

Abstract
Promoter recognition is the first and the most important step during gene expression. Our studies of the yeast (Saccharomyces cerevisiae) mitochondrial (mt) transcription machinery provide mechanistic understandings on the basic problem of how the mt RNA polymerase (RNAP) with the help of the initiation factor discriminates between promoter and non-promoter sequences. We have used fluorescence-based approaches to quantify DNA binding, bending, and opening steps by the core mtRNAP subunit (Rpo41) and the transcription factor (Mtf1). Our results show that promoter recognition is not achieved by tight and selective binding to the promoter sequence. Instead, promoter recognition is achieved by an induced-fit mechanism of transcription factor-dependent differential conformational changes in the promoter and non-promoter DNAs. While Rpo41 induces a slight bend upon binding both the DNAs, addition of the Mtf1 results in severe bending of the promoter and unbending of the non-promoter DNA. Only the sharply bent DNA results in the catalytically active open complex. Such an induced-fit mechanism serves three purposes: 1) assures catalysis at promoter sites, 2) prevents RNA synthesis at non-promoter sites, and 3) provides a conformational state at the non-promoter sites that would aid in facile translocation to scan for specific sites.
Keywords: DNA-Protein Interaction, Gene Transcription, RNA Polymerase, Transcription Initiation Factors, Transcription Promoter, DNA Bending and Melting, Promoter Recognition
Introduction
DNA-dependent RNA synthesis initiates with the specific binding of the RNA polymerase (RNAP)2 to its promoter. During this process of promoter selection, the RNAP is able to seek out active promoters within vast stretches of non-promoter sequences in the genome. The multi-subunit RNAPs of the bacteria, archaea, and eukarya rely on transcription factors for promoter selection and specific transcription (1–3) whereas the single subunits RNAPs of bacteriophages are able to carry out the same functions without any accessory factors (4). The RNAPs that transcribe the mitochondrial genomes are unique in that they are homologous to the single subunit RNAPs of bacteriophages, but they require one or more transcription factors for promoter-specific initiation (5–9). The transcription factors modulate various steps of initiation and play a key role in open complex formation (10, 11).
In this study, we have investigated the transcription machinery of the yeast (Saccharomyces cerevisiae) mitochondria, which consists of a nuclear encoded ∼153 kDa core subunit Rpo41 (12) and a ∼45 kDa transcription factor Mtf1 (13, 14). The two proteins are sufficient to catalyze transcription from the conserved nona-nucleotide promoter (5′-ATATAAGTA(+1)) of the yeast mitochondria that directs the synthesis of rRNA, tRNA, and respiratory chain protein mRNAs (15). Rpo41 cannot initiate specific transcription on duplex promoters without Mtf1 (13, 16, 17). However, when the DNA is negatively supercoiled or premelted, then Rpo41 can initiate specific transcription without Mtf1 (18). Based on these results, it was proposed that Rpo41 has the intrinsic ability to recognize the promoter. A similar conclusion was made for its homolog RNAP in the human mitochondria (19).
Mtf1 is regarded as a functional homolog of the sigma factor, as the two do not share sequence or structural similarity but play similar roles in transcription initiation (13, 14, 20). Mtf1 does not bind to the promoter DNA on its own, but it binds to the Rpo41-promoter complex aiding in the formation of the pre-initiation open complex by melting the promoter from −4 to +2 (11, 16). Recent studies indicate that Rpo41 and Mtf1 together contact the promoter within and immediately adjacent to the −10 to +5 region, and Mtf1 binds to the melted DNA region via its C-terminal residues (21, 22). The human analog of Mtf1, h-mtFB2, has similar properties (10).
To determine the role of the core subunit and the transcription factor and the mechanism of promoter selection, we have quantified the interactions of Rpo41 and Rpo41-Mtf1 with the promoter and non-promoter DNAs and characterized the protein-induced structural changes in these DNAs. Using a combination of fluorescence-based methods including fluorescence anisotropy, Förster resonance energy transfer (FRET), and 2-aminopurine (2-AP) fluorescence, we have measured DNA binding, bending, and melting. Our studies show that Rpo41 by itself does not discriminate between promoter and non-promoter DNA. Mtf1 is required, but differential tight binding to the promoter sequence is not the major mechanism of promoter selection by Rpo41-Mtf1. Instead, Rpo41-Mtf1 induces selective DNA bending in the promoter and unbending in the non-promoter DNA, based on which we propose that promoter selection is achieved by an induced-fit mechanism involving differential conformational changes. We show that the transcription factor Mtf1 plays an important role in modulating these DNA bending-melting conformational changes.
EXPERIMENTAL PROCEDURES
Proteins and Oligonucleotides
Expression and purification of recombinant Rpo41 and Mtf1 were reported previously (11). Expression plasmids were kind gifts from Professor Jaehning (23). Synthetic oligonucleotides were purchased from IDT and purified by urea-denatured polyacrylamide gel electrophoresis. Fluorescent labeling with tetramethylrhodamine (TMR) and Alexa Fluor 647 (A647) via a C6 amino linker on the 5′-end and separation of excessive unlabeled fluorescent dyes were performed as reported (24). The labeling ratio of dye to oligodeoxynucleotide strand was close to 1:1.
Transcription Assay
Transcription activity of Rpo41-Mtf1 on promoter and non-promoter DNA fragments was determined by the gel-based transcription assay (11). Briefly, the transcription reaction was initiated at 25 °C by adding a mixture of 100 μm ATP, 250 μm UTP, CTP, and GTP each, spiked with [α-32P]ATP or [γ-32P]ATP into a solution of Rpo41 (3 μm), Mtf1 (4 μm) and DNA fragments (4 μm) in the reaction buffer (50 mm Tris acetate, pH 7.5, containing 100 mm potassium glutamate, 10 mm magnesium acetate, 5 mm fresh DTT, 0.01% protein-grade Tween 20, and 5% glycerol). Reactions were stopped at 3 min with 400 mm EDTA and formamide dye (98% formamide, 0.025% bromphenol blue, 10 mm EDTA) and analyzed on 4 m urea contained 23% sequencing gel. The gel was exposed to a phosphor screen overnight and scanned on a Typhoon 9410 PhosphorImager instrument (Amersham Biosciences). The free ATP and RNA bands were quantified using ImageQuant.
