

Abstract
The propensity of α-synuclein to form amyloid plays an important role in Parkinson's disease. Three familial mutations, A30P, E46K, and A53T, correlate with Parkinson's disease. Therefore, unraveling the structural effects of these mutations has basic implications in understanding the molecular basis of the disease. Here, we address this issue through comparing details of the hydration of wild-type α-synuclein and its A53T mutant by a combination of wide-line NMR, differential scanning calorimetry, and molecular dynamics simulations. All three approaches suggest a hydrate shell compatible with a largely disordered state of both proteins. Its fine details, however, are different, with the mutant displaying a somewhat higher level of hydration, suggesting a bias to more open structures, favorable for protein-protein interactions leading to amyloid formation. These differences disappear in the amyloid state, suggesting basically the same surface topology, irrespective of the initial monomeric state.
Introduction
Parkinson's disease (PD)—like Alzheimer's disease (AD)—is a lingering neurodegenerative disease that induces degeneration of substantia nigra in the brain in people typically older than age 60. The loss of dopaminergic neurons causes movement disorders; common symptoms are tremor, rigidity, bradykinesia, and balance problems. PD is characterized by the presence of cytoplasmic neuronal deposits, so-called Lewy bodies (LBs) (1). The major component of LBs is α-synuclein, which forms ordered amyloid fibrils: in fact, α-synuclein immunoreactivity in LBs (2) is the pathological hallmark of PD (3). Besides wild-type protein, α-synuclein also has several inherited, familial mutations associated with PD, such as A53T (4), A30P (4,5), and E46K (6).
α-synuclein is a small, highly soluble, heat-stable protein (7,8) expressed at high levels in the brain, where its normal function is not fully understood (9–11). This 140-amino acid protein consists of three regions, 1), an amphipathic N-terminal region (residues 1–60), 2), the so-called NAC region (residues 61–95), and 3), the C-terminal region enriched in proline and acidic amino acids (residues 96–140). α-synuclein contains seven repeat regions (KTKEGV) (12), within which all familial mutations are found; A30P is located between the second and third repeat, A53T is between the fourth and fifth repeat, and E46K is within the fourth repeat region. Within the two repeats in the NAC region, no mutation has been detected to date that we know of. It was shown previously that mutation of E46 to alanine or lysine in position 46 and 83 increases the propensity of α-synuclein to polymerize (13) and glutamic acid residues within the repeats can have significant effects on modulating the assembly of α-synuclein fibrils.
The polymerization of wild-type and mutant α-synuclein is time- and concentration-dependent (14). The primary determinant of polymerization is the NAC region, whereas the two termini modulate filament formation (13). The wild-type protein and its mutants show different characteristics in many aspects, such as the rate of fibril formation and morphology (13–20), which underscores that these mutants are important in the development of PD. In vitro, A53T forms fibrils the fastest (19), A30P and E46K are somewhat slower, and wild-type is the slowest of all (13,18,20). The reasons for these observed differences are not clear, but they are probably related to the effect of mutations on both the solution structure of the protein and the interresidue interactions critical for amyloid formation. The latter is probably also manifested in distinct fibril morphology; wild-type fibrils form twisted filaments with a diameter of 10–15 nm, A30P mutants form straight filaments with a diameter of 11–16 nm, and A53T mutants form larger twisted filaments with a diameter of 16–19 nm (14). All filaments observed by circular dichroism (CD) and Fourier transform infrared (FTIR) spectroscopy show an antiparallel β-sheet character in all cases (16,17).
To elucidate the underlying structural aspects, the structure of monomeric α-synuclein was analyzed in terms of both global and local structural behavior. By means of a variety of techniques, the full-length protein is best described as a random coil in solution (8). Its hydrodynamic behavior has been studied by size-exclusion chromatography: based on an increased retention time, a slight compaction by E46K mutation was suggested (21). In small-angle x-ray scattering experiments, no significant difference in radius of gyration was noted between A30P and A53T mutants and the wild-type protein (19). In studies using paramagnetic relaxation enhancement, transient long-range interactions in the α-synuclein ensemble between the C-terminus and the central region of the protein were observed (22,23). These results suggest that fibril formation is inhibited by intramolecular interactions in the native structure, and that these interactions are greatly destabilized by A30P and A53T mutations (24), shifting the conformational ensemble of α-synuclein to a more open state that prevails under conditions that promote fibril formation (22).
