

Background: The AAA ATPase p97 mediates essential cellular processes, including ubiquitin-dependent protein degradation, and has been linked to human proteinopathies.
Results: VIM-containing cofactors bind as α-helical peptides with varying affinities to the N domain of p97.
Conclusion: VIM-containing cofactors compete with other cofactors recognizing the N domain, thus regulating p97 function.
Significance: Competitive binding is a crucial determinant in p97-cofactor interactions.
Keywords: ATPases, Endoplasmic Reticulum (ER), Protein Degradation, Ubiquitin, Ubiquitin Ligase, Ubiquitylation
Abstract
The AAA (ATPase associated with various cellular activities) ATPase p97, also referred to as valosin-containing protein (VCP), mediates essential cellular processes, including ubiquitin-dependent protein degradation, and has been linked to several human proteinopathies. p97 interacts with multiple cofactors via its N-terminal (p97N) domain, a subset of which contain the VCP-interacting motif (VIM). We have determined the crystal structure of the p97N domain in complex with the VIM of the ubiquitin E3 ligase gp78 at 1.8 Å resolution. The α-helical VIM peptide binds into a groove located in between the two subdomains of the p97N domain. Interaction studies of several VIM proteins reveal that these cofactors display dramatically different affinities, ranging from high affinity interactions characterized by dissociation constants of ∼20 nm for gp78 and ANKZF1 to only weak binding in our assays. The contribution of individual p97 residues to VIM binding was analyzed, revealing that identical substitutions do not affect all cofactors in the same way. Taken together, the biochemical and structural studies define the framework for recognition of VIM-containing cofactors by p97. Of particular interest to the regulation of p97 by its cofactors, our structure reveals that the bound α-helical peptides of VIM-containing cofactors overlap with the binding site for cofactors containing the ubiquitin regulatory X (UBX) domain present in the UBX protein family or the ubiquitin-like domain of NPL4 as further corroborated by biochemical data. These results extend the concept that competitive binding is a crucial determinant in p97-cofactor interactions.
Introduction
The type II AAA (ATPase associated with various cellular activities) ATPase p97 is a highly abundant protein with many roles in diverse biological processes, including endoplasmic reticulum-associated degradation (ERAD),2 ubiquitin fusion degradation (UFD), homotopic membrane fusion, autophagy, transcriptional control, and cell cycle regulation (1–4). Irrespective of the biological context, the general function of p97, also known as valosin-containing protein (VCP), and its yeast ortholog Cdc48 is to carry out mechanical work, a process that is fueled by ATP hydrolysis. Due to its involvement in several important cellular processes, it is not surprising that defects in p97 and its binding partners contribute to various diseases. p97 itself has been linked to cancer and a wide variety of neurodegenerative disorders, including Parkinson disease, Lewy body disease, Huntington disease, and spinocerebellar ataxia type III (Machado-Joseph disease) (5). Mutations in p97 cause inclusion body myopathy, Paget disease of the bone, and frontotemporal dementia (IBMPFD), a rare proteinopathy that mainly affects skeletal muscle, brain, and bone (6). p97 mutations also cause autosomal-dominantly inherited amyotrophic lateral sclerosis, a fatal neurodegenerative disease characterized by motor neuron degeneration (7).
p97 forms a homohexameric assembly where each subunit is composed of an N-terminal (p97N) domain and two hexameric ATPase domains referred to as D1 and D2, followed by an unstructured C terminus. The overall architecture of p97 can be described as two stacked hexameric rings formed by the D1 and D2 domains with the N domain on the outside of the D1 ring (8, 9).
Mammalian p97 binds to various cofactors, which define, at least in part, the biological processes in which this enzyme participates (10); the best understood examples are the heterodimeric UFD1-NPL4 complex, which specifies p97 participation in ERAD as well as other pathways, and the homotrimeric p47 protein, which recruits p97 to induce homotypic membrane fusion and involves it in autophagy. On a functional level the cofactors can be subdivided into substrate-recruiting and substrate-processing cofactors (11). Major substrate-recruiting cofactors determine the participation of p97 in different cellular pathways and bind in a mutually exclusive fashion to p97, whereas additional substrate-recruiting cofactors fine tune interactions within a given pathway (4). Substrate-processing cofactors typically display enzymatic activity, including E3 and E4 ubiquitin ligase, as well as deubiquitylation activities and primarily play a role in ERAD and UFD, where they regulate the fate of p97 substrates.
From a topological perspective, p97 cofactors can be grouped into N domain binding cofactors and a smaller group interacting with the C terminus (4, 10, 12). Cofactors binding to the C terminus of p97 include peptide:N-glycanase/UBA or UBX (PUB) domain-containing proteins, such as mammalian peptide:N-glycanase (13) as well as Ufd3 (PLAA in mammals), which interacts with the p97 C terminus via its PLAP, Ufd3p, Lub1p (PUL) domain (14). Among cofactors binding to the N domain, one can find several UBX-containing proteins, including p47, FAF1 (FAS-associated factor 1), and UBXD7, which are characterized by a common ubiquitin regulatory X (UBX) domain that adopts the same fold as ubiquitin (4). The UBX protein UBXD1 represents a special case because it contains both a PUB and a UBX domain; however, the latter does not interact with the N domain of p97 (15).
Besides the UBX domain and the ubiquitin-like domain (ULD) of NPL4, which both bind to the N domain of p97 at the interface between its two subdomains (16–18), three linear peptide-binding motifs have been identified in p97 cofactors (10, 19). These include the VCP-interacting motif (VIM), the VCP-binding motif, and the SHP box (also known as binding site 1 (BS1)). The VCP-binding motif is found in the E3 ligase HRD1, in the E4 ligase E4B, and in Ataxin-3. This motif is characterized by a repeat of four basic residues. The SHP box has been first identified in Shp1, the yeast ortholog of p47, and contains the sequence FpGXGQ(R/K)h (where p represents a polar residue and h is a non-polar residue), which is also found in UFD1. The VIM was originally identified in the E3 ubiquitin ligase gp78 (also referred to as the autocrine motility factor receptor (AMFR)) (20, 21) and small valosin-interacting protein (SVIP) (22), which has been proposed to act as an inhibitor of gp78-mediated ERAD (23). Earlier in vivo studies of p97 and gp78 revealed that the gp78-VIM is essential for recruitment of p97 to the endoplasmic reticulum and its subsequent function in ERAD (20). Recently, a VIM has also been identified in Vms1, the yeast homolog of ANKZF1 (ankyrin repeat and zinc finger domain-containing 1), a protein that has been implicated in mitochondrial protein degradation (24). From a structural perspective, it is interesting to note that residues belonging to the VIM are predicted to form an α-helix.
