

Abstract
Two recent studies mapped nucleosomes across the yeast and human genomes, teasing apart the relative contributions of DNA sequence and chromatin remodelers to nucleosome organization. These data suggest two emerging models: chromatin remodelers position nucleosomes around transcriptional start sites in yeast, while a few ‘locked’ nucleosomes may serve as barriers from which nucleosome arrays emanate in human genomes.
Packing eukaryotic genomes into high-order chromatin structures is critical for controlling most, if not all, processes derived from DNA. The minimal repeating unit of chromatin is the nucleosome, comprised of ∼147 basepairs wrapped around a histone octamer core (Richmond and Davey 2003). In comparison to ‘naked’ DNA, nucleosomal DNA is less accessible for DNA binding proteins, such as transcription factors, suggesting that the precise positioning and density of nucleosomes serves as a potent mechanism for controlling transcription and other DNA-templated processes (Li, Carey et al. 2007). It has been known for decades that nucleosomes are organized as non-random. regularly-spaced arrays, with the spacing between nucleosomes varying between different organisms and cell types (Van Holde 1988). However, only with the recent development of nucleosome mapping techniques on genome-wide scale has it become possible to determine global patterns of nucleosome positioning.
In past decade, several pioneering studies renewed interest in mapping nucleosomes by uncovering common themes of genomic nucleosome organization (Segal and Widom 2009). Using various model organisms, as well as human cells, these mapping studies found that nucleosome occupancy is relatively low at many enhancers, promoters, and transcription termination sites. Moreover, an array of highly-positioned nucleosomes surrounds transcription start sites (TSS) with positioning generally decreasing with distance from the TSS (Figure 1A). These findings suggest that non-random mechanisms promote the proper distribution of nucleosomes, which eventually allows for correct control of transcription initiation. Such mechanisms potentially include: intrinsic preference of histones for particular DNA sequences, statistical positioning (see below), competition with DNA binding proteins, post-translational modifications of histone, chromatin remodelers, the positioning of RNA polymerase (Pol), and higher-order chromatin folding.
Figure 1. Chromatin Remodelers and ‘Container’ Sequences in Nucleosome Organization.
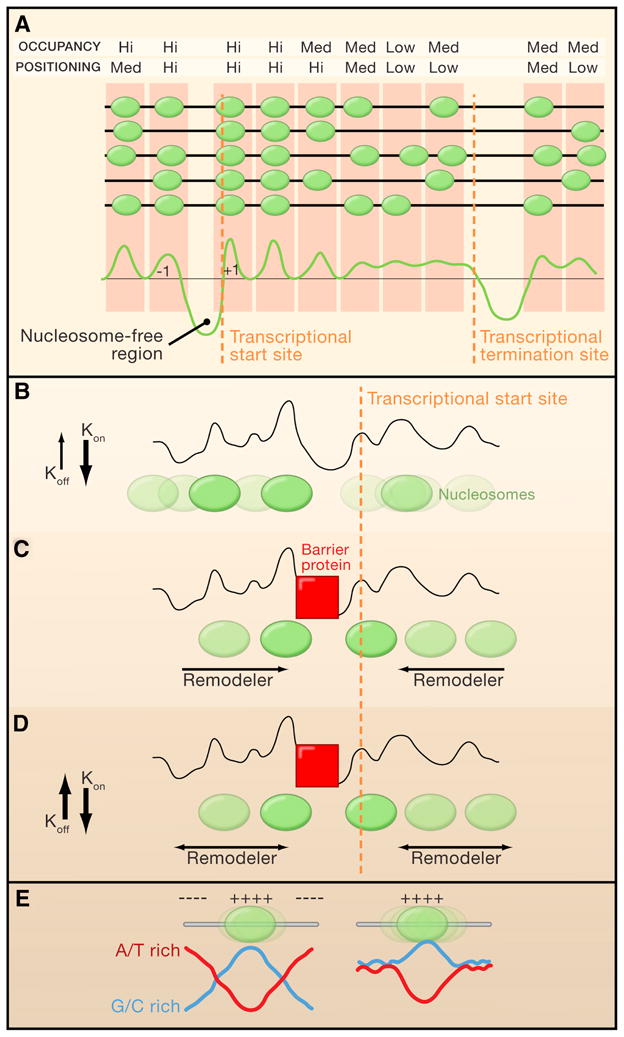
(A) Patterns of nucleosomes (green ellipses) at a hypothetical gene in Saccharomyces cerevisiae gene, with each dark grey line representing the same DNA sequence from different individual cells in the population. A graphical representation of the nucleosomes distribution is shown in the lower panel.