The structure of wild-type and mutant monomeric α-synuclein at the secondary-structure level was mostly studied by CD and NMR. NMR spectroscopy has shown that the amino terminal region prefers α-helical structure (25), which is strongly stabilized by binding of lipid micelles (7,26). The C-terminal region shows an unfolded structure. Observations using CD indicate that the helical content overall does not change upon mutation, with only subtle differences apparent between the wild-type and E46K mutant protein (21). NMR has shown that the local helical structure is disturbed by the A30P mutation, and an enhanced local extended structure is seen in the case of A53T (27). An AFM experiment addressing both global and local structural characteristics suggested that the primarily random-coil structure of α-synucelin shows a tendency toward β-type behavior in only 7% of the cases in the wild-type protein, but that this tendency increases to ∼38% upon A30P mutation (28).
In this article, we present results on the wild-type and A53T variants and on solutions containing no buffer agent or added salt. In all, these experiments suggest that familial mutations cause small but significant local and global perturbations in the structural ensemble of α-synuclein, which are probably relevant with respect to their enhanced amyloidogenic potential. Our study adds a new dimension to the structural interpretation of amyloid formation by close inspection of the hydration properties of wild-type α-synuclein and the A53T mutant using a combination of wide-line NMR and differential scanning calorimetry (DSC).
Materials and Methods
Purification and sample preparation of recombinant α-synuclein
Expression and purification of recombinant human wild-type and A53T mutant α-synuclein in a pRK-172-based expression system was performed as described (29). In sample preparation, the mass of lyophilized protein (without any further refinement) was measured and an appropriate amount of water was added to obtain the requested nominal concentrations, 50 mg/ml and 25 mg/ml. In NMR and DSC measurements, double-distilled water was used as a solvent. Both NMR and DSC measurements were carried out on three identical samples prepared independently. Amyloid was prepared by preincubating protein samples at 37°C for 24 h. Formation of amyloid was confirmed by measuring the fluorescence of 5 μM thioflavin T (ThT) added to an aliquot of the solution.
NMR spectrometry
The portion of mobile proton (water) fraction was directly measured by two 1H NMR methods, recording the free induction decay (FID) signal and Carr-Purcell-Meiboom-Gill (CPMG)-echo trains (in the temperature range where echoes could be detected at all). In some detail, the following pulse sequences have been applied for the FID signal (FID(t), i.e., the inversely Fourier-transformed spectrum (ω)) and the echo signals (t), which denote the time and frequency domains.
Response to the preparation pulse (Px′) is the FID signal. P represents a 90° pulse length and x′ the phase of that. All the protons in the sample contribute to the FID signal. The amplitude of this response at time zero is proportional to M0 = n0B0/T, where Bo is the magnetic induction, T is the absolute temperature, and n0 refers to the number of nuclear spins (protons) in the sample. Otherwise, all the following responses are proportional to the number (n0) of the contributing resonant nuclei.
Response to preparation pulses (Px′-τ-2Py′-2τ-2Py′-2τ-2Py′-2τ-…, τ) is the CPMG series. The only protons that contribute to the response signal are those for which the residual dipolar field (which is nearly zero because of the motional narrowing) is smaller than the inhomogeneous contribution to the line widths. In that case, only the mobile protons are detected. For details of our method, see studies by Tompa and colleagues (30,31) and the references therein. The CPMG echo-train also enables measurements of the spin-spin (transversal) relaxation rate (32).
Determination of the mobile water fraction is based on a comparison of the signal intensity or echo amplitude extrapolated to t = 0 with the corresponding values measured at a temperature where the whole sample is in the liquid state. Measurements were started at every temperature only after thermal equilibrium was established, i.e., when the NMR response became time-independent, usually after 10 min. The protein solutions showed no thermal hysteresis or sign of supercooling below the freezing point of bulk water. The data presented were obtained while samples were heating.
The effect of freezing on protein solutions was controlled by comparing NMR parameters obtained before and after a freeze-thaw cycle at temperatures above 0°C. We found that the freeze-thaw cycle caused no observable changes for the samples as far as the measured NMR parameters are concerned. The temperature was controlled by an open-cycle cryostat (Oxford Instruments, Oxfordshire, UK) with an uncertainty of smaller than ±1 K. 1H NMR measurements and data acquisition were accomplished using an SXP 4-100 NMR pulse spectrometer (Bruker, Billerica, MA) at a frequency of 82.6 MHz with a stability of better than ±10−6. This value represents the stability of the electromagnet, without any drift during measurements; the stability of frequency in our system is two orders of magnitude better. The spatial inhomogeneity of the magnetic field was 2 ppm. The data points in the figures are based on spectra recorded by averaging signals to reach a signal/noise ratio of 50. The number of NMR signals averaged was varied to achieve the desired signal quantity for each sample and for unfrozen water quantities. For the determination of the relative number of mobile protons, the sensitivity of the NMR spectrometer upon temperature change was controlled by measuring the length of the π/2 pulse to obtain reliable nuclear magnetization values (30). The extrapolation to zero time was done by fitting a stretched exponential (31). The hydration values are presented in units of g water/g protein.
