

Abstract
Neuromyelitis optica (NMO) is an inflammatory demyelinating disease that predominantly affects the optic nerves and spinal cord. Recombinant antibodies (rAbs) generated from clonally expanded plasma cells in an NMO patient are specific to AQP4 and pathogenic. We screened phage-displayed peptide libraries with these rAbs, and identified 14 high affinity linear and conformational peptides. The linear peptides shared sequence homologies with NMO autoantigen AQP4 on the extracellular surface. Competitive inhibition ELISA and immunocytochemistry demonstrated that these peptides represent epitopes of NMO autoantigen AQP4. Peptide epitopes/mimotopes may have potential uses for disease prognosis, monitoring, and therapy.
Keywords: Neuromyelitis optica, Peptide, Epitopes, Autoantibody, Autoantigen, Demyelination
1. Introduction
Neuromyelitis optica (NMO) is a severely debilitating autoimmune disease that predominantly attacks the optic nerve and spinal cord causing blindness and paralysis. A diagnostic hallmark of NMO is the presence of serum autoantibodies against AQP4 (AQP4-Ab) in most NMO patients. Prominent IgG and complement deposition in NMO lesions, a paucity of T cells, and beneficial effects of B cell depletion and plasma exchange suggest that the humoral response is important in the pathogenesis of NMO (Lucchinetti et al., 2002; Lennon et al., 2005; Kinoshita et al., 2009; Sabater et al., 2009) Furthermore, recent in vivo studies revealed that intracerebral injection of human AQP4-specific autoantibodies and complement produce NMO lesions in mice (Bradl et al., 2009; Saadoun et al., 2010).
In inflammatory and infectious CNS diseases, CD138+ plasma blasts in CSF are the primary source of intrathecal Ig production (Owens et al., 2007; Cepok et al., 2005. 2007). Using fluorescent activated cell sorting (FACS) and single-cell RT-PCR, we demonstrated the presence of clonally expanded plasma cells in NMO CSF with features of a T cell-dependent, antigen-targeted response. Consistent with the pathogenic role of AQP4 autoantibodies, our findings revealed that an intrathecal humoral immune response against AQP4 is evident at the onset of clinical disease. Furthermore, recombinant antibodies (rAbs) generated from clonally expanded plasma cells in NMO CSF are AQP4-specific and immunopathologic (Bennett et al., 2009). Herein, using NMO rAbs, we identified high affinity peptides from phage-displayed random peptide libraries, some of which represent epitopes of AQP4. Identification of high affinity peptides of NMO rAbs may improve diagnostics, and will likely determine the immunopathogenesis of disease. In addition, NMO B cell epitopes provide tools as protective epitopes (inhibitors) to compete for binding to pathogenic AQP4-Ab without triggering the complement cascade.
2. Materials and methods
2.1. Generation of NMO recombinant antibody
CD138 + plasma cell sorting and IgG heavy and light chain variable sequence amplification were performed as described (Ritchie et al., 2004; Bennett et al., 2009). All rAbs were constructed and generated as reported (Bennett et al., 2009).
2.2. Biopanning, phage titration and amplification
PhD.-12™ and PhD C7C Phage Display Peptide Libraries (New England BioLabs, Beverly, MA) were used for affinity selection of specific peptides. Panning was as described (Yu et al., 2006a) except that NMO rAb at a concentration of 10 μg/ml was coated to wells of Reacti-Bind™ Protein A-coated clear strip plates (Thermo Scientific, Rockford, IL), and antibodies in 50 μl Tris-buffed saline (TBS) were incubated overnight at 4 °C. Phage peptide libraries (1.5 × 1010 pfu) in 100 μl of TBST (TBS-0.5% Tween 20) were added to the wells and incubated overnight at 4 °C. Elution of bound phage was performed at 37 °C for 10 min (Yu et al., 2009). All NMO rAbs were panned against the phage libraries three times. If no positive phage were selected, two additional rounds of panning were conducted as described (Yu et al., 2009). Affinity-selected phage were titered after each round of panning. Phage titering and amplification were as described (Yu et al., 2006a).
2.3. ELISA
Unless specified, all ELISA were performed at room temperature.
2.3.1. Primary 96-well ELISA
Primary single-point 96-well ELISA was carried out as reported (Yu et al., 2006b). Individual plaques (12–24 plaques panned by each antibody) from the titration of each pan (except the first pan) were picked and placed into wells of U96 DeepWell™ plates (Agene Nunc International, Rochester, NY) containing 500 μl of a 1:10 dilution of OD 1 or 1:100 dilution of overnight culture of E coli ER 2738 in LB medium. Plates were covered with breathable sealing tape (NUNC). After 4.5 h of infection at 37 °C at 250 rpm, plates were centrifuged at 4000 rpm for 10 min at 4°C in a Sorvall RT 6000 refrigerated centrifuge. Phage supernatants were transferred to a new DeepWell™ plate and stored at 4 °C
For primary fast screening of potential positive phage, a microtiter plate (Corning) was coated with 50 μl (1 μg/ml) of antibody in 0.1 M carbonate buffer, pH 9.5, blocked with 3% BSA for 1 h and washed with TBST three times. Phage supernatants (50 μl) from the DeepWell™ plate were added directly to individual wells and incubated for 1 h with shaking at room temperature. Plates were washed with TBST 10 times for 1 min each time, followed by incubation with 50 μl of 1:500 dilution of horseradish peroxidase HRP/anti-M13 antibody conjugate (GE Healthcare) in 1% BSA/Tween 20 for 1 h. HRP/anti-M13 monoclonal antibody reacts specifically with the bacteriophage M13 major coat protein VIII. Plates were washed eight times with TBST, and bound phage were detected using HRP substrate ABTS (Vector Laboratories, Burlingame, CA). After incubation with substrate for 20–30 min, absorbance at 405 nm was determined spectrophotometrically with a Microplate Reader (Bio-Rad, Hercules, CA). All phage were screened in single point. BSA (3%)-coated wells served as negative controls.