Equilibrium Titration of Protein-DNA Binding
Fluorescence anisotropy measurements were carried out according to the published protocol (24) on a PTI QM-3 spectrofluorimeter mounted with Glen-Thomson calcite prism polarizers and a thermoelectrically controlled cell holder. Titration was conducted by adding concentrated Rpo41, or Mtf1, or their equimolar mixture to a solution of TMR-labeled duplex DNA (2–8 nm, with TMR label on the 5′-end of template strand) in the reaction buffer, and observed anisotropy values (robs) with excitation at 555 nm and emission at 580 nm were recorded. robs was plotted against protein concentrations, and was numerically fitted to Equation 1 to obtain the equilibrium dissociation constant (Kd),
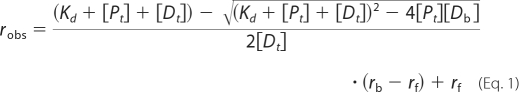 |
where rf and rb are the TMR anisotropy on free and protein-bound DNA, respectively.
In the heparin challenge assay, concentrated heparin (17–19 kDa, Sigma H3393) was added into a solution of 4 nm TMR-labeled RD-ds20 or −12/+8-mt20 DNAs that were saturated with Rpo41-Mtf1, and the replacement of promoter from the protein was monitored from the decrease in the fluorescence anisotropy.
Kinetics of Protein-DNA Association and Dissociation
Time-dependent fluorescence anisotropy changes of DNA-labeled TMR upon protein (Rpo41 or Rpo41-Mtf1) binding was performed on a T-format KinTek SF-2003 instrument according to the published protocol (25). Briefly, a solution of TMR-labeled ds-DNA (at a constant concentration between 80 and 100 nm) loaded in one syringe and the protein at various excessive concentrations loaded in another syringe were pushed to mix rapidly in the mixing chamber, which triggers the real-time measurement of fluorescence changes with excitation at 550 nm and emission longer than 570 nm over predefined time periods. The time-dependent anisotropy changes was fitted to Equation 2,
where rf and robs are the anisotropy values at starting and ending time points, respectively, A is the amplitude of the change, kobs is the observed rate constant, and t is the time. Fitting the dependence of k on excessive protein concentrations to Equation 3 reveals the association rate constant (kon),
where [P] is the protein concentration after mixing, and k− is the y-intercept.
The DNA dissociation rate constant was measured directly by rapidly mixing unlabeled DNA in a large excess with a preformed complex of TMR-labeled DNA and Rpo41 or Rpo41-Mtf1 (20 times excess) in the stopped-flow setup described earlier (25). The time course of fluorescence anisotropy change was fit to Eq. 2 where kobs correspond to koff.
FRET Assay
FRET assays were performed at 20 °C according to the published protocol of (ratio)A method (24, 26). FRET efficiencies (EFRET) between TMR labeled on the 5′ end of non-template and Alexa Fluor 647 (A647) on the 5′-end of template of the duplex were measured at a series of protein (Rpo41, Mtf1, or Rpo41-Mtf1) concentrations to assess the end-to-end distance changes according to the following formula,
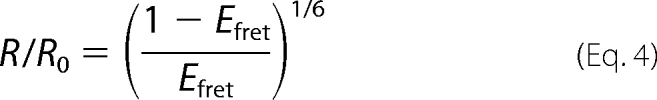 |
where Ro is the Förster radius of the donor-acceptor pair at which 50% of EFRET occurs (R0 = 66Å for TMR-A647) and R is the actual D-A distance measured from EFRET.
DNA Melting
Melting of duplex DNA was measured from 2-AP fluorescence changes (11). A mixture of Rpo41 and Mtf1 (1:1.2) was added to a solution of 150 nm 2-AP promoter or promoter mutant at 20 °C in the reaction buffer without Tween and glycerol, and the fluorescence intensity from 340 to 420 nm with excitation at 310 nm was measured and subtracted from background contribution by buffer and protein itself.
RESULTS
Specific Transcription from Promoter DNA
To determine the relative binding affinities of the Rpo41 and Mtf1 for the promoter and non-promoter sequences, we made several 20 bp DNAs that included a promoter DNA, a mutant promoter, and several non-promoter DNAs (Fig. 1A). We first tested each as substrate for transcription by Rpo41-Mtf1. Efficient RNA synthesis was observed only with the promoter DNA (Fig. 1B). RNA products from 2-mer to the expected 7–8 mer runoff RNAs (Fig. 1B, lane 1) were observed with the −12/+8-mt20 containing the 14 S rRNA promoter sequence from −12 to +8. The efficiency of total RNA synthesis from this minimal 20 bp promoter was comparable to the longer promoter templates (−25/+32-mt57 and −29/+32-mt61, Fig. 1A and supplemental Fig. S1, A and B). In a multi-round initiation reaction with ATP alone or ATP + 3′-dUTP, the production of 2-mer or 3-mer RNA on the short template was 1.5–3 fold less than on the two long templates (Fig. 1, C and D and supplemental Fig. S1, C and D). Considering, that the natural promoters in the yeast mt genome classified as strong and weak show 7–15-fold differences in their RNA synthesis activity (27), the transcriptional efficiency of the short −12/+8-mt20 promoter is comparable to the longer promoters. Thus, the short DNAs serve as good models for in vitro studies.
FIGURE 1.

Transcription on promoter and non-promoter DNAs. A, sequence of the DNA fragments used in this study and their transcription activities relative to the −12/+8-mt20. Sequences of long templates are shown in supplemental Fig. S1. B, transcription was measured at 25 °C with 4 μm DNA, 3 μm Rpo41-Mtf1, 100 μm ATP, and 250 μm each of GTP, UTP, and CTP spiked with [α-32P]ATP. Lanes show the RNA products resolved on a sequencing gel. Lane 5 is a control lane without the enzyme. C, transcription with 4 μm DNA, 3 μm Rpo41-Mtf1, 150 μm ATP, and 150 μm 3′dUTP spiked with [γ-32P]ATP. Lane 1 is control without the enzyme. D, top panel shows the amounts of 2 mer with ATP alone, and the bottom panel shows the amounts of 2–3 mers with ATP + 3′-dUTP.
