

Abstract
Burn injuries to extensive areas of the body are complicated by muscle catabolism. Elucidating the molecular mechanisms that mediate this catabolism may facilitate the development of a medical intervention. Here, we assessed the functional classification of genes that were differentially expressed in skeletal muscle following burn injury in 19 children (5.2±4.0 years of age), (64±15% total burn surface area, TBSA) relative to 13 healthy controls (11.9±6.0 years of age). Microarray analysis of samples taken within 10 days of burn injury revealed altered expression of a variety of genes, including some involved in cell and organelle organization and biogenesis, stress response, wound response, external stimulus response, regulation of apoptosis and intracellular signaling. The genes that encode peroxisome proliferator-activated receptors (PPARs; 3 isotypes PPARα, PPARγ and PPARδ also known as PPARß or PPARß/δ), which may serve as transcriptional nodal points and therapeutic targets for metabolic syndromes, were among those affected. In particular, expression of the main mitochondrial biogenesis factor PPARγ-1ß (or PGC-1ß) was downregulated (P<0.0001), while the expression of PPARδ was upregulated (P<0.001). Expression of PGC-1α, the closest homolog of PGC-1ß was upregulated (P=0.0037), and expression of the gene encoding mitochodrial uncoupling protein 2 (UCP2) was also upregulated (P=0.008). These results suggest that altered PPAR and mitochondrial gene expression soon after burn injury may lead to metabolic and mitochondrial dysfunction in human skeletal muscle.
Keywords: skeletal muscle, burn, trauma, mitochondria, mitochondrial, PGC-1ß, peroxisome proliferator-activated receptor δ, PGC-1α, uncoupling protein 2, microarray, genomics
Introduction
Almost two million people become burn victims in the US each year. A small percentage of these injuries are fatal, and all require immediate medical attention. While the clinical consequences for a patient with a large total body surface area (TBSA) burn injury are grave, the mechanisms mediating post-burn metabolic alterations and muscle catabolism remain uncertain.
When burn injury exceeds 30% of the TBSA, local inflammation generalizes into a severe systemic response metabolically characterized as muscle catabolism, leading to muscle wasting or cachexia (1). Burn injury is associated with anatomical, physiological, endocrinological and immunological alterations leading to general cachexia, and metabolic alterations appear to play a central role in this pathophysiological progression (2). While early burn wound excision has been shown to favorably influence the metabolic response and reduce catabolism (3), agents that could actually alter post-burn physiology would be powerful tools for ameliorating cachexia.
Recent nuclear magnetic resonance (NMR) and micro-array studies from our laboratory have indicated that burn injury causes mitochondrial dysfunction in skeletal muscle (4). Hence, post-burn cachexia may be due, at least in part, to alterations in the activities of molecules controlling mitochondrial function. Such alterations could potentially be achieved through the actions of peroxisome proliferator-activated receptors (PPARs; 3 isotypes PPARα, PPARγ and PPARδ also known as PPARß or PPARß/δ). PPARs are nuclear receptors that act as transcription factors and have been described as nodes of inputs and outputs involved in energy homeostasis (4). They are involved in the trans-criptional regulation of key metabolic pathways, such as lipid metabolism and insulin sensitivity, and accordingly have been shown to play roles in inflammation, diabetes, athero-sclerosis, obesity and hypertension (5,6). In light of their ability to act as transcriptional nodal points, PPARs have been implicated as therapeutic targets for metabolic syndromes (5). PPARα and PPARδ, in particular, are known to play roles in a number of physiological processes including cell growth, cell differentiation and cell death (7).