(B-D) Models for nucleosomes positioning by chromatin remodelers. The grey line represents a hypothetical sequence-based preference plot for nucleosome formation (higher peaks represent stronger positioning sequences). Color intensity directly correlates with positioning strength. Orange lines mark the position of a hypothetical transcriptional start sites (TSSs).
(B) Sequence preference. In the absence of additional nucleosome-positioning factors, DNA sequence preference is the only force driving nucleosome patterns. In this model, the high binding affinity between histones and DNA (high on rate Kon and low off rate Koff), restricts nucleosome fluidity.
(C) Directional remodeling. In the presence of cell extract and ATP, nucleasome-free regions are bound by DNA binding proteins that deplete nucleosomes and are likely to form functional barriers (red squares). In this model, chromatin remodelers directionally position nucleosomes against the barrier.
(D) Remodeler-induced fluidity. This model is the same as (C) except that remodelers increase nucleosome fluidity along the DNA, allowing statistical positioning of nucleosomes against the barrier (red squares).
(E) Schematic representation of a ‘container’ nucleosome positioning element. (++++) and (−−−−) mark sequences that favor or repel nucleosomes, respectively. The nucleosome on the left is strongly positioned because of the combination of a centered favoring sequence (G/C; rich-blue line) flanked by repelling sequence (A/T rich; red line). The nucleosome on the right is not strongly positioned because there are no repelling sequences around the favoring sequence.
One critical question is how do genomic sequences influence these patterns. Clearly, several general patterns of nucleosome occupancy (Figure 1A) depend significantly on the DNA sequence, especially the depletion of nucleosomes from transcription termination sites (TTSs) and to a lesser extent from TSSs (Kaplan, Moore et al. 2009). Nevertheless, nucleosome positioning (Figure 1A) seems to be less dependent on sequence (Zhang, Moqtaderi et al. 2009), supporting the idea that the precise location of a nucleosome in cells also involves on other cellular factors.
Nucleosome occupancy measures the density of nucleosomes at a specific genomic region in a population. Nucleosome positioning, in contrast, measures the extent to which a population of nucleosomes resists deviation from a specific location along DNA and describes the precise location of a nucleosome on DNA (Figure 1A). What sequence elements underlie nucleosome patterns? Studies in yeast have determined that tracts of adenosine-thymidine basepairs (i.e., poly dA:dT tracts), which are found in many yeast promoters, posses internal properties that deplete them of repressive nucleosomes, which in turn, enhances transcription in cells. Conversely elements with a high density of guanines-cytosine basepairs (i.e., G/C rich elements) tend to promote nucleosome formation (Iyer and Struhl 1995; Segal and Widom 2009). Another element purely based on DNA sequence is rotational positioning. DNA is wrapped around nucleosomes with dinucleotides of AA, TT, or TA approximately every 10 basepairs (i.e, a 10 basepair helical periodicity), reflecting these dinucleotides' tendency to optimize DNA bending by facing inwards to or outwards from the octamer core (Satchwell, Drew et al. 1986; Segal, Fondufe-Mittendorf et al. 2006; Zhang, Moqtaderi et al. 2009). Nevertheless, a more recent study from Zhang et al. (2011) showed that chromatin assembly in vitro fails to form arrays of highly positioned, evenly spaced nucleosomes that flank TSS, providing further support for the hypothesis that additional factors, beyond sequence, may play decisive roles in determining nucleosome positioning in vivo.