Almost no RNA synthesis was observed on the non-promoter DNAs, regardless of whether it contained the upstream sequence of the 14 S rRNA promoter from −25 to −10 (NP1-ds20), the +7 to +22 downstream sequence (NP2-ds20), or a random sequence with 50% AT bp (RD-ds20) (Fig. 1, A and B and supplemental Fig. S1B). Similarly, introduction of a single bp change of G:C to C:G at the −2 position in the promoter made the -2C:G-mt20 transcriptionally inactive (Fig. 1B, lane 2), which is consistent with the importance of the −2 bp reported in previous studies (28) and further demonstrating the high promoter specificity of Rpo41-Mtf1. Rpo41 alone showed no specific RNA products with any of the DNA fragments, which is also consistent with previous transcriptional studies (13, 17).
Rpo41 Binds Equally Well to Promoter and Non-promoter DNA
Next we asked if specific and efficient transcription from the promoter DNA results from its tight binding affinity. We first tested if Rpo41 alone binds the promoter and non-promoter sequences and if so whether it can discriminate between the two. We measured the equilibrium dissociation constant or the Kd values of the Rpo41 complexes using fluorescence anisotropy titrations. The DNAs were modified with TMR fluorophore, which was attached to 5′-end of one of the strands of the dsDNA. The TMR modified promoter DNA has an anisotropy of ∼0.18, which increased to ∼0.32 upon addition of Rpo41 (triangle in Fig. 2A), indicating that Rpo41 alone can bind to the promoter DNA. We did not observe a significant change in the fluorescence intensity (<5%) of the labeled fluorophore upon Rpo41 or Rpo41-Mtf1 binding, suggesting that the fluorophore at +8 does not have extensive interactions with the protein.
FIGURE 2.

Equilibrium binding of promoter and non-promoter DNAs by fluorescence anisotropy. The TMR-labeled -12/8-mt20 promoter (5 nm) in A and TMR-labeled RD-ds20 non-promoter (5 nm) in B was titrated with increasing Rpo41 (triangle), Mtf1 (square), Mtf1 after saturation with Rpo41 (blank circle), or with 1:1.2 ratio mixture of Rpo41 and Mtf1 (filled circle). Fluorescence anisotropy was measured after excitation at 555 nm and emission at 580 nm. Titration data were fit to Equation 1 (solid lines) to derive the Kd values listed in the table below with standard deviation errors from multiple independent measurements. C, heparin competition was monitored by fluorescence anisotropy of 5 nm TMR-labeled RD-ds20 (filled circle) or −12/+8-mt20 (blank circle) saturated with 5 nm Rpo41+Mtf1 (1:1.2) in the standard binding buffer at 20 °C. The adjacent table shows the heparin IC50 values.
The hyperbolic increase in fluorescence anisotropy with increasing Rpo41 was fit to equation 1 to derive a Kd of 58 nm for the −12/+8-mt20 promoter. The nanomolar value of the Kd indicates that Rpo41 binds to the promoter DNA with a high affinity. The lack of RNA synthesis is not because Rpo41 does not bind the promoter DNA.
Fluorescence anisotropy titrations were carried out with the non-promoter DNAs (triangles in Fig. 2B) and with the promoter mutant. Surprisingly, Rpo41 binds to all of these DNAs with Kd values that are similar to the promoter DNA and ranging from 48–66 nm (table in Fig. 2). Based on these results, we conclude that Rpo41 binds to all types of DNAs tightly, and it does not discriminate between promoter and non-promoter DNA at the initial DNA binding step.
Mtf1 Increases DNA Binding Affinity
To determine if Mtf1 confers differential binding to promoter versus non-promoter DNAs, we titrated the TMR-labeled promoter with a mixture of Rpo41 and Mtf1 (1:1.2) (Fig. 2, A and B, filled circle). Data fitting provided Kd of 0.2 nm for the promoter DNA complexed with the Rpo41-Mtf1. Thus, compared with Rpo41 alone, Rpo41-Mtf1 binds to the promoter with a ∼290-fold higher affinity. Doubling the Mtf1 concentration in the mixture of Rpo41 [Rpo41]:[MTF1] = 1:2.2) did not change the Kd value.
We also show that Mtf1 by itself does not bind to the promoter DNA (Fig. 2A, open squares). However, addition of a small amount of Mtf1 (∼10 nm) to a preformed promoter-Rpo41 complex decreases the fluorescence anisotropy from ∼0.32 to ∼0.29 (Fig. 2A, open circle). This is another indication that Mtf1 binds to the promoter-Rpo41 complex.
Similar titrations with the non-promoter DNAs provided Kd values between 0.7–1.3 nm for these DNAs (Fig. 2, see table), which indicates that Mtf1 stabilizes the binding of even the non-promoter DNAs but by 40–70-fold, which is lesser than the stabilization of the promoter DNA. When we compare the affinities of the Rpo41-Mtf1 for the promoter and non-promoter DNAs, we find that the promoter is bound more stably than the non-promoter by 3–6-fold. This difference between promoter and non-promoter binding translates into ΔΔG = ΔGpromoter − ΔGnonpromoter of 0.7–1.3 kcal/mol of binding energy, which is modest, and it suggests that differential affinities for the DNA is not the major mechanism of promoter selection.
We performed fluorescence anisotropy-based heparin challenge assays in which a complex of the promoter or the non-promoter DNA with Rpo41-Mtf1 was competed with increasing amounts of the polyanionic heparin (Fig. 2C). The results show that the non-promoter DNA complex is ∼5-times more sensitive to heparin challenge than the promoter complex (IC50 = 0.32 μg/ml for non-promoter versus 1.5 μg/ml for the promoter). This suggests that the nonspecific interactions with the non-promoter DNA most likely through the sugar-phosphate backbone of the DNA are tight, but they are easily challenged by heparin as opposed to the base specific interactions with the promoter DNA.