A recent study reported downregulation of the PPARγ coactivator 1ß (PGC-1ß) in a murine burn model (8), suggesting a novel mode of regulation for numerous complex biological programs, including metabolism by coactivator proteins (9,10). The first member of the PGC-1 family, PGC-1α, was identified from brown adipose tissue as a PPARγ-interacting protein (11). PGC-1ß was later identified as the closest homolog of PGC-1α; the two molecules share extensive sequence identity (12,13). PGC-1 coactivators play a critical role in the maintenance of glucose, lipid, and energy homeostasis and are thus likely to be involved in pathogenic conditions such as neurodegeneration, cardiomyopathy, obesity, diabetes (14), and, presumably, burn injury (8). PGC-1ß regulates numerous downstream genes including adenosine triphosphate (ATP) synthase and the H+ transporting mitochondrial F1 complex. It is thus suggested that the decreased ATP synthesis observed by NMR in burn victims may be the result of downregulation of these molecules (8). Furthermore, nuclear respiratory factors 1 and 2 (NRF-1 and NRF-2) are transcription factor targets of PGC-1α and ß and thus regulate the expression of some nuclear-encoded mito-chondrial genes (14). An interplay between PGC-1α and forkhead box O (FoxO) family transcription factors has been reported. In particular, decreased levels of PGC-1α in atrophied muscle has been suggested to facilitate FoxO-dependent cachexia (15). A possible interplay between PGC-1ß and FoxO has also been suggested (8).
The aim of the present study was to evaluate the functional classification of differentially expressed genes and to characterize the concerted expression of selected PPARs and associated genes, including PGC-1s and uncoupling proteins (UCPs), in the skeletal muscle of burned versus healthy control children. This study is based on the fundamental hypothesis that the effects of burn injury on expression of PPARs and related molecules such as PGC-1s may underlie the metabolic complications of burn injury through influences on mitochondrial function. Our study is discussed relative to potential implications for advancing mitochondrial molecular medicine.
Materials and methods
Human biopsy collection
Biopsies collected as part of the Glue grant on the genomics of inflammation (www.gluegrant.org) were used in this study. The review and analysis of the records was approved by the Subcommittee on Research of Massachusetts General Hospital, Boston, MA, USA.
Patient biopsies
As this study focused on gene expression soon after injury, time points that were 10 or more days post-burn were not used. A total of 24 patients were in the original study cohort, but data from 5 patients were excluded since the biopsies had been collected beyond this 10-day cutoff. Hence, the final analyzed group included 19 pediatric burn patients. Two samples were analyzed per subject, and only genomic data corresponding to the earliest biopsy time point were used in the analysis (n=19). The biopsies were collected 2.8±1.6 days post-burn (range 1-7 days post-burn). The patient mean age was 5.2±4.0 years (range 1-14 years). The mean TBSA affected was 64±15% (range 41-93%).
Control biopsies
Control biopsies were collected (2 samples per subject) from the skeletal muscle of 13 healthy subjects. The mean age of the control subjects was 11.9±6.0 years.
Extraction of RNA
Fresh specimens were immersed in 1 ml Trizol® (GibcoBRL, Invitrogen, Carlsbad, CA, USA). Each specimen was homogenized for 60 sec with a Brinkman Polytron 3000 before extraction of total RNA. Chloroform (200 μl) was added to each homogenized muscle sample and mixed with the sample by inverting the tube for 15 sec. After centrifugation at 12000 × g for 15 min, the upper aqueous phase was collected and precipitated by adding 500 μl isopropanol. Further centrifugation at 12000 × g for 10 min separated the RNA pellet, which was then washed with 500 μl of 70% ethanol and centrifuged at 7500 × g for 5 min prior to air drying. The pellet was re-suspended in 100 μl DEPC-H20. An RNeasy Kit (Qiagen, Germantown, MD, USA) was used to purify the RNA according to the manufacturer's protocol. Purified RNA was quantified by UV absorbance at 260 and 280 nm and stored at −70°C for later DNA microarray analysis.
Microarray hybridization
Biotinylated cRNA was generated with 10 μg of total cellular RNA according to the protocol outlined by Affymetrix Inc. (Santa Clara, CA, USA). The cRNA was hybridized onto Affymetrix MOE430A oligo-nucleotide arrays and stained, washed, and scanned according to the Affymetrix protocol.