DSC
Information about the structural and dynamic changes of protein and water as solvent can be deduced from the amount of heat absorbed or emitted by a sample subjected to controlled temperature change. Among calorimetric methods, DSC allows for precise determination of the enthalpy changes from the recorded temperature difference (31), ΔT, between the reference and the sample branches as , where K is the calorimeter constant. For the measurements, a heat-flux-type DSC 2920 cell (TA Instruments, Newcastle, DE) was used (33). The measurements were carried out at a controlled heating rate of 2 K·min−1.
Molecular dynamics simulation of hydration shell
For molecular dynamics (MD) simulations of the hydration shell of α-synuclein, we used 101 α-synuclein structures determined by a combination of paramagnetic relaxation enhancement NMR spectroscopy and ensemble MD simulations (23) as the best structural approximation of the disordered state of α-synuclein. For comparison, 10 monomeric globular structures (of which nine—PDB codes 1e20, 1e9h, 1nb0, 1nun, 1p5v, 1pk6, 1usu, 1xgw, and 1z2f—were comparable in size to α-synuclein, and one—1ndd—was a ubiquitinlike structure) were also selected. If the PDB structure did not contain hydrogen, the AddH tool of UCSF Chimera http://www.cgl.ucsf.edu/chimera/ (a visualization system for exploratory research and analysis (34)) was used to complete the structure. For modeling of the hydration shell, we used the solvateShell function of the sleap program (AmberTools 1.4 http://ambermd.org/) with a shell thickness of 2–8 Å (forcefield, leaprc.ff03.r1; water model, TIP3PBOX). After MD simulation, we used PERL scripts to calculate the number of water molecules in the hydration shell by calculating, for each water molecule, the distance between the water molecule (oxygen atom) and the nearest heavy atom of the protein, and determining the number of water molecules within a given distance range.
Results
In the first step, the hydration-versus-temperature curves for the wild-type and the mutant A53T α-synucleins at 25 mg/ml concentration was measured. The trends of these curves are very similar, but the hydration of the A53T variant shows systematically higher values than the wild-type α-synuclein (Fig. 1). The mobile water phase could be detected at −36 ± 1°C as the lowest temperature for the wild-type solution, and it appears between −43°C and −40°C for the A53T mutant. The hydration curves are essentially identical at a higher protein concentration, 50 mg/ml (Fig. 2). At 25 mg/ml, both the wild-type and the mutant A53T proteins have hydration-versus-temperature curves similar to that of the intrinsically disordered protein ERD10 (cf. (32,35)) and show striking contrast to the hydration curves of the globular proteins ubiquitin and bovine serum albumin (Fig. 3). The disordered tertiary structure results in hydration curves characterized by much steeper slopes and higher melting temperatures than are seen in a globular, i.e., ordered structure. The two-times-higher protein concentration (50 mg/ml vs. 25 mg/ml) resulted in reduced hydration levels and trends (Fig. 2), similar to the hydration curves of globular proteins ubiquitin and bovine serum albumin (Fig. 3). The hydration curves of the 50 mg/ml samples are almost temperature-independent up to −10°C, and the mobile water phase appears at ∼−50°C (wild-type α-synuclein at −53 ± 1°C, A53T mutant between −53°C and −48°C).
Figure 1.
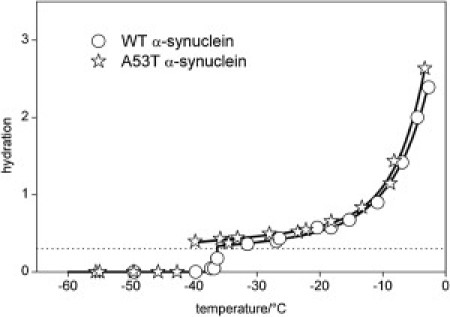
Temperature dependencies of hydration as measured by 1H NMR for 25 mg·cm−3α-synuclein dissolved in pure water of the wild and A53T variants. The dotted line is at the hydration level 0.3 g water/g protein; solid lines are guides to the eye.
Figure 2.
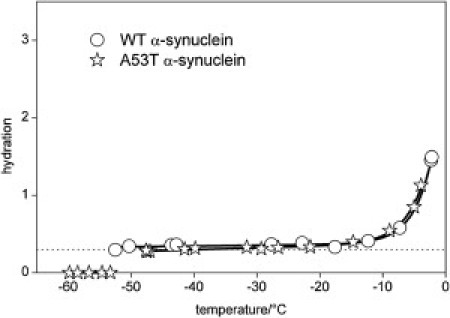
Temperature dependencies of hydration as measured by 1H NMR for 50 mg·cm−3α-synuclein dissolved in pure water of the wild and A53T variants. The dotted line is at the hydration level 0.3 g water/g protein; solid lines are guides to the eye.
