

Abstract
Objective
Myocardin is a cardiac- and smooth muscle-specific transcription factor that potently activates the expression of downstream target genes. Previously, we demonstrated that overexpression of myocardin inhibited the proliferation of smooth muscle cells (SMCs). Recently, myocardin was reported to induce the expression of microRNA-1 (miR-1) in cardiomyocytes. In this study, we investigate whether myocardin induces miR-1 expression to mediate its inhibitory effects on SMC proliferation.
Methods and Results
Using T-REx inducible system expressing myocardin in human vascular SMCs, we found that overexpression of myocardin resulted in significant induction of miR-1 expression and inhibition of SMC proliferation, which was reversed by miR-1 inhibitors. Consistently, introduction of miR-1 into SMCs dramatically inhibited their proliferation. We have isolated spindle-shaped and epithelioid human SMCs and demonstrated that spindle-shaped SMCs were more differentiated and less proliferative. Correspondingly, spindle-shaped SMCs had significantly higher expression levels of both myocardin and miR-1 than epithelioid SMCs. We identified Pim-1, a serine/threonine kinase, as a target gene for miR-1 in SMCs. Western blot and luciferase reporter assays further confirmed that miR-1 targets Pim-1 directly. Furthermore, neointimal lesions of mouse carotid arteries display down-regulation of myocardin and miR-1 with up-regulation of Pim-1.
Conclusion
Our data demonstrate that miR-1 participates myocardin-dependent SMC proliferation inhibition.
Keywords: microRNA-1, myocardin, Pim-1, proliferation, vascular smooth muscle cells
Introduction
During the early embryonic stages of vasculogenesis, smooth muscle cells (SMCs) are highly proliferative and migratory. However, in adult blood vessels, SMCs become quiescent and express a repertoire of contractile proteins necessary for the contractile function of fully differentiated SMCs 1. Interestingly, SMCs, unlike cardiac and skeletal myocytes, are not terminally differentiated, and are capable of regaining their highly proliferative and migratory characteristics under certain conditions such as vascular injury 2. Expression of a majority of SMC marker genes is known to be dependent upon CArG boxes in their promoters/enhancers. Serum response factor (SRF) is essential for the regulation of muscle specific genes through its interaction with the muscle-enriched SRF co-factor myocardin 3. Intriguingly, many SRF cofactors are antagonistic in action, therefore involved in regulating phenotypic switching of SMCs between proliferation and differentiation 1, 4, 5. In addition, heterogeneity of vascular SMCs has been well established in many species with potential pathophysiological significance in vascular system 2, 6. Spindle-shaped human vascular SMCs are more differentiated and less proliferative than epithelioid SMCs 2, 6. We have recently reported that myocardin is able to repress SMC proliferation by functionally antagonizing the NF-κB signaling pathway 7. SMC proliferation is a complicated process regulated by numerous signaling mediators. For instance, Pim-1, an oncogenic serine/threonine kinase, is reported to promote proliferation of cultured SMCs and neointimal hyperplasia 8. However, it remains unknown whether Pim-1 or other signaling molecules could be targeted in myocardin inhibition of SMC proliferation.
Recently, the involvement of microRNAs (miRNAs) in phenotypic modulation of SMCs has been reported. microRNA-21 (miR-21) 9, miR-2410, miR-2511 and miR-221/22212, 13 are reported to modulate SMC proliferation. Importantly, myocardin has been reported to induce the expression of the smooth muscle-enriched miRNA gene, miR-143/14514–18. miR-143/145 promotes contractile phenotype of vascular SMCs through regulating myocardin-dependent expression of SMC differentiation markers and cytoskeletal dynamics14–18. However, whether myocardin induces miRNAs to mediate its anti-proliferative effects in SMC remains largely unknown.
microRNA-1 (miR-1) is a critical mediator of cell proliferation and differentiation in cardiac 19, 20 and skeletal muscles 21. In particular, it was reported that miR-1 was highly enriched in cardiac and skeletal muscle cells as compared with non-striated muscle tissues during animal development and in adults 19–21. miR-1 promotes cardiac and skeletal muscle gene expression and muscle differentiation, in part, by repressing the key transcriptional regulators HDAC421 or hand222. Conversely, it has been shown that tissue-specific expression of miR-1 is dependent on SRF in the heart 22 and MyoD family of transcription factors in skeletal muscle 21, 23.
Given that SRF was necessary for miR-1 expression in myocardium 22 and myocardin is also highly expressed in smooth muscle cells 3, we hypothesize that myocardin induces the expression of miR-1 in vascular SMCs, thereby contributing to myocardin regulation of SMC proliferation and differentiation. In the current study, we report that myocardin induces miR-1 expression in SMCs, and induced miR-1 expression consequently inhibits SMC proliferation. We identify Pim-1 as one of the target genes of miR-1 in SMCs and show decreases in myocardin and miR-1 and increase in Pim-1 in neointimal lesions of mouse carotid arteries. Our studies therefore provide a novel molecular mechanism by which myocardin and miRNAs are involved in the control of SMC proliferation and differentiation.
Materials and Methods
Reagents
RPMI 1640, bromodeoxyuridine (BrdU), propidium iodide (PI) and antibodies for Flag tag and β-actin were obtained from Sigma–Aldrich Canada Ltd (Oakville, Canada). Fetal bovine serum (FBS), 1% penicillin and streptomycin, Alamar Blue reagent and PureLink™ miRNA Isolation Kit were from Invitrogen Canada Inc (Burlington, Canada). RNeasy plus Mini kit, the primer sets of myocardin and QuantiTect SYBR Green PCR Kit were purchased from Qiagen (Mississauga, Canada). TaqMan® MicroRNA Reverse Transcription Kit, the primers of miR-1 and U6B, and universal PCR Master Mix were purchased from Applied Biosystems (Foster City, CA). miR-1 mimic, miRNA negative control, miR-1 inhibitor and miRNA hairpin inhibitor negative control were purchased from Dharmacon (Lafayette, CO). Antibodies for myocardin (M-16) and blocking peptide, Pim-1 (12H8), HDAC4 (A-4) and Ras homolog enriched in brain (Rheb) (C-19) and RIPA buffer were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). GAPDH antibody was from Cell Signaling Technology, Inc. (Danvers, MA).
Cell culture, cloning of spindle-shaped and epithelioid phenotypes and transient transfection
Human aortic SMCs (CRL-1999) were purchased from ATCC. Human aortic SMCs harboring the tetracycline-regulated T-REx™ system (Invitrogen) to express myocardin were established and cultured as previously described 7. Two SMC phenotypes of spindle-shaped and epithelioid SMCs were cloned using a limiting dilution cloning method as previously described24,25. To transfect miRNAs into human aortic SMCs, cells were plated in 96-well plates (4,000 cells per well) for overnight in the presence of 10% serum, followed by transfection with miRNAs using Lipofectamine 2000 (Invitrogen) for 5 h as instructed by the manufacturer. The miRNA transfection efficiency in all different cells was above 95% as monitored by paralleled transfections with Cy3-conjugated negative control siRNA (Invitrogen). Then, cells were cultured in fresh medium for further assays.