2.3.2 Phage peptide ELISA
To confirm positive phage results from single-point 96-well ELISA, analyses were repeated with normalized amounts of phage. NMO rAb (50 μl, 1 μg/ml) and irrelevant antibody as negative control were coated in duplicate in wells of an ELISA plate overnight at 4 °C Wells were blocked with 3% BSA for 1 h and 5×109 selected phage were added to each well. The phage detection procedure was the same as described above. ELISA was repeated at least once. Error bars represent standard deviation.
2.3.3. Dose-response and competitive Inhibition ELISA
For dose–response ELISA, serial 2- or 4-fold phage dilutions were added to wells coated with respective panning rAb and an isotype control rAb as negative control. Phage detection was performed as described above. For inhibition ELISA, rAbs (10 μg/ml) were pre-incubated with corresponding serial two-fold phage dilutions in 50 μl of 1% BSA-TBST and with irrelevant phage for 30 min at room temperature. The mixture was added to wells of ELISA plates containing the same phage bound to anti-M13 antibody coated the night before at 4 °C. The inhibited rAb was then detected with a 1:500 dilution of anti-Flag antibody (HRP-conjugated goat anti-ECS, Bethyl Laboratories, Montgomery, TX) followed by ABTS color development as described above.
2.3.4. Peptide inhibition assay
All peptides were synthesized by EZBiolab (http://www.ezbiolab.com). Each peptide contained the desired number of amino acids followed by a C terminal peptide sequence GGGC, which is the spacer between the peptide and the phage minor coat protein pIII (Table 1). The peptides were dissolved in dimethylformamide (final concentration 10 μg/ml) and stored at −80 °C. For peptide inhibition ELISA, serial two-fold dilutions of synthetic peptides were incubated with NMO rAb 58 (10 μg/ml) at room temperature for 30 min. Peptide-rAb mixtures were added to wells pre-coated with phage 58-C8. The detection of bound rAb was the same as described above.
Table 1.
Synthetic peptides used for inhibition assays.
| Name | Peptide sequence |
|---|---|
| #58-C8 (16 aa) | NTLWML(1–6)QRANYAGGGC |
| AQP4-C8(19 aa) | QAFWKAVTAEFLAML(32–46)GGGC |
| AQP4-loop A (15 aa) | GGTEKPLPVDM(60–70)GGGC |
2.4. Immunoblots
NuPAGE Bis-Tris Mini Gels (Invitrogen, Carlsbad, CA) were used for phage SDS-PAGE analysis with 1X MOPS SDS Running Buffer. Phage peptides (1 × 1010/well) in TBS were denatured and reduced by incubation with 1 × protein sample buffer containing β-mercaptoethanol (Pierce Biotechnology, Rockford, IL) at 95 °C for 10 min. Duplicate gels were eletrophoresed for 50 min under a constant voltage of 200 V. The gel was then electroblotted onto a PVDF membrane (Bio-Rad) for 60 min under constant voltage of 15 V using Trans-Blot® Semi-Dry Cell (Bio-Rad). After blocking with 1× casein/TBS (Vector Laboratories) containing 0.1% Tween 20 for 1 h, the filter was incubated with corresponding rAb at 1 μg/ml for 1 h in 1 × casein/TBS buffer with 0.1 % Tween 20. A 1:5000 dilution of peroxidase-conjugated goat anti-human IgG (H + L) (Sigma, St. Louis, MO) was used to detect rAb binding. After incubation with each antibody, membranes were washed once with TBST for 15 min, then four more times for 5 min each. The membrane was incubated with SuperSignal® West Femto Maximum Sensitivity chemiluminescent substrate (Pierce) as recommended by the manufacturer. For detection of phage pill protein, duplicate filters were incubated with a 1:25,000 dilution of anti-M13 pill monoclonal antibody (New England BioLabs). Anti-M13 pill monoclonal antibody recognizes nonreduced and denatured reduced forms of bacteriophage M13 minor coat protein pill or pill fusions with peptides. A 1:25,000 dilution of goat anti-mouse IgG peroxidase conjugate (Vector lab) was used as secondary antibody followed by chemiluminescent detection with Immobilon (Millipore).
2.5. Immunocytochemistry
To further investigate whether AQP4 peptides compete with native epitopes of AQP4 for NMO Ab binding, we performed indirect immunocytochemistry. AQP4 expressing Fischer rat thyroid (FRT) epithelial cells grown on cover slips were stained with NMO rAb 58 (50μg/ml) pre-incubated with phage peptide 58-C8 (3.5 × 1010), followed by incubation of secondary antibody Alexa 488 tagged goat anti-human antibody (10 μg/ml). Cells were fixed and counterstained with DAPI.