Genomic data analysis
Two specimens from each subject were analyzed. Data files consisted of scanned images of probes hybridized with RNA extracted from the skeletal muscle specimens isolated at the specified times from burned or control subjects. These data files were converted to cell intensity files (.CEL files) with the Microarray suite 5.0 (MAS, Affymetrix). The data were scaled to a target intensity of 500, and all possible pair-wise array comparisons of the replicates to normal control mice were performed for each time point (i.e., four combinations when two arrays from each time point were compared to the two arrays hybridized to RNA from control mice) using an MAS 5.0 change call algorithm. Probe sets that had a signal value difference greater than 100, and in which one of the two samples being compared was not called ‘absent’, were scored as differentially modulated when i) the number of change calls in the same direction were at least 3, 4 and 6 when the number of comparisons were 4, 6 and 9, respectively; and ii) the other comparisons were unchanged. Such scoring is designed to partially compensate for biological stochasticity and technical variation. Based on the ratios of 100 genes determined by Affymetrix to be invariant in most conditions tested, an additional constraint of a minimum ratio of 1.65 was applied to control the known false positives at 5%.
Statistical analysis
The focus of this analysis was the small-N set of PPARs and associated (PGC-1 and UCP) genes. For this type of analysis, the number of multiple comparisons is small. For this reason, correction for multiple comparisons to control the family-wise error rate (FWER) using a Bonferroni procedure was not applied (FWER/Bonferroni is a very conservative error control procedure aiming to control the probability of even a single type I error for all multiple comparisons performed). Instead, a t-test was used under the assumption that the error rate for false positives is not significantly altered due to the small number of comparisons.
Results
Burn injury affects the functional classification of gene expression in human skeletal muscle
Fig. 1 shows the transcriptome profiles of differentially expressed genes involved in key regulatory pathways at early time points following burn injury in 19 children with ≥30% TBSA and in 13 controls. Expression levels of a variety of genes were downregulated, including many involved in cell and organelle organization and biogenesis, stress response, wounding response, external stimulus response, regulation of apoptosis and intracellular signaling.
Figure 1.
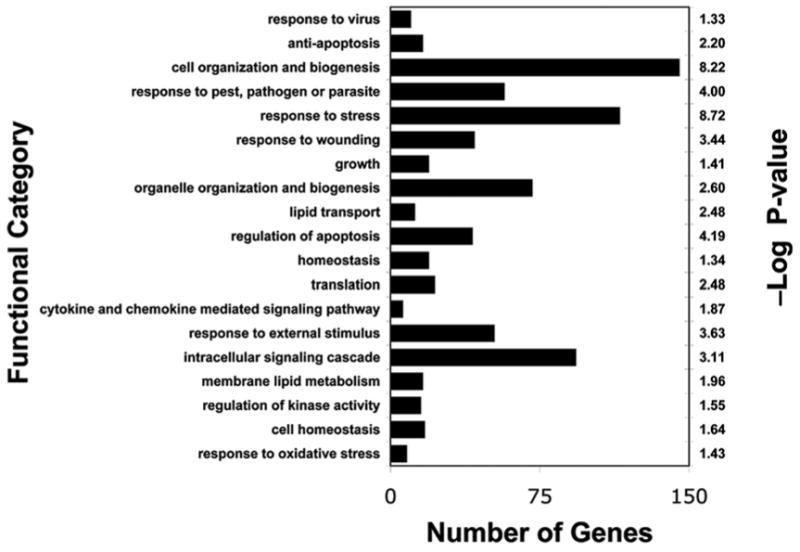
Functional classification of differentially downregulated genes in skeletal muscle biopsies of 19 burned children relative to those of 13 healthy control subjects. Genes showing significant differential expression (cutoff of 5% false discovery rate and 1.4-fold change) were grouped based on Gene Ontology functional classification. Bars indicate the number of downregulated genes (*P<0.05; **P<0.01).
Burn injury downregulates expression of PGC-1β and upregulates expression of PGC-α
Expression of PGC-1ß and PGC-1α differed in burned children relative to that in the controls (Fig. 2). PGC-1ß expression was significantly downregulated whereas that of PGC-1α showed a significant compensatory increase of a lesser magnitude.