Western blot analysis
Western blot analysis was performed as described previously 26. Briefly, protein was extracted from cells with RIPA buffer and equal amounts of protein from each sample (20 μg) were separated by 9% SDS-PAGE and transferred to nitrocellulose membranes. The membranes were then incubated with primary antibodies and appropriate horseradish peroxidase-coupled secondary antibodies. The chemiluminescence signals were detected with ECL Detection kit (GE healthcare, Piscataway, NJ). The densitometric analysis was conducted with ImageMaster (Pharmacia Biotech, Uppsala, Sweden). To detect endogenous myocardin in cultured SMCs, the membranes were pre-blocked with PBS-T (PBS with 0.15% (v/v) Tween-20) containing 5% skim milk overnight at 4 °C, followed by incubation at room temperature for 1 h with myocardin (M-16, sc-21561) antibody (1:200 diluted in PBS-T). After washing, the membranes were subsequently incubated at room temperature for 1 h with horseradish peroxidase-coupled donkey anti-goat secondary antibody (sc-2020, Santa Cruz, 1:4000 diluted in PBS-T containing 0.5% skim milk). The chemiluminescence signals were detected with ECL Plus Western Blotting Detection Reagents (GE healthcare).
Northern blot analysis
Northern blot analysis was performed as described previously 21. Briefly, total RNA was extracted from cells with TRIzol (Invitrogen). 40 μg of each RNA sample (5 μg in skeletal muscle control sample) was processed with polyethylene glycol to remove large-sized RNAs and to facilitate the detection of miRNAs. The miR-1 probe sequence is 5′-TACATACTTCTTTACATTCCA -3′. Results in Supplemental Fig II was exposed overnight, whereas results in Fig. 1C were exposed for 5 days.
Figure 1. Overexpression of myocardin induces miR-1 expression in human SMCs.
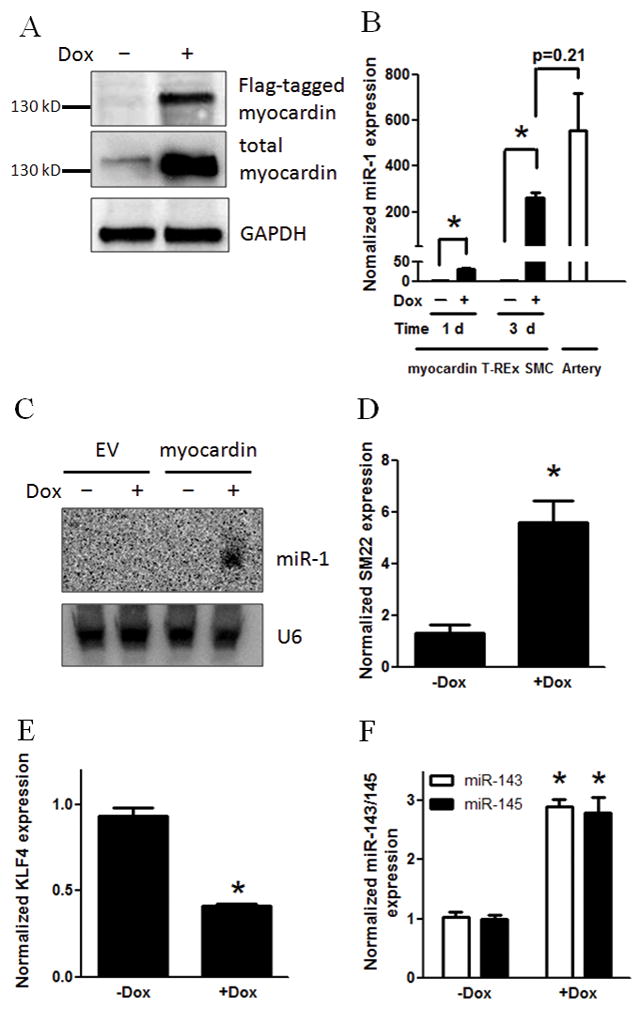
Human vascular SMCs harboring T-REx system for Flag-tagged myocardin expression or an empty vector (EV) were treated with doxycycline (Dox) for 1 d (A, B, D–F) or 3 d (B, C). A: Protein levels of Flag-tagged and total myocardin in myocardin T-REx cells were determined with Flag tag antibody or myocardin M16 antibody, respectively, in Western blot analysis. GAPDH served as a loading control. B: miR-1 levels in myocardin T-REx SMCs with Dox treatment for 1 d or 3 d and in intact mouse carotid artery were determined by quantitative real-time PCR assays. C: miR-1 levels in myocardin and EV T-REx SMCs with Dox treatment for 3 d were determined by Northern blotting assays. U6 serves as a loading control. D–F: SM22 mRNA level (D), KLF4 mRNA level (E) and miR-143/145 level (F) in myocardin T-REx cells with Dox treatment for 1 d were determined by quantitative real-time PCR assays. The y-axis indicates the miRNA and mRNA expression levels normalized by U6B and GAPDH, respectively. * indicates significant difference compared with the control (p < 0.01, n=3).
Cell proliferation assay
Cells were plated in 96-well plates (4,000 cells per well) for overnight and then transfected with miRNAs as described above. After 48 h, Alamar Blue (10% v/v) was added for additional 3 h before fluorescence was measured in triplicates for each sample with a fluorescence plate reader with excitation and emission at 560 and 590 nm, respectively.
Analysis of miRNA expression
miRNAs from human vascular SMCs or mouse arteries were isolated with PureLink miRNA Isolation Kit (Invitrogen). The expression levels of mature miR-1, miR-143, miR-145 and U6B were measured using the Applied Biosystems TaqMan MicroRNA Assays system. Real time PCR was performed in triplicate from each sample using universal PCR Master Mix in the iCycle iQ real-time PCR detection system (Bio-Rad, Mississauga, Canada). iQ Software 3.1 software (Bio-Rad) was used for data analyses. U6B expression was used to normalize the expression of miR-1, miR-143 and miR-145. The 2Δ-Ct method was used to analyze real-time PCR data.
Analysis of mRNA expression
Total RNA was isolated from human vascular SMCs or mouse arteries using the RNeasy plus Mini kit (Qiagen). cDNA was synthesized using iScript cDNA Synthesis Kit (Bio-Rad). The mRNA levels of myocardin, Pim-1, SM22 and GAPDH was evaluated by real-time PCR as previously described (Chen et al., 2008). Briefly, real time PCR was performed in triplicate for each sample using QuantiTect SYBR Green PCR Kit (Qiagen) in the iCycle iQ real-time PCR detection system (Biorad, USA). The GAPDH primer set was obtained from PrimerDesign Ltd (Southampton, UK), and myocardin, Pim-1 and SM22 primer sets from Qiagen. The cycling conditions were: 15 min initial activation step at 95 °C; then 94°C for 15 s, annealing at 55 °C for 30 s, and 72°C for 30 s, repeated for 40 cycles; finally the melting curve analysis was performed. Each assay included a no-template negative control and a no-reverse-transcription negative control.