2.6. Sequence alignment
Single-stranded phage DNA was purified and sequenced to deduce amino acid sequence of the peptide. Consensus peptides were identified by sequence alignment using ClustalW (http://www.ebijc.uk/clustalw/). Phage peptides with the highest affinity for NMO rAbs were aligned to the human AQP4 M1 isoform (NP001641.1).
3. Results
3.1. Positive phage peptides that reacted with NMO recombinant antibodies were identified by panning phage displayed random peptide libraries
A total of 11 NMO rAbs were generated by co-expressing the paired IgG heavy and light-chain variable (V) region sequences of clonally expanded CD138+ plasma cells in mammalian tissue culture cells (Yu et al., 2006a; Bennett et al., 2009). The NMO rAbs were then studied for epitope specificity. A streamlined protocol was used to determine phage peptide specificity after affinity selection. Briefly, 12–24 individual phage plaques from each pan (except the first pan) were amplified in U96 deep well plates, and their reactivity to panning antibody was determined by single point ELISA (96-well ELISA) using crude phage culture without further purification (Yu et al., 2006b). Potential positive clones were confirmed by duplicate ELISA with IgG1 isotype control rAb or preimmune human IgG control. DNA from only positive phage clones were purified and sequenced. Each selected phage was named by its panning rAb followed by the location of the well from which the phage was initially identified (Yu et al., 2010).
NMO rAb 58 identified three unique peptides after three rounds of panning at 4 °C incubation; rAb 31 did not select any linear peptides at either temperature, but did select seven circular peptides at 4 °C Circular peptides were selected from Ph.D.-C7C circular library. The peptide is flanked by a pair of cysteine residues, which are oxidized during phage assembly to a disulfide linkage, resulting in the displayed peptides being presented to the target as loops. All remaining NMO rAbs were used to select phage peptides only at 4 °C incubation, but failed to identify any positives. For these rAbs, 4th and 5th pans were carried out using a fast panning protocol (Yu et al., 2009), by which NMO rAbs 53 and 168 each selected high affinity peptides (Fig. 1). Peptides selected by the remaining 7 rAbs did not bind to panning rAbs specifically (no high absorbance value) in ELISA (data not shown). High affinity peptides were those with a high absorbance values in ELISA (at least three times higher compared to isotype control rAbs or pre-immune human IgG). Overall, 11 NMO rAbs were applied for phage peptide panning, and four selected high affinity peptides (Fig. 1). Importantly, consensus peptides were isolated with each of the four NMO rAbs.
Fig. 1.
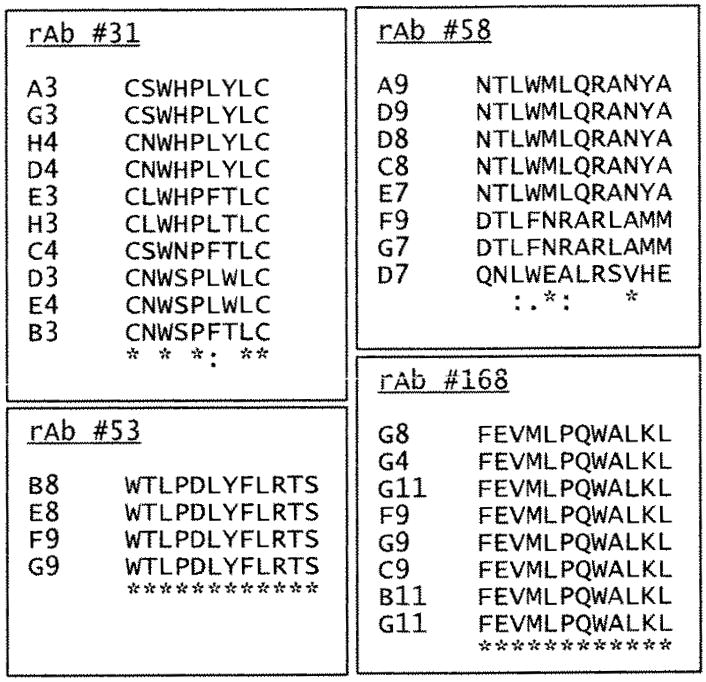
Peptides identified by panning phage-displayed random peptide libraries with 4 NMO rAbs. The peptides are aligned to show consensus sequences. The symbol “*” indicates identical residues. The symbol “:” indicates conserved residues, and the symbol “.” indicates semi-conserved residues. Circular peptides were identified by NMO rAb #31. All remaining peptides were obtained from 12-mer phage libraries.
3.2. NMO rAbs identified high affinity phage peptides
To further characterize affinity-selected peptides, dose-response and competitive inhibition assays were performed. Three representative linear phage peptides selected by rAbs 58, 53 and 168 were used. Each peptide bound to panning rAb, but not to irrelevant rAb in a dose-dependent manner (Fig. 2). To confirm that the selected phage peptides bound specifically to panning rAbs in solution phase, competitive inhibition ELISA assays were used. Phage peptide in solution phase competitively inhibited binding of panning rAb to the same peptide coated in ELISA wells in a dose-dependent manner, while an irrelevant phage peptide did not inhibit binding (Fig. 3). We further compared the binding affinity of peptides to NMO rAbs at 4 °C and 37 °C, and found that peptides panned by rAbs 58 and 53 showed higher binding affinities to their panning rAbs at 4 °C than that at 37 °C, but not to peptides panned with rAbs 31 and 168 (data not shown).