Statistical Positioning Theory
One model that is frequently used to explain how arrays of well-positioned nucleosomes form is statistical positioning (Fedor, Lue et al. 1988). This model relies on two key assumptions: 1) the existence of a ‘barrier’ element that locally prevents nucleosome formation and fluidity; and 2) that nucleosomes can freely move bidirectionally along barrier-free DNA. Statistical positioning predicts an array of positioned nucleosomes emanating from the barrier, decreasing in positioning with distance from the barrier simply because of the statistical probability of a nucleosome to occupy a certain position away from the barrier. Nucleosome-free regions by nature should potentially serve as barriers, yet statistical positioning of nucleosomes around nucleosome-free regions (both TSS and TTS) is not apparent in vitro. Moreover, in cells nucleosome phasing is more apparent around TSS than TTS, despite similar depletion of nucleosomes at both sites.
However, a few issues may impede the statistical positioning of nuclesomes in vitro: 1) nucleosomes-free regions might not serve as functional barriers, such that unstable nucleosomes may form on these region but fail to accumulate there due to their intrinsic instability; and 2) stable nucleosome on a preferred sequence could be relatively ‘locked’ in place due to a low dissociation rate. The second concern is supported by the observation that sequence largely guides assembly of nucleosome in vitro (Kaplan, Hughes et al. 2010; Zhang, Wippo et al. 2011; Zhang, Moqtaderi et al. 2009). However, chromatin assembly in the presence of the chromatin remodeler ACF can override sequence preference probably by increasing nucleosome fluidity (Zhang, Moqtaderi et al. 2009). One important role for nucleosome depletion around TSSs is to promote binding of non-histone proteins or complexes to DNA. It is possible that such binding is required to convert a nucleosomes-free region into an effective barrier, which in turn, allows statistical positioning of nucleosomes. Although this idea has not been directly tested on the genome-wide scale, binding of the GAGA transcription factor to a heat shock gene in Drosophila increases nucleosome positioning in vitro (Tsukiyama, Becker et al. 1994).
Yeast Genome: Remodelers Pack Nucleosomes Against Promoters
These issues led Zhang and colleagues (2011) to test what mechanisms beyond DNA sequence are responsible for nucleosome positioning around TSSs (Zhang, Wippo et al.). Specifically, whether ATP-dependent chromatin remodelers can override DNA sequence preference to reproduce patterns of nucleosomes as observed in cells. Four families of ATP-dependent chromatin remodelers are currently known. All families share a similar ATPase domain and use ATP hydrolysis to alter nucleosomes composition, structure, and positioning (Clapier and Cairns 2009). Moreover, specific remodelers have been shown to influence genome wide nucleosome positioning in vivo (Whitehouse, Rando et al. 2007; Hartley and Madhani 2009), and ATP was shown to be important for nucleosome positioning at a specific gene in vitro (Tsukiyama, Becker et al. 1994).
Zhang et al. assembled nucleosomes in vitro on yeast DNA in the presence of a crude cell extract as a source of chromatin-associated proteins. Yet, this extract failed to significantly improve nucleosome positioning around TSSs, suggesting that the concerted action of intrinsic DNA sequence elements and cognate DNA-binding proteins is not sufficient for promoting correct nucleosome positioning in vitro. Moreover, assuming that the cell extract allowed the formation of more rigid barriers due to the binding of protein complexes at nucleosome-free regions, this result supports the idea that sequence, rather than statistical positioning, likely guides nucleosomes positioning in vitro.
However, a breakthrough insight came when the researchers included ATP in the cell extract. Remarkably, this combination dramatically improved nucleosome positioning and phasing (i.e appearance of an array of evenly spaced nucleosome) around TSSs, strongly suggesting that ATP-dependent chromatin remodeling complexes are key factors for nucleosome positioning in yeast cells. Moreover, nucleosomes-free regions proximal to the TSS-proximal became significantly more pronounced and nearly indistinguishable from chromatin isolated from cells. These findings agree with previous studies that suggested a role for the RSC chromatin remodeling complex in nucleosome depletion at promoters (Hartley and Madhani 2009). Moreover, these findings support the general idea that ATP-dependent remodelers can override histones' preferences for particular DNA sequences establishing nucleosome positioning patterns that can, in turn, influence transcription (Cairns 2009).