Luciferase assay
The luciferase reporter plasmid containing human Pim-1 3′UTR sequence is a generous gift of Dr. Kalpana Ghoshal (The Ohio State University, Columbus, OH) 27. The mutation of putative miR-1 seed sequence in Pim-1 3′UTR was conducted using QuikChange Site-directed Mutagenesis kit (Stratagene). The reporter plasmids, together with miR-1 or miRNA negative control, were transfected into cells using Lipofectamine 2000 (Invitrogen), and luciferase activity was measured with the Luciferase assay kit (Promega).
BrdU incorporation assay
BrdU incorporation assay was performed and analyzed as described previously 24. Briefly, cells grown on glass coverslips were labeled with 10 μM BrdU for 60 min. After fixed in 80% and 100% ethanol, cells were permeabilized in 0.25% Triton X-100 in PBS (pH 7.4) for 20 min, incubated with 4N hydrochloric acid for 20 min, neutralized with sodium borate (pH 8.5) for 2~3 min, pre-blocked with 1% BSA and 0.1% Tween-20 in PBS for 30 min, and then incubated with anti-BrdU antibody (BD Biosciences) and Alexa Fluor 488-conjugated secondary antibody (Invitrogen). The nuclei were counterstained with PI in the presence of RNase A. Cells were analyzed for their BrdU uptake with a laser scanning cytometer (LSC), and the BrdU incorporation rate was expressed as the percentage of BrdU-positive cells in total scanned cells.
Neointimal lesion formation through ligation of mouse carotid arteries
C57BL/6 male mice (6–8 week, 19–21 g) were purchased from the Laboratory Animal Center of the Academy of Military Medical Sciences of China. Seven and five mice were used for extraction of miRNA and mRNA, respectively. The left common carotidartery was dissected and ligated near the carotid bifurcation as previously described 28. 15 d after ligation, both left and right carotid arteries (to serve as negative controls) were sampled and then examined for vascular wall morphology using H & E staining or stored in 0.5 ml RNAlater, followed by extraction of miRNA (7 mice) or mRNA (5 mice) as described above. All animal studies were approved by and performed according to the Guidelines for the care and Use of laboratory Animals in Nankai University (A5521-01), which strictly conforms to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85-23, revised 1996).
Data analysis
Data were presented as the mean ± SEM. Statistically significant differences were evaluated by Student’s t test when two groups were compared and by ANOVA when three or more groups were compared. The Mann-Whitney U non-parametric analysis was used for data that were not normality and homogeneity of variance. P < 0.05 was considered significant.
Results
Myocardin induces miR-1 expression in human vascular SMCs
Myocardin is a potent transcriptional co-activator for smooth muscle-specific gene expression 3, 5, and also strongly represses the proliferation of SMCs when overexpressed 7, 29. We have previously generated a stable cell line through T-REx tetracycline-regulated system to overexpress Flag-tagged myocardin in human aortic SMCs 7. Addition of 1 μg/ml doxycycline (Dox) into culture medium induced the expression of myocardin as detected by Western blot analysis using antibodies against either the Flag tag or myocardin (Fig. 1A), consistent with our prior report 7. The specificity of myocardin antibody (M-16, Santa Cruz) was confirmed using blocking peptide and myocardin siRNA. Pre-incubation with the blocking peptide made the band at the same position as detected by Flag tag antibody to disappear (Supplemental Fig. IA). Furthermore, we used myocardin siRNA to ensure the specificity of myocardin (M-16) antibody to detect endogenous myocardin protein. With protein lysates from cultured human aortic SMCs, myocardin (M-16) antibody specifically detected a band at the identical position as the overexpressed Flag-tagged myocardin protein (Supplemental Fig. IB). Of note, the blot signal was reduced upon transfection with myocardin siRNA as compared to transfection with a control siRNA, confirming the specificity of the myocardin (M-16) antibody for endogenous myocardin in SMCs (Supplemental Fig. IB). The specificity of the M-16 antibody was further demonstrated by myocardin peptides, in which addition of myocardin peptide used to generate myocardin M-16 antibody completely blocked the signal of overexpressed myocardin in Western blot (Supplemental Fig. IA).
We then examined the myocardin induction of miR-1 using reverse-transcription-real-time PCR. Our results showed that mature miR-1 level increased ~30 and ~ 250 folds in response to Dox treatment for 1 d and 3 d, respectively, in myocardin inducible SMCs, but not in control cells with empty vectors (Fig. 1B and data not shown). Of note, after 3 d Dox induction, the miR-1 level in cultured SMCs became comparable to that in intact carotid artery (Fig. 1B). Subsequently, we used Northern blot assays to confirm the expression of mature miR-1 in response to myocardin induction. As anticipated, the mature miR-1 was detected in myocardin inducible SMCs after 3 d treatment with Dox, but not in the control cells with empty vector (Fig. 1C and Supplemental Fig. II).
Intriguingly, the induction of miR-1 upon myocardin overexpression was even greater than that of many established myocardin target genes. As shown in Fig. 1D–F, myocardin overexpression resulted in a ~5-fold increase in mRNA expression of SM22 (Fig. 1D), ~3-fold increase in miR-143 and miR-145 levels (Fig. 1F). A decrease in the mRNA level of KLF4, a negative regulator of SMC contractile phenotype 30, was also observed (Fig. 1E). Therefore, our data demonstrated that myocardin is capable of inducing miR-1 expression in human vascular SMCs.
Expression of myocardin and miR-1 in two phenotypes of human SMCs
Myocardin is an essential inducer of contractile phenotype of SMCs 5, 31. To investigate the physiological significance of miR-1 in myocardin-dependent contractile phenotype, we sought to examine the potential difference of myocardin and miR-1 expression in two phenotypes of human SMCs. Using limiting dilution method as previously described 6, 26, we cloned two subpopulations from primary cultured human aortic SMCs: spindle-shaped (Fig. 2A) and epithelioid SMCs (Fig. 2B). To characterize the phenotypic difference of SMCs, we showed that epithelioid SMCs are more proliferative than spindle-shaped SMCs as indicated by BrdU incorporation rates in the presence of 10% serum (Fig. 2C), and that spindle-shaped SMCs are more differentiated than epithelioid SMCs as indicated by higher expression levels of smooth muscle contractile marker gene, such as SM22 (Fig. 2D). In addition, epithelioid SMCs exhibited a nonsignificant higher mRNA level of KLF4 than spindle-shaped SMCs (Fig. 2E). Therefore, the characteristics of two phenotypes of human aortic SMCs are consistent with previous demonstrations of these two typical phenotypes of human SMCs 2, 6, 26.
Figure 2. Characterization of spindle-shaped and epithelioid phenotypes of human vascular SMCs.