Fig. 2.
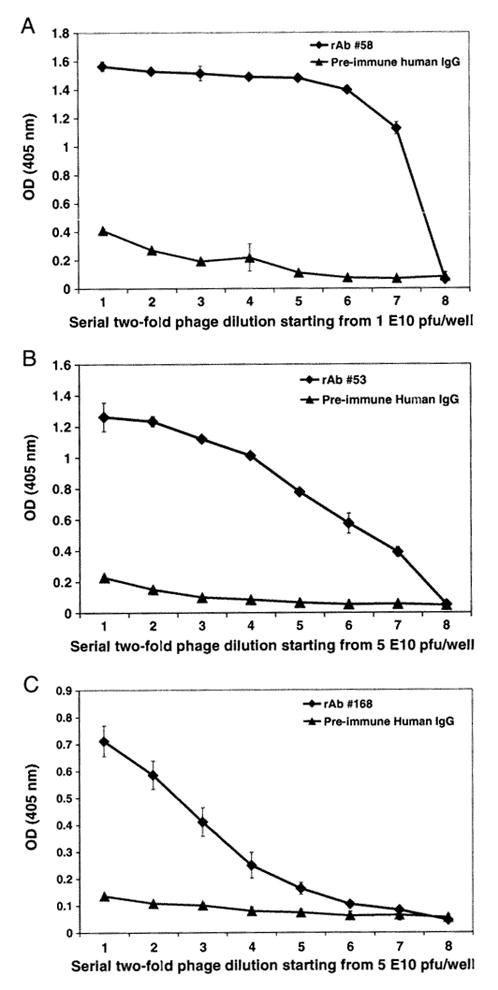
Phage peptides bind to NMO rAbs in a dose-dependent manner. NMO rAbs (1 μg/ml) were coated onto wells of ELISA plates. Serial two-fold dilutions of representative phage peptides were added to wells containing each of the 3 panning rAbs (A: phage 58-C8 to rAb 58; B: phage 53-E8 to rAb 53; C: phage 168-B11 to rAb 168). Bound phage were detected by HRP-conjugated anti-M 13 antibody followed by ABTS color development. Pre-immune human IgG served as negative control. The data represents the mean values of duplicate experiments.
Fig. 3.
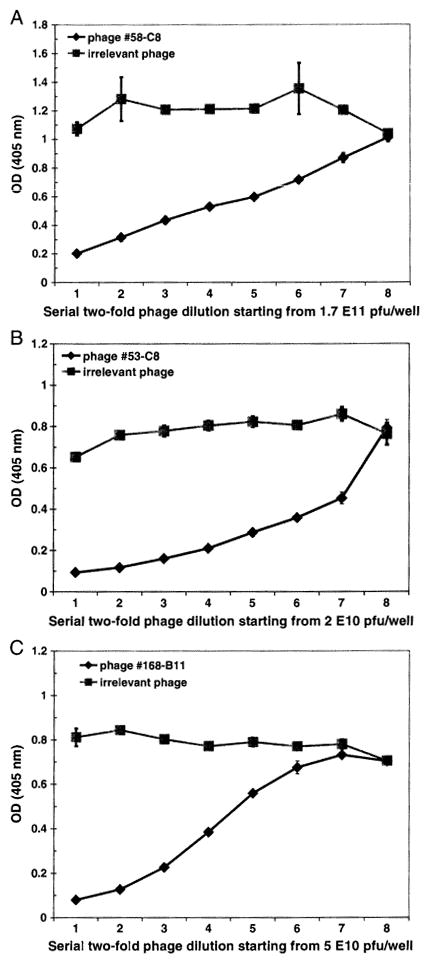
Competitive inhibition ELISA confirms binding specificity of phage peptides. Phage peptides (−1 × 1010) were added to ELISA wells coated with anti M13 antibody, NMO rAb (10 μg/ml) pre-incubated with serial 2-fold dilutions of phage were added to each well. Bound rAb was detected by HRP-conjugated anti-Flag antibody. Binding of phage peptide to NMO rAb in solid phase was inhibited by the same phage in solution in a dose-dependent manner, but not by an irrelevant phage peptide (A: phage 58-C8 to rAb 58; B: phage 53-C8 to rAb 53; C: phage 168-B11 to rAb 168).
3.3. Both linear and conformational epitopes were identified by NMO rAbs
To determine the conformational nature of the phage peptides, we examined their binding specificity and linearity to corresponding NMO rAbs in Western blot under reducing conditions. Both rAbs 58 and 168 bound to the corresponding peptides (64 kD band) that resulted from fusion of the peptide to the pill minor coat protein of M13 phage (Fig. 4A, lanes 1 and 3), while rAb 53 failed to recognize its corresponding peptide (Fig. 4A, lane 2). Western blot results indicated that peptides recognized by rAbs 58 and 168 represent linear epitopes, while peptides selected by rAb 53 corresponded to a conformational epitope that was sensitive to denaturation. The positive control of anti-pill antibody incubated with a duplicate filter showed the minor coat protein of the 64 kD band for all three phage proteins (Fig. 4B).