Figure 2.
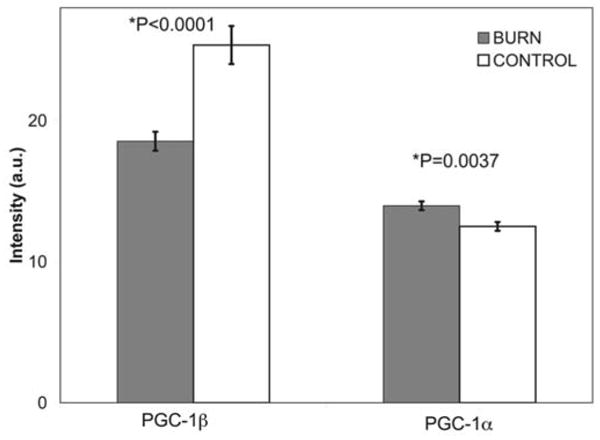
PGC-1ß and PGC-1α gene mRNA expression in muscle tissue from patients who suffered 30% TBSA burns. Gray columns represent gene expression from 19 burned children and white columns represent gene expression from 13 control subjects. PGC-1ß expression was significantly downregulated at early time points post-burn. There was a concurrent compensatory increase in PGC-1α expression. (*t-test, 2-tailed, equal variances).
Burn injury regulates expression of PPARα PPARδ and UCP2 in human skeletal muscle
As shown in Fig. 3, expression of PPAR genes was affected by the presence of burn injury. Note that PPARα gene expression was downregulated in burn victim tissue, while that of the PPARδ gene was upregulated. UCP2 gene expression was also upregulated (Fig. 3). Other uncoupling proteins were not differentially expressed.
Figure 3.
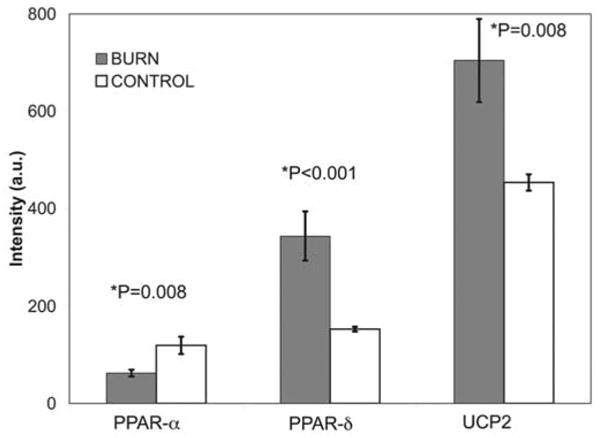
PPARα, PPARδ and UCP2 transcription in tissue from burn patients. Gray columns represent gene expression from 19 burned children and white columns represent gene expression from 13 control subjects. PGC-1α·expression was significantly downregulated at early time points post-burn. There was a concurrent significant compensatory increase in PGC-1δ expression. The expression of UCP2 was also significantly increased. (*t-test, 2-tailed, unequal variances).
Discussion
The present study demonstrated a distinct pattern of functional classifications of genes differentially expressed in burn victim tissue specimens relative to that in uninjured controls. Our microarray experiments revealed significant down-regulation of a great variety of genes in skeletal muscle soon after burn trauma. Downregulated genes included those encoding proteins involved in cell and organelle organization and biogenesis, wound response, external stimulus response, regulation of apoptosis, intracellular signaling and stress response, including ability to deal with oxidative stress (16). It is our view that the pattern of altered transcription observed can be attributed in large part to the concerted action of PPARs and PGC-1s.
Burn trauma was associated with downregulation of PPARs and PGC-1ß expression and with upregulation of PGC-1α and UCP2 expression. This pattern of altered expression leads to mitochondrial uncoupling and thus dysfunction in human burn injury as suggested previously in murine studies (4,8). The presently observed downregulation of PGC-1ß transcription, which may be an upstream regulatory mechanism involving mitochondrial uncoupling, coincided with downregulation of ATP synthase and other oxidative phosphorylation genes. This type of response is accompanied by a concomitant reduction in ATP synthesis rate, which can lead to skeletal muscle cell apoptosis (17,18), and thus may underlie muscle wasting (2).