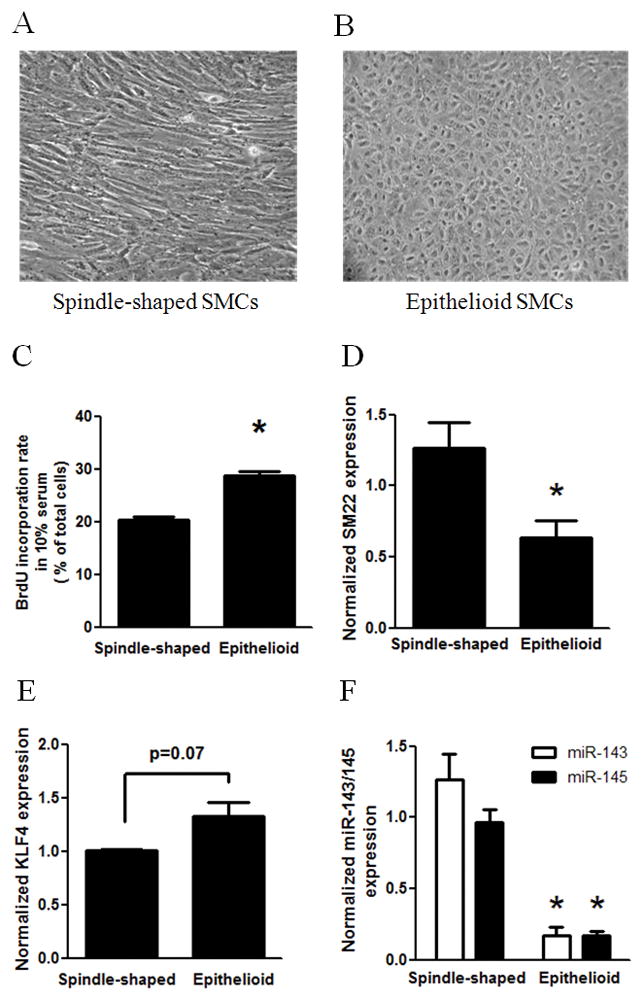
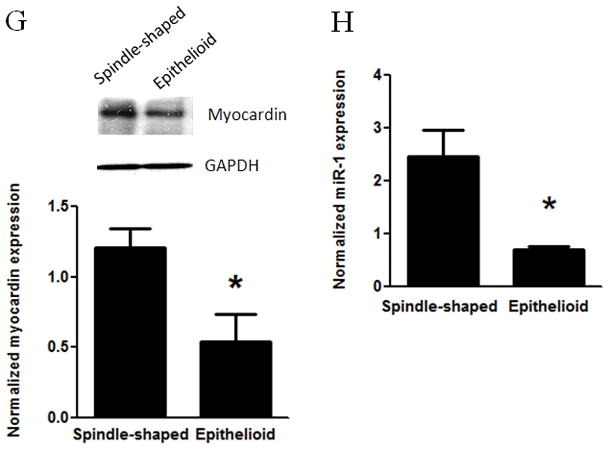
Spindle-shaped and epithelioid SMCs were cloned from primarily cultured human aortic SMCs using limiting dilution. A and B: Micrographs of cloned spindle-shaped (A) and epithelioid (B) SMCs were taken using phase contrast microscopy. C: The proliferative activities in 10% serum of spindle-shaped and epithelioid SMCs were measured using the BrdU incorporation assay with LSC analysis. D–F: SM22 mRNA level (D), KLF4 mRNA level (E) and miR-143/145 level (F) were determined in quantitative PCR assays. G: Protein level of myocardin was determined with myocardin antibody in Western blot analysis, and normalized by GAPDH. Insert was the result of a representative Western blot. H: miR-1 level was determined using PCR. * indicates significant difference compared with spindle-shaped SMCs (p < 0.05, n=4).
We examined the expression of myocardin and miR-1 in spindle-shaped and epithelioid SMCs using Western blot analysis and real-time PCR, respectively. As shown in Fig. 2G, myocardin is expressed at significant lower level in epithelioid SMCs than in spindle-shaped cells. This observation is consistent with the role of myocardin in promoting SMC differentiation 3, 5 and inhibiting cell proliferation 7, 29. Importantly, our results revealed that expression levels of mature miR-1 were significantly lower in epithelioid SMCs than spindle-shaped cells (Fig. 2H). Similarly, we found that the expression of miR-143 and miR-145 (Fig. 2F) was lower in epithelioid SMCs. Taken together, these data suggest that endogenous myocardin also regulates the expression of miR-1. Our results also suggest that myocardin-dependent miR-1 expression may contribute to lower activity of proliferation and the phenotypic heterogeneity of human vascular SMCs.
miR-1 mediates myocardin-dependent inhibition of SMC proliferation
Given that miR-1 could enhance cardiac and skeletal muscle differentiation and inhibit muscle cell proliferation as well as its induction by myocardin in differentiated SMCs 21, 22, we hypothesized that miR-1 may contribute to myocardin-mediated inhibitory effects of SMC proliferation. To test this hypothesis, we first examined the effects of miR-1 inhibitor on myocardin-dependent inhibition of SMC proliferation. Human vascular SMCs harboring T-REx inducible system for overexpression of myocardin were transfected with miR-1 inhibitor (1 nM) or miRNA hairpin inhibitor negative control for 5 h, followed by addition of Dox for myocardin induction. After 48 h, cell proliferation was measured using Alamar Blue assays as described in Materials and Methods. Our results showed that myocardin inhibited the proliferation of SMCs (Fig. 3), consistent with our previous report 7. Importantly, transfection with miR-1 inhibitor, but not the negative control, significantly reversed the proliferation inhibition induced by myocardin (Fig. 3). Transfection with miR-1 inhibitor or its negative control did not significantly affect the proliferation of SMCs in the absence Dox-induced myocardin overexpression (data not shown), suggesting that miR-1 functions downstream of myocardin to regulate SMC proliferation.
Figure 3. Reversal of the inhibitory effect of myocardin on cell proliferation by miR-1 inhibitor.
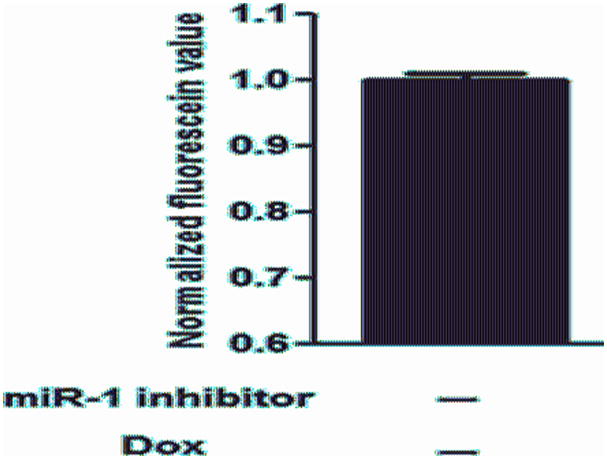
Human vascular SMCs with T-REx system for myocardin expression were first transfected with miR-1 inhibitor (1 nM) or miRNA hairpin inhibitor negative control for 5 h, followed by induction with Dox for 48 h. Cell proliferation was measured by the Alamar Blue assay. The y-axis shows the normalized fluorescein value. * indicates significant difference compared with the control cells with transfection of inhibitor negative control and without Dox induction (p < 0.01, n=5). ** indicates significant difference compared with cells with transfection of inhibitor negative control and with Dox induction (p < 0.01, n=5).