Fig. 4.
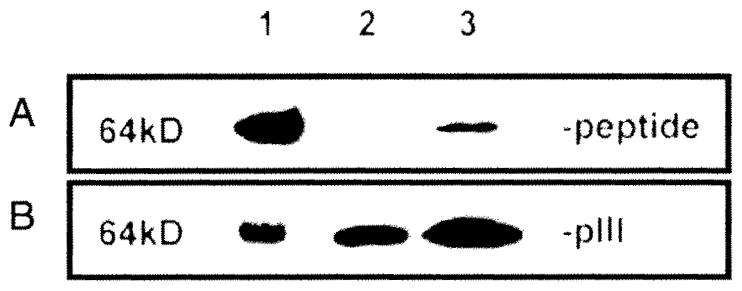
Western blot demonstrates that two NMO rAbs selected linear peptide epitopes. Purified phage peptides (1010 pfu) were separated on a 4–12% gradient SDS-PAGE, blotted to a PVDF membrane and probed with panning NMO rAb at 1 μg/ml. Phage peptide 58-C8 and 168-B11 (at 64 kD) were recognized by corresponding rAb 58 and 168 respectively (A, lanes 1 and 3), indicating that they represent linear epitopes; rAb 53 did not recognize panned phage 53-E8 (lane 2), suggesting that the epitope may be conformational. A duplicate membrane incubated with anti-pill antibody revealed a 64 kD minor coat protein pill with all three phage peptides (B).
3.4. Peptides identified by NMO rAbs represent AQP4 epitopes
We first performed a sequence alignment analysis by comparing each of the three peptides with human AQP4 protein using clustal W (http://www.ebi.ac.uk/Tools/cIustalw). Peptides panned by both rAbs 58 and 53 shared sequence homology with NMO auto-antigen AQP4 sequence in the N-terminal loop A area, and peptides panned by rAb 168 shared sequence homology at a region close to the predicted loop E extracellular area (Fig. 5).
Fig. 5.
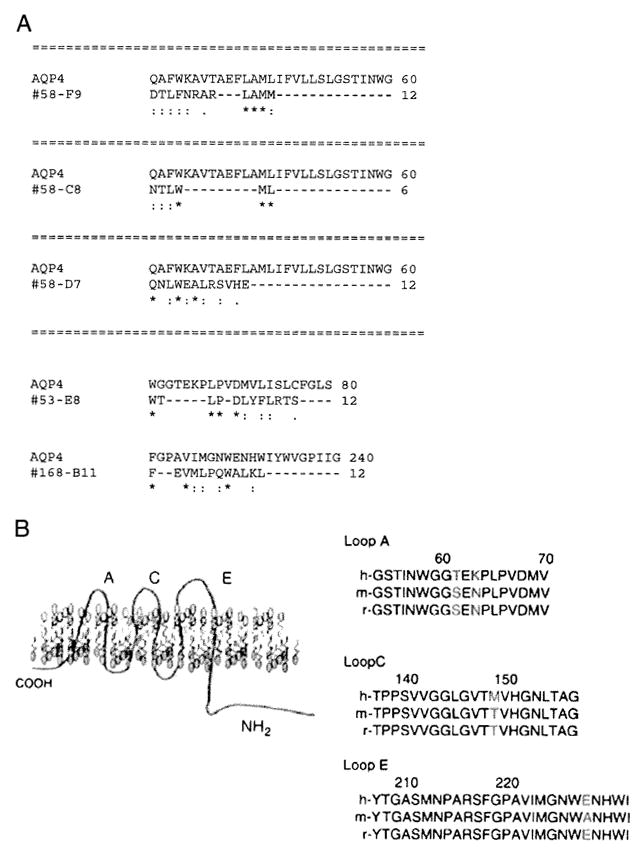
NMO rAbs identified AQP4 epitopes. A. NMO rAb peptides shared sequence homologies with AQP4. Peptides identified by three NMO rAbs were aligned to the AQP4 sequence using Clustal W2. The numbers flanking the sequences indicate the amino acid positions. Symbols “*”, “:” and “.” indicate identical, conserved and semi-conserved residues respectively. Peptides selected by rAbs 53 and 58 aligned to regions dose to the AQP4 epitope Loop A (aa 60–70), whereas peptides by rAb 168 shared sequence homology with epitope Loop E (as 226–230). B. Predicted locations AQP4 epitopes. The positions of amino acid difference Between human, mouse and rat are shown in red (Graber et al., 2008).
3.4.1. NMO rAb phage peptides inhibited binding of AQP4 to NMO rAb in FRT cells
We performed indirect immunocytochemistry using AQP4 expressing FRT epithelial cells. FRT cells stained with rAb 58 pre-absorbed by phage 58-C8 had very low background or no AQP4 staining compared to control cells incubated with rAb 58 alone. Both FRT-AQP4-M1 and FRT-AQP4-M23 cells showed similar staining for rAb 58 and rAb 58 plus phage peptide 58-C8 (Fig. 6).