An early DNase I footprinting study had used partially purified Rpo41 and Mtf1 and identified a protection region extending from −15 to +15 on the promoter, which is significantly larger than the template used in this work (16). Because of the low purity of Rpo41 and Mtf1 in the mitochondrial extracts used and undetermined protein concentrations and their ratios to DNA, it is possible that the footprint resulted from multiple proteins on the DNA, as we find that Rpo41-Mtf1 can bind to specific and nonspecific sites with similar binding affinities. Longer promoter templates (e.g. from −25 to +20) did not increase the binding affinity for the Rpo41-Mtf1-template complex (data not shown). In fact, we observed multiple Rpo41-Mtf1-DNA complexes on the longer templates.
Mtf1 Stabilizes the Dynamic Rpo41-DNA Complex by Decreasing the Off-rate
According to the relationship: Kd = koff/kon, where koff and kon are the dissociation and association rate constants of the complex, respectively, Mtf1 may stabilize the Rpo41-DNA complex by decreasing the koff or by increasing the kon or both. We determined the koff and kon from the time-dependent changes in fluorescence anisotropy in the following stopped-flow mixing set-up (25). Fluorescence anisotropy changes were measured in millisecond to second time scale after mixing TMR-labeled DNA with the Rpo41 or with an equimolar mixture of Rpo41 + Mtf1 (Fig. 3A). Under the conditions in Fig. 3, B and C, promoter and non-promoter DNAs bind to Rpo41 or Rpo41-Mtf1 at nearly identical rate constants (kobs) of 46–60 s−1. The kobs was measured at various Rpo41 or Rpo41-Mtf1 concentrations, and from the slopes of kobs versus [protein], kon values ranging from 2.5–3.6 × 108 m−1 · s−1 were determined (Fig. 3, C and E). The similar diffusion-limited kon values for promoter and random DNAs indicate that Rpo41 and Rpo41-Mtf1 do not differentiate between promoter and non-promoter sequences at the initial binding step.
FIGURE 3.

Stopped-flow kinetics of promoter and non-promoter DNA binding. A, TMR-labeled DNA from one syringe of the stopped-flow instrument was mixed with Rpo41 or Rpo41+Mtf1 at various concentrations from second syringe. Fluorescence emission was measured in parallel and perpendicular orientation of polarizer relative to polarized excitation. B, time courses of TMR anisotropy showing the rapid increase upon mixing of 120 nm Rpo41 (black) or 1:1 mixture of Rpo41 and Mtf1 (gray) with 40 nm TMR-labeled −12/+8-mt20. Data were fit to a single exponential function to obtain the observed rate constant kobs = 46 s−1 for Rpo41 binding or 60 s−1 for Rpo41+Mtf1 binding. C, rate of binding (kobs) to −12/+8-mt20 is plotted against increasing Rpo41 (blank circle) or Rpo41-Mtf1 (filled circle). The data were fit to a linear equation to obtain the kon values listed in the table below. D, association kinetics of the TMR-labeled non-promoter DNA RD-ds20 measured similarly as in A provided kobs = 56 s−1 in the presence of Rpo41 and 57 s−1 in the presence of Rpo41 and Mtf1. E, concentration dependence of kobs against [Rpo41] (blank circle) or Rpo41-Mtf1 (filled circle) provided the kon values listed in the table below, with data uncertainties showing the range of two or more independent measurements.
To measure the dissociation rate constant, koff, we designed a chase experiment in which a preformed complex of Rpo41 or Rpo41-Mtf1 with the TMR-labeled DNA was mixed with a 10-fold molar excess of the unlabeled DNA. In this experiment, the dissociation rate of the labeled DNA can be measured from the decrease in fluorescence anisotropy, because once the labeled DNA dissociates from the protein-DNA complex, the protein rebinds with a higher probability to the unlabeled DNA present in excess. The off-rate measurements show that Rpo41 alone forms a dynamic complex with both the promoter and the non-promoter DNA and has a fast koff of 16–18 s−1 (Fig. 4, B and D, and the associated table). This is probably the reason why a stable Rpo41-DNA complex was not detected in previous non-equilibrium assays (16).
FIGURE 4.

Kinetics of DNA dissociation. A, unlabeled −12/+8-mt20 (1 μm) from one syringe of the stopped-flow instrument was mixed with a pre-formed complex of 100 nm Rpo41 or Rpo41-Mtf1 and 40 nm TMR-labeled −12/+8-mt20 or RD-ds20 from a second syringe. The time-dependent decrease in fluorescence anisotropy of TMR-labeled DNAs was fit to a single exponential equation to obtain the koff values listed in the table. Dissociation kinetics of −12/+8-mt20 -Rpo41 (B), −12/+8-mt20-Rpo41-Mtf1 (C), RD-ds20 -Rpo41 (D), and RD-ds20-Rpo41-Mtf1 (E) are shown. The data uncertainties show the range of at least two independent measurements.
When Mtf1 was present, the koff of the promoter DNA decreased from 16–18 s−1 to 0.02 s−1 (Fig. 4C) and that of the non-promoter DNA decreased from 16–18 s−1 to 0.07–0.09 s−1 (Fig. 4E and the associated table). Thus, off-rate of the promoter DNA decreases by ∼800-fold in the presence of Mtf1 and off-rate of the non-promoter DNA decreases by ∼200-fold, which accounts for the lower Kd of the DNA complex in the presence of Mtf1. Similarly, the 4-fold lower off-rate from the promoter DNA as compared with the non-promoter DNA accounts for the 3–6-fold tighter affinity of the Rpo41-Mtf1 for the promoter DNA.