Next, we investigated whether miR-1 is sufficient to inhibit SMC proliferation. It is well known that phenotypically different SMCs have different proliferative responses to various stimuli, in which the epithelioid SMCs are more proliferative than spindle-shaped SMCs 2, 6. We examined the effects of miR-1 on the proliferation of these two phenotypes of human SMCs. The cells were transfected with either miR-1 mimic or negative control miRNA (50 nM) for 48 h, followed by determination of cell proliferation with Alamar Blue assays and BrdU incorporation assays. Indeed, transfection of miR-1 led to a modest, but statistically significant reduction in cell proliferation in both spindle-shaped (Fig. 4A) and epithelioid SMCs (Fig. 4B). In addition, a greater inhibitory effect of miR-1 mimic was observed for proliferation of epithelioid SMCs (~15% reduction) than that of spindle-shaped cells (~5% reduction). Furthermore, BrdU incorporation analyses confirmed the decrease in cell proliferation of both phenotypes upon miR-1 transfection (Fig. 4C and 4D). Together, our data demonstrate that miR-1 is sufficient to inhibit cell proliferation in SMCs.
Figure 4. Overexpression of miR-1 inhibits the proliferation of SMCs.
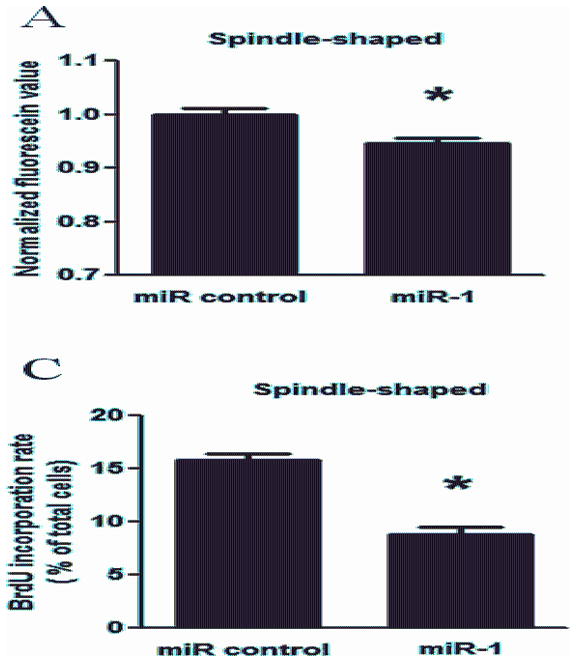
The proliferation of spindle-shaped (A, C) or epithelioid SMCs (B, D) was measured after transfection with miR-1 (50 nM) or miRNA negative control for 48 h by Alamar Blue assay (A, B) and the BrdU incorporation assay with LSC (C, D). The y-axis of panel A or B indicates the normalized fluorescein value. The y-axis of panel C or D indicates the percentage of BrdU-positive cells among total cells in LSC analysis. * indicates significant difference compared with cells transfected with miRNA negative control (p < 0.05, n=5).
Pim-1 is a target gene of miR-1 in human vascular SMCs
To further reveal the molecular mechanism underlying miR-1-mediated inhibition of SMC proliferation, we attempted to identify miR-1 target genes in human SMCs. Several miR-1 target genes have been reported to mediate miR-1-dependent effects on cell proliferation and/or differentiation 21, 22, which include Pim-1, an oncogenic serine/threonine kinase 27, HDAC4, a histone deacetylase mediating cell proliferation-differentiation in skeletal muscle 21, hand2, a member of bHLH transcription factor involving cardiomyocyte development 22, and Rheb, a GTPase essential for cell growth regulation 32. We evaluated the effects of miR-1 on the expression levels of Pim-1, HDAC4, hand2 and Rheb in human SMCs. Western blot analysis showed that transfection of miR-1 significantly decreased the expression level of endogenous Pim-1 protein (Fig. 5A & B). Additionally, there was no significant decrease in the mRNA level of Pim-1 upon miR-1 transfection, although a tendency for the decreased expression of Pim-1 was observed in spindle-shaped SMCs (Fig. 5C). In contrast, the protein expression levels of HDAC4, hand 2 and Rheb were not significantly changed by miR-1 in both spindle-shaped and epithelioid SMCs (Fig. 5D and data not shown).
Figure 5. Downregulation of Pim-1 by miR-1 in human vascular SMCs.
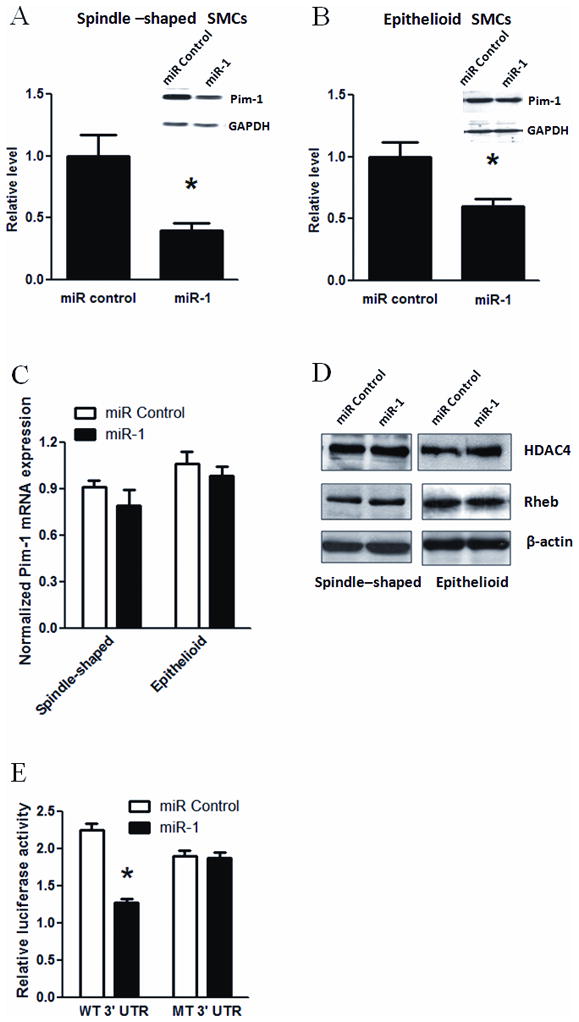
A and B: Spindle-shaped (A) and epithelioid (B) SMCs were transfected with miR-1 or miRNA negative control for 48 h, followed by protein extraction and Western blot analysis using antibodies for Pim-1 and GAPDH. The y-axis indicates the expression levels of Pim-1 protein in spindle-shaped (A) and epithelioid (B) after normalized with GAPDH signals. Inserts in A and B were representative Western blots. * indicates significant difference compared with miRNA negative control (p < 0.01, n=3). C: Quantitative PCR assay showed Pim-1 mRNA level in spindle-shaped and epithelioid SMCs transfected with miR-1 or miRNA negative control. The y-axis indicates the expression levels of Pim-1 mRNA after normalization with GAPDH signals (n=3). D: Representative Western blots from 3 independent experiments showed the effects of miR-1 transfection on the expression levels of HDAC4 and Rheb in two phenotypes. β-actin served as loading control. E: Luciferase reporter assay validated Pim-1 as a target of miR-1. The reporter mRNA contained luciferase ORF and Pim-1 3′ UTR sequence with (MT) or without (WT) mutation of putative miR-1 seed sequence. The y-axis indicates the relative luciferase activity. * indicates significant difference compared with miRNA control (p < 0.05, n=4).
