

Abstract
T cell hyperactivation and complement consumption are prominent features of the immunopathology of systemic lupus erythematosus. Although complement activation is secondary to autoantibodies that form immune complexes (ICs), the trigger for alterations in human peripheral blood T cells is poorly understood. To study the impact (on T cells) of several types of preformed ICs and terminal complement complex, also referred to as C5b-9, we incubated these immune reactants with peripheral blood naive CD4+ T cells as well as Jurkat cells and analyzed their effects on cellular behavior. We first assembled the C5b-9 in situ on the membrane and observed its assembly primarily on a single site where it promoted aggregation of membrane rafts and recruitment of the CD3 signaling complex. However, C5b-9 alone did not initiate proliferation or commencement of downstream signaling events associated with T cell activation. When T cells were treated with ICs together with nonlytic C5b-9, changes associated with T cell activation by possible antigen engagement then occurred. T cell antigen receptor signaling proteins, including ζ-chain, ZAP-70, Syk, Src, and Lck, were phosphorylated and organized in a synapse-like structure. The cytoskeleton formed F-actin spindles and a distal pole complex, resulting in a bipolar distribution of phosphorylated ezrin-radixin-moesin and F-actin. Furthermore, ICs and nonlytic C5b-9 induced T cell proliferation and IFN-γ production. These results raise the possibility that ICs and the nonlytic C5b-9 modulate T cell-mediated responses in systemic lupus erythematosus and other related chronic inflammatory disorders.
Keywords: Complement System, Cytoskeleton, Interferon, Signaling, T Cell
Introduction
T cell activation is the first step for the generation of effector T cells. The contact between peptide-MHC on antigen-presenting cells (APCs)2 with the T cell antigen receptor (TCR) on naive or antigen-experienced cells leads to the formation of filamentous actin (F-actin) and organizes the microtubule cytoskeleton to transport intracellular vesicles, cell surface receptors, and co-stimulatory molecules forming the immunological synapse (IS). Such structures are also formed on NK cells and CD8+ T lymphocytes to initiate their effector functions (1, 2). Thus far, formation of such structures with multiple protein complexes of disease plasma has not been reported.
The complement system is a key participant in the innate immune-mediated inflammatory responses (3). Complement opsonized immune complexes (ICs) are sequestered by follicular dendritic cells in lymphoid follicles. Uptake of these ICs by follicular dendritic cells enhances antigen presentation to T follicular helper (TFH) cells in germinal centers and lowers the threshold for B cell activation, resulting in terminal differentiation of long lived plasma cells and memory B cells (4–7). Although the modulation of T cell responses by the complement system has been suggested, a role for ICs and complement in directly affecting T cell activation has not been reported previously (8, 9).
In IC-mediated diseases, infections, and rheumatic diseases, complement activation triggers sequential assembly of the terminal complement C5b, C6, and C7 (C5b-7), which then inserts into the membrane and subsequently binds to C8 and C9 to form trans-membrane channels (10). Cells resist C5b-9-mediated lytic death by its removal from the membrane by exocytosis within minutes of its deposition, leaving nonlytic C5b-9 on the cell membrane (11, 12). In addition, C5b-9 in the plasma binds to clustrin and vitronectin to form cytolytically inactive C5b-9.
Cytolytically inactive C5b-9 induces expression of adhesion molecules, cytokine production, and vascular leakage in endothelial cells (13, 14). Also, deposition of nonlytic C5b-9 is more common on host cells and is considered a more physiologically relevant event that triggers a number of signaling events such as the following: 1) synthesis of eicosanoids, interleukin (IL)-1β, and tumor necrosis factor (TNF)-α; 2) platelet activation; 3) release of growth factors; 4) increase in diacylglycerol and ceramides; 5) activation of membrane phospholipases, and 6) apoptosis (15–20). These studies suggest that in the state of moderate complement activation, the nonlytic C5b-9 acts as a signaling protein complex. This led us to explore the role of nonlytic C5b-9 in T cell activation.
In this study, we explore the role of nonlytic C5b-9 purified from serum and in situ C5b-9 assembled from purified late complement proteins in T cell activation. The in situ C5b-9 represents both lytic and nonlytic C5b-9, where lytic C5b-9 is removed by endocytosis or ectocytosis leaving nonlytic C5b-9 on the membrane (11, 21, 22).
ICs that activate the complement system are observed in SLE patients along with hyperactivation of T cells associated with an increase in calcium levels (23). Freshly isolated T cells from SLE patients demonstrate aggregation of membrane rafts (MRs) that may contribute to T cell activation (24). We previously observed fully assembled C5b-9 on ICs in SLE patients (25). In addition, we observed the presence of low affinity FcγRIIIA on CD4+ T cells. This led us to investigate interactions among nonlytic C5b-9, ICs, and human naive CD4+ T cells.
Here, we show a phenomenon that has not been observed previously, C5b-9 forms a synapse on human naive CD4+ T cells. This synapse facilitates the deposition of ICs, which leads to co-receptor engagement and downstream signaling in T cells such as phosphorylation of Lck, ζ-chain, ZAP-70, and Src in microclusters. During this process, T cells polarize by sequestering of the TCR signaling proteins and organizing cytoskeletal elements in a supramolecular activation cluster-like structures (26). Cells treated with ICs and nonlytic C5b-9 demonstrated the production of IFN-γ and cell proliferation. In vivo such events may contribute to the disease progression. This study provides the first link among ICs, complement activation and T cells. The T cell activation from C5b-9 and ICs by altering TCR signal strength may circumvent checkpoints that maintain peripheral immune tolerance (27).
EXPERIMENTAL PROCEDURES
Tissue culture media, fetal bovine serum (FBS), and culture supplies were from Invitrogen. We purchased antibodies from several vendors, including Cell Signaling Technologies (phospho-Src Tyr-416, phospho-SykTyr-525/526, and phospho-Zap-70Tyr-319), Santa Cruz Biotechnology, Santa Cruz, CA (phospho-LckTyr-192 and CD3ζ chain), Sigma (phosphotyrosine, clone PY20), and DakoCytomation, Denmark (neo-epitope of C9, clone aE11). Secondary conjugates were from Jackson ImmunoResearch, and Alexa Fluor® 488 and Alexa Fluor® 594 labeling kits were from Invitrogen. The purified complement proteins C5b-6, C7, C8, and C9 were from Complement Technologies (Tyler, TX), and ovalbumin, anti-ovalbumin, and other reagents were from Sigma or Fisher.
Human Naive CD4+ T Cells and Cell Lines
Jurkat cells (J.RT3) were obtained from American Type Culture Collection. They were propagated in RPMI supplemented with 10% FBS. Human naive CD4+ cells were isolated from peripheral blood lymphocytes. The peripheral blood lymphocyte fraction was separated from 20 ml of heparinized blood using Histopaque (Sigma). The adherent monocytes were removed by plating the cells in a Nunc culture dish at 37 °C in 5% CO2 for 12 h. The nonadherent cell population was labeled using a mixture of antibody mixture from naive CD4+ T cell isolation kit (Miltenyi Biotec, Germany). The naive CD4+ T cells were then separated using a magnetic assisted cell separation column. The purity of the isolated cells was analyzed using flow cytometry by staining for CD4 and CD45RA (naive CD4+ T cells markers). These cells were 93–96% pure based on the CD4+ CD45RA+ staining. The purified cells were over 97% viable in all the preparation using vital dye trypan blue.
Preparation of Ovalbumin-Anti-ovalbumin ICs (Ova-ICs)
Affinity-purified anti-ovalbumin was mixed with purified chicken ovalbumin at a molar ratio of 1:0.5 and 1:4 in 250 μl of phosphate-buffered saline (PBS). The mixture was kept at 37 °C for 1 h and then at 4 °C overnight. This mixture was then centrifuged at 15,000 × g for 30 min, and the supernatant was collected. Protein estimation was carried out, and the Ova-ICs were then stored at −70 °C.
Purification of ICs from SLE Patients
The ICs from SLE patients (SLE-ICs) were purified from plasma using an affinity matrix as described previously (25, 28). In brief, 1.5 ml of plasma was passed over the affinity resin under gravity at room temperature (RT); thereafter, the resin was washed with 25 column volumes of PBS. During the wash step, resin was stirred multiple times, and proteins were monitored in the eluate. ICs were eluted using 0.1 m glycine-HCl (pH 3.5). The eluted material was neutralized using 1 m Tris base. High salt from the ICs preparation was removed by buffer exchange, and ICs were then stored in aliquots at −70 °C.