Such processes may occur in response to the increased levels of cytokines (e.g., TNFα in systemic burns) (19) causing dysregulation of PPARs and PGC-1s, as observed in liver cells (20). These findings thus suggest a more general role for PPARs and PGC-1s in human skeletal muscle metabolism, implicating them in inflammation, diabetes, and obesity, and are in agreement with previous findings in murine models of burn injury (4,8). On the other hand, skeletal muscle PGC-1α and PPARδ gene expression was significantly lower in diabetic patients (21). Rosiglitazone treatment has been shown to restore PGC-1α and PPARδ levels while improving insulin sensitivity and muscular oxidative capacity (21).
Our noteworthy finding of PGC-1ß downregulation is consistent with prior data in mice (8) and may explain the concurrent downregulation of mitochondrial biogenesis observed (Fig. 1) inasmuch as PGC-1ß, like PGC-1α, is a major mitochondrial biogenesis factor (22). The observed upregulation of PGC-1α may be related to dysregulation of the thyroid hormone triiodothyronine (T3), which controls PGC-1α expression (23). Meanwhile, PPARα, which has a biogenesis effect on mitochondria (24), was downregulated in burn injury tissue. Since PGC-1α regulation involves both NRF-1 and T3 (23,24), its upregulation in burn injury tissue may also be an effect of T3 dysregulation, in addition to or rather than, an indirect effect of burn injury via cytokines or reactive oxygen species (ROS).
Downregulation of PGC-1ß has been linked to insulin resistance through Irs1 (8) and Akt (25) in mice. Both of these mechanisms may be operating in human burns. In the latter case, inhibition of Akt phosphorylation prevents subsequent Akt-mediated phosphorylation of FoxO1, 3 and 4, which would normally suppress atrogen expression and proteolysis. In addition, Akt would normally contribute to protein synthesis via mTOR, but Akt attenuation leads to inhibition of protein synthesis and cachexia (26). These findings, together with recent published reports (15,25,27), suggest that these two mechanisms, which may involve an increase in FoxO3 expression, may account for the muscle wasting observed in burned patients (8).
Given that PGC-1ß has recently been recognized as an activator of UCPs (14,22,28), its downregulation would be expected to cause UCP downregulation rather than the upregulation observed in this study (Fig. 3). Therefore, the question of whether PGC-1ß has a role in mitochondrial uncoupling induced by UCP2 in burn trauma needs to be investigated further using in vivo assessment by NMR, which is superior to electric potential measurements across the inner mitochondrial membrane (29). Regardless, UCP2 expression may be stimulated by PGC-1α, and UCPs seem to greatly reduce mitochondrial production of ROS (14). Thus, we propose that the downregulation of PGC-1ß observed herein may leave the muscle unprotected against damage caused by ROS, which would likely be further exacerbated by the burn. PGC-1α-mediated induction of UCP2 expression may then compensate for the fact that UCP3 is not differentially expressed.
We report two novel findings in this study, namely the downregulation of PPARα and the upregulation of PPARδ in burn-injured skeletal muscle. PPARδ-mediated reduction in PPARγ expression (30) may explain the presently observed downregulation of PGC-1ß. A direct effect of burn injury on PGC-1ß expression, however, cannot be ruled out (8). Incidentally, agents that elevate PPARs and PGC-1s have been reported to improve mitochondrial biogenesis and skeletal muscle function (31). Hence, we posit that such remedies may be helpful for burn injury.
Our demonstration of PPAR and PGC-1 induction may have direct clinical relevance. Recent clinical studies indicated that PGC-1α agonists can alleviate post-burn insulin resistance (32,33) and prevent burn-induced damage in remote organs (34). Furthermore, since PPARs are transcriptional nodal points, they have been suggested as potential therapeutic targets for metabolic syndromes (5,35). We are thus pursuing research to investigate the mechanism of this burn-induced aberration in metabolic control in an effort to understand burn pathology well enough to design treatments to prevent and/or minimize the extent of burn damage. Such studies may provide new insights into the biology of burn injury, and their results could lead to advances in burn treatment that will reduce the morbidity and mortality of burn victims and improve their quality of life.