In order to understand the rules governing recognition of the VIM by p97, we have determined the co-crystal structure of the p97N domain in complex with a gp78-derived peptide coupled with a detailed structure-function analysis of additional VIM-containing cofactors. This allows us to derive general rules for VIM binding by p97 and for how this interaction is related to the binding of additional p97 cofactors.
EXPERIMENTAL PROCEDURES
Cloning and Site-directed Mutagenesis
The following constructs were used in this study: human full-length p97 (amino acids (aa) 1–806) and p97N (aa 1–187) (18); p97N + linker (p97NL, aa 1–208) (pProEx_HTA, Invitrogen, KasI/HindIII, N-terminal His tag); human p97ND1 (aa 1–480) and p97ND1-R155H (18); human UFD1 and NPL4 (18); human full-length UBXD1 (aa 1–441) and UBXD1N (aa 1–150) (pET28M-SUMO1, EMBL Heidelberg, BamHI/XhoI, N-terminal His-SUMO tag); human SVIP (pET28a, NdeI/XhoI, N-terminal His tag); human SenP2 (pET28a) (25); GST-tagged constructs in pETM30 (EMBL Heidelberg, NcoI(BspI/PciI)/XhoI, N-terminal His-GST tag): human full-length SVIP (aa 1–77) and truncations (aa 1–35, 1–39, 15–35, 15–39, 18–35, and 18–39); human gp78 cytosolic domain (gp78C, aa 308–634); human FAF1UBX (aa 568–650).
For site-directed mutagenesis of UBXD1 (A59L/R62A, K198E/N201D, and 52EA53/52RR53), p97ND1 (D35A, D35N, V38A, F52A, R53A, I70A, L72A, Y110A, Y143A, and I175A), and SVIP (R32A), the QuikChange® II site-directed mutagenesis kit from Agilent was used. Unmodified as well as N-terminally biotinylated (Biotin-Ahx) human peptides were purchased from Genscript and Panatecs: gp78 (622VTLRRRMLAAAAERRLQKQ640), ANKZF1 (653ALSDREKRALAAERRLAA670), SVIP (18LEEKRAKLAEAAERRQKE35), and UBXD1 (49TNEAQMAAAAALARLEQKQSR69).
Protein Expression and Purification
SenP2 expression and purification was performed as described (25). Proteins were expressed in E. coli BL21(DE3) RIL cells (Novagen) by induction with 0.1–0.5 mm IPTG at an A600 of 0.6 overnight at 16 °C and were purified in 50 mm Tris, pH 8, 150 mm KCl, 5% (v/v) glycerol, 5 mm MgCl2 buffer (buffer A), and 5 mm β-mercaptoethanol by metal affinity chromatography (Ni-TED, Macherey & Nagel) and size exclusion chromatography (Superdex 200 or Superose 6, GE Healthcare). Prior to size exclusion chromatography, full-length p97, p97NL, and its N domain were dialyzed at 4 °C overnight in the presence of tobacco etch virus protease (1:50) followed by metal affinity chromatography to remove uncleaved proteins as well as His-tagged tobacco etch virus protease.
SUMO-UBXD1, SUMO-UBXD1N, and variants were expressed in E. coli BL21(DE3) RIL cells (Novagen) by induction at an A600 of 0.4–0.6 with 1 mm IPTG overnight at 25 °C. They were purified in 20 mm Tris, pH 8, 500 mm NaCl buffer, and 10 mm β-mercaptoethanol by metal affinity chromatography (Ni-TED, Macherey & Nagel) and dialyzed at 4 °C overnight against 50 mm Tris, pH 8, 50 mm NaCl, and 10 mm β-mercaptoethanol (SUMO-UBXD1) or 100 mm HEPES, pH 7.0, 50 mm NaCl, and 10 mm β-mercaptoethanol (SUMO-UBXD1N) followed by anion (Mono Q 10/100 GL, GE Healthcare) (SUMO-UBXD1) or cation exchange chromatography (Mono S 10/100 GL, GE Healthcare) (SUMO-UBXD1N). After cleavage of the SUMO tag with SenP2 protease (1:250, 2 h, 4 °C), proteins were applied to a Superdex 75 10/30 GL column (GE Healthcare) equilibrated in 50 mm Tris, pH 8, 150 mm KCl, and 5 mm β-mercaptoethanol to remove the SUMO tag and SenP2 protease. All proteins were concentrated to ∼10 mg/ml by ultrafiltration (Vivaspin, Sartorius), shock-frozen, and stored at −80 °C.
Crystallization
For crystallization of the p97 N-terminal (p97N) domain in complex with the gp78 peptide, p97N was incubated with the peptide at a molar ratio of 1:2 (0.5 mm p97N and 1 mm peptide) for 1 h at 4 °C. Crystals were grown by vapor diffusion in hanging drops containing equal volumes of protein in 50 mm Tris, pH 8.0, 150 mm KCl, and 5 mm dithiothreitol (DTT) and a reservoir solution consisting of 18–22% (w/v) polyethylene glycol 3000, 0.2 m NaCl, and 100 mm Tris, pH 7.0, equilibrated against the reservoir solution.
Data Collection and Structure Determination
Crystals were cryoprotected by soaking in mother liquor containing 15% (v/v) glycerol and flash-cooled in liquid nitrogen, and data collection was performed at 100 K. Data were collected at beamline BL 14.1 (BESSY, Berlin) and processed using iMosflm and Scala (26, 27). Data collection statistics are summarized in Table 1. For subsequent calculations, the CCP4 suite and Phenix were utilized (28, 29) with exceptions as indicated. The structure was solved by molecular replacement using Phaser (30) with the N domain of p97 (Protein Data Bank entry 1S3S) as search model. The data were merohedrally twinned with a twin fraction of 0.26–0.27 (Britton analysis, H-test), and the structure was refined with Phenix incorporating the twin law h, −h −k, −l and TLS refinement in all cycles. Solvent molecules were automatically added with Phenix. The figures were generated with PyMOL (available on the World Wide Web).