Purification of Fluid Phase Pre-assembled C5b-9
Fluid phase C5b-9 was isolated from pooled normal sera as described previously (29, 30). In brief, 200 mg of zymosan A was added to 20 ml of serum, mixed, and incubated at 37 °C for 1 h. The C5b-9 was then precipitated by adding 7% polyethylene glycol 6000 weight by volume, and the mixture was incubated overnight at 4 °C. C5b-9-containing fractions were then pelleted by centrifugation at 43,000 × g for 45 min. The pellet was washed again with 7% polyethylene glycol. The precipitate was dissolved in PBS, and the protein content was measured. Thereafter, the mixture was fractionated over a SuperoseTM 6 YK column (GE Healthcare). The fractions were analyzed for the presence of polymerized C9 by monitoring the generation of C9 neo-epitope using a monoclonal antibody (clone aE11) in an ELISA system. The fractions containing C5b-9 were pooled and concentrated, and the protein was estimated. The preparation was stored at −70 °C. The nonlytic dose for C5b-9 was determined by incubating 1 × 106 Jurkat cells with 0.25, 1.25, 2.5, and 5 μg of protein and measuring apoptosis and necrosis using Vybrant Apoptosis Assay 3 (Eugene, OR) as per the manufacturer's suggested protocol. This titration was also carried out in the presence of 2.0 and 5.0 μg of Ova-IC. At a concentration of 2.5 μg of C5b-9 in the presence of ICs after a 2-h interval, cells remain viable. Subsequently, we used 2.5 μg of C5b-9 (nonlytic) in the presence of 1 μg of purified ICs in all experiments unless otherwise noted.
Localization of C5b-9 on Naive CD4+ T Cells and Jurkat Cells Using Alexa Fluor® 594-labeled C9
Purified C9 was labeled with Alexa Fluor® 594 as per the manufacturer's recommendation. For in situ deposition of the C5b-9, a total of 2 × 106 cells were washed with RPMI and plated onto 24-well culture plates. After a 4-h incubation in plain RPMI, the cells were incubated with purified C5b-6 (2 μg), C7 (1 μg), C8 (2 μg), and C9-Alexa Fluor® 594 (2.5 μg) to form C5b-9 (in situ). These conditions were used for in situ deposition of C5b-9 in all subsequent experiments. Cell aliquots at 0.75, 1.5, 3, 4, and 24 h were removed, washed with cold PBS, and fixed in 3% formaldehyde for 30 min and then mounted on slides for examination. The cells with C5b-9 deposits remained viable as observed with trypan blue dye prior to fixation. The viability was also assessed using Vybrant assay kit (Invitrogen). The cells were observed for the formation of C5b-9 using a confocal and fluorescent microscope. The percentages of cells with deposits were calculated by counting the cells with and without C5b-9 deposits from at least three fields in three independent experiments. The images were captured using both ×200 and 630.
Co-localization of Ova-ICs and AHG with in Situ C5b-9 Deposits on Naive CD4+ T Cells and Jurkat Cells
To co-localize ICs and C5b-9, Jurkat or naive CD4+ T cells were treated with labeled Ova-ICs or AHG, 5 μg of total protein for 1 × 106 cells during assembly of in situ C5b-9. Control cells were treated with equivalent amounts of labeled ICs or labeled C9. Ova-ICs were formed in vitro by incubating the ovalbumin with anti-ovalbumin antibody labeled with Alexa Fluor® 488 (Invitrogen). AHG was also labeled with Alexa Fluor® 488. The Ova-ICs with antibody to antigen molar ratio of 1:4 were used for membrane deposition, because this ratio demonstrated the most consistent phosphorylation of proteins of the TCR pathway in the Western blot analysis. After treatment of cells with Ova-ICs or AHG and in situ C5b-9, cells were washed and images were captured using either fluorescent or confocal microscope.
Co-localization of MRs and C5b-9 on Human Naive CD4+ T Cells
The cells were starved for 4 h in plain RPMI, before treatment. Human naive CD4+ cells or Jurkat cells were treated with either nonlytic C5b-9 or in situ C5b-9 in the presence of SLE-ICs (2 μg) or Ova-ICs (2 μg). Post-treatment, the cells were harvested and washed with cold PBS and resuspended in 0.1% BSA/PBS. To 1 × 106 cells, a total of 0.2 μg of cholera toxin B (CTB) conjugated with FITC was added and allowed to incubate for 20 min in an ice bath (31). In control cells during the in situ C5b-9 formation, C8 protein was omitted. Thereafter, these cells were washed and mounted on slides using SlowFade Gold antifade reagent with DAPI. These cells were then examined as described previously. The cell images were captured at ×400 and 630.
Staining for F-actin and Phosphorylated Ezrin, Radixin, and Moesin (pERM)
To observe the cytoskeletal polarization, we stained cells for F-actin and pERM (32). In 24-well plates, 2 × 106 cells were treated with 2 μg of purified SLE-ICs or Ova-ICs with 2.5 μg of nonlytic C5b-9 for 2 h. After a 2-h treatment, the medium was replaced with RPMI supplemented with 10% FBS and incubated at 37 °C in 5% CO2. For staining, the cells were harvested at 4- and 12-h intervals. The cells were washed with cold PBS and fixed in cold 3% formaldehyde prepared in PBS for 10 min and then permeabilized for 10 min by keeping in 90% ice-cold methanol at −20 °C. After washing again, the cells were stained using a 1:200 dilution of anti-pERM antibody prepared in blocking buffer obtained from Cell Signaling Technologies (Danvers, MA) for 1 h. After washing two times with PBS, the cells were again washed and treated at RT for 1 h with 1:100 dilution of anti-rabbit Alexa Fluor® 594 prepared in blocking buffer. Following pERM staining, the cells were stained with phalloidin-Alexa Fluor® 488, according to the manufacturer's recommendation. The cells were observed with the appropriate filter in the fluorescent microscope, and images were captured at ×200, 400, or 630. For scoring cells with cytoskeletal changes, images from three fields were examined in each experiment. The percentages of cells exhibiting the changes were calculated by counting total cells and cell with changes.
Co-localization of CD3 with in Situ C5b-9 Deposits on Naive CD4+ T Cells and Jurkat Cells
Cells were treated with purified complement proteins C5b-6, C7, C8, and Alexa Fluor® 594-labeled C9 to form in situ C5b-9. These cells were fixed with 3% formaldehyde for 15 min at RT. For detection of CD3, after two washes cells were incubated with anti-CD3-FITC conjugate in 1% BSA/PBS for 30 min. The cells were washed and mounted on a slide and examined for C5b-9 and CD3 with a fluorescent microscope, and images were captured at ×200 and 630.
Confocal Microscopy
Fluorescence was analyzed using an Olympus confocal laser-scanning microscope (Olympus FV1000, BX61). Multiple channel imaging was employed to analyze conjugates emitting fluorescence at wavelengths of 405, 488, and 594 nm. To visualize the cells, we used DAPI for staining nuclei and differential interference contrast (DIC) image. Cellular distribution of proteins in cells was observed by capturing images in horizontal optical sections from 0.22 to 0.55 μm in vertical steps throughout the whole height of the representative cell. In some circumstances, the optical zoom was used to visualize and capture the cellular details in the images.
Tyrosine Phosphorylation of TCR Signaling Proteins
Briefly, 1 × 106 Jurkat cells were washed and suspended in 1 ml of RPMI and plated in 24-well culture plates. Cells were then starved for 4 h in plain RPMI prior to treatment. These cells were treated with nonlytic C5b-9 alone, ICs alone, or in combination with the ICs and nonlytic C5b-9. Purified nonlytic C5b-9 (2.5 μg) and ICs purified from two SLE patients at two concentrations of SLE-ICs, one at 1 μg and the other at 4.5 μg, were used. In addition, Ova-ICs at antibody to antigen molar ratios of 1:0.5 designated as Ova-IC(1) and molar ratio of 1:4 designated as Ova-IC(2) were also used. These ratios were selected to generate ICs in either antibody or antigen excess (33, 34). At the end of the incubation period, cells were removed from the culture dish, washed with ice-cold PBS, and lysed with buffer containing 20 mm Tris (pH 8.0), 137 mm NaCl, 5 mm Na2EDTA, 10% (v/v) glycerol, 1% (v/v) Triton X-100, 1 mm EGTA, 10 mm sodium fluoride, 1 mm PMSF, 1 mm aprotinin, 1 mm leupeptin, and 10 mm Na3VO4. The cell lysates were vortexed, briefly sonicated, and then subjected to centrifugation at 13,500 rpm in a microcentrifuge (Eppendorf 5424). The protein quantities were estimated using a commercial protein assay kit (Bio-Rad), and a total of 50 μg of protein was mixed with SDS-PAGE 4× loading buffer containing 50 mm DTT. The samples were electrophoresed using NuPAGE 4–12% gradient gel and transferred to Immobilon PVDF membrane (Millipore). The blots were incubated with blocking buffer composed of 3% BSA at RT. Thereafter, blots were washed with Tris-buffered saline containing Tween 20 (TBS-T), and the membranes were probed with anti-phosphotyrosine monoclonal antibody (PY20) followed by anti-mouse-HRP antibody, and the blots were developed with chemiluminescent substrate (Millipore).