Figure 3.
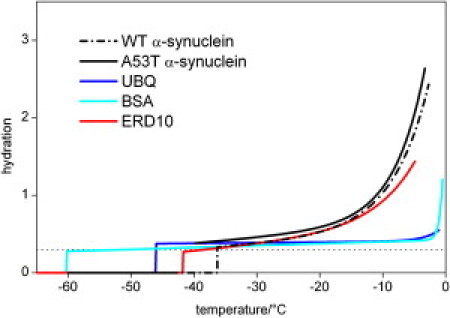
Comparison of the temperature dependencies of hydration as measured by 1H NMR of the 25 mg·cm−3α-synuclein dissolved in pure water (wild and A53T variants) and the globular (ubiquitin and bovine serum albumin) or intrinsically disordered (ERD10) proteins. The measured data points are represented by smooth lines for the sake of clarity. The dotted line is at the hydration level 0.3 g water/g protein.
These results raise the question of how the amyloid fibers behave in this system. The hydration of the amyloid fibrils shows a thermal trend different from those of both the intrinsically disordered and the globular proteins (Fig. 4). The low-temperature detection limits of the mobile water phases are as low as in the case of the globular proteins. There are no temperature-independent regions of hydration. The hydration values are low compared to the other proteins studied. The hydration of the α-synuclein molecules is roughly two times that of the corresponding amyloid fibers (Fig. 4). At temperatures below −20°C, the difference between the mobile water phases around the amyloid fibrils and the corresponding α-synuclein molecules is 1.5–2 times larger for A53T than for the wild-type variant. The enthalpy changes measured by DSC compared to that of water (Fig. 5) show marked differences between wild-type α-synuclein and its amyloid. The enthalpy changes for the amyloid sample are almost identical to those seen for the globular protein ubiquitin.
Figure 4.
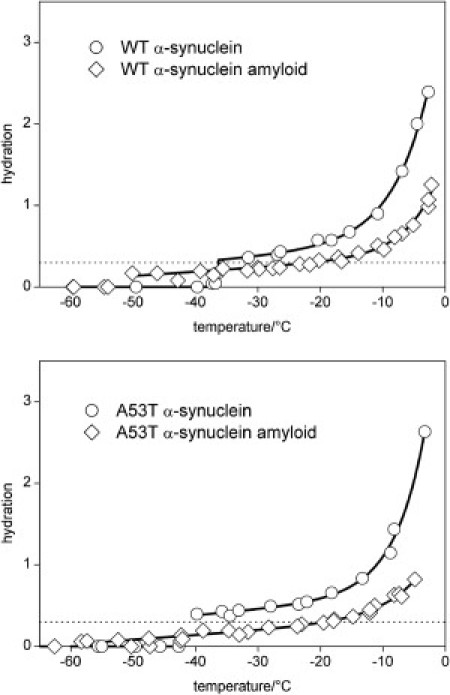
Comparison of the temperature dependencies of hydration as measured by 1H NMR of α-synuclein and its amyloid dissolved in pure water (wild–type (upper) and A53T (lower); protein concentration is 25 mg·cm−3 in each case). The dotted line is at the hydration level 0.3 g water/g protein; solid lines serve as guides to the eye.
Figure 5.
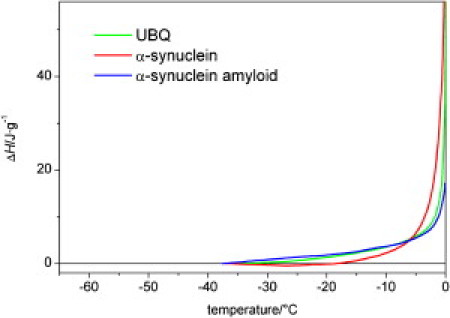
Enthalpy changes for wild-type α-synuclein, its amyloid, and the globular protein ubiquitin dissolved in pure water.
To bridge our hydration measurements with previous structural studies, we investigated the monomeric wild-type α-synuclein and 10 different globular proteins using MD simulations of 101 representative structures of the α-synuclein ensemble (23). These structures were derived by performing NMR measurements on spin-labeled α-synuclein, incorporating the resulting long-range distance constraints into ensemble MD simulations. In our case, MD simulations were used to place water molecules around the 101 structures, which were then analyzed for distribution as a function of distance from the protein surface, as given in Materials and Methods. In short, we used the solvateShell function of the sleap program to place water molecules, calculated their distance from the nearest heavy atom of the protein, and then determined the number of water molecules within a given distance range.