TABLE 1.
Data collection and refinement statistics
| Data collection | |
| Wavelength (Å) | 0.9184 |
| Resolution (Å)a | 35.12–1.8 (1.9–1.8) |
| Space group | P32 |
| Cell dimensions | |
| a = b, c (Å) | 70.2, 74.8 |
| α = β, γ (degrees) | 90, 120 |
| Unique reflections | 38,133 |
| 〈I/σ(I)〉a,b | 7.5 (2.4) |
| Completeness (%)a | 100 (100) |
| Redundancya | 3.8 (3.8) |
| Rsyma,c | 0.106 (0.492) |
| Refinement statistics | |
| Resolution limits | 35.12–1.8 |
| No. of reflections | 38,107 |
| No. of protein/ligand/solvent atoms | 2,902/4/381 |
| Rcryst (Rfree)d | 0.147 (0.171) |
| Root mean square deviations | |
| Bond lengths (Å) | 0.006 |
| Bond angles (degrees) | 1.057 |
| Average B-factors (Å2) | |
| Protein (p97N/gp78) | 32.2/32.8 |
| Solvent molecules | 39.0 |
| Ramachandran statistics (%)e | |
| Favored | 95.8 |
| Allowed | 3.9 |
| Outliers | 0.3 |
a Numbers in parentheses refer to the respective highest resolution data shell in the data set.
b 〈I/σI〉 indicates the average of the intensity divided by its S.D. value.
c Rsym = 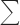 hkli|Ii − 〈I〉|/hkli〈I〉, where Ii is the ith measurement and 〈I〉 is the weighted mean of all measurements of I.
hkli|Ii − 〈I〉|/hkli〈I〉, where Ii is the ith measurement and 〈I〉 is the weighted mean of all measurements of I.
d Rcryst = ‖Fo| − |Fc‖/|Fo|, where Fo and Fc are the observed and calculated structure factor amplitudes. Rfree, same as Rcryst for 5% of the data randomly omitted from the refinement.
e Ramachandran statistics indicate the fraction of residues in the favored (98%), allowed (>99.8%), and disallowed regions of the Ramachandran diagram, as defined by MolProbity (45).
In Vitro Binding Assays
For pull-down assays, biotinylated peptides or GST-tagged proteins were immobilized on Strep-Tactin-Sepharose beads (IBA BioTAGnology) or glutathione-Sepharose beads (GSH, Novagen). In all experiments, 10 μl of beads were incubated with 12.5 μm biotinylated peptides or 1 μm GST-tagged proteins in 200 μl of PBS buffer, pH 7.4, with 0.1% (v/v) Triton X-100 at 4 °C for 1 h. p97ND1 alone was included as control. After centrifugation (1250 × g, 30 s) beads were washed four times with 400 μl of binding buffer. Unless otherwise stated, equimolar amounts of purified p97ND1 and variants in a total volume of 200 μl of binding buffer were added to immobilized peptides or GST-tagged proteins and treated in the same way as in the first step. Immobilized proteins were directly analyzed by 15% (v/v) SDS-PAGE. Competition experiments were carried out in buffer A in the same way. GST-UBX or GST-gp78C (2.5 μm) was incubated either with 5 μm p97ND1 plus 1 μm UFD1-NPL4 or 2.5 μm p97ND1 in the absence or presence of increasing amounts of His-SVIP, as indicated, and pulled down with GSH beads. Immobilized proteins were directly analyzed by 18% (v/v) SDS-PAGE.
For isothermal titration calorimetry (ITC) proteins were extensively dialyzed against 50 mm Tris, pH 8, 5% (v/v) glycerol, 150 mm KCl, 5 mm MgCl2, and 1 mm β-mercaptoethanol, followed by degassing. In all experiments, 75–150 μm UBXD1/variants or SVIP was titrated as the ligand into the sample cell containing 5–10 μm p97 or variants. A volume of 10 μl of ligand was added at a time with a total number of 30 injections, resulting in a final molar ratio of ligand to protein varying between 4:1 and 5:1. All experiments were performed using a VP-ITC instrument (MicroCal, GE Healthcare) at 25 °C. ITC data were corrected for heats of dilution of the protein solution. Corrected data were analyzed with a single-site binding model using software supplied by the ITC manufacturer and nonlinear least-squares fitting to calculate the dissociation constant (KD).
Biolayer interferometry (BLI) was performed using an Octet Red System (Forté Bio) using streptavidin-coated biosensors. Peptides and analytes were diluted in PBS buffer, pH 7.4, containing 1 mg/ml BSA and 0.1% (v/v) Tween 20. Streptavidin biosensors were coated with biotinylated peptides by incubation for 600 s in a 100 nm peptide solution followed by a wash step (1000 s) with buffer. The biosensors were then immersed in analyte solution (10–200 nm) for 300 s to allow association and subsequently transferred into buffer to follow dissociation for 1000 s. All steps were performed at 30 °C and 1000 rpm in a 96-well plate containing 200 μl of solution in each well. From changes in light interference over time upon binding and dissociation, the kinetic rate constants kon and koff and the equilibrium dissociation constant KD were derived using a 1:1 binding model (ForteBio OctetRED Evaluation software version 6.3).
The interaction of p97 with UBXD1 was also determined by a native gel mobility shift assay. p97 (5 μm) in buffer A was incubated with increasing amounts of UBXD1 and variants (0.5–25 μm) for 1 h on ice. Bound and unbound forms were separated on a 4% (v/v) native gel. Finally, complex formation between p97 and UBXD1 was analyzed by analytical size exclusion chromatography (Superdex 200, GE Healthcare) in buffer A. p97 (50 μm) was incubated with UBXD1 at a 1:0.7 molar ratio for 1 h on ice.