Importantly, the ability of the cell extract and ATP to promote an in vivo-like nucleosome patterns was independent of other nucleotides, arguing that active transcription and DNA replication per se are not predominant mechanisms for controlling nucleosome organization in cells. Thus, together these data support a model in which DNA binding proteins, ATP-dependent chromatin remodeling complexes, and DNA sequences that promote nucleosomes-free regions (e.g., dA:dT tracts) act in concert to promote statistical positioning of nucleosomes.
Another prediction of the statistical positioning model is that the distance between adjacent nucleosomes should inversely correlate with nucleosome density. However, Zhang et al. found that employing different ratios of histones to DNA during chromatin assembly did not significantly alter the spacing between nucleosomes at TSSs, again arguing against statistical positioning.
Moreover, because it is reasonable to assume that nucleosomes are relatively free to move bidirectionaly between barriers in the presence of remodelers, statistical positioning should result in relatively uniform distribution of nucleosomes. Yet, at a lower histone to DNA ratio, nucleosomes were relatively more depleted from nucleosome-free regions and gene bodies compared to the +1 nucleosome position (i.e the first nucleosome downstream to the TSS). Together, these experiments led Zhang and colleagues to suggest the intriguing possibility that ATP dependent remodelers work directionally to pack nucleosomes against a functional barrier at promoters (Figure 1B, C).
One alternative explanation is that ATP-dependent remodelers promote fluidity of nucleosomes, which then allows statistical positioning of nucleosomes against functional barriers (that are cell extract dependent), without the need for directional remodeling (Figure 1D). This model is consistent with the finding that chromatin assembly in vitro by the ATP-dependent remodeler ACF shows less sequence-guided positioning than chromatin assembly by salt dialysis (Zhang, Moqtaderi et al. 2009). Inside cells, histones are constantly and rapidly exchanged at promoters, regardless of whether transcription is occurring or not (Dion, Kaplan et al. 2007). Thus, the rapid cycles of eviction and deposition of histones near promoters by ATP dependent machineries may also contribute to histone positioning. Regardless of the uncertainties in the precise mechanistic details, it is becoming clear that ATP-dependent chromatin remodelers play pivotal roles in nucleosome positioning around TSSs genome-wide.
Human Genome: “Container” Sequences Position Nucleosomes
How relevant are the rules in yeast to more complex mammalian genomes? Most likely, DNA sequences alone cannot solely determine nucleosome positioning in mammalian cells because classic studies in the chromatin literature demonstrated that different cells derived from the same organism (hence sharing a common genome) exhibit different average nucleosome spacing (Van Holde 1988). Now a new study by Valouve et al. (2011) brings us one step closer to understanding nucleosome pattern formation in primary human cells.
Using ultra deep sequencing, the authors characterized the dynamics of nucleosome positioning in granulocytes and CD4+ and CD8+ T cells from a single human donor. In agreement with previous observations, the spacing between nucleosomes was different between cell types. Moreover, internucleosomal linker DNA was shorter at actively transcribed genes compared to repressed genes in both T cells and granulocytes. In agreement with this finding, examination of different epigenetic chromatin states also revealed an intimate relationship between chromatin states and internucleosomal spacing. Specifically, mono-methylation of lysine 4 together with acetylation of lysine 27 on H3 (i.e., H3K4me1-H3K27ac), two histone modifications associated with euchromatic enhancers and active promoters, showed the shortest internucleosomal linker DNA (∼30bp) whereas nucleosomes associated with repressive, heterochromatic histone modifications (H3K9me3 and H3K27me3) showed the largest spacing (∼58bp).
Given the important role remodelers play in nucleosome organization (Zhang, Wippo et al.; Cairns 2009), one intriguing possibility is that different combinations of histone post-translational modifications recruit specific remodelers that in turn, enforce particular nucleosome patterns. In support of this hypothesis, BPTF (bromodomain PHD finger transcription factor), a subunit of the NURF ATP-dependent chromatin remodeling complex, is recruited to chromatin by the combination of H3K4me2/3 together with H4K16ac(Ruthenburg, Li et al.). Additionally, changes in the levels of linker histone H1 can also affect spacing (Fan, Nikitina et al. 2005). Similar to yeast cells, nucleosomes purified from human cells showed distinct nucleosome phasing, whereas the in vitro reconstituted nucleosomes did not (Valouev, Johnson et al.). However, the lack of phasing observed in vitro could be due, in part, to the low ratio of histone to DNA used for the nucleosome reconstitution.