Using this approach, we found the same qualitative and quantitative differences between the hydration shells of ordered and disordered proteins that were reported in earlier experimental work (35) also observed here by NMR at 25 mg/ml. The hydration shell within 5 Å contains exactly the same number of water molecules/residue around both ordered and disordered proteins as in the first hydration shell found by solid-phase NMR (Fig. 6 A). We found earlier in our measurements that the melting point of the first hydration shell is lower in globular proteins (see, e.g., Tompa and co-workers (31,32). We also found with MD simulations that the distance distribution of water molecules from the protein in a 5-Å shell (Fig. 6 B) is significantly different between the globular domains and α-synuclein (unpaired t-test, p = 0.0077). This difference means that the first hydration shell around globular proteins is more compact (more water molecules can be found near to the protein), which may also explain our previous finding (35) related to the melting point of the first hydration shell. We hypothesized that the dominant protein environment of water molecules can lower the melting point of the hydration shell more around globular proteins than around disordered proteins with dominant water environment.
Figure 6.
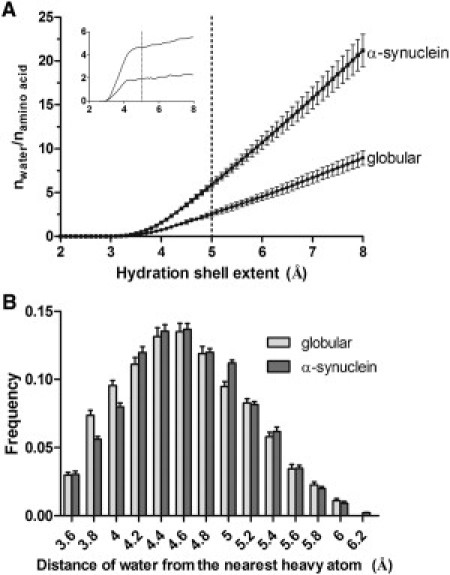
Molecular dynamics simulation of the hydration shell on globular domains and α-synuclein models. (A) Number of water molecules normalized to the number of residues as a function of hydration shell extent. The hydration shell was modeled using the sleap module of AmberTools on 10 selected globular domain and 101 α-synuclein structures (see Methods). The dotted line corresponds to the first hydration shell water number measured by NMR. (Inset) The first derivates of the curves. (B) Frequency distribution of the distance of water molecules (oxygen atom) from the nearest protein heavy atom in the 5-Å-extent hydration shell.
Discussion
Previous structural studies on α-synuclein (19,22,23) have addressed structural details of the largely disordered protein that are of importance for amyloid formation in PD. In these studies, it has always been implied that an important readout of observed structural differences is the level and heterogeneity of hydration of the protein. Because familial mutants of α-synuclein, which show subtle differences in structure from the wild-type protein, are more prone to aggregation (18,20,24), we expected that these differences might also be manifested in hydration, which is thus critical in amyloidogenic propensity of the protein. In this article, we demonstrate that wide-line NMR combined with DSC and MD can provide insights about such details of protein hydration.
We found that the α-synuclein and other proteins studied have an overall hydration value close or equal to 0.3 g water/g protein, typical for proteins in general (36). Furthermore, the hydration curves have features characteristic of intrinsically disordered proteins (Fig. 3). The thermal trends of the hydration curves for wild-type protein and A53T mutant are very similar at the lower protein concentration, 25 mg/ml (Fig. 1). It is important to note that we could detect a small but systematic difference between the A53T variant and the wild-type protein in the actual amounts of the mobile water phase, as well as in the lowest temperature limits where mobile water phases could be detected (Fig. 1). This clearly suggests a bias for more open structures, with the potential of binding more water molecules in the case of the mutant. In principle, this observation agrees with prior observations that in the wild-type protein there is a transient α-helical structure (27) and long-range interactions (23), which are partially released by the A53T mutation and thus give way to more extended structures (22,27). The compaction that results from the underlying intramolecular interactions may have a role in keeping the aggregation-prone NAC region away from unwanted intermolecular interactions, as suggested recently for intrinsically disordered proteins in general (37).We have demonstrated here that such a structural transition is manifested in an increased level of hydration.