Western Analysis of Lck, Syk, Src, and Zap-70
To investigate the activation of the TCR complex, the membrane blots prepared as above were probed for phospho-Lck (Tyr192/Ser194), phospho-Syk (Tyr525/526), phospho-Src (Tyr416), and phospho-Zap-70 (Tyr319). We used primary antibodies at a dilution of 1:5000 in 3% BSA prepared in TBS-T. Next, the blots were treated with an appropriate dilution of the secondary antibodies conjugated to HRP enzyme, and blots were developed as above. The same blots were re-probed using anti-pSyk, anti-pLck, anti-pSrc, and anti-pZap-70. After Western blot analysis, the membranes were stripped and stained with Coomassie Blue R-250 stain, and the stained membranes were scanned, and images were analyzed using ImageJ software (National Institutes of Health, Bethesda) to compare the intensity of protein loading for each lane.
Co-localization of TCR Signaling Proteins
The purified naive CD4+ T cells were plated in RPMI with 10% FBS supplemented with 50 ng/ml of IL-2 and allowed to grow for 2 days. Thereafter, the cells were harvested, washed two times with plain RPMI 1640 medium, and starved for 4 h. Naive CD4+ T cells (1 × 106) were treated with 2.5 μg/ml nonlytic C5b-9 in the presence of 1 μg/ml ICs. The cells were harvested at 1- and 2-h intervals, washed twice with cold PBS, and fixed for 15 min with 3% paraformaldehyde in PBS at RT. After washing, cells were permeabilized as described previously. The cells were then washed and treated for 1 h with blocking buffer composed of 1% BSA and 5% species-specific serum. These cells were then treated overnight with an appropriate dilution of primary antibodies at 4 °C. Subsequently, cells were stained with appropriate secondary antibody conjugated with either Alexa Fluor® 488 or Alexa Fluor® 594 diluted in blocking buffer. After 1 h of incubation, cells were washed three times and mounted on glass slides with antifade reagent containing DAPI from Invitrogen. The cells were examined in a minimum of three fields in every experiment. Confocal imaging was performed as described earlier using the Olympus Fluoview 1000 microscope.
Cell Proliferation
Cells were loaded using carboxyfluorescein diacetyl succinimidyl ester (CFSC) as per the manufacturer's suggested protocol (Invitrogen). Immediately after CFSC loading, cells were kept in RPMI supplemented with 10% FBS for 30 min at 37 °C. These cells were then washed three times with 15 ml of plain RPMI and incubated further for 4 h. 1 × 106 cells were then treated with 2.5 μg of nonlytic C5b-9, 1 μg of SLE-ICs, or a combination of C5b-9 and ICs for 2 h in plain RPMI. Thereafter, 50,000 cells were plated in an individual well of a 48-well culture plate with RPMI supplemented with 10% FBS. The cells were harvested at 24 and 48 h and analyzed in flow cytometer (SLRII, BD Biosciences) for carboxyfluorescein diacetate succinimidyl ester dilution. For positive control, 1 × 106 cells/1 ml were stimulated using 5 μl of phytohemagglutinin (Invitrogen). The data were analyzed using proliferation node of FlowJo software (Treestar, Ashland, OR). The dead cells were excluded from the analysis by SSC and FSC gating.
Measurement of IFN-γ
Purified naive CD4+ T cells at a density of 1 × 106/ml were starved in plain RPMI for 4 h. Then 1 ml of cells were treated with 2.5 μg of nonlytic C5b-9, 1 μg of SLE-ICs, or a combination of nonlytic C5b-9 and ICs for 2 h. Control cell did not receive any treatment. After treatment, the media were replaced with complete media supplemented with 20 ng of IL-2, and 0.5 × 106 cells were seeded in 96-well plate. For positive control, 1 μg of plate-bound anti-CD3 and 0.5 μg of anti-CD28 antibodies were used. The supernatants were harvested at 48-h intervals for IFN-γ measurement. IFN-γ was measured using an ELISA kit as per the manufacturer's recommendation (PeproTech).
RESULTS
General Experimental Approach
Purified human peripheral blood naive CD4+ T cells or Jurkat cells were treated with nonlytic C5b-9 and ICs. In addition, in situ C5b-9 was assembled using purified complement proteins. Two types of ICs, including the in vitro-formed Ova-ICs or SLE-ICs, were used to treat both cell types in combination with either nonlytic or in situ C5b-9. AHG was also utilized as IC substitute. The effect of these two players, ICs and C5b-9, were characterized relative to events associated with T cell activation and IS formation, including mobility of MRs, cytoskeletal changes, phosphorylation of TCR signaling proteins, cell proliferation, and IFN-γ production. Similar results were obtained with all three types of ICs and two forms of C5b-9.
Pattern of C5b-9 Deposition
To address a possible role of nonlytic C5b-9 in T cell activation, we assembled the in situ C5b-9 on naive CD4+ T cells and Jurkat T cells using purified human terminal complement pathway proteins (C5b-6, C7, C8, and Alexa Fluor® 594-labeled C9). We observed that in both cell types over a period of 1–2 h, the deposited C5b-9 formed a single large cluster on the cell membrane (Fig. 1A and supplemental Video 1). C9 alone or the assembly of in situ C5b-9 in the absence of C8 did not demonstrate this pattern (Fig. 1B, left panel, and supplemental Video 2). The in situ C5b-9 could be visualized as early as 45 min from the start time of treatment of cells with complement proteins. A total of 30–50% of the cells showed C5b-9 deposition as scored by examination of three independent optical fields (Fig. 1B). By 3–4 h, these deposits had grown substantially, occupying ∼5–15% of the cell membrane (Fig. 1A, right panel). All initial experiments were done using Jurkat cells, and afterward these findings were also confirmed using naive CD4+ T cells.
FIGURE 1.

A, in situ C5b-9 assembled from purified complement proteins (image captured at ×630 and enlarged in Photoshop). C5b-9 deposit formed at a single site on CD4+ T cell (red). Time was 45 min (left panel) and 3 h (right panel) after addition of complement proteins. Image represents one of five experiments. B, in situ C5b-9 shown on multiple naive CD4+ T cells at 90-min intervals. From left to right, cells were treated with C9 alone (left panel); C5b-9 formed with purified C5b-6, C7, C8, and labeled C9 (2nd from left), C5b-9 with phase contrast image of cells (2nd from right) at ×200. Confocal DIC image of cells showing C5b-9 deposit captured at ×600 with 2.3× optical zoom (right panel). Arrows point to the in situ C5b-9 deposits. Image represents 1 of 10 experiments.
Co-localization of ICs with in Situ C5b-9
To assess the effect of ICs on in situ C5b-9 deposition, we followed in parallel the binding of Ova-ICs labeled with Alexa Fluor® 488. The simultaneous treatment of cells with labeled Ova-ICs and in situ-formed C5b-9 showed co-deposition of both complexes on naive CD4+ T cells (Fig. 2) and Jurkat cells (data not shown). Similar co-deposition of in situ C5b-9 was observed with Alexa Fluor® 488-labeled AHG (supplemental Fig. S1) and SLE-ICs (data not shown). The presence of C5b-9 was required to clearly visualize the AHG or ICs on the cells.
FIGURE 2.

Co-localization of in situ C5b-9 with Alexa Fluor® 488-labeled Ova-ICs on naive CD4+ T cell. Cells were treated with labeled Ova-ICs in the presence of in situ C5b-9 for 45 min. Arrows point to the C5b-9 and ICs (×630). Image represents one of five experiments.
MRs Aggregation Triggered by Nonlytic and in Situ C5b-9
To further investigate the relevance of C5b-9 deposition relative to T cell function, we analyzed the behavior of MRs. MRs are rich in cytoskeletal proteins and participate in the formation of IS. In activated T cells, as part of IS formation, signaling proteins are phosphorylated and organize themselves in central and peripheral supramolecular activation clusters. This structural organization is accomplished by the formation of branched actin filaments coupled to retrograde F-actin (35). During this process, multichain oligomers of the TCR become associated with the MRs (36). We employed the β subunit of CTB conjugated with FITC to visualize MRs. CTB binds to GM1 ganglioside, a major constituent of MRs, and is routinely used for identifying these membrane structures (37).
Nonlytic deposition of C5b-9 on the T cell membrane triggered lateral diffusion of MRs that resulted in their aggregation below the C5b-9 on both naive CD4+ T cells (Fig. 3A and supplemental Video 3) and Jurkat cells (data not shown). This pattern was observed with in situ C5b-9, as well as with the preassembled nonlytic C5b-9 (Fig. 3, A and B, and supplemental Video 4). Cells incubated with ICs alone did not show MRs aggregation (Fig. 3C and supplemental Video 5). Omission of C8 during in situ C5b-9 assembly abrogated MRs aggregation (supplemental Video 2). Simultaneous addition of ICs with the C5b-9 did not alter this pattern. These results show that the C5b-9 deposition alone is sufficient to cause MR aggregation.
