 10.1016/0006-291x(78)91602-9. [DOI] [PubMed] [Google Scholar]
- 11.Bragadin M., Pozzan T., Azzone G.F. Kinetics of Ca2+ carrier in rat liver mitochondria. Biochemistry. 1979;18:5972–5978. doi: 10.1021/bi00593a033. [DOI] [PubMed] [Google Scholar]
- 12.McCormack J.G., Browne H.M., Dawes N.J. Studies on mitochondrial Ca2+-transport and matrix Ca2+ using Fura-2-loaded rat heart mitochondria. Biochim. Biophys. Acta. 1989;973:420–427. doi: 10.1016/s0005-2728(89)80384-6. [DOI] [PubMed] [Google Scholar]
- 13.Wan B., LaNoue K.F., Scaduto R.C., Jr. Regulation of citric acid cycle by calcium. J. Biol. Chem. 1989;264:13430–13439. [PubMed] [Google Scholar]
- 14.Vinogradov A., Scarpa A. The initial velocities of calcium uptake by rat liver mitochondria. J. Biol. Chem. 1973;248:5527–5531. [PubMed] [Google Scholar]
- 15.Scarpa A., Graziotti P. Mechanisms for intracellular calcium regulation in heart. I. Stopped-flow measurements of Ca2+ uptake by cardiac mitochondria. J. Gen. Physiol. 1973;62:756–772. doi: 10.1085/jgp.62.6.756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wingrove D.E., Amatruda J.M., Gunter T.E. Glucagon effects on the membrane potential and calcium uptake rate of rat liver mitochondria. J. Biol. Chem. 1984;259:9390–9394. [PubMed] [Google Scholar]
- 17.Magnus G., Keizer J. Minimal model of β-cell mitochondrial Ca2+ handling. Am. J. Physiol. 1997;273:C717–C733. doi: 10.1152/ajpcell.1997.273.2.C717. [DOI] [PubMed] [Google Scholar]
- 18.Nguyen M.H., Dudycha S.J., Jafri M.S. The effects of Ca2+ on cardiac mitochondrial energy production is modulated by Na+ and H+ dynamics. Am. J. Physiol. Cell Physiol. 2007;292:2004–2020. doi: 10.1152/ajpcell.00271.2006. [DOI] [PubMed] [Google Scholar]
- 19.Dash R.K., Beard D.A. Analysis of cardiac mitochondrial Na+-Ca2+ exchanger kinetics with a biophysical model of mitochondrial Ca2+ handling suggests a 3:1 stoichiometry. J. Physiol. 2008;586:3267–3285. doi: 10.1113/jphysiol.2008.151977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dash R.K., Qi F., Beard D.A. A biophysically based mathematical model for the kinetics of mitochondrial calcium uniporter. Biophys. J. 2009;96:1318–1332. doi: 10.1016/j.bpj.2008.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bazil J.N., Buzzard G.T., Rundell A.E. Modeling mitochondrial bioenergetics with integrated volume dynamics. PLOS Comput. Biol. 2010;6:e1000632. doi: 10.1371/journal.pcbi.1000632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pradhan R.K., Qi F., Dash R.K. Characterization of membrane potential dependency of mitochondrial Ca2+ uptake by an improved biophysical model of mitochondrial Ca2+ uniporter. PLoS ONE. 2010;5:e13278. doi: 10.1371/journal.pone.0013278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bose S., French S., Balaban R.S. Metabolic network control of oxidative phosphorylation: multiple roles of inorganic phosphate. J. Biol. Chem. 2003;278:39155–39165. doi: 10.1074/jbc.M306409200. [DOI] [PubMed] [Google Scholar]
- 24.Wu F., Yang F., Beard D.A. Computer modeling of mitochondrial tricarboxylic acid cycle, oxidative phosphorylation, metabolite transport, and electrophysiology. J. Biol. Chem. 2007;282:24525–24537. doi: 10.1074/jbc.M701024200. [DOI] [PubMed] [Google Scholar]
- 25.Nicholls D.G. Mitochondria and calcium signaling. Cell Calcium. 2005;38:311–317. doi: 10.1016/j.ceca.2005.06.011. [DOI] [PubMed] [Google Scholar]
- 26.Läuger P. Ion transport through pores: a rate-theory analysis. Biochim. Biophys. Acta. 1973;311:423–441. doi: 10.1016/0005-2736(73)90323-4. [DOI] [PubMed] [Google Scholar]
- 27.Läuger P., Neumcke B. Theoretical analysis of ion conductance in lipid bilayer membranes. Membranes. 1973;2:1–59. [PubMed] [Google Scholar]
- 28.Keener J.P., Sneyd J. Springer; New York: 1998. Mathematical Physiology. [Google Scholar]
- 29.Pradhan R.K., Beard D.A., Dash R.K. A biophysically based mathematical model for the kinetics of mitochondrial Na+-Ca2+ antiporter. Biophys. J. 2010;98:218–230. doi: 10.1016/j.bpj.2009.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Beard D.A. A biophysical model of the mitochondrial respiratory system and oxidative phosphorylation. PLOS Comput. Biol. 2005;1:e36. doi: 10.1371/journal.pcbi.0010036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jung D.W., Panzeter E., Brierley G.P. On the relationship between matrix free Mg2+ concentration and total Mg2+ in heart mitochondria. Biochim. Biophys. Acta. 1997;1320:310–320. doi: 10.1016/s0005-2728(97)00036-4. [DOI] [PubMed] [Google Scholar]
- 32.Cox D.A., Matlib M.A. A role for the mitochondrial Na+-Ca2+ exchanger in the regulation of oxidative phosphorylation in isolated heart mitochondria. J. Biol. Chem. 1993;268:938–947. [PubMed] [Google Scholar]
- 33.Brini M. Ca2+ signaling in mitochondria: mechanism and role in physiology and pathology. Cell Calcium. 2003;34:399–405. doi: 10.1016/s0143-4160(03)00145-3. [DOI] [PubMed] [Google Scholar]
- 34.Bers D.M. Calcium cycling and signaling in cardiac myocytes. Annu. Rev. Physiol. 2008;70:23–49. doi: 10.1146/annurev.physiol.70.113006.100455. [DOI] [PubMed] [Google Scholar]
- 35.Rizzuto R., Marchi S., Pinton P. Ca2+ transfer from the ER to mitochondria: when, how and why. Biochim. Biophys. Acta. 2009;1787:1342–1351. doi: 10.1016/j.bbabio.2009.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.