Rpo41 Bends Promoter and Non-promoter DNA Equally
An earlier study had suggested that the promoter DNA is bent by the yeast mitochondrial RNA polymerase fractions (29). To investigate whether DNA bending is caused by Rpo41 alone or by Rpo41-Mtf1 and to quantify the extent of DNA bending, we used the approach of FRET. One end of the dsDNA was attached to the fluorescent donor TMR and the other to the acceptor A647 and the FRET efficiency (EFRET) between the fluorophores was determined from sensitized acceptor fluorescence using the (ratio)A method (26). The EFRET of the free 20-bp promoter DNA was ∼0.30, and upon addition of Rpo41, the EFRET increased indicating DNA bending by Rpo41 (Fig. 5A, triangle). To determine the degree of DNA bending, we measured the maximum EFRET by titrating the promoter DNA with increasing Rpo41. The titration provided a maximum EFRET of ∼0.45 and a Kd of ∼29 nm, which is similar to that obtained from the fluorescence anisotropy titration. The maximum EFRET indicates that Rpo41 binding shortens the end-to-end distance in the DNA by ∼8 Å. Assuming that the DNA adopts a kink-based bent structure in the Rpo41-DNA complex, a DNA bending angle of ∼52° was calculated (24).
FIGURE 5.

DNA bending measurements of promoter and non-promoter DNAs by FRET. Donor-acceptor doubly labeled DNA (20 nm) was titrated with Mtf1 (blank circle), Rpo41 (red triangle), or a mixture of Rpo41+Mtf1 (1:1.2) (black filled circle), or titrated with Mtf1 following Rpo41 saturation (green square). EFRET was calculated at each point as described under “Experimental Procedures.” A, FRET measurements on −12/+8-mt20. B, FRET measurements on RD-ds20. C, FRET measurements on promoter mutant −2C:G-mt20. D, FRET efficiencies of free DNA (black bars) and in a complex with Rpo41 (red bars) or Rpo41-Mtf1 (green bars). The yellow bars show the FRET efficiencies at the highest protein concentration used. Standard errors from multiple measurements are shown. Error bars show the standard deviation for −12/+8-mt20 and RD-mt20 from multiple independent experiments or the range from two measurements for other DNA constructs.
Similarly, the EFRET of the non-promoter and mutant promoter increased upon addition of Rpo41 to the same value as observed with the promoter DNA (Fig. 5, B and C). These results indicate that Rpo41 alone induces modest bending in all DNAs, irrespective of the DNA sequence. Thus, Rpo41 by itself does not discriminate between promoter and non-promoter sequences either at the binding or at the DNA bending step.
Differential DNA Bending by Mtf1
Mtf1 by itself does not bind DNA, and hence it does not cause any observable change in the FRET signal (Fig. 5A, blank circle). However, adding Mtf1 to the promoter-Rpo41 complex causes a dramatic increase in EFRET (Fig. 5A, square). This large increase in EFRET was confirmed by titrating the promoter with increasing Rpo41-Mtf1 (1:1.2 ratio). The EFRET increased from an initial value of ∼0.30 to a maximum value of ∼0.74 in a hyperbolic manner that fit to Kd of 0.3 nm (Fig. 5A, filled circle). The almost equal sensitivity of FRET and anisotropy for DNA binding of Rpo41 or Rpo41-Mtf1 suggests an insignificant “propeller effect” or restriction of the tumbling of tethered dyes. Because the same limiting value of EFRET is observed in two types of titrations with different order of additions, it indicates that there is no preferential order of binding Mtf1 and Rpo41 to the DNA.
The increase in EFRET from 0.45 in the presence of Rpo41 alone to ∼0.74 with Rpo41-Mtf1 indicates that Mtf1 induces an additional shortening of the end-to-end distance in the promoter DNA by ∼13 Å and an overall shortening of ∼21 Å. Assuming a single central kink-based bent structure, we estimate that the final bending angle is ∼89° for the DNA in complex with Rpo41-Mtf1. This is similar to the DNA bending angle of ∼80–90° observed in the T7 RNAP initiation open complex (24, 30) and ∼55–80° for the E. coli RNAP initiation open complex (31, 32). Adding the initiating nucleotide ATP (200 μm) did not cause an additional change in the FRET (EFRET of 0.75, Fig. 5A, diamond).
EFRET measurements with the non-promoter DNAs provided very different results. Addition of Mtf1 to the non-promoter DNA pre-bound to Rpo41 did not increase the EFRET as observed with the promoter DNA (Fig. 5, B and D). Instead, we observed a progressive decrease in EFRET from its initial value of ∼0.43 to a low value of ∼0.3, which is the EFRET of the free DNA. Titration of the non-promoter DNA with increasing concentration of Rpo41-Mtf1 showed an initial increase in EFRET from ∼0.29–0.30 to ∼0.42–0.45 value that corresponds to the small bend induced by Rpo41 alone. Higher concentrations of Rpo41-Mtf1 resulted in a decrease in EFRET from ∼0.42–0.45 to ∼0.3. These results indicate that Mtf1 causes an unbending of the non-promoter DNA bound to the Rpo41. The exact mechanism of unbending is unclear.
The sharp bending of the promoter DNA induced was not observed in the -2C:G mutant promoter. Titration of the mutant promoter with Rpo41-Mtf1 increased EFRET to the value observed with Rpo41 alone, but unlike the non-promoter DNA, a subsequent decrease in EFRET was not observed with the mutant promoter (Fig. 5C). This indicates that the mutant promoter remains modestly bent in the presence of Rpo41-Mtf1. Thus, promoter, non-promoter, and the -2C:G mutant promoter assume different conformational states when bound to Rpo41-Mtf1.