The similarity of the hydration-versus-temperature curves for wild-type and A53T mutant α-synuclein at the higher concentration, 50 mg/ml (Fig. 2), probably signals the tendency of the protein to undergo amyloid formation, which brings it to a uniform structural state irrespective of the initial structural ensemble. The same conclusion was previously drawn from observations made by a range of structural techniques (18,19). It is also worthy of note that these hydration curves resemble that of globular proteins but retain some characteristics of disordered proteins (Fig. 2), i.e., their thermal behavior is different from both the intrinsically disordered and globular proteins. That is, the amyloid fibrils are surrounded by a lower amount of mobile water phase than monomeric proteins (Figs. 3 and 4) and exhibit a heterogeneous solvent-accessible surface in terms of protein-water interactions. This behavior suggests that even in the highly ordered aggregated/amyloid state α-synuclein has some residual structural disorder. Such structural disorder in amyloid has already been demonstrated by different techniques, such as hydrogen-deuterium exchange (38), solid-state NMR (39), and site-directed spin-labeling (40) for α-synuclein, as well as for other proteins (41,42). This phenomenon is an extension of fuzziness, i.e., structural disorder of intrinsically disordered proteins in the bound state (43). The enthalpy changes measured relative to water also differentiate between the monomeric protein and the amyloid fibril samples, but do not show much difference between the amyloid fibril and the globular protein ubiquitin (Fig. 5). The quantitative relevance of all these observations is underlined by MD simulations, which show that the measured behavior corresponds to the first hydration layer of the structural ensemble of the protein. Thus, the evolution with temperature of the hydrate layer of α-synuclein observed using our combination of techniques corresponds to that observed by solution NMR (23).
In this sense, our combination of techniques is appropriate to provide qualitative and quantitative information on water molecules in contact with the protein molecule. Because the formation of amyloid results from changes in the fine balance of intermolecular and intramolecular interactions of proteins, both of which critically depend on the presence, binding, and dynamics of water molecules in the hydration shell, we suggest that a detailed description of hydration is a good proxy for addressing the amyloidogenic character of proteins. Our approach sets a precedent for such studies, and suggests a potential way to address the deleterious molecular events that occur in structural transitions of α-synuclein in Parkinson's disease.
Acknowledgments
We are grateful to Prof. Christopher Dobson (Department of Chemistry, University of Cambridge) for providing the PDB files of the 101 different α-synuclein structures describing the disordered ensemble.
The authors acknowledge support from the Hungarian Academy of Sciences. This work was also supported by a Korean-Hungarian Joint Laboratory grant from Korea Research Council of Fundamental Science and Technology (KRCF), and by an FP7 Marie Curie Initial Training Network grant (No. 264257, IDPbyNMR) and an FP7 Infrastructures grant (No. 261863, BioNMR) from the European Commission.
References
- 1.Galvin J.E., Lee V.M., Trojanowski J.Q. Pathobiology of the Lewy body. Adv. Neurol. 1999;80:313–324. [PubMed] [Google Scholar]
- 2.Spillantini M.G., Schmidt M.L., Goedert M. α-Synuclein in Lewy bodies. Nature. 1997;388:839–840. doi: 10.1038/42166. [DOI] [PubMed] [Google Scholar]
- 3.Forno L.S. Neuropathology of Parkinson's disease. J. Neuropathol. Exp. Neurol. 1996;55:259–272. doi: 10.1097/00005072-199603000-00001. [DOI] [PubMed] [Google Scholar]
- 4.Polymeropoulos M.H., Lavedan C., Nussbaum R.L. Mutation in the α-synuclein gene identified in families with Parkinson's disease. Science. 1997;276:2045–2047. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- 5.Krüger R., Kuhn W., Riess O. Ala30Pro mutation in the gene encoding α-synuclein in Parkinson's disease. Nat. Genet. 1998;18:106–108. doi: 10.1038/ng0298-106. [DOI] [PubMed] [Google Scholar]
- 6.Zarranz J.J., Alegre J., de Yebenes J.G. The new mutation, E46K, of α-synuclein causes Parkinson and Lewy body dementia. Ann. Neurol. 2004;55:164–173. doi: 10.1002/ana.10795. [DOI] [PubMed] [Google Scholar]