FIGURE 3.

A, co-localization of MRs with in situ C5b-9 on naive CD4+ T cells. Cells stained with CTB for MRs after 2 h of in situ C5b-9 assembly (×630). Cells show aggregation of MRs below the in situ C5b-9 deposits. Arrows point to aggregated MRs and C5b-9. Image represents 1 of 10 experiments. B, MR aggregation in naive CD4+ T cell upon treatment with nonlytic C5b-9 and Ova-ICs for 2 h. Cells stained with CTB-FITC show aggregation of MRs. Confocal image captured at ×630 with ×2.3 optical zoom. Arrows point to aggregated MRs. Merge shows MRs on DIC image. Image represents 1 of 10 experiments. C, naive CD4+ T cells treated with Ova-ICs for 2 h and stained for MRs as in B. MRs aggregation was not observed.
CD3 Localizes with in Situ C5b-9
We next analyzed the behavior of the CD3 signaling complex with in situ C5b-9-treated CD4+ T cells. Within 45 min following the assembly of in situ C5b-9, the CD3 signaling complex moved from a uniform membrane distribution to form aggregate below the in situ C5b-9 deposit (Fig. 4). ICs had no effect on this event. Cells lacking in situ C5b-9 deposit did not show CD3 aggregation (upper cell in Fig. 4). Both CD3 and MRs co-localized with the in situ C5b-9 deposits.
FIGURE 4.

Naive CD4+ T cells with in situ C5b-9 deposit stained for CD3. CD3 co-localizes with C5b-9 (arrows point to CD3 and C5b-9). A cell without in situ C5b-9 deposition shows a uniform pattern of staining without clustering of CD3 (upper cell). A cell treated with Ova-ICs also shows uniform staining (far right panel). Image was captured at ×630 after 2 h of C5b-9 formation. Image represents one of two experiments.
ICs and C5b-9 Treatment Lead to the Formation of Distal Pole Complex (DPC) and Uropods
Based on co-localization of CD3 and MRs with the in situ C5b-9 deposits, we reasoned that the nonlytic C5b-9 could be activating the T cells. Activation of T cells is associated with polarization and phosphorylation of TCR signaling proteins. Polarization of T cells is achieved through actin polymerization by forming F-actin. The ERM complex proteins ezrin and moesin tether transmembrane and cytoplasmic proteins to these actin filaments. As these events unfold, there is concomitant migration of proteins not directly participating in TCR signaling to the opposite pole. This pole is marked by accumulation of phosphorylated ezrin (Thr567), radixin (Thr564), and moesin (Thr558) (pERM) proteins, discs-large homologue-1 (DLG1), and LFA antigens and is termed as DPC (38, 39).
The treatment of cells with nonlytic C5b-9 and ICs led to the formation of F-actin filaments and cellular redistribution of pERM. Both naive CD4+ T cells (Fig. 5, A and B, and supplemental Video 6) and Jurkat cells (supplemental Video 7) developed F-actin filaments. A bipolar distribution of F-actin was observed (supplemental Fig. S3). Treatment of cells with either the nonlytic C5b-9 or ICs alone was insufficient to trigger these polarization events (supplemental Videos 8 and 9). Counting cells from three microscopic fields revealed that ∼35% of cells treated with the nonlytic C5b-9 and ICs formed a bipolar F-actin complex and showed migration of pERM to the DPC. The initial migration of pERM to the DPC could be observed as early as 4 h (Fig. 5A), and this polarization was complete by 12 h (Fig. 5B). The nonlytic C5b-9- and IC-treated cells also formed a uropod where both F-actin and pERM preferentially accumulated (Fig. 5B). The pERM did not pack as tightly as the F-actin, being primarily observed below the polarized side. These events, including the accumulation of pERM in uropods in response to nonlytic C5b-9 and ICs, are consistent with T cell activation phenomena.
FIGURE 5.

A, naive CD4+ T cells stained for F-actin with phalloidin (green) and pERM (red). Cell image is 4 h post-treatment with nonlytic C5b-9 and Ova-ICs. Migration of pERM and F-actin formation is observed. Images were captured at ×630. Image represents one of two experiments. B, naive CD4+ T cells stained for F-actin (green) and pERM (red) (confocal z-series sequential images) after 12 h of treatment with nonlytic C5b-9 and SLE-ICs. Cells show complete migration of pERM and formation of F-actin and uropods. Uropod is marked by arrows (upper panel from left to right). Uropods show accumulation of pERM and F-actin. Lower panel, cells as in upper panel on DIC image background. Images represent one of five experiments.
Phosphorylation and Co-localization of TCR Signaling Proteins
We next examined the phosphorylation of proteins known to associate with TCR signaling using Western blotting (40). Ova-ICs or SLE-ICs, in conjunction with the nonlytic C5b-9, led to the phosphorylation of six proteins in the molecular mass range of 50–74 kDa (Figs. 6A and 7A). Nonlytic C5b-9 or ICs alone did not trigger these phosphorylation events. Minor differences in the phosphorylation patterns between Ova-ICs and SLE-ICs were observed. One was the stronger phosphorylation of a 74-kDa protein, likely Lck, in Ova-IC-treated cells (Fig. 6A). Phosphorylation of a 110-kDa protein was also observed, likely to be the p110α subunit of PI3K (41). The 36-kDa adaptor protein, linker for activation of T cells (LAT), shifted to 38 kDa, because of the phosphorylation of all of six available sites compared with four such sites in the inactive 36-kDa LAT protein (42). Phosphorylation of ZAP-70 and Lck, two early TCR signaling proteins, was observed in response to both Ova-ICs and SLE-ICs treatment (Figs. 6B and 7B). In addition, phosphorylation of Syk and Src was observed. These phosphorylation events are consistent with T cell activation.
FIGURE 6.
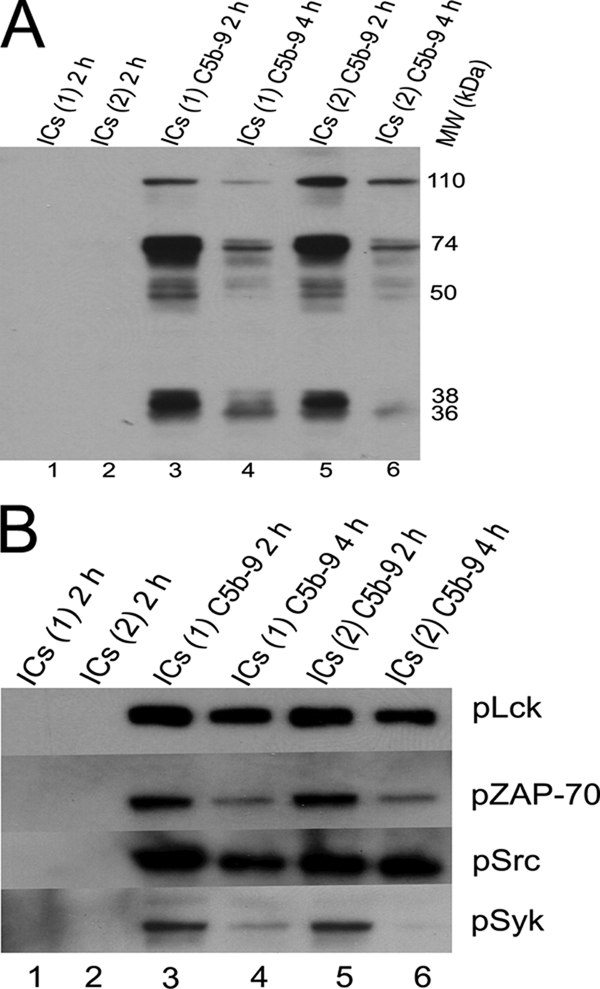
A, Western blot analysis of cell lysates probed with an anti-PY20 antibody. Jurkat cells were treated with nonlytic C5b-9 and two Ova-ICs preparations, antibody to antigen ratio of 1:0.5 marked ICs (lane 1) and at ratio of 1:4 marked ICs (lane 2) for 2 and 4 h. ICs and nonlytic C5b-9-treated cells at 2 h (lanes 3 and 5) show phosphorylation of several proteins, including those of 38, 50–74, and 110 kDa. ICs or C5b-9 alone did not show phosphorylation of these proteins. Figure represents one of two experiments. B, Western blot analysis of cells treated as described in A. Blots were probed for phosphorylated Lck, ZAP-70, Src, and Syk. All four proteins are phosphorylated in response to Ova-ICs and nonlytic C5b-9 together. Cells treated for 2 h show enhanced phosphorylation in comparison with 4-h intervals. Figure represents one of two experiments.
FIGURE 7.