Fig. 6.
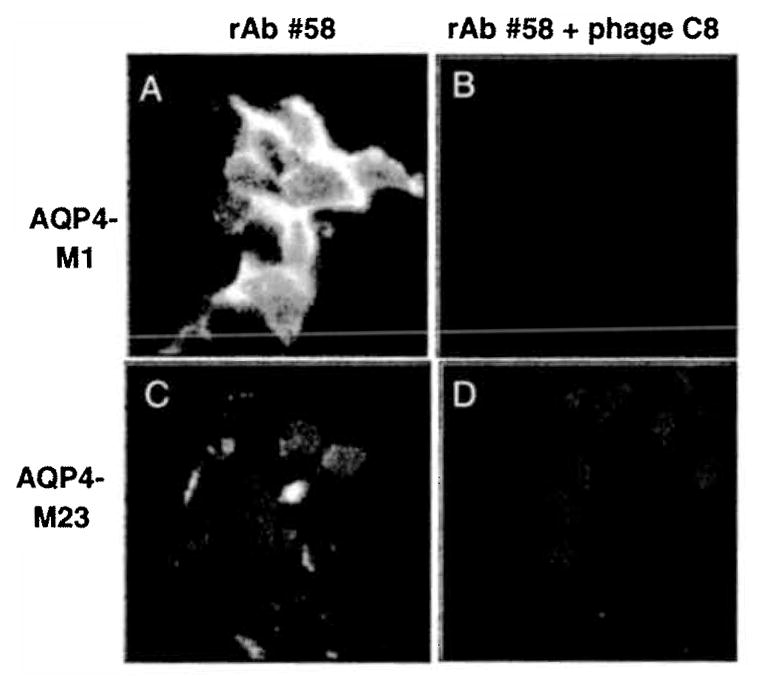
NMO rAb phage peptides inhibited binding of AQP4 to NMO rAb in Fisher rat thyroid cells. FRT epithelial cells transfected with both M1 and M23 AQP4 isoforms were used to study phage inhibition. NMO rAb 58 (50 μg/ml) pre-incubated with phage peptide 58-C8 (3.5 × 1010) (Fig. 1) were added to FRT cells grown on cover slips on ice for 20 min, followed by Alexa 488 tagged goat anti-human antibody (10 μg/ml). Stained cells were fixed by PFA, counterstained with DAPI and examined microscopically. A, FRT-AQJM-M1 stained with rAb #58 only; B, FRT-AQP4-M1 stained with rAb #58 plus phage peptide 58-C8; C, FRT-AQP4-M23 stained with rAb #58 only; D. FRT-AQP4-M23 stained with rAb #58 plus phage peptide 58-C8.
3.4.2. Competitive inhibition ELISA confirms that the NMO rAb peptide competes with AQP4 epitopes for antibody binding
To further investigate whether these peptides functionally compete with native epitopes of AQP4, we obtained commercially synthesized representative peptides containing the desired number of amino acids followed by a C terminal peptide sequence GCCC, which is the spacer between the peptide and the phage pill minor protein (Table 1). We synthesized AQP4-C8 peptide (16-mer) to which phage peptide 58-C8 aligned, as well as peptide sequence of AQP4-loop A as predicted by Graber et al. (2008) (Fig. 7), since our peptide 58-C8 aligned to the region close to loop A of AQP4. Competitive inhibition ELISA revealed that synthetic peptide AQP4-C8 competed with phage peptide 58-C8 for binding to rAb 58 in a dose-dependent manner. Importantly, synthetic peptide AQP4-C8 had greater binding affinity to rAb 58 than the predicted AQP4 epitope Loop A region (Fig. 7).
Fig. 7.
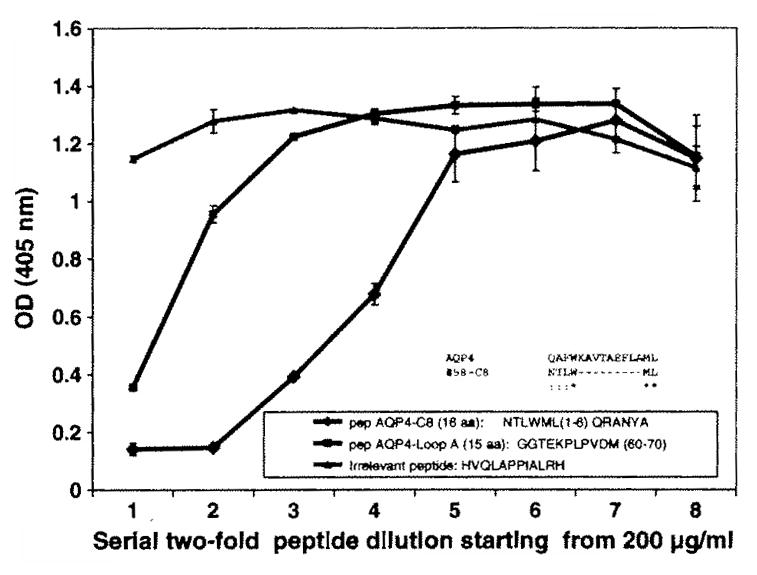
Competitive inhibition EUSA confirms that NMO rAb peptide competes with AQP4 epitopes for antibody binding. Phage peptide 58-C8 (~1 × 1010) were added to EUSA wells pre-coated with anti-M 13 antibody. NMO rAb 58 (10 μg/ml) pre-incubated with serial 2-fold dilutions of synthetic peptides were added to coated phage. Bound rAb was detected by HRP-conjugated anti-Flag antibody. Binding of phage peptide 58-C8 to rAb 58 in the solid phase was inhibited by synthetic peptides AQP4-C8 and AQP4-Loop A in solution, but not by an irrelevant peptide. Peptide AQP4-C8, a 16-mer synthetic peptide of AQP4, shared sequence homology with phage peptide 58-C8; peptide AQP4-Loop A, a 15-mer synthetic peptide of Loop A region, was predicted to be the AQP4 epitope (Graber et al., 2008).