The present findings help to pave the way to a better understanding of the physiological and pathological processes that accompany systemic burn trauma and suggest that further research exploring the role of PPARs, PGC-1s and UCPs in a variety of inflammatory circumstances in which agonists may have protective effects should be pursued. Since PPARs are controllable factors in most tissues and respond to common signaling pathways, they are promising pharmaceutical targets. To this end, elucidating the role of PPARs and related molecules in burn-associated muscle catabolism through physiological and molecular genetic analyses should significantly advance mitochondrial molecular medicine.
Acknowledgments
This work was supported in part by a Shriner's Hospital for Children research grant (no. 8893) to A. Aria Tzika, a National Institute Institutes of Health (NIH) Center Grant (P50GM021700) to Ronald G. Tompkins and by an NIH Center Grant to the Stanford Genome Technology Center. We also thank Dr Ann Power Smith of Write Science Right for editorial assistance.
References
- 1.Sheridan RL, Tompkins RG. What's new in burns and metabolism. J Am Coll Surg. 2004;198:243–263. doi: 10.1016/j.jamcollsurg.2003.11.007. [DOI] [PubMed] [Google Scholar]
- 2.Yu YM, Tompkins RG, Ryan CM, Young VR. The metabolic basis of the increase in energy expenditure in severely burned patients. JPEN J Parenter Enteral Nutr. 1999;23:160–168. doi: 10.1177/0148607199023003160. [DOI] [PubMed] [Google Scholar]
- 3.Barret JP, Herndon DN. Modulation of inflammatory and catabolic responses in severely burned children by early burn wound excision in the first 24 hours. Arch Surg. 2003;138:127–132. doi: 10.1001/archsurg.138.2.127. [DOI] [PubMed] [Google Scholar]
- 4.Padfield KE, Astrakas LG, Zhang Q, et al. Burn injury causes mitochondrial dysfunction in skeletal muscle. Proc Natl Acad Sci USA. 2005;102:5368–5373. doi: 10.1073/pnas.0501211102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brown JD, Plutzky J. Peroxisome proliferator-activated receptors as transcriptional nodal points and therapeutic targets. Circulation. 2007;115:518–533. doi: 10.1161/CIRCULATIONAHA.104.475673. [DOI] [PubMed] [Google Scholar]
- 6.Dressel U, Allen TL, Pippal JB, Rohde PR, Lau P, Muscat GE. The peroxisome proliferator-activated receptor beta/delta agonist, GW501516, regulates the expression of genes involved in lipid catabolism and energy uncoupling in skeletal muscle cells. Mol Endocrinol. 2003;17:2477–2493. doi: 10.1210/me.2003-0151. [DOI] [PubMed] [Google Scholar]
- 7.Genini D, Carbone GM, Catapano CV. Multiple interactions between peroxisome proliferator-activated receptors and the ubiquitin-proteasome system and implications for cancer pathogenesis. PPAR Res. 2008;2008:195065. doi: 10.1155/2008/195065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tzika AA, Mintzopoulos D, Padfield K, et al. Reduced rate of adenosine triphosphate synthesis by in vivo31P nuclear magnetic resonance spectroscopy and downregulation of PGC-1ß in distal skeletal muscle following burn. Int J Mol Med. 2008;21:201–208. [PubMed] [Google Scholar]
- 9.Spiegelman BM, Heinrich R. Biological control through regulated transcriptional coactivators. Cell. 2004;119:157–167. doi: 10.1016/j.cell.2004.09.037. [DOI] [PubMed] [Google Scholar]