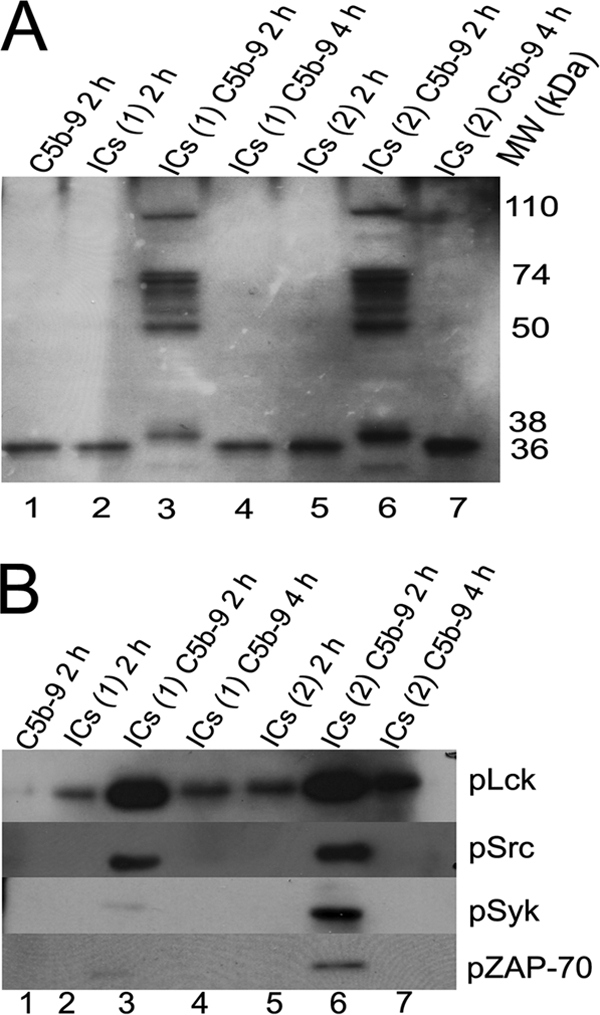
A, Western blot analysis of cell lysates probed with an anti-PY20 antibody. Jurkat cells were treated with nonlytic C5b-9 and SLE-ICs purified from two patients marked ICs(1) and ICs(2). Cells treated with SLE-ICs together with nonlytic C5b-9 (lanes 3 and 6) show phosphorylation of at least six proteins from 50 to 74 kDa. Proteins at 36, 38, and 110 kDa were also phosphorylated. Figure represents one of two experiments. B, Western blot analysis of cells treated as described in A. Blots were probed for phosphorylated Lck, ZAP-70, Src, and Syk. SLE-ICs and nonlytic C5b-9-treated cells after 2 h show phosphorylation of all four signaling proteins. The strongest phosphorylation was observed for Lck followed by Src and then Syk and ZAP-70. Figure represents one of two experiments.
Following co-incubation of cells with nonlytic C5b-9 and ICs, TCR signaling proteins appeared in microclusters in Jurkat (data not shown) and naive CD4+ T cells (Fig. 8, A and B). In such cells, these proteins were phosphorylated and migrated toward the C5b-9 deposits, accumulating by 1 h below and around the C5b-9. In these microclusters the intensity of phosphorylation of signaling proteins increased up to 2 h, which then slowly declined. Co-localization of the phosphorylated form of the ζ-chain and ZAP-70 along with the C5b-9 deposits (Fig. 8A and supplemental Videos 10 and 11) was observed. The co-receptor signaling protein Lck also co-localized with the phosphorylated ζ-chain (supplemental Fig. S4). Furthermore, pLck formed a cluster roughly 4 μm in diameter (supplemental Fig. S3), which is similar in size to those observed in T cells activated with peptide-MHC (43). Treatment with the nonlytic C5b-9 alone or ICs alone did not phosphorylate the ζ-chain or ZAP-70 (Fig. 8A, far right panel, and supplemental Video 12) or cause Lck cluster formation.
FIGURE 8.

A, naive CD4+ T cells stained for pζ-chain (green) and pZAP-70 (red), following treatment with nonlytic C5b-9 and SLE-ICs. Nuclei were stained with DAPI. Shown are the pζ-chain (left), pZAP-70 (2nd left), and merge of pζ-chain and pZAP-70 (2nd right). Both proteins are present in peripheral microclusters and co-localize by 1 h of treatment. IC-treated cells do not show pζ-chain and pZAP-70 (right panel) (enhanced using Photoshop to observe microclusters of pζ and/or pZAP-70). Confocal image at ×630. Figure represents one of five experiments. B, naive CD4+ T cells stained for pLAT (red) and pζ-chain (green), treated with SLE-ICs and nonlytic C5b-9. Both proteins are present in microclusters and co-localize after 1 h of treatment (upper panels). Merge of pLAT and pζ-chain on DIC image (upper far right panel) and nuclei stained with DAPI (lower far left panel). Nonlytic C5b-9 treated (lower central panel) and SLE-ICs treated (far right panel). Nonlytic C5b-9 or ICs alone did not phosphorylate either LAT or ζ-chain. Confocal image is at ×630. Figure represents one of five experiments.
LAT and the Sh2-binding leukocyte phosphoprotein of 76 kDa (SLP-76) are recruited to the MRs and are crucial for T cell activation (42, 44–46). Phosphorylation of LAT by ZAP-70 is an early event in T cell activation that contributes to the assembly of signaling scaffolds. In cells treated together with ICs and nonlytic C5b-9, confocal microscopy revealed co-localization of phosphorylated forms of LAT with the ζ-chain (Fig. 8B and supplemental Video 13). Cells treated with either ICs (supplemental Video 14) or nonlytic C5b-9 alone did not phosphorylate LAT (Fig. 8B, bottom right two panels). Overall, these results are similar to the events observed in T cells activated by the peptide-MHC complex (47).
ICs and Nonlytic C5b-9 Induce T Cell Proliferation
To further evaluate the relevance of the T cell activation signal emanating from ICs and nonlytic C5b-9, we asked whether this signal is sufficient to induce proliferation of the CD4+CD45RA+ T cells. The CFSE-loaded cells, upon treatment with nonlytic C5b-9 and ICs together, showed proliferation (Fig. 9). The microscopic examination of the cells treated with ICs and nonlytic C5b-9, revealed a small number of large foci, whereas phytohemagglutinin-treated cells showed a large number of smaller foci (supplemental Fig. S5). In the phytohemagglutinin-treated group, almost all cells responded to signal, and after a limited number of cell divisions, cell growth was arrested. In contrast, a smaller number of cells that received the nonlytic C5b-9 and ICs continue to divide until the time of examination, thus forming large colonies (supplemental Fig. S5). Previous reports using non-T cells have also demonstrated cellular proliferation in response to nonlytic C5b-9 (48, 49).
FIGURE 9.

Proliferation of naive CD4+ T cells in response to treatment with nonlytic C5b-9 and SLE-ICs, observed by CFSC loading. Cells treated with nonlytic C5b-9 and SLE-ICs together show proliferation as observed by dilution of CFSC label as secondary peaks. Data were analyzed with FlowJo software (proliferation node). Figure represents one of three experiments.
Production of IFN-γ
The IFN-γ is the signature cytokine of TH1 phenotype that is associated with a proinflammatory response and autoimmune disease pathology (50). To further substantiate a role for ICs and nonlytic C5b-9 in T cell-mediated immune pathology, we measured secretion of IFN-γ by naive CD4+ T cells following 48 h of treatment with various stimuli. The untreated cells did not demonstrate secretion of IFN-γ. A very low level of IFN-γ production was observed following treatment of T cells with only nonlytic C5b-9 (15 pg/ml). The cells treated with SLE-ICs showed significant production of IFN-γ (360 pg/ml), which was further enhanced by the presence of nonlytic C5b-9 (506 pg/ml). The SLE-ICs and nonlytic C5b-9-treated cells demonstrated a 33-fold increase in IFN-γ production when compared with nonlytic C5b-9 treatment alone. Cells treated with anti-CD3 and anti-CD28 also secreted IFN-γ (127 pg/ml) (Fig. 10). Similar results were obtained in two additional experiments. These findings are similar to a previous report in which the production of C-C and C-X-C chemokines in response to C5b-9 was enhanced by the presence of ICs during treatment (51).
FIGURE 10.

IFN-γ production in response to nonlytic C5b-9 and SLE-ICs. Levels of IFN-γ produced by naive CD4+ T cells, untreated and treated with nonlytic C5b-9, SLE-ICs, nonlytic C5b-9 with SLE-ICs, and anti-CD3 with anti-CD28 antibodies are shown. Maximum level of IFN-γ was produced by cells treated with SLE-ICs and nonlytic C5b-9. Figure represents one of three experiments.