Only the Sharply Bent DNA Is Melted
To determine if DNA bending leads to DNA melting, the adenine at -4NT in the promoter and the -2C:G mutant promoter was replaced with the fluorescent analog 2-aminopurine (2-AP). Previous studies had shown that 2-AP at -4NT undergoes a large increase in fluorescence intensity upon addition of Rpo41-Mtf1, but not with the Rpo41 alone (11). These results were reproduced with the titration of 2AP-substituted promoter with a mixture of Rpo41 and Mtf1 (1:1.2 molar ratio) (Fig. 6). Titration of the 2AP modified -2C:G mutant promoter with Rpo41-Mtf1, on the other hand, showed only a slight fluorescence increase. These results indicate that the mutant promoter, which is modestly bent upon binding Rpo41-Mtf1, is not melted. On the other hand, the promoter, which is sharply bent by Rpo41-Mtf1, is melted efficiently. Thus, sharp DNA bending and melting are coupled, as observed in T7 RNAP (24). It is interesting that a single bp transversion from G:C to C:G at position -2 abolishes DNA bending and melting, which is required for the formation of competent pre-initiation open complex. This explains the important role of -2 G:C bp of the promoter in initiating transcription (28).
FIGURE 6.

DNA melting measured by 2-AP fluorescence. The 2-AP base (black star in the cartoon) was placed at -4NT in the −12/+8-mt20 (150 nm, filled triangles) and in the -2C:G -mt20 (150 nm, blank triangles). The ratio of 2-AP fluorescence intensity (F) at the individual Rpo41-Mtf1 concentrations to that of the free 2-AP DNA fragments (F0) is representative of DNA melting.
DISCUSSION
In this report, we have investigated how the transcription machinery of the yeast mitochondria consisting of the catalytic subunit Rpo41 and the transcription factor Mtf1 selectively initiates transcription from the promoter sequence and discriminate against the non-promoter sequences.
Our equilibrium DNA binding studies show that Rpo41 binds to both the promoter and non-promoter sequences with similar affinities. Based on these results, we propose that Rpo41 by itself does not make sequence-specific contacts with the duplex DNA, or if such interactions are made, they do not make a significant energetic contribution to the promoter selection. However, Rpo41 is known to catalyze promoter-specific transcription on pre-melted promoters (18). Therefore, it appears that Rpo41 can make sequence-specific contacts with the promoter only when the DNA is melted. Thus, on the duplex mitochondrial genome, Rpo41 by itself cannot distinguish between promoter and non-promoter sequences.
Mtf1 stabilizes promoter binding by ∼300-fold primarily by decreasing the off-rate of the promoter complex. Surprisingly, Mtf1 had a stabilizing effect on the non-promoter complexes as well, although by only 40–70-fold. Thus, the difference in the binding affinities of Rpo41-Mtf1 for the promoter and non-promoter DNAs is only 3–6-fold. This indicates that differential DNA binding affinity is not the major mechanism of promoter selection by the yeast mitochondrial RNA polymerase.
This feature of the yeast mtRNAP is different from the phage T7 RNAP, which binds to the promoter ∼105-order tighter than to non-promoter sequences (24, 33). Similarly, Mtf1 is different from σ70, which stabilizes the promoter complex by decreasing the off-rate and destabilizes the nonspecific complex by increasing the off-rate increase (34). A recent gel-shift study of Schizosaccharomyces pombe mtRNAP showed that the spRpo41 binds to the promoter DNA on its own; however, there was a significant increase in the shifted complex in the presence of spMtf1 (35), which is consistent with our results. This study also showed that the spRpo41-Mtf1 failed to bind mutant promoters. Thus, it appears that the spRpo41-Mtf1 can differentiate promoters from the non-promoters at the binding step; however, it is also possible that complexes with the non-promoter DNAs did not survive the gel-shift assay, because they dissociated due to their faster off-rates. Whether spRpo41-Mtf1 can distinguish between promoter and non-promoter sequences at the binding step needs to be rigorously tested using equilibrium binding assays, as used in our studies.
Both Rpo41 and Rpo41-Mtf1 bind tightly to the non-promoter DNAs, but these DNAs are not transcribed (Fig. 1B). Hence, tight DNA binding does not translate into specific catalysis. Rpo41 alone bends both promoter and non-promoter DNAs by ∼52°, which interestingly is similar to the bending angle in the closed complex of T7 RNAP (24). Differential DNA bending in promoter, mutant promoter, and non-promoter DNAs was observed only in the presence of Mtf1. In the promoter DNA, Mtf1 addition causes an added bending in the DNA from ∼52° to ∼90°. This sharply bent DNA resembles the DNA in the open complex of T7 RNAP (24). The non-promoter DNA, on the other hand, was unbent. Interestingly, when the promoter contained a single transversion at the −2 position, the DNA remained modestly bent in the presence of Rpo41-Mtf1. Our recent studies show that promoter bending can be measured by single molecule FRET and was observed with Rpo41-Mtf1 (54). These studies revealed DNA bending/unbending transitions indicating that this process is dynamic and Mtf1 increased the bending transitions while suppressing the unbending transitions.
By inducing different conformational changes in the promoter, non-promoter, and mutant promoter (Fig. 7), Rpo41-Mtf1 is able to distinguish between specific and nonspecific DNAs. Only the sharply bent DNA is melted as measured by 2-aminopurine fluorescence increase. Thus, DNA bending and melting are coupled processes in the yeast mitochondrial RNAP, similar to T7 RNAP (24). The dramatic effect of a single base pair change indicates a cooperative mechanism of promoter selection whereby all base-specific interactions are required to sharply bend and melt the DNA. Our results indicate that transcription factor-enhanced DNA bending and melting rather than protein-DNA binding plays an important role in transcription efficiency and loss of an induced conformational change is responsible for the reduction or inactivation of transcription efficiency documented for sequence-mutated promoter variants (28, 29, 36, 37).
FIGURE 7.

Differential conformational changes in promoter selection by mitochondrial RNA polymerase. Schemes show that Rpo41 (blue) by itself binds the promoter (A), mutant promoter (B), and non-promoter (C) with similar Kd values and induces similar DNA bends in all DNAs. No specific structural interactions between the RNAP and DNA are implied by the cartoons. Mtf1 (pink) binding strengthens DNA binding, but only the promoter DNA is sharply bent and melted, whereas the mutant is slightly bent, and the non-promoter remains unbent. The Kd values shown here are based on the equilibrium titration assays.