RESULTS
Functional Studies of Human VIM-containing Proteins
gp78, ANKZF1, and SVIP have been shown to interact via a VIM with p97 (20, 22, 24). The binding sequence of gp78 to p97 has been mapped to a short sequence stretch containing two arginine-rich sequences separated by a hydrophobic amino acid stretch, including several conserved alanine residues, that is located at the extreme C terminus of gp78 (19). A simple sequence alignment of the VIM of gp78 with ANKZF1 and SVIP is shown in Fig. 1A. To classify the binding strength of VIM-containing proteins, interaction studies with p97ND1 were performed. Initial streptavidin pull-down assays (Fig. 1B) using N-terminally biotinylated peptides covering the VIM revealed a strong binding for gp78 and ANKZF1, whereas no interaction could be detected for SVIP.
FIGURE 1.

Characterization of the p97 interaction with human VIM proteins. A, sequence alignment of previously characterized human proteins with a proposed VIM. Strictly conserved amino acids are highlighted with a black background, and similar amino acids are shown with a gray background. The α-helix of gp78 is indicated as a cylinder above the sequences. Streptavidin pull-down assays (B) and biolayer interferometry (C) involving biotinylated VIM peptides and WT p97ND1 are shown. D, ITC studies of p97 variants with full-length SVIP and SVIP-R32A. E, GST pull-down assays of full-length SVIP, SVIP-R32A, and truncated versions interacting with p97ND1 at a 1:0.5 molar ratio.
To further characterize these interactions, biolayer interferometry (Fig. 1C) was used, which allowed us to measure the kinetic rate constants for association and dissociation and to calculate the equilibrium dissociation constant KD. In biolayer interferometry, any change in the number of molecules bound to the biosensor tip causes a shift in the interference pattern that can be measured in real time (31). Again, for SVIP no interaction could be detected (Fig. 1C). Analysis of the gp78 and ANKZF1 interaction revealed similarly high affinities with dissociation constants of 21.3 ± 0.3 and 16.3 ± 0.2 nm, respectively. The kinetic rate constants were also identical within error limits (for gp78, kon = 1.4 ± 0.1 × 105 m−1 s−1 and koff = 2.9 ± 0.2 × 10−3 s−1; for ANKZF1, kon = 1.7 ± 0.4 × 105 m−1 s−1 and koff = 2.7 ± 0.4 × 10−3 s−1). However, the observed biexponential binding behavior indicates a more complex binding kinetic, and only the first 300 s could be fitted to a 1:1 model. Whether the slower phase of the association curves truly represented an inherent step in complex formation rather than an artifact attributable to a nonspecific binding step, such as aggregation or interaction with the matrix, is currently not known. One explanation for the biphasic association could be the hexameric architecture of p97, which counteracts the monovalent binding to the peptide that forms the basis for a quantitative kinetic analysis. Alternatively, a two-state binding model is conceivable involving conformational changes in p97 upon binding. Finally, mass transport and rebinding events cannot be ruled out either.
The absence of binding by the SVIP VIM peptide despite a high degree of sequence homology to the gp78 and ANKZF1 VIM suggested that additional sequences are required for interaction. SVIP is composed of an N-terminal domain (aa 1–39) and a C-terminal domain (aa 40–77), and earlier pull-down experiments identified the p97 binding site in the N-terminal domain of SVIP (22) containing the proposed VIM consensus sequence (20), which, however, has not been experimentally verified so far. ITC of full-length SVIP with p97ND1 (Fig. 1D) resulted in a KD of 0.67 ± 0.03 μm and a binding stoichiometry of n = 0.81 ± 0.01, consistent with the binding of six SVIP molecules to the p97 hexamer (6:6). An alanine mutant of the highly conserved Arg32 located in the VIM almost completely abolished interaction, which confirms that the SVIP VIM is crucial for the interaction with p97 (Fig. 1D). In contrast to p97ND1, only a weak association with the p97 N-terminal domain was observed, in agreement with published GST pull-down assays (22). However, already p97N plus the flexible linker (p97NL) that connects the N domain to the adjacent D1 domain revealed an enhanced affinity (KD of 5.92 ± 0.04 μm), indicating that the linker contributes to the interaction.
To further investigate the involvement of the N-D1 linker as well as the D1 domain in SVIP interaction, ITC studies were performed with the p97ND1-R155H variant, the most common IBMPFD-associated mutation (6). IBMPFD mutations are located at the interface of the N domain and the D1 domain, resulting in a reduced affinity for ADP that is usually bound to the D1 domain (32). Recent structural studies revealed that IBMPFD variants undergo a nucleotide-induced conformational change of the N domain (up and down conformation) in the presence of the non-hydrolyzable ATP analog ATPγS (32), and an unbalanced cofactor binding to p97 has been suggested as a key pathological feature of IBMPFD (33). Consistent with these observations, the affinity of SVIP is significantly reduced by a factor of about 7 (KD 4.76 ± 0.04 μm) for the R155H variant of p97ND1 with bound ATPγS.
Interestingly, a closer examination of the SVIP VIM sequence in comparison with the gp78 motif revealed that the proposed α-helix is predicted to be longer in SVIP, both at the N and the C terminus, which might affect binding. Therefore, smaller SVIP fragments (Fig. 1E) were analyzed in a GST pull-down assay. Indeed, our studies revealed that additional residues N-terminal and, more importantly, C-terminal of the VIM signature motif are necessary for a high affinity interaction. The GST-tagged construct, which is identical to the biotinylated peptide described above (aa 18–35), revealed a largely reduced binding, and the fragment comprising amino acids 15–35 already shows a significantly reduced binding. These data indicate that these residues are required for binding, presumably due to a stabilization of the helix, which would be consistent with a role of Asp17 and Arg39 as N- and C-terminal helix-capping residues.
The Structural Basis for the p97-VIM Interaction
To elucidate the molecular basis of VIM recognition by p97, we determined the crystal structure of the p97N domain in complex with a gp78-derived peptide encompassing residues 622–640, which are located at the extreme gp78 C terminus. Earlier pull-down experiments describing the gp78 VIM binding motif reported that a GST fusion of the gp78 VIM only interacts with p97ND1 and not with the isolated N domain (20). In contrast, pull-down and biolayer interferometry experiments using a biotinylated gp78 VIM peptide revealed that gp78 interacts also with the p97N domain (data not shown), and subsequently we could co-crystallize p97N with a non-biotinylated gp78 peptide. Given the results with SVIP, it is conceivable that the affinity of the gp78 VIM peptide would be even higher for p97ND1.