To examine how DNA sequences contribute to nucleosome positioning, Valouev and colleagues focused on a relatively small subset of highly positioned nucleosomes. Interstingly, they could not detect the 10 basepair periodicity of dinucleotides found in yeast nucleosomes. Rather, they found that DNA sequences at the center of highly positioned nucleosomes were enriched for G/C nucleotides whereas the flanking sequences were more A/T rich. This led to a model in which nucleosome ‘repelling’ sequences (A/T rich) encompass nucleosome-favoring sequences (ie., G/C rich) to serve as a ‘container’ that promotes the accurate positioning of a nucleosomes (Figure 1E). Importantly, Valouev et al. estimated that < 20% of the nucleosomes are highly positioned in vivo, suggesting a significant, but limited, contribution of container sequences to the overall nucleosome pattern. However, because ‘container nucleosomes’ are strongly positioned, they can potentially serve as functional barriers to promote sequence independent positioning of adjacent nucleosomes by statistical positioning or by ATP-dependent chromatin remodelers.
Thus, a global view is emerging that chromatin purified from cells, but not when assembled in vitro, exhibits an array of relatively well-positioned nucleosomes emanating from strongly positioned container nucleosomes. If container nucleosomes can function as a barrier, the fact that they do not promote nucleosome phasing in vitro argues against barrier-induced statistical positioning. It will be interesting to test whether the addition of chromatin remodelers can reconstitute the in vivo nucleosome positioning patterns seen around “container” nucleosomes.
Whereas DNA sequence rich with G/C basepairs promote nucleosome occupancy in vitro, Valouev et al. found that in cells CpG tetranucleotides are relatively depleted in nucleosomes, and CpG islands (i.e genomic regions that contain high frequency of CpG dinucleotides) are relatively nucleosome free (Valouev, Johnson et al.). This raises the intriguing possibility that CpG islands may function in human cells similarly to poly dA:dT tracts in yeast to promote nucleosome depletion at promoters, although by different mechanisms (i.e in a sequence independent for CpG islands versus a sequence dependent manner for dA:dT tracts). It is possible that the combined action of chromatin remodelers and sequence specific binding proteins, which compete with nucleosomes for binding to specific DNA sequences, promote nucleosome depletion at CpG rich promoters. Such protein-bound, nucleosome-depleted promoters can in turn serve as functional barriers to promote positioning of nucleosome.
In agreement with this hypothesis, the binding pattern of poised RNA polymerase II at active promoters suggests that it could potentially serve as a barrier for nucleosome positioning around TSSs. Moreover, the binding sites for the chromatin insulator CTCF and the transcriptional repressor NRSF/REST are depleted of nucleosomes and flanked by an array of highly positioned nucleosomes only in chromatin purified from cells, but not from in vitro reconstituted chromatin. Here again, it will be of interest to examine directly the contribution of ATP-dependent chromatin remodelers to nucleosome positioning in human cells. Similar to the findings by Zhang and colleagues in yeast cells, the data from Valouev and colleagues support the hypothesis that additional factors beyond DNA sequence and barrier-induced statistical positioning play pivotal roles in determining nucleosome positioning in human cells.
One key question is what is the biological relevance of the variation observed in global nucleosome organization (reflected by changes in the length of internucleosomal linker DNA) between cell types and functional chromatin elements? Specific remodelers can space nucleosomes differently, and the precise remodeler signature can influence H1 deposition (Lusser, Urwin et al. 2005). It is intriguing to speculate that different linker lengths are the result of nucleosomes positioned by various chromatin remodelers. Thus, changes in DNA binding factors may translate into changes in the epigenetic landscape, dictated by modifications in DNA and histone proteins. These epigenetic landscapes, in turn, recruit specific remodelers that impose nucleosome patterns, which then either favor the activation or the repression of transcription. It will be interesting to see if this general view is supported by computational analysis of histone and DNA modifications, nucleosome positioning, and binding sites of transcriptional enhancers and repressors. In addition, it is also possible that certain nucleosomal rearrangements are either compatible with or repressive towards transcription by RNA Pol II, and such nucleosome patterns may play additional roles in transcriptional regulation such as defining exon boundaries (Kornblihtt, Schor et al. 2009) and prevention of cryptic transcription. More research is needed in order to address these key outstanding questions, but as in real estate, it seems in chromatin, what matters is “location, location, location.”