To further confirm that Pim-1 was a direct target gene of miR-1 in SMCs, we employed the luciferase reporter assay using the Pim-1 3′ UTR luciferase reporter gene. Our results showed that overexpression of miR-1 significantly inhibited the activity of Pim-1 3′ UTR luciferase reporter gene (Fig. 5E). More specifically, mutations introduce into the miR-1 seed sequences resulted in loss of inhibition by miR-1 (Fig. 5E). Together, our results demonstrated that Pim-1 is a direct regulatory target of miR-1 in SMCs. Given that Pim-1 is known to stimulate the proliferation of SMCs 8, our results provide an explanation for miR-1-mediated SMC proliferation repression.
Down-regulation of miR-1 expression in neointima formation of mouse carotid arteries
To further study the expression of myocardin and miR-1 in SMCs in vivo, we performed carotid artery ligation to induce neointimal lesions in mice, a well-established experimental procedure to study in vivo SMC proliferation 28. 2 weeks after the ligation, carotid arteries were harvested for tissue cross-sectioning and extraction of mRNA and miRNA. Consistent with previous reports, significant neointima formation was detected in ligated carotid arteries through H & E staining (Fig. 6A, uninjured and 6B, injured). Real-time PCR results showed that miR-1 expression was significantly down-regulated in neointimal lesions compared with uninjured arteries (Fig. 6C), accompanied by significant down-regulation of myocardin (Fig. 6D). We also observed a modest, but statistically significant increase of Pim-1 mRNA in neointima lesion (Fig. 6E). Taken together, our studies suggested that the regulatory cascade of myocardin-miR-1-Pim-1 is likely involved in the regulation of vascular SMC proliferation, both in vitro and in vivo.
Figure 6. Downregulation of myocardin and miR-1 in neointima lesions in mouse carotid arteries.

Neointima lesions were created by ligation of carotid arteries of mice for 15 d as described in Materials and Methods. A and B: Micrographs of arterial cross-sections with H & E staining with (B, injured) or without (A, control) ligation. M, media; NI, neointima; L, lumen. C–E: Control and injured arterial segments were harvested for extraction of miRNA or mRNA, followed by measuring the expression of miR-1 (C), myocardin (D) or Pim-1 (E) using PCR. The expression levels of miR-1 and myocardin or Pim-1 were normalized by U6B and GAPDH, respectively. * indicates significant difference compared with control group (p < 0.001, n=7 for miR-1; p < 0.05, n=5 for myocardin and Pim-1).
Discussion
In current study, we have revealed that the expression of miR-1 is induced by myocardin in human vascular SMCs and miR-1 mediates the inhibitory effects of myocardin on SMC proliferation. This study has provided a novel mechanism underlying myocardin-induced inhibition of SMC proliferation. Our results are consistent with the role of myocardin and miR-1 in growth and proliferation of cardiomyocytes and skeletal myoblasts as previously reported 19–22.
The expression and functions of miR-1 were not previously reported in vascular SMCs. Earlier studies using Northern blot revealed the enriched expression of miR-1 in cardiac and skeletal muscle 21, 22. Moreover, recent reports with more sensitive real-time PCR assay have documented the presence of miR-1 in non-muscle tissues, including mouse cerebral cortex and lung 33. Most importantly, we revealed for the first time that expression of miR-1 is dramatically induced by myocardin in vascular SMCs, which was confirmed using Northern blot assays. Such miR-1 induction by myocardin is actually echoing previous finding that SRF controls the expression of miR-1 in cardiac muscle.
Myocardin overexpression for 3 d substantially induced the expression of miR-1 in cultured SMCs to an extent comparable to that in intact artery. Although miR-1 level in SMCs did not reach the level as observed in adult skeletal muscle (see Supplemental Fig. II), it is sufficient to mediate, at least in part, the function of myocardin in smooth muscle cells
We found a correlation between differential expression of myocardin and miR-1 and proliferative capacity in spindle-shaped and epithelioid phenotypes of human SMCs, which further supports the physiological relevance of myocardin induction of miR-1 in the innate heterogeneity of proliferative capacity of vascular SMC. The functional importance of myocardin and miR-1 in SMCs is further strengthened by our in vivo finding that miR-1 and myocardin expression is down-regulated in neointima lesions using a mouse carotid artery ligation model.
One of the major functions of myocardin in SMCs is to induce differentiation and contractile phenotype 3, 5, 31, associated with an increase in the expression of contractile marker genes 5, 31. Conversely myocardin was also shown to inhibit SMC proliferation 7. It is notable that miR-1 was induced by myocardin even greater (~30 fold) than those of well-established myocardin targets including SM22 (~6 fold), miR-143/145 (~3 fold), suggesting that the expression of miR-1 may contribute to the cellular effects of myocardin in SMCs. The role of miR-1 in myocardin-induced SMC contraction is currently investigated in our lab (Jiang Y, X-L Zheng, unpublished observation). In the present study, we have revealed that miR-1 mediates myocardin-dependent inhibition of SMC proliferation. Our conclusion is based on three lines of evidences: 1) miR-1 inhibitor partially reverses the inhibitory effects of myocardin on SMC proliferation, 2) exogenous miR-1 inhibits SMC proliferation and 3) miR-1 directly down-regulates the expression of target gene Pim-1, which promotes SMC proliferation.
Our data that miR-1 inhibits SMC proliferation are in line with its role in cardiomyocytes, skeletal myoblasts and lung cancer cells 21, 22, 27. More importantly, the roles of miR-1 in myocardin-dependent cellular effects have important physiological relevance to vascular smooth muscle functions, given that myocardin is a master regulator of contractile phenotype of SMCs 5, 31. The phenotypic plasticity is an integral feature of mature SMCs and distinctive phenotypes of SMCs exist in normal blood vessel wall 1, 2, 6. Our data suggest that differential expression levels of myocardin and miR-1 may contribute to the innate diversity of SMC phenotype, providing a potential explanation for the lower proliferation rate of spindle-shape SMC than its epithelioid counterpart. Furthermore, since expression of myocardin and miR-1 are suppressed during neointima formation, deregulation of myocardin-miR-1 signaling could facilitate SMC proliferation in some pathophysiological context, for instance restenosis after angioplasty.