- 7.Davidson W.S., Jonas A., George J.M. Stabilization of α-synuclein secondary structure upon binding to synthetic membranes. J. Biol. Chem. 1998;273:9443–9449. doi: 10.1074/jbc.273.16.9443. [DOI] [PubMed] [Google Scholar]
- 8.Weinreb P.H., Zhen W., Lansbury P.T., Jr. NACP, a protein implicated in Alzheimer's disease and learning, is natively unfolded. Biochemistry. 1996;35:13709–13715. doi: 10.1021/bi961799n. [DOI] [PubMed] [Google Scholar]
- 9.Iwai A., Masliah E., Saitoh T. The precursor protein of non-Aβ component of Alzheimer's disease amyloid is a presynaptic protein of the central nervous system. Neuron. 1995;14:467–475. doi: 10.1016/0896-6273(95)90302-x. [DOI] [PubMed] [Google Scholar]
- 10.Jakes R., Spillantini M.G., Goedert M. Identification of two distinct synucleins from human brain. FEBS Lett. 1994;345:27–32. doi: 10.1016/0014-5793(94)00395-5. [DOI] [PubMed] [Google Scholar]
- 11.Maroteaux L., Campanelli J.T., Scheller R.H. Synuclein: a neuron-specific protein localized to the nucleus and presynaptic nerve terminal. J. Neurosci. 1988;8:2804–2815. doi: 10.1523/JNEUROSCI.08-08-02804.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Uéda K., Fukushima H., Saitoh T. Molecular cloning of cDNA encoding an unrecognized component of amyloid in Alzheimer disease. Proc. Natl. Acad. Sci. USA. 1993;90:11282–11286. doi: 10.1073/pnas.90.23.11282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Greenbaum E.A., Graves C.L., Giasson B.I. The E46K mutation in alpha-synuclein increases amyloid fibril formation. J. Biol. Chem. 2005;280:7800–7807. doi: 10.1074/jbc.M411638200. [DOI] [PubMed] [Google Scholar]
- 14.Giasson B.I., Uryu K., Lee V.M. Mutant and wild type human α-synucleins assemble into elongated filaments with distinct morphologies in vitro. J. Biol. Chem. 1999;274:7619–7622. doi: 10.1074/jbc.274.12.7619. [DOI] [PubMed] [Google Scholar]
- 15.Conway K.A., Harper J.D., Lansbury P.T. Accelerated in vitro fibril formation by a mutant α-synuclein linked to early-onset Parkinson disease. Nat. Med. 1998;4:1318–1320. doi: 10.1038/3311. [DOI] [PubMed] [Google Scholar]
- 16.Conway K.A., Harper J.D., Lansbury P.T., Jr. Fibrils formed in vitro from α-synuclein and two mutant forms linked to Parkinson's disease are typical amyloid. Biochemistry. 2000;39:2552–2563. doi: 10.1021/bi991447r. [DOI] [PubMed] [Google Scholar]
- 17.El-Agnaf O.M., Jakes R., Wallace A. Effects of the mutations Ala30 to Pro and Ala53 to Thr on the physical and morphological properties of α-synuclein protein implicated in Parkinson's disease. FEBS Lett. 1998;440:67–70. doi: 10.1016/s0014-5793(98)01419-7. [DOI] [PubMed] [Google Scholar]
- 18.Li J., Uversky V.N., Fink A.L. Effect of familial Parkinson's disease point mutations A30P and A53T on the structural properties, aggregation, and fibrillation of human α-synuclein. Biochemistry. 2001;40:11604–11613. doi: 10.1021/bi010616g. [DOI] [PubMed] [Google Scholar]
- 19.Li J., Uversky V.N., Fink A.L. Conformational behavior of human α-synuclein is modulated by familial Parkinson's disease point mutations A30P and A53T. Neurotoxicology. 2002;23:553–567. doi: 10.1016/s0161-813x(02)00066-9. [DOI] [PubMed] [Google Scholar]
- 20.Narhi L., Wood S.J., Citron M. Both familial Parkinson's disease mutations accelerate α-synuclein aggregation. J. Biol. Chem. 1999;274:9843–9846. doi: 10.1074/jbc.274.14.9843. [DOI] [PubMed] [Google Scholar]
- 21.Fredenburg R.A., Rospigliosi C., Lansbury P.T., Jr. The impact of the E46K mutation on the properties of α-synuclein in its monomeric and oligomeric states. Biochemistry. 2007;46:7107–7118. doi: 10.1021/bi7000246. [DOI] [PubMed] [Google Scholar]
- 22.Bertoncini C.W., Jung Y.S., Zweckstetter M. Release of long-range tertiary interactions potentiates aggregation of natively unstructured α-synuclein. Proc. Natl. Acad. Sci. USA. 2005;102:1430–1435. doi: 10.1073/pnas.0407146102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dedmon M.M., Lindorff-Larsen K., Dobson C.M. Mapping long-range interactions in α-synuclein using spin-label NMR and ensemble molecular dynamics simulations. J. Am. Chem. Soc. 2005;127:476–477. doi: 10.1021/ja044834j. [DOI] [PubMed] [Google Scholar]
- 24.Bertoncini C.W., Fernandez C.O., Zweckstetter M. Familial mutants of α-synuclein with increased neurotoxicity have a destabilized conformation. J. Biol. Chem. 2005;280:30649–30652. doi: 10.1074/jbc.C500288200. [DOI] [PubMed] [Google Scholar]