Acknowledgments
“We dedicate this piece to the memory of Professor Jon Widom, whose work guided this field forward in ways that will have lasting impact. His wisdom and spirit will be missed but not forgotten.”
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Cairns BR. Nature. 2009;461(7261):193–198. doi: 10.1038/nature08450. [DOI] [PubMed] [Google Scholar]
- Clapier CR, Cairns BR. Annu Rev Biochem. 2009;78:273–304. doi: 10.1146/annurev.biochem.77.062706.153223. [DOI] [PubMed] [Google Scholar]
- Dion MF, Kaplan T, et al. Science. 2007;315(5817):1405–1408. doi: 10.1126/science.1134053. [DOI] [PubMed] [Google Scholar]
- Fan Y, Nikitina T, et al. Cell. 2005;123(7):1199–1212. doi: 10.1016/j.cell.2005.10.028. [DOI] [PubMed] [Google Scholar]
- Fedor MJ, Lue NF, et al. J Mol Biol. 1988;204(1):109–127. doi: 10.1016/0022-2836(88)90603-1. [DOI] [PubMed] [Google Scholar]
- Hartley PD, Madhani HD. Cell. 2009;137(3):445–458. doi: 10.1016/j.cell.2009.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer V, Struhl K. EMBO J. 1995;14(11):2570–2579. doi: 10.1002/j.1460-2075.1995.tb07255.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan N, Hughes TR, et al. Genome Biol. 2010;11(11):140. doi: 10.1186/gb-2010-11-11-140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan N, Moore IK, et al. Nature. 2009;458(7236):362–366. doi: 10.1038/nature07667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornblihtt AR, Schor IE, et al. Nat Struct Mol Biol. 2009;16(9):902–903. doi: 10.1038/nsmb0909-902. [DOI] [PubMed] [Google Scholar]
- Li B, Carey M, et al. Cell. 2007;128(4):707–719. doi: 10.1016/j.cell.2007.01.015. [DOI] [PubMed] [Google Scholar]
- Lusser A, Urwin DL, et al. Nat Struct Mol Biol. 2005;12(2):160–166. doi: 10.1038/nsmb884. [DOI] [PubMed] [Google Scholar]
- Richmond TJ, Davey CA. Nature. 2003;423(6936):145–150. doi: 10.1038/nature01595. [DOI] [PubMed] [Google Scholar]
- Ruthenburg AJ, Li H, et al. Cell. 145(5):692–706. doi: 10.1016/j.cell.2011.03.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satchwell SC, Drew HR, et al. J Mol Biol. 1986;191(4):659–675. doi: 10.1016/0022-2836(86)90452-3. [DOI] [PubMed] [Google Scholar]
- Segal E, Fondufe-Mittendorf Y, et al. Nature. 2006;442(7104):772–778. doi: 10.1038/nature04979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal E, Widom J. Curr Opin Struct Biol. 2009;19(1):65–71. doi: 10.1016/j.sbi.2009.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal E, Widom J. Trends Genet. 2009;25(8):335–343. doi: 10.1016/j.tig.2009.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukiyama T, Becker PB, et al. Nature. 1994;367(6463):525–532. doi: 10.1038/367525a0. [DOI] [PubMed] [Google Scholar]
- Valouev A, Johnson SM, et al. Nature. 474(7352):516–520. doi: 10.1038/nature10002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Holde KE. Chromatin. New York: Springer-Verlag; 1988. [Google Scholar]
- Whitehouse I, Rando OJ, et al. Nature. 2007;450(7172):1031–1035. doi: 10.1038/nature06391. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Moqtaderi Z, et al. Nat Struct Mol Biol. 2009;16(8):847–852. doi: 10.1038/nsmb.1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Wippo CJ, et al. Science. 2011;332(6032):977–980. doi: 10.1126/science.1200508. [DOI] [PMC free article] [PubMed] [Google Scholar]