DNA bending has been observed in almost all transcription initiation complexes from bacteriophage, bacteria, archaea, and eukaryotes (38, 39). T7 RNAP by itself can bend the promoter DNA to cause spontaneous melting in a region from −4 to +2, similar to what is observed with Rpo41-Mtf1 (25). DNA bending of bacterial promoters requires the presence of both the core RNAP subunit and the accessory sigma factor (40). Transcription factors induced DNA bending near the TATA element is an essential step for the recruitment of archaeal RNAP or eukaryotic RNAP II to the promoter (41, 42). However, transcription factor-enhanced differential conformational changes of the DNA as a mechanism for promoter recognition has not been demonstrated for these transcription systems.
It will be interesting to see whether the mammalian mitochondrial systems employ such a factor-dependent induced-fit mechanism for promoter recognition. Recently, Shutt et al. (9) proposed that the human mtRNAP consisting of the core catalytic enzyme, POLRMT, and the transcription factor TFB2M is a two component complex, which is similar to the yeast Rpo41-Mtf1 complex. Like Rpo41, POLRMT appears to have some intrinsic promoter recognition besides its primary polymerization activity (19). Similarly like Mtf1, TFB2M is involved in DNA melting by binding and stabilizing the single-stranded DNA adjacent to the transcription start site (10). To achieve specific transcription initiation on human mitochondrial promoters LSP and HSP1, cooperative actions from both proteins are required to recognize promoter and form a competent pre-initiation complex. However, there are no data directly addressing DNA binding and distortion (bending and melting) by POLRMT and TFB2M.
DNA bending as a mechanism to differentiate between specific and nonspecific sequences is observed in many DNA binding proteins such as EcoRV endonuclease, lac repressor-operator, and p53-DNA complexes. In these proteins, similar to the Rpo41-Mtf1, DNA is bent in specific complexes and not bent in nonspecific complexes (43–47). MutS is peculiar in that it appears to bend DNA at nonspecific sites but not at specific sites (48), and λ Cro protein bends DNA at both specific and nonspecific sites (49). We propose that this type of induced-fit mechanism whereby specific sites are selected by differential DNA bending is analogous to the induced-fit transition mechanism of enzymes, where conformational differences dictate enzyme specificity (50). In the case of transcription, the unbent non-promoter DNA is optimal for RNA polymerase translocation during the search for promoter sites on the genome whereas the sharply bent promoter DNA structure is an intermediate that resembles the transition state toward the formation of the catalytically active open complex. The energetic cost of bending the DNA may be compensated by additional interactions of the active site with the bent DNA structure and/or the resulting single-stranded DNAs, which makes promoter opening to proceed spontaneously.
The search for the promoter on the mitochondrial genome is likely to be a complex process involving several factors. In vivo the mitochondrial DNA is coated with an HMG-like protein, such as Abf2 in yeast and TFAM in human (51). Abf2 is a DNA packaging protein that binds DNA in a phase of 15–30 bp excluding A-tract sequences (52, 53). The yeast mitochondrial promoter sequence is AT-rich and its selective exposure would significantly reduce the search region and increase the accessibility of the promoter sites to the RNA polymerase. As opposed to Rpo41 alone, which dissociates more easily from the DNA, the Rpo41-Mtf1 complex binds DNA more tightly; thus, a productive search for promoter along the DNA can occur by sliding without frequent dissociation. The ease of DNA bending may be used as a check mark for promoter-specific sites.
Supplementary Material
Acknowledgment
We thank Swaroopa Paratkar for providing purified Rpo41 and Mtf1 proteins.
This research was supported, in whole or in part, by National Institutes of Health Grant GM51966 (to S. S. P.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Fig. S1.
- RNAP
- RNA polymerase
- FRET
- Förster resonance energy transfer
- 2-AP
- 2-aminopurine.
REFERENCES
- 1. Reznikoff W. S., Siegele D. A., Cowing D. W., Gross C. A. (1985) Annu. Rev. Genet. 19, 355–387 [DOI] [PubMed] [Google Scholar]
- 2. Verrijzer C. P., Tjian R. (1996) Trends Biochem. Sci. 21, 338–342 [PubMed] [Google Scholar]
- 3. Soppa J. (1999) Mol. Microbiol 31, 1295–1305 [DOI] [PubMed] [Google Scholar]
- 4. McAllister W. T. (1993) Cell Mol. Biol. Res. 39, 385–391 [PubMed] [Google Scholar]
- 5. Masters B. S., Stohl L. L., Clayton D. A. (1987) Cell 51, 89–99 [DOI] [PubMed] [Google Scholar]
- 6. Fisher R. P., Clayton D. A. (1988) Mol. Cell Biol. 8, 3496–3509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Falkenberg M., Gaspari M., Rantanen A., Trifunovic A., Larsson N. G., Gustafsson C. M. (2002) Nat. Genet. 31, 289–294 [DOI] [PubMed] [Google Scholar]
- 8. Matsushima Y., Garesse R., Kaguni L. S. (2004) J. Biol. Chem. 279, 26900–26905 [DOI] [PubMed] [Google Scholar]
- 9. Shutt T. E., Lodeiro M. F., Cotney J., Cameron C. E., Shadel G. S. (2010) Proc. Natl. Acad. Sci. U.S.A. 107, 12133–12138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sologub M., Litonin D., Anikin M., Mustaev A., Temiakov D. (2009) Cell 139, 934–944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tang G. Q., Paratkar S., Patel S. S. (2009) J. Biol. Chem. 284, 5514–5522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Levens D., Lustig A., Rabinowitz M. (1981) J. Biol. Chem. 256, 1474–1481 [PubMed] [Google Scholar]