The 1.8 Å crystal structure was solved by molecular replacement using the N domain of the p97ND1 crystal structure as a search model (Protein Data Bank entry 1S3S) and was refined to a crystallographic R-factor of 14.7% and a free R-factor of 17.1% (Table 1). The structure revealed two protein complexes in the asymmetric unit, which can be superimposed with root mean square deviations of 0.43 Å (p97N) and 0.72 Å (gp78). The structure of p97N in complex with the gp78 peptide revealed no significant conformational changes compared with the p97N apo structure (Protein Data Bank entry 3QQ7) as reflected by root mean square deviations of 0.64 and 0.78 Å following superposition for the two p97N molecules present in the asymmetric unit of the complex crystals. Electron density corresponding to residues 622–639 of the gp78 peptide could be unambiguously identified. An unbiased SIGMAA weighted 2Fo − Fc map calculated after omission of the peptide from the model and subsequent refinement is shown in Fig. 2A.
FIGURE 2.

Structure of p97N in complex with the gp78 VIM. A, SIGMAA weighted 2Fo − Fc omit electron density map of the gp78 peptide (colored according to atom type with carbon atoms in green) contoured at 1 times the root mean square deviation with a surface representation of the N domain in gray. B, ribbon and surface representation of p97N (gray) in complex with the gp78 peptide (green) in two different orientations.
As described earlier (8, 9, 18), the p97N domain (residues 10–186) contains two subdomains linked by a flexible linker of six amino acids: an N-terminal double ψ β-barrel (referred to as either Nn or Na) and a C-terminal α + β domain (Nc or Nb) with a central four-stranded antiparallel β-sheet (Fig. 2B). The complex structure reveals that the peptide forms an α-helix, as previously predicted for this motif. This helix interacts with the N domain, specifically at the interface between its subdomains, and engages in several critical interactions (Figs. 2 and 3). Due to the helical nature of this peptide, the important residues are located primarily on the face of the helix pointing toward the N domain. Both polar/charged (22%/41% of the total interface) residues and non-polar residues (37%) contribute to the interface.
FIGURE 3.

Structural and functional studies of the p97N-gp78 peptide interaction. A, view into the binding interface. Residues involved in binding are shown in stick representation using the same color code for carbon atoms as in Fig. 2. The dashed lines indicate hydrogen bonds. B, surface representation of the binding pocket of p97N. gp78 residues involved in complex formation are shown in stick representation. The conformational change of Arg53 identified in the p97N-gp78 complex structure is indicated with carbon atoms in magenta (p97N-gp78) and orange (apo-p97N, Protein Data Bank entry 3QQ7). Shown are streptavidin pull-down assay (C) and biolayer interferometry (D) of WT p97ND1 and biotinylated gp78 peptides (WT and variants).
The gp78 α-helix inserts into a hydrophobic pocket, which is restricted at its base by a salt bridge formed between Arg144 and Asp35 (Nη2–Oδ2, 3.2 Å; Nη2–Oδ1, 2.9 Å) connecting the Nn and Nc subdomains. p97 recognizes the VIM α-helix mainly via loop elements from both subdomains. Participating residues from gp78 include Met628, Leu629, Ala630, Ala632, Ala633, and Leu637 as well as the side chains of Arg625, Arg626, Glu634, and Arg636, which are located in the hydrophobic p97 pocket formed by residues Val38, Ile70, Leu72, Tyr110, Tyr143, and Ile175 (Fig. 3A). In addition to these hydrophobic contacts, several main chain-side chain and side chain-side chain hydrogen bonds are observed, including Arg625, Arg626, Glu634, and Arg636 of gp78 (Arg625-Nη1–Val108-O (2.8 Å), Arg625-Nη2–Val108-O (2.9 Å), Arg626-Nη1–Arg53-O (3.0 Å), Arg636-Nϵ–Asp35-Oδ1 (2.8 Å), Arg636-Nη1–Asp35-Oδ1 (2.9 Å), Arg636-Nη2–Ala142-O (3.0 Å), Glu634-Oϵ1–Arg53-Nη1 (2.7 Å), and Glu634-Oϵ2–Arg53-Nϵ (3.0 Å)). Compared with apo-p97N, the side chain of Arg53 significantly alters its conformation and covers the helical gp78 (Fig. 3B). The three arginines are not only involved in electrostatic interactions but also provide valuable hydrophobic contacts via their long aliphatic side chains to Ile175 (Arg625), Phe52 (Arg626), and Ala142 as well as Arg144 (Arg636).
Analysis of the p97-gp78 VIM Interface
Biolayer interferometry revealed that the p97-gp78 interaction is completely abolished in the presence of 500 mm NaCl and is reduced by a factor of about 5 in the presence of 5% (v/v) glycerol (data not shown). Glycerol is known to prevent protein aggregation by inhibiting protein unfolding and by stabilizing aggregation-prone intermediates through preferential interactions with hydrophobic surface regions that favor amphiphilic interface orientations of glycerol (34). This suggests that glycerol blocks the hydrophobic p97 binding surface, thus inhibiting interaction with gp78. These data show that both hydrophobic and electrostatic interactions contribute to the p97N-gp78 interface.
To determine the functional significance of the molecular interactions observed in the complex, a detailed mutational analysis was carried out. Streptavidin pull-down assays and biolayer interferometry revealed that all three gp78 arginines (Arg625, Arg626, and Arg636) are important, with Arg636 being the most critical. Interestingly, among the remaining arginines, it is not the conserved N-terminal Arg626 but the non-conserved Arg625 that is more important for this interaction. An exchange of Glu634 to leucine increased the affinity (Fig. 3, C and D); a leucine at this position is predicted to form favorable strong hydrophobic interactions with Leu72, a residue critical in peptide binding (see below), and the aliphatic side chain atoms of Arg53 in p97. Based on the crystal structure, the side chain of Arg53 covers the peptide-binding site, suggesting a strong interaction (Fig. 3B). Unexpectedly, the R53A variant largely retained the ability to interact with the gp78 peptide (see below), indicating that the salt bridge between Glu634 and Arg53 that is responsible for this conformational change is not crucial for interaction with the peptide (Fig. 4A). This is in agreement with the stronger binding of the gp78 E634L variant described above.