- 10.Handschin C, Spiegelman BM. Peroxisome proliferator-activated receptor gamma coactivator 1 coactivators, energy homeostasis, and metabolism. Endocr Rev. 2006;27:728–735. doi: 10.1210/er.2006-0037. [DOI] [PubMed] [Google Scholar]
- 11.Puigserver P, Wu Z, Park CW, Graves R, Wright M, Spiegelman BM. A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell. 1998;92:829–839. doi: 10.1016/s0092-8674(00)81410-5. [DOI] [PubMed] [Google Scholar]
- 12.Kressler D, Schreiber SN, Knutti D, Kralli A. The PGC-1-related protein PERC is a selective coactivator of estrogen receptor alpha. J Biol Chem. 2002;277:13918–13925. doi: 10.1074/jbc.M201134200. [DOI] [PubMed] [Google Scholar]
- 13.Lin J, Puigserver P, Donovan J, Tarr P, Spiegelman BM. Peroxisome proliferator-activated receptor gamma coactivator 1beta (PGC-1beta), a novel PGC-1-related transcription coactivator associated with host cell factor. J Biol Chem. 2002;277:1645–1648. doi: 10.1074/jbc.C100631200. [DOI] [PubMed] [Google Scholar]
- 14.Lin J, Handschin C, Spiegelman BM. Metabolic control through the PGC-1 family of transcription coactivators. Cell Metab. 2005;1:361–370. doi: 10.1016/j.cmet.2005.05.004. [DOI] [PubMed] [Google Scholar]
- 15.Sandri M, Lin J, Handschin C, et al. PGC-1alpha protects skeletal muscle from atrophy by suppressing FoxO3 action and atrophy-specific gene transcription. Proc Natl Acad Sci USA. 2006;103:16260–16265. doi: 10.1073/pnas.0607795103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Parihar A, Parihar MS, Milner S, Bhat S. Oxidative stress and anti-oxidative mobilization in burn injury. Burns. 2008;34:6–17. doi: 10.1016/j.burns.2007.04.009. [DOI] [PubMed] [Google Scholar]
- 17.Yasuhara S, Perez ME, Kanakubo E, et al. Skeletal muscle apoptosis after burns is associated with activation of proapoptotic signals. Am J Physiol Endocrinol Metab. 2000;279:E1114–E1121. doi: 10.1152/ajpendo.2000.279.5.E1114. [DOI] [PubMed] [Google Scholar]
- 18.Astrakas LG, Goljer I, Yasuhara S, et al. Proton NMR spectroscopy shows lipids accumulate in skeletal muscle in response to burn trauma-induced apoptosis. FASEB J. 2005;19:1431–1440. doi: 10.1096/fj.04-2005com. [DOI] [PubMed] [Google Scholar]
- 19.Maass DL, Hybki DP, White J, Horton JW. The time course of cardiac NF-kappaB activation and TNF-alpha secretion by cardiac myocytes after burn injury: contribution to burn-related cardiac contractile dysfunction. Shock. 2002;17:293–299. doi: 10.1097/00024382-200204000-00009. [DOI] [PubMed] [Google Scholar]
- 20.Kim MS, Sweeney TR, Shigenaga JK, et al. Tumor necrosis factor and interleukin 1 decrease RXRalpha, PPARalpha, PPARgamma, LXRalpha, and the coactivators SRC-1, PGC-1 alpha, and PGC-1beta in liver cells. Metabolism. 2007;56:267–279. doi: 10.1016/j.metabol.2006.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mensink M, Hesselink MK, Russell AP, Schaart G, Sels JP, Schrauwen P. Improved skeletal muscle oxidative enzyme activity and restoration of PGC-1 alpha and PPAR beta/delta gene expression upon rosiglitazone treatment in obese patients with type 2 diabetes mellitus. Int J Obes. 2007;31:1302–1310. doi: 10.1038/sj.ijo.0803567. [DOI] [PubMed] [Google Scholar]
- 22.St-Pierre J, Lin J, Krauss S, et al. Bioenergetic analysis of peroxisome proliferator-activated receptor gamma coactivators 1alpha and 1beta (PGC-1alpha and PGC-1beta) in muscle cells. J Biol Chem. 2003;278:26597–26603. doi: 10.1074/jbc.M301850200. [DOI] [PubMed] [Google Scholar]