DISCUSSION
The major observation in this study is the activation of human peripheral blood naive CD4+ T cells by the nonlytic C5b-9 and ICs. To begin C5b-9 formation, C5 is cleaved to C5a and C5b. C5b then initiates the formation of C5b-9 by binding C6, C7, and C8, followed by up to 16 C9 proteins. C5b-9 present in fluid phase is associated with clusterin and vitronectin and is unable to form trans-membrane channels. In contrast, the in situ-formed C5b-9 is capable of transiently inserting into the membrane. Although most of the in situ C5b-9 behaves like nonlytic, a small fraction is able to transiently insert in the membrane and trigger Ca2+ flux. Previous reports have shown that the cytolytically inactive C5b-9 activates endothelial cells and procoagulant activities in these cells (52). We show that both in situ and nonlytic C5b-9 trigger aggregation of MRs and cytoskeletal changes, and in situ C5b-9 co-localizes with ICs and CD3. Nonlytic C5b-9 triggered signaling events, cell proliferation, and production of IFN-γ.
Recently, C5a has been shown to facilitate T cell activation in a murine system (9) and Th17 cell differentiation in an arthritic mouse model (53). In this study, we demonstrate a potential role of the nonlytic C5b-9 complex in T cell activation. C5b-9 and ICs are commonly present in humoral autoimmunity and mediate disease pathology (54, 55). Employing these two natural immunoreactions instead of antibodies to cross-link T cell receptors, we demonstrate activation of naive CD4+ T cells. Interaction of ICs and nonlytic C5b-9 with the T cells could occur predominantly in secondary germinal centers and endothelial venules, where complement activation products and ICs are present along with naive CD4+ T cells (56, 57).
C5b-9 Binding to T Cells
Aggregation of CD3 and MRs with in situ C5–9 are events similar to those observed during TCR engagement by peptide-MHC complex. Monomeric C5b-9 binds to as many as 918 phospholipid molecules, whereas the dimer binds up to 1460 phospholipid molecules (58). C5b-9 at different stages of assembly on the cell membrane is also capable of physically re-organizing the lipid bilayer (59) and thereby creating conformational changes in the membrane proteins. We hypothesize that the initial binding of C5b-9 to membrane phospholipids creates a hydrophobic zone on the T cell membrane. This further attracts additional molecules of C5b-9 and C9, resulting in the growth of the primary deposit (Fig. 1). This was most evident based on the increasing intensity of the C9 label within in situ C5b-9 over time.
MRs Engagement
MRs are microdomains rich in cholesterol and sphingolipid that float in membrane phospholipids. They play a central role in T cell activation through protein sorting and membrane trafficking (36), as well as participate in the spatial organization of the structures involved in cell activation (60, 61). In activated T cells, TCR-CD3 complexes are translocated to the MRs where they cluster at the IS. The phase separation of MRs and glycerophospholipids in the cell membrane allows for a high degree of MR lateral mobility (62). T cells isolated from SLE patients demonstrate aggregation of MRs. Aggregation of MRs has been implicated in disease pathogenesis. We observed aggregation of MRs in cells from assembly of in situ C5b-9 as well as with preassembled nonlytic C5b-9. Ligation of MRs with CTB triggers MR aggregation and accelerates disease progression in the lupus mouse model (63). In contrast, the disruption of MR aggregation by methyl-β-cyclodextrin delayed the appearance of disease (64).
The aggregation of the MRs by C5b-9 and their co-localization with the TCR signaling proteins suggests a role for C5b-9 in T cell hyperactivation and its possible contribution to altered CD3-TCR signaling observed in SLE (23). The ICs formed with autoantibodies triggers activation of the classical complement pathway resulting in the formation of C5b-9. Elevated levels of C5b-9 are observed in plasma and urine of SLE patients (55). In the peptide-MHC-activated T cells, MRs are polarized toward the uropod initially and thereafter cycle toward and accumulate at the leading edge of T cells (65). We noted similar changes in naive CD4+ T cells after treatment with ICs and nonlytic C5b-9. We presume that this lateral movement of MRs by the C5b-9 is triggered by binding of C5b-9 to glycerophosphates (62). Ligation of CD28 lowers the threshold for T cell activation by recruiting the MRs to the site of interaction between T cells and APCs (66). Nonlytic C5b-9 may thus support a similar co-activation. Human naive CD4+ T cells treated with ICs and nonlytic C5b-9 in the presence of anti-CD3 show increased expression of CD25 and glucocorticoid-induced tumor necrosis factor receptor.3 Hence, we propose that the complement system and ICs trigger T cell activation using the same membrane structures that are used during peptide-MHC presentation.
Involvement of CD3
The TCR-CD3 clustering upon peptide-MHC recognition results in conformational changes that are necessary for cell activation (67). The physical reorganization of the cell membrane is central to all three proposed models of TCR activation, i.e. multimerization, conformational change, or co-receptor heterodimerization (68). The binding of C5b-9 dimers to membrane phospholipids by altering the cell polarity could trigger clustering of CD3, resulting in its aggregation leading to the conformational changes in TCR.
Nonlytic C5b-9 and ICs Binding Mediated Cellular Events
Cytoskeletal polarization observed during TCR activation is supported by formation of the F-actin that is also responsible for the formation of microtubule-organizing center (39). Microtubule-organizing center formation leads to the organization of IS, a key requirement for T cell activation. Such cytoskeletal changes were observed in cells treated with nonlytic C5b-9 and ICs (60). We found an absolute requirement of both C5b-9 and ICs for triggering of the changes associated with T cell activation.
Nonlytic C5b-9 in Other Cell Types
In aortic endothelial cells, the nonlytic C5b-9 induces cell cycle activation through the PI3K/Akt pathway (48). Deposition of nonlytic C5b-9 on target cells during acute and chronic inflammation induces hydrolysis of plasma membrane phospholipids and activation of heterotrimeric G proteins (69). Other activities associated with nonlytic C5b-9 include cytokine synthesis, proto-oncogene activation, and mitotic signaling (70, 71). In addition, it activates Ras, Raf-1, extracellular signal-regulated kinase (ERK-1), and cytokine synthesis in the JY25 B cell line (72) as well as proliferation (48). Taken together, these results indicate that the nonlytic C5b-9 is capable of influencing cell function by reorganization of signaling molecules on the cell membrane. In these cited studies, complement activation to form C5b-9 was achieved using antibodies directed to cell surface antigens thereby forming in situ ICs (48, 71). However, in our studies we separated these two potential signaling events.
IC Binding to T Cells
The binding of ICs observed to the T cell membrane is mediated via Fc receptors (FcR). C5b-9 by triggering capping and/or aggregation increases local receptor density, thus facilitating the IC binding (Fig. 2). Although the presence of the FcR on human CD4+T cells is controversial (73), increased expression of FcγR and FcμR chain receptors on T cells in multiple sclerosis has been reported (74). In addition, FcR-bearing cells are increased upon allogenic activation (75). Also, in SLE T cells, the expression of the CD3ζ chain is decreased, although the expression of FcRγ chain is up-regulated, and it populates the CD3-TCR complex (76) and MRs (77). Binding of AHG via FcR in mouse thymocytes and T cells has been observed (78). These previous studies and the IC binding observed in this report suggest the presence of low affinity FcR on T cells. In our system, ICs play a crucial role in TCR activation, because in their absence the nonlytic C5b-9 treatment alone did not lead to cell polarization or phosphorylation of TCR signaling proteins. The binding of labeled AHG to T cells was blocked by an affinity-purified anti-FcγRIIIB and a monoclonal antibody that recognizes both FcγRIIIA and FcγRIIIB. In addition, FcRIIIγA/B gene-specific transcripts were amplified from naive CD4+ T cells, and DNA sequencing confirmed them to be FcRγIIIA/B.4 This suggests that ICs and AHG binding utilized low affinity FcγRIII receptors present on CD4+ T cells.
Formation of Synaptic Structures Similar to IS
T cell activation is associated with the formation of a specialized signaling structure known as the IS. Antigen presentation by APCs to T cells triggers formation of TCR microclusters, which originate in the periphery and migrate to the central supramolecular association cluster. The cells treated with nonlytic C5b-9 and ICs showed a similar type of microclusters, where the phosphorylated TCR signaling proteins localize. Similar IS-like structures have also been observed in activated NK cells (2, 26). Confocal images of cells treated with ICs and in situ C5b-9 demonstrated the formation of dense structures at a single site similar to those observed in activated T and NK cells. Synapse-like structure formation by the nonlytic C5b-9 suggests a role for complement in reorganizing the cell membrane thus influencing the lymphocyte function.