We previously reported that myocardin physically interacts with NF-κB p65 subunit so as to inhibit NF-κB-dependent gene transcription and cell proliferation 7. Our current study, in which we show that myocardin also reduces proliferation through myocardin-induced miRNA, has provided a complementary mechanism for myocardin inhibition of SMC proliferation. The inhibitory effect of miR-1 on SMC proliferation is, at least in part, through the down-regulation of the expression of Pim-1 gene encoding an oncogenic serine/threonine kinase, which is required for injury-induced neointima formation and SMC proliferation 8. Intriguingly, the interdependence between Pim-1 expression and NF-κB signaling has been demonstrated in the context of chemoresistance of prostate cancer 34 and CD40-dependent proliferation of B cells 35. It will be interesting to investigate the interplay between miR-1-Pim-1 cascade and NF-κB signaling in SMC proliferation under the control of myocardin.
In summary, we demonstrated myocardin induction of miR-1 inhibits SMC proliferation, probably through down-regulation of Pim-1. It will be important to determine whether myocardin-miR-1-Pim-1 pathway could serve as therapeutic target in proliferative vascular diseases.
Supplementary Material
Acknowledgments
X.L.Z. is recipient of Senior Scholarship Award from Alberta Heritage Foundation for Medical Research. Z-P Huang is a Postdoctoral Fellow and DZ Wang is an Established Investigator of the American Heart Association.
Sources of Funding
This work was supported by an operating grant from the Canadian Institute of Health Research (RT734076 to X.L.Z.) and by National Institutes of Health (R01 HL085635 to D.Z.W). Funding to pay the Open Access publication charges for this article was provided by the Canadian Institute of Health Research.
Footnotes
Disclosures
None declared.
References
- 1.Owens GK, Kumar MS, Wamhoff BR. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol Rev. 2004;84:767–801. doi: 10.1152/physrev.00041.2003. [DOI] [PubMed] [Google Scholar]
- 2.Hao H, Gabbiani G, Bochaton-Piallat ML. Arterial smooth muscle cell heterogeneity: implications for atherosclerosis and restenosis development. Arterioscler Thromb Vasc Biol. 2003;23:1510–1520. doi: 10.1161/01.ATV.0000090130.85752.ED. [DOI] [PubMed] [Google Scholar]
- 3.Wang D, Chang PS, Wang Z, Sutherland L, Richardson JA, Small E, Krieg PA, Olson EN. Activation of cardiac gene expression by myocardin, a transcriptional cofactor for serum response factor. Cell. 2001;105:851–862. doi: 10.1016/s0092-8674(01)00404-4. [DOI] [PubMed] [Google Scholar]
- 4.Wang Z, Wang DZ, Hockemeyer D, McAnally J, Nordheim A, Olson EN. Myocardin and ternary complex factors compete for SRF to control smooth muscle gene expression. Nature. 2004;428:185–189. doi: 10.1038/nature02382. [DOI] [PubMed] [Google Scholar]
- 5.Wang Z, Wang DZ, Pipes GC, Olson EN. Myocardin is a master regulator of smooth muscle gene expression. Proc Natl Acad Sci U S A. 2003;100:7129–7134. doi: 10.1073/pnas.1232341100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li S, Fan YS, Chow LH, Van Den Diepstraten C, van Der Veer E, Sims SM, Pickering JG. Innate diversity of adult human arterial smooth muscle cells: cloning of distinct subtypes from the internal thoracic artery. Circ Res. 2001;89:517–525. doi: 10.1161/hh1801.097165. [DOI] [PubMed] [Google Scholar]
- 7.Tang RH, Zheng XL, Callis TE, Stansfield WE, He J, Baldwin AS, Wang DZ, Selzman CH. Myocardin inhibits cellular proliferation by inhibiting NF-kappaB(p65)-dependent cell cycle progression. Proc Natl Acad Sci U S A. 2008;105:3362–3367. doi: 10.1073/pnas.0705842105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Katakami N, Kaneto H, Hao H, Umayahara Y, Fujitani Y, Sakamoto K, Gorogawa S, Yasuda T, Kawamori D, Kajimoto Y, Matsuhisa M, Yutani C, Hori M, Yamasaki Y. Role of pim-1 in smooth muscle cell proliferation. J Biol Chem. 2004;279:54742–54749. doi: 10.1074/jbc.M409140200. [DOI] [PubMed] [Google Scholar]
- 9.Ji R, Cheng Y, Yue J, Yang J, Liu X, Chen H, Dean DB, Zhang C. MicroRNA expression signature and antisense-mediated depletion reveal an essential role of MicroRNA in vascular neointimal lesion formation. Circ Res. 2007;100:1579–1588. doi: 10.1161/CIRCRESAHA.106.141986. [DOI] [PubMed] [Google Scholar]
- 10.Chan MC, Hilyard AC, Wu C, Davis BN, Hill NS, Lal A, Lieberman J, Lagna G, Hata A. Molecular basis for antagonism between PDGF and the TGFbeta family of signalling pathways by control of miR-24 expression. EMBO J. 29:559–573. doi: 10.1038/emboj.2009.370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kuhn AR, Schlauch K, Lao R, Halayko AJ, Gerthoffer WT, Singer CA. MicroRNA expression in human airway smooth muscle cells: role of miR-25 in regulation of airway smooth muscle phenotype. Am J Respir Cell Mol Biol. 42:506–513. doi: 10.1165/rcmb.2009-0123OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Davis BN, Hilyard AC, Nguyen PH, Lagna G, Hata A. Induction of microRNA-221 by platelet-derived growth factor signaling is critical for modulation of vascular smooth muscle phenotype. J Biol Chem. 2009;284:3728–3738. doi: 10.1074/jbc.M808788200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu X, Cheng Y, Zhang S, Lin Y, Yang J, Zhang C. A necessary role of miR-221 and miR-222 in vascular smooth muscle cell proliferation and neointimal hyperplasia. Circ Res. 2009;104:476–487. doi: 10.1161/CIRCRESAHA.108.185363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Boettger T, Beetz N, Kostin S, Schneider J, Kruger M, Hein L, Braun T. Acquisition of the contractile phenotype by murine arterial smooth muscle cells depends on the Mir143/145 gene cluster. J Clin Invest. 2009;119:2634–2647. doi: 10.1172/JCI38864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Elia L, Quintavalle M, Zhang J, Contu R, Cossu L, Latronico MV, Peterson KL, Indolfi C, Catalucci D, Chen J, Courtneidge SA, Condorelli G. The knockout of miR-143 and -145 alters smooth muscle cell maintenance and vascular homeostasis in mice: correlates with human disease. Cell Death Differ. 