FIGURE 4.

The p97 N-terminal domain represents a conserved VIM-interacting domain. A, top, sequence alignment of the gp78, ANKZF1, SVIP, and UBXD1 (identified in the accompanying paper by Stapf et al. (46)) VIM. Residues involved in p97N-gp78 interaction are labeled with stars, and residues of the VIM signature motif RX5AAX2R are highlighted with red backgrounds. Bottom, streptavidin pull-down assays involving biotinylated VIM peptides (gp78, ANKZF1, and UBXD1) and GST pull-down assay involving GST-tagged SVIP (aa 15–39) and p97ND1 WT and variants. B, surface representation of the p97 VIM interaction site. Residues colored in red represent the core region of the binding domain, which is essential for the respective VIM interaction, and residues colored in orange and yellow contribute moderately to the interaction, whereas residues colored in green are not involved in the respective interaction.
For p97, these studies revealed that the hydrophobic residues Val38, Ile70, Leu72, Tyr143, and Ile175 located at the base of the p97 binding pocket contribute significantly to gp78 binding, which on this side is mediated mainly by Leu629 and Ala633, whereas Tyr110 of p97 is not important for interaction. An exchange to alanine of Arg53 as well as Phe52, which contact Arg626 and Glu634 of gp78, only leads to a moderate decrease in affinity (Fig. 4 and supplemental Fig. S1). Besides these hydrophobic interactions, the most prominent contacts between p97 and gp78 involve the combination of electrostatic and hydrophobic interactions mediated by the gp78 arginine residues Arg625, contacting Val108 and Ile175, and Arg636, engaging with Asp35, Ala142, and Arg144 (Fig. 3A). The accompanying paper by Stapf et al. (46) demonstrates that Asp35 is crucial for the VIM-p97 interaction in vivo by analyzing the interaction of Vms1, the yeast homolog of ANKZF1, and Cdc48.
Molecular Discrimination between Different VIM Proteins
The paper by Stapf et al. (46) also presents a detailed bioinformatic search, which identified additional VIM proteins that previously had not been recognized as members of this family of p97 cofactors. Interestingly, their studies indicate that VIM-containing cofactors can be grouped into two subfamilies: (i) “RAAR” proteins, which contain the consensus sequence RX5AAX2R with two conserved arginine residues separated by a hydrophobic amino acid stretch including two conserved alanine residues, as found in gp78, ANKZF1, and SVIP, and (ii) “AAR” proteins, which contain the consensus sequence AAX2R but lack the first conserved arginine, as found, for example, in the UBX protein UBXD1. In vitro as well as in vivo interaction studies by Stapf et al. (46) confirmed that UBXD1 indeed binds via this modified VIM motif to p97.
To investigate whether an arginine N-terminal of the highly conserved AAX2R VIM motif is essential for a subset of VIM proteins and to identify the specific molecular details for VIM recognition, the binding behavior of VIM proteins from both subclasses was analyzed by pull-down assays. ANKZF1, which shares 67% amino acid identity in the VIM with gp78 and exhibits a similarly high binding affinity, showed significantly more pronounced binding defects compared with gp78. The p97 Asp35, Val38, Ile70, Leu72, and Tyr143 alanine variants completely abolished binding; the F52A, R53A, and I175A variants showed a ∼3-fold reduced binding; and Tyr110 again was not involved in binding (Fig. 4). The data obtained via pull-down assays could be confirmed by biolayer interferometry (supplemental Fig. S1). Because in all VIM proteins, the AAX2R signature motif is highly conserved, probably resulting in a similar binding mode with the arginine (Arg636 of gp78) involved in electrostatic interactions (Asp35), hydrogen bonds (main chain of Ala142), and hydrophobic interactions, not much rotational flexibility of the α-helical VIM binding motif appears to be possible. Arg625 of gp78, which makes major contributions to the p97 interaction by providing both electrostatic and hydrophobic contacts, is replaced by an aspartic acid in ANKZF1, and this residue is predicted to not be involved in interactions. Nevertheless, both gp78 and ANKZF1 interact with similarly high affinities with p97. The reason for this might be that Leu629 of gp78 is replaced by an arginine in ANKZF1 that would be within hydrogen-bonding distance of the side chain of Thr56 and the carbonyl oxygen of Gly54. This aliphatic side chain could provide favorable hydrophobic interactions with Ile175. Furthermore, the exchange of gp78 Met628 by a lysine residue could result in a cation-π-stacking interaction with the aromatic side chain of Tyr143 as well as a hydrogen bond with the Tyr143 hydroxyl group.
The UBXD1 VIM revealed only a low binding affinity in pull-down assays, and no interaction was detectable with concentrations up to 50 μm by biolayer interferometry (data not shown). This weak interaction is probably the consequence of the presence of only one arginine, namely the one corresponding to Arg636 of gp78 that is available for binding. Furthermore, eight of 10 hydrophobic interactions involve only alanine residues. Consequently most of the p97 alanine variants already completely abolished the interaction with the UBXD1 peptide (Fig. 4). The F52A variant, which provides hydrophobic contacts to the first arginine found in the VIM consensus sequence RX5AAX2R of gp78 and ANKZF1 revealed no effect in UBXD1 binding, and the V38A and R53A mutants still showed significant binding. However, the I175A mutant, which also completely abolishes binding, would not be within 5 Å of the UBXD1 binding motif, indicating that despite the presence of the VIM AAX2R signature sequence, there must be differences in the translation/rotation of this helical interaction motif.
Although an analysis of the p97ND1 mutants using a GST-fused SVIP peptide covering amino acids 15–39 revealed some variations in the contribution of individual residues on the p97N domain surface, there is generally good agreement between SVIP and the other VIM cofactors, in particular ANKZF1 and UBXD1.
In summary, these comparative binding studies clearly show that all VIM proteins analyzed here bind to the same hydrophobic p97N interdomain cleft composed of Asp35, Val38, Ile70, Leu72, and Tyr143. However, differences due to variances in the amino acid composition are observed that translate into changes in affinity and might trigger a slightly altered orientation of the α-helix, thus resulting in a slightly altered binding specificity.