Summary and Hypothesis
We are unaware of previous reports demonstrating the effects of nonlytic C5b-9 on the activation of naive CD4+ T cells, with or without ICs. The findings reported here were surprising to us and raise many questions. The T cell repertoire selection is based on the strength of the TCR signal generated upon binding of peptide-MHC complexes presented by APC. In addition, T cell signal strength is a key determinant of T cell tolerance (27, 79). Hence, the activation of downstream signaling of TCR by nonlytic C5b-9 and ICs has the potential to modulate the peripheral tolerance by contributing to the strength of the primary TCR signal generated by peptide-MHC. The dwell time of the interaction between TCR and peptide-MHC complexes is influenced by co-receptors such as CD28. Signal emanating from C5b-9 and ICs could either alter the amount or the site of phosphorylation of immunoreceptor tyrosine-based activation motif in the CD3 complex or change the dwell time to modulate the T cell selection (80). A weak T cell activation signal generated by ICs and nonlytic C5b-9 in the absence of peptide-MHC engagement could lead to escape of T cells from deletion. In addition, by adding to the strength of the TCR signal that is received by effector T cells could increase their resistance to suppression (81).
The ICs and nonlytic C5b-9-mediated phosphorylation of signaling proteins point to the involvement of classical TCR signaling events and a possible role for Syk. We observed phosphorylation of Syk in ICs and nonlytic C5b-9-treated cells, possibly due to FcRγ chain engagement, upon ligation of FcR by ICs (Figs. 6B and 7B). This was also confirmed by confocal microscopy and Western blotting of anti-FcRγIIIA/B immunoprecipitates. The CD4+ T cells from SLE bind to labeled AHG and demonstrate the presence of FcγRIIIA/B.4 The strong phosphorylation of Lck, along with that of ζ-chain, ZAP-70, and LAT and their co-localization, points to participation of traditional downstream TCR-signaling elements.
We speculate that in the setting of infections in which antibody and complement are key to pathogen clearance, the presence of ICs bearing complement fragments would facilitate T cell activation. Once antigen is cleared by the immune response, IC formation and complement activation cease. However, in the setting of humoral autoimmunity and persistent inflammation secondary to chronic antigenemia, T cell activation by ICs and nonlytic C5b-9 could perpetuate undesirable immune responses. Bystander T cell activation, which can be induced by cross-linking of membrane-bound receptor and certain cytokines, is implicated in disease pathology (82). ICs and nonlytic C5b-9 either by engaging components of TCR or by triggering the bystander T cell activation could influence T cell responses. These results also provide insight into the possible mechanisms that are responsible for altered T cell responses observed in autoimmunity (23). The results presented may also partly explain the difficulties targeting the T cell surface markers for therapeutic interventions such as TGN 1412 (anti-CD28). The T cell responses described in this report outline several new avenues as to how the complement system and ICs could modulate T cell responses.
Acknowledgments
We thank Drs. Claudia Kemper (Medical Research Council, London, UK), Andrey Shaw (Washington University, St. Louis), and Andrew C. Chan (Genentech) for critically reading the manuscript and their valuable suggestions. We especially thank Dr. John P. Atkinson for guidance and manuscript revision (Washington University, St. Louis).
This work was supported by The Campbell-Avery Charitable Trust, The Dorr Family Charitable Trust, and Lupus/Juvenile Research Group of St. Louis, MO.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S5 and Videos 1–14.
A. K. Chauhan and T. L. Moore, unpublished observations.
Chauhan, A. K., and Moore, T. L. (2011) Clin. Exp. Immunol., in press.
- APC
- antigen-presenting cell
- CTB
- cholera toxin B
- DIC
- differential interference contrast
- DPC
- distal pole complex
- IC
- immune complex
- IS
- immunological synapse
- MR
- membrane raft
- Ova-IC
- ovalbumin-anti-ovalbumin immune complex
- pERM
- phosphorylated ezrin-radixin-moesin
- p
- phosphorylated
- SLE-IC
- immune complex from SLE patient
- TCR
- T cell antigen receptor
- CFSC
- carboxyfluorescein diacetyl succinimidyl ester
- FcR
- Fc receptor
- AHG
- aggregated human γ-globulin.
REFERENCES
- 1. Friedl P., den Boer A. T., Gunzer M. (2005) Nat. Rev. Immunol. 5, 532–545 [DOI] [PubMed] [Google Scholar]
- 2. Orange J. S. (2008) Nat. Rev. Immunol. 8, 713–725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mastellos D., Germenis A. E., Lambris J. D. (2005) Curr. Drug Targets Inflamm. Allergy 4, 125–127 [DOI] [PubMed] [Google Scholar]
- 4. Dempsey P. W., Allison M. E., Akkaraju S., Goodnow C. C., Fearon D. T. (1996) Science 271, 348–350 [DOI] [PubMed] [Google Scholar]
- 5. Fang Y., Xu C., Fu Y. X., Holers V. M., Molina H. (1998) J. Immunol. 160, 5273–5279 [PubMed] [Google Scholar]
- 6. Fearon D. T., Carter R. H. (1995) Annu. Rev. Immunol. 13, 127–149 [DOI] [PubMed] [Google Scholar]
- 7. Vinuesa C. G., Sanz I., Cook M. C. (2009) Nat. Rev. Immunol. 9, 845–857 [DOI] [PubMed] [Google Scholar]
- 8. Kemper C., Atkinson J. P. (2007) Nat. Rev. Immunol. 7, 9–18 [DOI] [PubMed] [Google Scholar]
- 9. Strainic M. G., Liu J., Huang D., An F., Lalli P. N., Muqim N., Shapiro V. S., Dubyak G. R., Heeger P. S., Medof M. E. (2008) Immunity 28, 425–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hammer C. H., Nicholson A., Mayer M. M. (1975) Proc. Natl. Acad. Sci. U.S.A. 72, 5076–5080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Morgan B. P., Dankert J. R., Esser A. F. (1987) J. Immunol. 138, 246–253 [PubMed] [Google Scholar]
- 12. Moskovich O., Fishelson Z. (2007) J. Biol. Chem. 282, 29977–29986 [DOI] [PubMed] [Google Scholar]
- 13. Bossi F., Fischetti F., Pellis V., Bulla R., Ferrero E., Mollnes T. E., Regoli D., Tedesco F. (2004) J. Immunol. 173, 6921–6927 [DOI] [PubMed] [Google Scholar]
- 14. Casarsa C., De Luigi A., Pausa M., De Simoni M. G., Tedesco F. (2003) Eur. J. Immunol. 33, 1260–1270 [DOI] [PubMed] [Google Scholar]
- 15. Shin M. L., Rus H. G., Niculescu F. I. (1996) in Biomembranes (Lee A. G. ed) pp. 119–146, JAI Press, Greenwich, CT [Google Scholar]
- 16. Morgan B. P. (1989) Biochem. J. 264, 1–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Benzaquen L. R., Nicholson-Weller A., Halperin J. A. (1994) J. Exp. Med. 179, 985–992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Niculescu F., Rus H., Shin S., Lang T., Shin M. L. (1993) J. Immunol. 150, 214–224 [PubMed] [Google Scholar]
- 19. Cybulsky A. V., Salant D. J., Quigg R. J., Badalamenti J., Bonventre J. V. (1989) Am. J. Physiol. 257, F826–F836 [DOI] [PubMed] [Google Scholar]
- 20. Fishelson Z., Attali G., Mevorach D. (2001) Mol. Immunol. 38, 207–219 [DOI] [PubMed] [Google Scholar]
- 21. Pilzer D., Gasser O., Moskovich O., Schifferli J. A., Fishelson Z. (2005) Springer Semin. Immunopathol. 27, 375–387 [DOI] [PubMed] [Google Scholar]
- 22. Green H., Goldberg B. (1960) Ann. N.Y. Acad. Sci. 87, 352–362 [DOI] [PubMed] [Google Scholar]
- 23. Crispín J. C., Kyttaris V. C., Terhorst C., Tsokos G. C. (2010) Nat. Rev. Rheumatol. 6, 317–325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jury E. C., Kabouridis P. S., Flores-Borja F., Mageed R. A., Isenberg D. A. (2004) J. Clin. Invest. 113, 1176–1187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chauhan A. K., Moore T. L. (2006) Clin. Exp. Immunol. 145, 398–406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Grakoui A., Bromley S. K., Sumen C., Davis M. M., Shaw A. S., Allen P. M., Dustin M. L. (1999) Science 285, 221–227 [DOI] [PubMed] [Google Scholar]