2009;16:1590–1598. doi: 10.1038/cdd.2009.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cheng Y, Liu X, Yang J, Lin Y, Xu DZ, Lu Q, Deitch EA, Huo Y, Delphin ES, Zhang C. MicroRNA-145, a novel smooth muscle cell phenotypic marker and modulator, controls vascular neointimal lesion formation. Circ Res. 2009;105:158–166. doi: 10.1161/CIRCRESAHA.109.197517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xin M, Small EM, Sutherland LB, Qi X, McAnally J, Plato CF, Richardson JA, Bassel-Duby R, Olson EN. MicroRNAs miR-143 and miR-145 modulate cytoskeletal dynamics and responsiveness of smooth muscle cells to injury. Genes Dev. 2009;23:2166–2178. doi: 10.1101/gad.1842409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cordes KR, Sheehy NT, White MP, Berry EC, Morton SU, Muth AN, Lee TH, Miano JM, Ivey KN, Srivastava D. miR-145 and miR-143 regulate smooth muscle cell fate and plasticity. Nature. 2009;460:705–710. doi: 10.1038/nature08195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhao Y, Ransom JF, Li A, Vedantham V, von Drehle M, Muth AN, Tsuchihashi T, McManus MT, Schwartz RJ, Srivastava D. Dysregulation of cardiogenesis, cardiac conduction, and cell cycle in mice lacking miRNA-1–2. Cell. 2007;129:303–317. doi: 10.1016/j.cell.2007.03.030. [DOI] [PubMed] [Google Scholar]
- 20.van Rooij E, Sutherland LB, Qi X, Richardson JA, Hill J, Olson EN. Control of stress-dependent cardiac growth and gene expression by a microRNA. Science. 2007;316:575–579. doi: 10.1126/science.1139089. [DOI] [PubMed] [Google Scholar]
- 21.Chen JF, Mandel EM, Thomson JM, Wu Q, Callis TE, Hammond SM, Conlon FL, Wang DZ. The role of microRNA-1 and microRNA-133 in skeletal muscle proliferation and differentiation. Nat Genet. 2006;38:228–233. doi: 10.1038/ng1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhao Y, Samal E, Srivastava D. Serum response factor regulates a muscle-specific microRNA that targets Hand2 during cardiogenesis. Nature. 2005;436:214–220. doi: 10.1038/nature03817. [DOI] [PubMed] [Google Scholar]
- 23.McCarthy JJ, Esser KA. MicroRNA-1 and microRNA-133a expression are decreased during skeletal muscle hypertrophy. J Appl Physiol. 2007;102:306–313. doi: 10.1152/japplphysiol.00932.2006. [DOI] [PubMed] [Google Scholar]
- 24.Gui Y, Zheng XL. Epidermal growth factor induction of phenotype-dependent cell cycle arrest in vascular smooth muscle cells is through the mitogen-activated protein kinase pathway. J Biol Chem. 2003;278:53017–53025. doi: 10.1074/jbc.M309640200. [DOI] [PubMed] [Google Scholar]
- 25.Blaes N, Bourdillon MC, Daniel-Lamaziere JM, Michaille JJ, Andujar M, Covacho C. Isolation of two morphologically distinct cell lines from rat arterial smooth muscle expressing high tumorigenic potentials. In Vitro Cell Dev Biol. 1991;27A:725–734. doi: 10.1007/BF02633218. [DOI] [PubMed] [Google Scholar]
- 26.Zheng XL, Yuan SG, Peng DQ. Phenotype-specific inhibition of the vascular smooth muscle cell cycle by high glucose treatment. Diabetologia. 2007;50:881–890. doi: 10.1007/s00125-006-0543-6. [DOI] [PubMed] [Google Scholar]
- 27.Nasser MW, Datta J, Nuovo G, Kutay H, Motiwala T, Majumder S, Wang B, Suster S, Jacob ST, Ghoshal K. Down-regulation of micro-RNA-1 (miR-1) in lung cancer. Suppression of tumorigenic property of lung cancer cells and their sensitization to doxorubicin-induced apoptosis by miR-1. J Biol Chem. 2008;283:33394–33405. doi: 10.1074/jbc.M804788200. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 28.Kumar A, Lindner V. Remodeling with neointima formation in the mouse carotid artery after cessation of blood flow. Arterioscler Thromb Vasc Biol. 1997;17:2238–2244. doi: 10.1161/01.atv.17.10.2238. [DOI] [PubMed] [Google Scholar]
- 29.Milyavsky M, Shats I, Cholostoy A, Brosh R, Buganim Y, Weisz L, Kogan I, Cohen M, Shatz M, Madar S, Kalo E, Goldfinger N, Yuan J, Ron S, MacKenzie K, Eden A, Rotter V. Inactivation of myocardin and p16 during malignant transformation contributes to a differentiation defect. Cancer Cell. 2007;11:133–146. doi: 10.1016/j.ccr.2006.11.022. [DOI] [PubMed] [Google Scholar]
- 30.King KE, Iyemere VP, Weissberg PL, Shanahan CM. Kruppel-like factor 4 (KLF4/GKLF) is a target of bone morphogenetic proteins and transforming growth factor beta 1 in the regulation of vascular smooth muscle cell phenotype. J Biol Chem. 2003;278:11661–11669. doi: 10.1074/jbc.M211337200. [DOI] [PubMed] [Google Scholar]
- 31.Long X, Bell RD, Gerthoffer WT, Zlokovic BV, Miano JM. Myocardin is sufficient for a smooth muscle-like contractile phenotype. Arterioscler Thromb Vasc Biol. 2008;28:1505–1510. doi: 10.1161/ATVBAHA.108.166066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sayed D, Hong C, Chen IY, Lypowy J, Abdellatif M. MicroRNAs play an essential role in the development of cardiac hypertrophy. Circ Res. 2007;100:416–424. doi: 10.1161/01.RES.0000257913.42552.23. [DOI] [PubMed] [Google Scholar]
- 33.Mishima T, Mizuguchi Y, Kawahigashi Y, Takizawa T. RT-PCR-based analysis of microRNA (miR-1 and -124) expression in mouse CNS. Brain Res. 2007;1131:37–43. doi: 10.1016/j.brainres.2006.11.035. [DOI] [PubMed] [Google Scholar]
- 34.Zemskova M, Sahakian E, Bashkirova S, Lilly M. The PIM1 kinase is a critical component of a survival pathway activated by docetaxel and promotes survival of docetaxel-treated prostate cancer cells. J Biol Chem. 2008;283:20635–20644. doi: 10.1074/jbc.M709479200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhu N, Ramirez LM, Lee RL, Magnuson NS, Bishop GA, Gold MR. CD40 signaling in B cells regulates the expression of the Pim-1 kinase via the NF-kappa B pathway. J Immunol. 2002;168:744–754. doi: 10.4049/jimmunol.168.2.744. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.