SVIP Is an Efficient Competitor of p97N Domain Cofactors
Proteins recognizing the p97N domain are well known to bind in a mutually exclusive manner. The best studied p97N cofactor is p47, which competes with UFD1-NPL4, SVIP, and UBXD1 for p97 binding (15, 22, 35, 36). However, p97 does not always simply interact with only one protein; p97 is known to form ternary or even higher order protein complexes with varying stoichiometries. Recently, we could identify a ternary p97 complex composed of UFD1-NPL4 and the UBX protein FAF1 with a stoichiometry of 6:1:1 (18). In contrast, our analysis with SVIP indicates a 6:6 stoichiometry, which indicates that no additional cofactors can bind. Accordingly, SVIP has not been shown so far to co-exist with other proteins, thus suggesting that SVIP might efficiently disrupt the association of proteins with p97. In order to analyze the mutually exclusive binding of SVIP, competition pull-down experiments were performed. In the absence of competitor, GST-tagged FAF1UBX domain, UFD1-NPL4, and p97ND1 form a stable ternary complex (Fig. 5A); however, the addition of increasing amounts of SVIP disrupted this ternary complex already at a 2-fold molar excess.
FIGURE 5.

p97N cofactors interact in a mutually exclusive manner in pull-down competition experiments. A, GST-FAF1UBX was incubated with p97ND1-UFD1-NPL4 in the absence or presence of increasing amounts of His-SVIP as indicated. B, GST-gp78C was incubated with p97ND1 in the absence or presence of increasing amounts of His-SVIP as indicated.
The comparative binding data of VIM proteins shown above indicate that proteins with a VIM binding motif bind in a mutually exclusive manner to the p97N domain. Furthermore, SVIP has been described as an inhibitor of gp78-mediated ERAD (23), thus suggesting that both compete for the same binding site. In order to analyze whether there is a competition between different VIM proteins, which has not been demonstrated so far, a competition pull-down experiment was carried out with the GST-tagged cytosolic domain of gp78 (gp78C) and His-tagged SVIP. This experiment revealed that already a 5-fold molar excess efficiently prevented the interaction of gp78C with p97ND1 (Fig. 5B). In summary, these competition experiments clearly show that the VIM protein SVIP very efficiently competes with other p97N cofactors, irrespective of whether they interact via a UBX domain (FAF1), a ULD (NPL4), or a VIM (gp78).
Bipartite Interaction of UBXD1 with p97
UBXD1, although belonging to the UBX family of p97 cofactors, does not interact with p97 via its UBX domain (15, 37) because the conserved FPR signature common to p97-interacting UBX domains is replaced with SGG in UBXD1. Instead this cofactor utilizes, like p47 and UFD1-NPL4, a so-called bipartite binding mechanism (15). In p47 and UFD1-NPL4, this involves binding either via the UBX domain of p47 or the ULD of NPL4 and a Shp box (p47 or UFD1) to the N domain of p97, and hence both cofactors compete for p97 binding in vitro (35, 36). p47 binds as a trimer where each monomer is associated with two adjacent p97 protomers (38). In contrast, only one UFD1-NPL4 heterodimer binds to two adjacent protomers of a p97 hexamer (39).
UBXD1 is composed of an N-terminal domain that displays no homology to known structures, a central PUB domain, and the C-terminal UBX domain (Fig. 6A). It interacts via its PUB domain with the C terminus of p97 and contacts the N domain with its VIM (15, 46). To define this bipartite interaction involving a VIM in more detail, ITC was used to determine the affinity of the interaction between full-length UBXD1 and full-length p97 as well as the stoichiometry of this complex. Despite the bipartite p97 recognition, we determined only a moderate affinity of ∼3.5 μm (KD = 3.4 ± 0.9 μm) for this complex. A binding stoichiometry of n = 0.47 ± 0.05 is consistent with the binding of three UBXD1 molecules to the p97 hexamer (6:3) (Fig. 6A). This would be similar to p47, which is known to interact as a trimer with p97 with a reported binding stoichiometry of n = 0.5 (18, 38). A similar binding stoichiometry would be consistent with the observed mutually exclusive binding of p47 and UBXD1 (15).
FIGURE 6.

Functional studies of the p97-UBXD1 interaction. A, top, domain architecture of UBXD1 and p97. The interactions of the UBXD1 VIM with the N domain and the UBXD1 PUB domain with the C terminus of p97 are indicated. The non-functional UBX domain is colored in black. Bottom, ITC studies of full-length p97 with WT UBXD1, a UBXD1 VIM knock-out (58AAX2R62 58ALX2A62), and a UBXD1 PUB knock-out (K198E/N201E). B, analytical size exclusion chromatography of p97-UBXD1. p97 was incubated with UBXD1 in a 1:0.7 molar ratio. The indicated elution fractions were analyzed by SDS-PAGE followed by Coomassie Brilliant Blue staining.
Site-directed mutagenesis was used to study the contribution of both binding motifs/domains to the interaction. Binding of UBXD1 to p97 using a UBXD1 mutant with a replacement of the highly conserved VIM 58AAX2R62 by 58LAX2A62 almost completely abolished this interaction (Fig. 6A). Similar results were obtained with the isolated N-terminal domain of UBXD1 (aa 1–150) (data not shown). The interaction of the PUB domain of peptide:N-glycanase with the C terminus of p97 is strictly dependent upon an arginine and an asparagine residue (13), which correspond to Lys198 and Asn201 of UBXD1. Furthermore, a quintuple PUB domain mutant of UBXD1 (N184A/K193A/Y194A/K198A/N201A) has been shown to abolish the interaction of UBXD1 with p97 (15). A double K198E/N201D variant of the UBXD1 PUB domain displayed a largely reduced affinity (Fig. 6A). Native gel shift experiments with increasing amounts of UBXD1 WT and variants showed complete complex formation for the WT already at a 1:1 molar ratio, whereas for the variants, 1:2 (VIM variant) and 1:5 ratios (PUB variant) were necessary (data not shown). This observation is consistent with a decreased affinity of the variants. The in vitro interaction of WT UBXD1 with p97 was further analyzed by analytical size exclusion chromatography (Fig. 6B). Analysis of the p97-UBXD1 binary complex by SDS-PAGE also indicates a 6:3 stoichiometry.