- 27. Holst J., Wang H., Eder K. D., Workman C. J., Boyd K. L., Baquet Z., Singh H., Forbes K., Chruscinski A., Smeyne R., van Oers N. S., Utz P. J., Vignali D. A. (2008) Nat. Immunol. 9, 658–666 [DOI] [PubMed] [Google Scholar]
- 28. Low J. M., Chauhan A. K., Gibson D. S., Zhu M., Chen S., Rooney M. E., Ombrello M. J., Moore T. L. (2009) Proteomics Clin. Appl. 3, 829–840 [DOI] [PubMed] [Google Scholar]
- 29. Accardo-Palumbo A., Triolo G., Casiglia D., Sallì L., Giardina E., Triolo G. (1993) J. Immunol. Methods 163, 169–172 [DOI] [PubMed] [Google Scholar]
- 30. Morgan B. P., Daniels R. H., Williams B. D. (1988) Clin. Exp. Immunol. 73, 473–478 [PMC free article] [PubMed] [Google Scholar]
- 31. Leitinger B., Hogg N. (2002) J. Cell Sci. 115, 963–972 [DOI] [PubMed] [Google Scholar]
- 32. Valitutti S., Dessing M., Aktories K., Gallati H., Lanzavecchia A. (1995) J. Exp. Med. 181, 577–584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mannik M. (1987) J. Rheumatol. 14, Suppl, 13, 35–42 [PubMed] [Google Scholar]
- 34. Wener M. H., Mannik M., Schwartz M. M., Lewis E. J. (1987) Medicine 66, 85–97 [DOI] [PubMed] [Google Scholar]
- 35. Burkhardt J. K., Carrizosa E., Shaffer M. H. (2008) Annu. Rev. Immunol. 26, 233–259 [DOI] [PubMed] [Google Scholar]
- 36. Dykstra M., Cherukuri A., Pierce S. K. (2001) J. Leukocyte Biol. 70, 699–707 [PubMed] [Google Scholar]
- 37. Heyningen S., Van (1974) Science 183, 656–657 [DOI] [PubMed] [Google Scholar]
- 38. Cullinan P., Sperling A. I., Burkhardt J. K. (2002) Immunol. Rev. 189, 111–122 [DOI] [PubMed] [Google Scholar]
- 39. Billadeau D. D., Nolz J. C., Gomez T. S. (2007) Nat. Rev. Immunol. 7, 131–143 [DOI] [PubMed] [Google Scholar]
- 40. Abraham R. T., Weiss A. (2004) Nat. Rev. Immunol. 4, 301–308 [DOI] [PubMed] [Google Scholar]
- 41. Deane J. A., Fruman D. A. (2004) Annu. Rev. Immunol. 22, 563–598 [DOI] [PubMed] [Google Scholar]
- 42. Zhang W., Sloan-Lancaster J., Kitchen J., Trible R. P., Samelson L. E. (1998) Cell 92, 83–92 [DOI] [PubMed] [Google Scholar]
- 43. Varma R., Campi G., Yokosuka T., Saito T., Dustin M. L. (2006) Immunity 25, 117–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Barda-Saad M., Braiman A., Titerence R., Bunnell S. C., Barr V. A., Samelson L. E. (2005) Nat. Immunol. 6, 80–89 [DOI] [PubMed] [Google Scholar]
- 45. Bunnell S. C., Kapoor V., Trible R. P., Zhang W., Samelson L. E. (2001) Immunity 14, 315–329 [DOI] [PubMed] [Google Scholar]
- 46. Harder T., Kuhn M. (2000) J. Cell Biol. 151, 199–208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Blanchard N., Di Bartolo V., Hivroz C. (2002) Immunity 17, 389–399 [DOI] [PubMed] [Google Scholar]
- 48. Fosbrink M., Niculescu F., Rus V., Shin M. L., Rus H. (2006) J. Biol. Chem. 281, 19009–19018 [DOI] [PubMed] [Google Scholar]
- 49. Dashiell S. M., Rus H., Koski C. L. (2000) Glia 30, 187–198 [DOI] [PubMed] [Google Scholar]
- 50. Viallard J. F., Pellegrin J. L., Ranchin V., Schaeverbeke T., Dehais J., Longy-Boursier M., Ragnaud J. M., Leng B., Moreau J. F. (1999) Clin. Exp. Immunol. 115, 189–195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Czermak B. J., Lentsch A. B., Bless N. M., Schmal H., Friedl H. P., Ward P. A. (1999) Am. J. Pathol. 154, 1513–1524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Tedesco F., Pausa M., Nardon E., Introna M., Mantovani A., Dobrina A. (1997) J. Exp. Med. 185, 1619–1627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Hashimoto M., Hirota K., Yoshitomi H., Maeda S., Teradaira S., Akizuki S., Prieto-Martin P., Nomura T., Sakaguchi N., Köhl J., Heyman B., Takahashi M., Fujita T., Mimori T., Sakaguchi S. (2010) J. Exp. Med. 207, 1135–1143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Marmont A. M. (1980) Lancet 1, 1088 [DOI] [PubMed] [Google Scholar]
- 55. Chiu Y. Y., Nisihara R. M., Würzner R., Kirschfink M., de Messias-Reason I. J. (1998) J. Investig. Allergol. Clin. Immunol. 8, 239–244 [PubMed] [Google Scholar]
- 56. Zwirner J., Felber E., Schmidt P., Riethmüller G., Feucht H. E. (1989) Immunology 66, 270–277 [PMC free article] [PubMed] [Google Scholar]
- 57. Mellbye O. J., Shen Y., Høgåsen K., Mollnes T. E., Førre O. (1996) Clin. Rheumatol. 15, 441–447 [DOI] [PubMed] [Google Scholar]
- 58. Podack E. R., Biesecker G., Müller-Eberhard H. J. (1979) Proc. Natl. Acad. Sci. U.S.A. 76, 897–901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Esser A. F., Kolb W. P., Podack E. R., Müller-Eberhard H. J. (1979) Proc. Natl. Acad. Sci. U.S.A. 76, 1410–1414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Viola A., Gupta N. (2007) Nat. Rev. Immunol. 7, 889–896 [DOI] [PubMed] [Google Scholar]
- 61. He H. T., Marguet D. (2008) EMBO Rep. 9, 525–530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Brown D. A., London E. (2000) J. Biol. Chem. 275, 17221–17224 [DOI] [PubMed] [Google Scholar]
- 63. Mitchell J. S., Kanca O., McIntyre B. W. (2002) J. Immunol. 168, 2737–2744 [DOI] [PubMed] [Google Scholar]
- 64. Deng G. M., Tsokos G. C. (2008) J. Immunol. 181, 4019–4026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Gómez-Móuton C., Abad J. L., Mira E., Lacalle R. A., Gallardo E., Jiménez-Baranda S., Illa I., Bernad A., Mañes S., Martínez-A. C. (2001) Proc. Natl. Acad. Sci. U.S.A. 98, 9642–9647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Alegre M. L., Frauwirth K. A., Thompson C. B. (2001) Nat. Rev. Immunol. 1, 220–228 [DOI] [PubMed] [Google Scholar]
- 67. Minguet S., Swamy M., Alarcón B., Luescher I. F., Schamel W. W. (2007) Immunity 26, 43–54 [DOI] [PubMed] [Google Scholar]
- 68. van der Merwe P. A. (2001) Immunity 14, 665–668 [DOI] [PubMed] [Google Scholar]
- 69. Niculescu F., Rus H., Shin M. L. (1994) J. Biol. Chem. 269, 4417–4423 [PubMed] [Google Scholar]
- 70. Rus H., Niculescu F., Badea T., Shin M. L. (1997) Immunopharmacology 38, 177–187 [DOI] [PubMed] [Google Scholar]
- 71. Niculescu F., Rus H. (2001) Immunol. Res. 24, 191–199 [DOI] [PubMed] [Google Scholar]
- 72. Niculescu F., Rus H., van Biesen T., Shin M. L. (1997) J. Immunol. 158, 4405–4412 [PubMed] [Google Scholar]
- 73. Nimmerjahn F., Ravetch J. V. (2008) Nat. Rev. Immunol. 8, 34–47 [DOI] [PubMed] [Google Scholar]
- 74. Merrill J. E., Biberfeld G., Landin S., Sidén A., Norrby E. (1980) Clin. Exp. Immunol. 42, 345–354 [PMC free article] [PubMed] [Google Scholar]
- 75. Stout R. D., Herzenberg L. A. (1975) J. Exp. Med. 142, 611–621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Enyedy E. J., Nambiar M. P., Liossis S. N., Dennis G., Kammer G. M., Tsokos G. C. (2001) Arthritis Rheum. 44, 1114–1121 [DOI] [PubMed] [Google Scholar]
- 77. Krishnan S., Nambiar M. P., Warke V. G., Fisher C. U., Mitchell J., Delaney N., Tsokos G. C. (2004) J. Immunol. 172, 7821–7831 [DOI] [PubMed] [Google Scholar]
- 78. Santana V., Turk J. L. (1975) Immunology 28, 1173–1178 [PMC free article] [PubMed] [Google Scholar]
- 79. Eichmann K. (1995) Immunol. Lett. 44, 87–90 [DOI] [PubMed] [Google Scholar]
- 80. Palmer E., Naeher D. (2009) Nat. Rev. Immunol. 9, 207–213 [DOI] [PubMed] [Google Scholar]
- 81. Baecher-Allan C., Brown J. A., Freeman G. J., Hafler D. A. (2001) J. Immunol. 167, 1245–1253 [DOI] [PubMed] [Google Scholar]
- 82. Bangs S. C., McMichael A. J., Xu X. N. (2006) Trends Immunol. 27, 518–524 [DOI] [PubMed] [Google Scholar]