- 25.Eliezer D., Kutluay E., Browne G. Conformational properties of α-synuclein in its free and lipid-associated states. J. Mol. Biol. 2001;307:1061–1073. doi: 10.1006/jmbi.2001.4538. [DOI] [PubMed] [Google Scholar]
- 26.Chandra S., Chen X., Südhof T.C. A broken α-helix in folded α-synuclein. J. Biol. Chem. 2003;278:15313–15318. doi: 10.1074/jbc.M213128200. [DOI] [PubMed] [Google Scholar]
- 27.Bussell R., Jr., Eliezer D. Residual structure and dynamics in Parkinson's disease-associated mutants of α-synuclein. J. Biol. Chem. 2001;276:45996–46003. doi: 10.1074/jbc.M106777200. [DOI] [PubMed] [Google Scholar]
- 28.Sandal M., Valle F., Samorì B. Conformational equilibria in monomeric α-synuclein at the single-molecule level. PLoS Biol. 2008;6:e6. doi: 10.1371/journal.pbio.0060006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.van Raaij M.E., Segers-Nolten I.M., Subramaniam V. Quantitative morphological analysis reveals ultrastructural diversity of amyloid fibrils from α-synuclein mutants. Biophys. J. 2006;91:L96–L98. doi: 10.1529/biophysj.106.090449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tompa K., Bánki P., Vasáros J. Diffusible and residual hydrogen in amorphous Ni(Cu)–Zr–H alloys. J. Alloy. Comp. 2003;350:52–55. [Google Scholar]
- 31.Tompa K., Bánki P., Tompa P. Interfacial water at protein surfaces: wide-line NMR and DSC characterization of hydration in ubiquitin solutions. Biophys. J. 2009;96:2789–2798. doi: 10.1016/j.bpj.2008.11.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bokor M., Csizmók V., Tompa K. NMR relaxation studies on the hydrate layer of intrinsically unstructured proteins. Biophys. J. 2005;88:2030–2037. doi: 10.1529/biophysj.104.051912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kamasa P., Bokor M., Tompa K. DSC approach for the investigation of mobile water fractions in aqueous solutions of NaCl and Tris buffer. Thermochim. Acta. 2007;464:29–34. [Google Scholar]
- 34.Pettersen E.F., Goddard T.D., Ferrin T.E. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 35.Tompa P., Bánki P., Tompa K. Protein-water and protein-buffer interactions in the aqueous solution of an intrinsically unstructured plant dehydrin: NMR intensity and DSC aspects. Biophys. J. 2006;91:2243–2249. doi: 10.1529/biophysj.106.084723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.García de la Torre J. Hydration from hydrodynamics. General considerations and applications of bead modelling to globular proteins. Biophys. Chem. 2001;93:159–170. doi: 10.1016/s0301-4622(01)00218-6. [DOI] [PubMed] [Google Scholar]
- 37.Uversky V.N. Intrinsically disordered proteins may escape unwanted interactions via functional misfolding. Biochim. Biophys. Acta. 2011;1814:693–712. doi: 10.1016/j.bbapap.2011.03.010. [DOI] [PubMed] [Google Scholar]
- 38.Del Mar C., Greenbaum E.A., Woods V.L., Jr. Structure and properties of α-synuclein and other amyloids determined at the amino acid level. Proc. Natl. Acad. Sci. USA. 2005;102:15477–15482. doi: 10.1073/pnas.0507405102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Heise H., Hoyer W., Baldus M. Molecular-level secondary structure, polymorphism, and dynamics of full-length α-synuclein fibrils studied by solid-state NMR. Proc. Natl. Acad. Sci. USA. 2005;102:15871–15876. doi: 10.1073/pnas.0506109102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen M., Margittai M., Langen R. Investigation of α-synuclein fibril structure by site-directed spin labeling. J. Biol. Chem. 2007;282:24970–24979. doi: 10.1074/jbc.M700368200. [DOI] [PubMed] [Google Scholar]
- 41.Tompa P. Structural disorder in amyloid fibrils: its implication in dynamic interactions of proteins. FEBS J. 2009;276:5406–5415. doi: 10.1111/j.1742-4658.2009.07250.x. [DOI] [PubMed] [Google Scholar]
- 42.Alexa A., Tompa P., Friedrich P. Mutual protection of microtubule-associated protein 2 (MAP2) and cyclic AMP-dependent protein kinase II against μ-calpain. J. Neurosci. Res. 1996;44:438–445. doi: 10.1002/(SICI)1097-4547(19960601)44:5<438::AID-JNR4>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 43.Tompa P., Fuxreiter M. Fuzzy complexes: polymorphism and structural disorder in protein-protein interactions. Trends Biochem. Sci. 2008;33:2–8. doi: 10.1016/j.tibs.2007.10.003. [DOI] [PubMed] [Google Scholar]