4. Discussion
A diagnostic hallmark of NMO is the presence of autoantibodies directed against AQP4 (AQP4-Ab) in ~70% of patients. About 30% of NMO patients are AQP4-Ab seronegative, and the antigens in these patients are unknown. Identification of high affinity peptides that bind to NMO rAbs will not only improve diagnosis, but also is likely to provide critical information about the immunopathogenesis of disease. In addition, some high affinity peptides that bind to NMO rAbs could potentially be used to inhibit the immunopathogenesis of NMO autoantibody. Finally, peptides identified by rAbs in AQP4-Ab seronegative patients can be used to find novel NMO antigens using a bioinformatics approach.
We previously demonstrated that 6/11 rAbs generated from clonally expanded plasma cells in a patient with NMO were AQP4-specific and mediated both AQP4-directed antibody-dependent cellular toxicity and complement-mediated lysis (Bennett et al., 2009). In this study, using these 11 rAbs to pan phage-displayed peptide libraries, we identified 14 high affinity phage peptides that reacted with four NMO rAbs. Twelve of these 14 peptides were selected by rAbs 58 and 31 with three rounds of traditional panning, and two more peptides were obtained by rAbs 53 and 168 using a combination of three traditional rounds of panning followed by two additional pans using a fast panning protocol developed in our lab (Yu et al., 2009). Peptides selected by the remaining 7 rAbs did not bind specifically to panning rAbs with high affinity (high absorbance value) when evaluated by EUSA (data not shown). The rigor of panning procedures may have generated selection of peptides with higher binding affinities to NMO rAbs.
All 14 peptides were selected by incubation of rAbs with phage-displayed peptides libraries at 4°C. We also found that peptides panned with rAbs 58 and 53 had higher binding affinities at 4 °C than at 37 °C (data not shown), indicating that lower temperatures confer greater stability of molecular interactions. The equilibrium association constant of human IgG1 binding to the Fc receptor is increased at lower temperature (Shopes, 1995), suggesting that temperature dependence is driven largely by enthalpic forces, and that a small but positive entropic contribution to free energy leads to a tighter complex at lower temperature.
B cell epitopes can be linear or conformational. For each affinity selected phage peptide, we determined whether it was linear or conformational. Linear, but not conformational epitopes are recognized by rAbs in Western blotting under denaturing conditions. Our findings revealed that both linear and conformational epitopes of NMO rAbs were identified (Fig. 4). To determine whether the phage peptides represent epitopes of AQP4, sequence alignment analysis, indirect immunocytochemistry and competitive inhibition ELISA were performed. Peptide sequence alignment with AQP4 revealed that peptides selected by rAbs 58, 53 and 168 shared sequence homology with AQP4. Although rAb 168 did not bind to AQP4 transfected cells (Bennett et al., 2009), its peptide shared sequence homology to Loop E, suggesting that it may represent an AQP4 epitope. Furthermore, indirect immunocytochemistry and competitive inhibition ELISA showed that phage peptides panned by rAb 58 functionally competed with epitopes of AQP4. Binding of rAb 58 to FRT cells expressing native AQP4-M1 and M23 was inhibited by peptide 58-C8, revealing that the peptide and native AQP4 share the same conformation. Taken together, phage peptides panned by rAb 58 represent AQP4 epitopes.
In summary, we have demonstrated that panning phage-displayed random peptide libraries with rAbs generated from the CSF of patients with NMO can successfully identify high affinity peptides that represent AQP4 epitopes. These epitopes can be used to study the immunopathogenesis of disease produced by antibodies directed against AQP4, and to determine novel NMO antigens in AQP4-Ab seronegative patients using a bioinformatics approach.
Acknowledgments
This work was supported by Public Health Service grants AG032958 and AG006127 (D.G.). We thank Marina Hoffman for editorial assistance and Cathy Allen for manuscript preparation.