- 13. Jang S. H., Jaehning J. A. (1991) J. Biol. Chem. 266, 22671–22677 [PubMed] [Google Scholar]
- 14. Schubot F. D., Chen C. J., Rose J. P., Dailey T. A., Dailey H. A., Wang B. C. (2001) Protein Sci. 10, 1980–1988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Biswas T. K., Edwards J. C., Rabinowitz M., Getz G. S. (1985) Proc. Natl. Acad. Sci. U.S.A. 82, 1954–1958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Schinkel A. H., Groot Koerkamp M. J., Tabak H. F. (1988) EMBO J. 7, 3255–3262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Xu B., Clayton D. A. (1992) Nucleic Acids Res. 20, 1053–1059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Matsunaga M., Jaehning J. A. (2004) J. Biol. Chem. 279, 44239–44242 [DOI] [PubMed] [Google Scholar]
- 19. Gaspari M., Falkenberg M., Larsson N. G., Gustafsson C. M. (2004) EMBO J. 23, 4606–4614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Shadel G. S., Clayton D. A. (1995) Mol. Cell Biol. 15, 2101–2108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Paratkar S., Patel S. S. (2010) J. Biol. Chem. 285, 3949–3956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Savkina M., Temiakov D., McAllister W. T., Anikin M. (2010) J. Biol. Chem. 285, 3957–3964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Matsunaga M., Jang S. H., Jaehning J. A. (2004) Protein Expr. Purif. 35, 126–130 [DOI] [PubMed] [Google Scholar]
- 24. Tang G. Q., Patel S. S. (2006) Biochemistry 45, 4936–4946 [DOI] [PubMed] [Google Scholar]
- 25. Tang G. Q., Patel S. S. (2006) Biochemistry 45, 4947–4956 [DOI] [PubMed] [Google Scholar]
- 26. Clegg R. M. (1992) Methods Enzymol. 211, 353–388 [DOI] [PubMed] [Google Scholar]
- 27. Biswas T. K. (1990) Proc. Natl. Acad. Sci. U.S.A. 87, 9338–9342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Biswas T. K., Getz G. S. (1986) J. Biol. Chem. 261, 3927–3930 [PubMed] [Google Scholar]
- 29. Schinkel A. H., Groot Koerkamp M. J., Teunissen A. W., Tabak H. F. (1988) Nucleic Acids Res. 16, 9147–9163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yin Y. W., Steitz T. A. (2002) Science 298, 1387–1395 [DOI] [PubMed] [Google Scholar]
- 31. Rippe K., Guthold M., von Hippel P. H., Bustamante C. (1997) J. Mol. Biol. 270, 125–138 [DOI] [PubMed] [Google Scholar]
- 32. Rivetti C., Guthold M., Bustamante C. (1999) EMBO J. 18, 4464–4475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bandwar R. P., Patel S. S. (2002) J. Mol. Biol. 324, 63–72 [DOI] [PubMed] [Google Scholar]
- 34. Record M. T., Reznikoff J. W. S., Craig M. L., McQuade K. L., Achlax P. J. (1996) Escherichia coli RNA Polymerase (Eσ70), Promoters, and the Kinetics of the Steps of Transcription Initiation, 2nd Ed., ASM Press, Washington D.C [Google Scholar]
- 35. Jiang H., Sun W., Wang Z., Zhang J., Chen D., Murchie A. I. (2011) Nucleic Acids Res. 39, 5119–5130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Biswas T. K., Getz G. S. (1990) J. Biol. Chem. 265, 19053–19059 [PubMed] [Google Scholar]
- 37. Biswas T. K., Ticho B., Getz G. S. (1987) J. Biol. Chem. 262, 13690–13696 [PubMed] [Google Scholar]
- 38. Coulombe B., Burton Z. F. (1999) Microbiol Mol. Biol. Rev. 63, 457–478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Pérez-Martin J., de Lorenzo V. (1997) Annu. Rev. Microbiol 51, 593–628 [DOI] [PubMed] [Google Scholar]
- 40. Heumann H., Ricchetti M., Werel W. (1988) EMBO J. 7, 4379–4381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bartlett M. S. (2005) Curr. Opin. Microbiol 8, 677–684 [DOI] [PubMed] [Google Scholar]
- 42. Lee T. I., Young R. A. (2000) Annu. Rev. Genet. 34, 77–137 [DOI] [PubMed] [Google Scholar]
- 43. Winkler F. K., Banner D. W., Oefner C., Tsernoglou D., Brown R. S., Heathman S. P., Bryan R. K., Martin P. D., Petratos K., Wilson K. S. (1993) EMBO J. 12, 1781–1795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kalodimos C. G., Biris N., Bonvin A. M., Levandoski M. M., Guennuegues M., Boelens R., Kaptein R. (2004) Science 305, 386–389 [DOI] [PubMed] [Google Scholar]
- 45. Nagaich A. K., Appella E., Harrington R. E. (1997) J. Biol. Chem. 272, 14842–14849 [DOI] [PubMed] [Google Scholar]
- 46. Jen-Jacobson L. (1997) Biopolymers 44, 153–180 [DOI] [PubMed] [Google Scholar]
- 47. Albright R. A., Matthews B. W. (1998) Proc. Natl. Acad. Sci. U.S.A. 95, 3431–3436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wang H., Yang Y., Schofield M. J., Du C., Fridman Y., Lee S. D., Larson E. D., Drummond J. T., Alani E., Hsieh P., Erie D. A. (2003) Proc. Natl. Acad. Sci. U.S.A. 100, 14822–14827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Erie D. A., Yang G., Schultz H. C., Bustamante C. (1994) Science 266, 1562–1566 [DOI] [PubMed] [Google Scholar]
- 50. Post C. B., Ray W. J., Jr. (1995) Biochemistry 34, 15881–15885 [DOI] [PubMed] [Google Scholar]
- 51. Chen X. J., Butow R. A. (2005) Nat. Rev. Genet. 6, 815–825 [DOI] [PubMed] [Google Scholar]
- 52. Diffley J. F., Stillman B. (1991) Proc. Natl. Acad. Sci. U.S.A. 88, 7864–7868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Diffley J. F., Stillman B. (1992) J. Biol. Chem. 267, 3368–3374 [PubMed] [Google Scholar]
- 54. Kim H., Tang G. Q., Patel S. S., Ha T. (2011) Nucleic Acids Res. doi: 10.1093/nar/gkr736 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.