- 23.Weitzel JM, Iwen KA, Seitz HJ. Regulation of mitochon-drial biogenesis by thyroid hormone. Exp Physiol. 2003;88:121–128. doi: 10.1113/eph8802506. [DOI] [PubMed] [Google Scholar]
- 24.Psarra AM, Sekeris CE. Nuclear receptors and other nuclear transcription factors in mitochondria: regulatory molecules in a new environment. Biochim Biophys Acta. 2008;1783:1–11. doi: 10.1016/j.bbamcr.2007.10.021. [DOI] [PubMed] [Google Scholar]
- 25.Sugita H, Kaneki M, Sugita M, Yasukawa T, Yasuhara S, Martyn JA. Burn injury impairs insulin-stimulated Akt/PKB activation in skeletal muscle. Am J Physiol Endocrinol Metab. 2005;288:E585–E591. doi: 10.1152/ajpendo.00321.2004. [DOI] [PubMed] [Google Scholar]
- 26.Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell. 2006;124:471–484. doi: 10.1016/j.cell.2006.01.016. [DOI] [PubMed] [Google Scholar]
- 27.Zhang Q, Carter EA, Ma BY, White M, Fischman AJ, Tompkins RG. Molecular mechanism(s) of burn-induced insulin resistance in murine skeletal muscle: role of IRS phosphorylation. Life Sci. 2005;77:3068–3077. doi: 10.1016/j.lfs.2005.02.034. [DOI] [PubMed] [Google Scholar]
- 28.Puigserver P, Rhee J, Lin J, et al. Cytokine stimulation of energy expenditure through p38 MAP kinase activation of PPARgamma coactivator-1. Mol Cell. 2001;8:971–982. doi: 10.1016/s1097-2765(01)00390-2. [DOI] [PubMed] [Google Scholar]
- 29.Cline GW, Vidal-Puig AJ, Dufour S, Cadman KS, Lowell BB, Shulman GI. In vivo effects of uncoupling protein-3 gene disruption on mitochondrial energy metabolism. J Biol Chem. 2001;276:20240–20244. doi: 10.1074/jbc.M102540200. [DOI] [PubMed] [Google Scholar]
- 30.Desvergne B, Michalik L, Wahli W. Transcriptional regulation of metabolism. Physiol Rev. 2006;86:465–514. doi: 10.1152/physrev.00025.2005. [DOI] [PubMed] [Google Scholar]
- 31.Shen W, Hao J, Tian C, et al. A combination of nutriments improves mitochondrial biogenesis and function in skeletal muscle of type 2 diabetic Goto-Kakizaki rats. PLoS ONE. 2008;3:e2328. doi: 10.1371/journal.pone.0002328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cree MG, Zwetsloot JJ, Herndon DN, et al. Insulin sensitivity and mitochondrial function are improved in children with burn injury during a randomized controlled trial of fenofibrate. Ann Surg. 2007;245:214–221. doi: 10.1097/01.sla.0000250409.51289.ca. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cree MG, Newcomer BR, Herndon DN, et al. PPAR-alpha agonism improves whole body and muscle mitochondrial fat oxidation, but does not alter intracellular fat concentrations in burn trauma children in a randomized controlled trial. Nutr Metab. 2007;4:9. doi: 10.1186/1743-7075-4-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sener G, Sehirli AO, Gedik N, Dulger GA. Rosiglitazone, a PPAR-gamma ligand, protects against burn-induced oxidative injury of remote organs. Burns. 2007;33:587–593. doi: 10.1016/j.burns.2006.10.381. [DOI] [PubMed] [Google Scholar]
- 35.Yumuk VD. Targeting components of the stress system as potential therapies for the metabolic syndrome: the peroxisome-proliferator-activated receptors. Ann NY Acad Sci. 2006;1083:306–318. doi: 10.1196/annals.1367.019. [DOI] [PubMed] [Google Scholar]