References
- Bennett JL, Lam C, Kalluri SR, Saikali P, Bautista K, Dupree C, Glogowska M, Case D, Antel JP, Owens CP, Gilden D, Nessler S, Stadelmann C, Hemmer B. Intrathecal pathogenicanti-aquaporin-4 antibodies in early neuromyelitis optica Ann. Neurol. 2009;66:617–629. doi: 10.1002/ana.21802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradl M, Misu T, Takahashi T, Watanabe M, Mader S, Reindl M, Adzemovic M, Bauer J, Berger T, Fujihara K, Itoyama Y, Lassmann H. Neuromyelitis optica; pathogenicity of patient immunoglobulin in vivo. Ann Neurol. 2009;66:630–643. doi: 10.1002/ana.21837. [DOI] [PubMed] [Google Scholar]
- Cepok S, Rosche B, Grummel V, Vogel F, Zhou D, Sayn J, Sommer N, Hartung HP, Hemmer B. Short-lived plasma blasts are the main B cell effector subset during the course of multiple sclerosis. Brain. 2005;128:1667–1676. doi: 10.1093/brain/awh486. [DOI] [PubMed] [Google Scholar]
- Cepok S, von Geldern G, Nolting T, Grummel V, Srivastava R, Zhou D, Hartung HP, Adams O, Arendt G, Hemmer B. Viral load determines the B-cell response in the cerebrospinal fluid during human immunodeficiency virus infection. Ann Neurol. 2007;62:458–467. doi: 10.1002/ana.21195. [DOI] [PubMed] [Google Scholar]
- Graber DJ, Levy M, Kerr D, Wade WF. Neuromyelitis optica pathogenesis and aquaporin 4. J Neuroinflammation. 2008;5:22. doi: 10.1186/1742-2094-5-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinoshita M, Nakatsuji Y, Kimura T, Moriya M, Takata K, Okuno T, Kumanogoh A, Kajiyama K, Yoshikawa H, Sakoda S. Neuromyelitis optica: Passive transfer to rats by human immunoglobulin. Biochem Biophys Res Commun. 2009;386:623–627. doi: 10.1016/j.bbrc.2009.06.085. [DOI] [PubMed] [Google Scholar]
- Lennon VA, Kryzer TJ, Pittock SJ, Verkman AS, Hinson SR. IgG marker of optic-spinal multiple sclerosis binds to the aquaporin-4 water channel. J Exp Med. 2005;202:473–477. doi: 10.1084/jem.20050304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucchinetti CF, Mandler RN, McGavern D, Bruck W, Gleich G, Ransohoff RM, Trebst C, Weinshenker B, Wingerchuk D, Parisi JE, Lassmann R. A role for humoral mechanisms in the pathogenesis of Devic’s neuromyelitis optica. Brain. 2002;125:1450–1461. doi: 10.1093/brain/awf151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owens GP, Winges KM, Ritchie AM, Edwards S, Burgoon MP, Lehnhoff L, Nielsen K, Corboy J, Gilden DH, Bennett JL. VH4gene segments dominate the intrathecal humoral immune response in multiple sclerosis. J Immunol. 2007;179:6343–6351. doi: 10.4049/jimmunol.179.9.6343. [DOI] [PubMed] [Google Scholar]
- Ritchie AM, Gilden DH, Williamson RA, Burgoon MP, Yu X, Helm K, Corboy JR, Owens GP. Comparative analysis of the CD19+ and CD138+ cell antibody repertoires in the cerebrospinal fluid of patients with multiple sclerosis, j. Immunol. 2004;173:649–656. doi: 10.4049/jimmunol.173.1.649. [DOI] [PubMed] [Google Scholar]
- Saadoun S, Waters P, Bell BA, Vincent A, Verkman AS, Papadopoulos MC. Intra-cerebral injection of neuromyelitis optica immunoglobulin G and human complement produces neuromyelitis optica lesions in mice. Brain. 2010;133:349–361. doi: 10.1093/brain/awp309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabater L, Giralt A, Boronat A, Hankiewicz K, Blanco Y, Llufriu S, Alberch J, Graus F, Saiz A. Cytotoxic effect of neuromyelitis optica antibody (NMO-IgG) to astrocytes: an in vitro study. J Neuroimmunol. 2009;215:31–35. doi: 10.1016/j.jneuroim.2009.07.014. [DOI] [PubMed] [Google Scholar]
- Shopes B. Temperature-dependent binding of human IgG1 to a human high affinity Fc receptor. Mol Immunol. 1995;32:315–378. doi: 10.1016/0161-5890(94)00155-t. [DOI] [PubMed] [Google Scholar]
- Yu X, Gilden DH, Ritchie AM, Burgoon MP, Keays KM, Owens GP. Specificity of recombinant antibodies generated from multiple sclerosis cerebrospinal fluid probed with a random peptide library. J Neuroimmunol. 2006a;172:121–131. doi: 10.1016/j.jneuroim.2005.11.010. [DOI] [PubMed] [Google Scholar]
- Yu X, Owens GP, Gilden DH. Rapid and efficient identification of epitopes/mimotopes from random peptide libraries. J Immunol Methods. 2006b;316:67–74. doi: 10.1016/j.jim.2006.08.006. [DOI] [PubMed] [Google Scholar]
- Yu X, Barmina O, Burgoon M, Gilden D. Identification of measles virus epitopes using an ultra-fast method of panning phage-displayed random peptide libraries. J Virol Methods. 2009;156:169–173. doi: 10.1016/j.jviromet.2008.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu X, Gilden D, Schambers L, Barmina O, Burgoon M, Bennett JI, Owens GP. Peptide reactivity between multiple sclerosis (MS) CSF IgG and recombinant antibodies generated from clonally expanded plasma cells in MS CSF. J Neuroimmunol. 2010 doi: 10.1016/jjneuroim. 2010.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]