As mentioned above, the sequence of the UBXD1 VIM is different from that found in gp78 because it lacks the positively charged residues located N-terminal to the alanine stretch of the binding motif. The two arginines at this position are important for the interaction of gp78 with p97 but are replaced by glutamate (Glu52) and alanine (Ala53) in UBXD1. Exchange of Arg-Arg to Glu-Ala is predicted to result in the loss of hydrogen bonds as well as a weaker hydrophobic interaction due to the missing arginine side chains. Surprisingly, replacement of UBXD1 52EA53 with 52RR53 did not increase the affinity of UBXD1 to p97 as analyzed by ITC (data not shown), which indicates that positively charged amino acids N-terminal to the alanine stretch are not important for all VIM proteins.
Taken together, these data demonstrate that the VIM and PUB domain of UBXD1 both contribute to p97 binding, with the PUB domain apparently making a stronger contribution. The binding stoichiometry is consistent with a p97 hexamer binding to three UBXD1 monomers. This binding mode is unique to p97 cofactors in that the two major cofactor-binding sites of p97, the N domain and the C terminus, are simultaneously contacted by a cofactor. Presumably, this simultaneous interaction will restrict the conformational variability of p97.
DISCUSSION
Binding Mechanisms Involved in p97N Cofactor Diversity
The assembly of p97 cofactors is regulated by multiple mechanisms, including mutually exclusive binding, conformational changes induced by ATP binding/hydrolysis or hierarchical binding, or even a combination of different mechanisms, thus resulting in complexes that differ in stoichiometry, symmetry, and quaternary arrangement.
It has been proposed that ATP binding/hydrolysis triggers conformational changes of the p97N domain, which are important for cofactor protein binding or release (40). Based on structural as well as biochemical studies of IBMPFD mutants, nucleotide-induced conformational changes in the p97N domain, the main binding site for cofactors, could be demonstrated (32), and altered cofactor interactions could be shown in vitro and in vivo (33, 41, 42). The VIM protein SVIP described in this study, which requires for high affinity binding also the N-D1 linker and adjacent D1 domain, has a significantly impaired binding to the IBMPFD mutant R155H in the ATP-bound state. A structural model of the gp78 peptide with p97ND1 revealed that the C terminus of the VIM is in close contact with the D1 domain as well as the flexible linker connecting the N and D1 domains (supplemental Fig. S2A). Based on the assumption that the SVIP VIM binds in the same way as the gp78 peptide, the additional C-terminal residues of the SVIP VIM contributing to an elongated helix compared with gp78 would contact the D1 domain, and an involvement of the flexible linker connecting the N and D1 domains is also possible. Due to the nucleotide-induced conformational changes of the N domain, this interaction is disrupted (supplemental Fig. S2B). Recently, it could also be shown that the interaction of UBXD1 and p97 is impaired in IBMPFD mutants (42).
It has been reported that some cofactors that target identical or overlapping sites on p97 bind in a mutually exclusive manner, including the substrate-recruiting cofactors p47 and UFD1-NPL4 (35, 36) as well as the VIM proteins (15, 20). The crystal structure of p97N in complex with the VIM of gp78 described in this study together with the p97N-FAF1UBX structure (18) and an NMR-derived model of the NPL4 N-terminal ULD in complex with p97N (16) allows for a comparison of how cofactors interacting with the N domain recognize p97. Although there is no sequence similarity between the VIM binding motif and the UBX domain of FAF1 or the ULD of NPL4, the binding sites partially overlap, and all bind to the same general area, including the hydrophobic interdomain cleft of the p97N domain (Fig. 7A). This demonstrates that an N domain and hence a p97 monomer can only interact with one of these proteins at any given time.
FIGURE 7.

Competitive cofactor binding to p97N. A, ribbon representations together with the molecular surface of p97N in complex with the VIM of gp78 (this study, colored in green), FAF1UBX (Protein Data Bank entry 3QQ8, colored in gold) and NPL4ULD (Protein Data Bank entry 2PJH, colored in blue). B, ribbon representations of p97 in complex with FAF1UBX (Protein Data Bank entry 3QQ8) and gp78 (this study) with p97N in magenta (FAF1UBX complex) and gray (gp78 complex), FAF1UBX in gold, and gp78 in green. Two orientations that differ by a 90° rotation are shown. C, close-up view showing the common p97N binding surface important for FAF1UBX and gp78 interaction. Key residues involved in the respective interaction are shown in a stick representation using the same color code for carbon atoms as in A.
However, in many cases, p97 does not simply interact with a single cofactor, and higher order complexes composed of several cofactors exist (4, 43). Recently, we could demonstrate that p97 assembles UFD1-NPL4 and the UBX proteins FAF1 or UBXD7 in a hierarchical manner, with UFD1-NPL4 association being a prerequisite for UBX protein binding (18). Only one UFD1-NPL4 heterodimer and one UBX monomer have been identified in this complex with a 6:1:1 stoichiometry, thus potentially allowing further cofactors to bind to non-occupied N domains.
Some p97 cofactors contain more than one p97-binding site, resulting in a bipartite interaction, as shown for UFD1-NPL4, p47, and UBXD1 (15, 35, 36). The presence of multiple binding modules on a single protein affords diverse interacting modes with p97, thus adapting it to its various cellular functions. At the same time, bipartite interactions restrict access of other cofactors to p97. The bipartite interaction of UBXD1 described here is predicted to prevent p97 binding to the classical cofactors UFD1-NPL4 and p47 (15). At the same time, it would also block access to cofactors recognizing the C terminus of p97.
The Structural Basis for Mutually Exclusive Cofactor Binding to p97
UBX-domain-containing cofactors represent the largest group of p97 binding partners (4). Our recent crystal structure of p97N in complex with the UBX domain of FAF1 revealed that residues from connecting loops of both subdomains of p97 contribute to the p97-FAF1 interface (18). These residues contact the surface of the UBX β-sheet in the region around β-strands 1, 3, 4, and 5. However, the crucial determinants of UBX domain binding to the N domain of p97 are provided by the UBX 579R … 619FPR621 signature motif. The FPR motif adopts a cis-proline configuration, which allows the aromatic ring of Phe619 to insert into the hydrophobic interdomain cleft of p97N. Both arginines deeply stretch into the p97-binding pocket to anchor the UBX domain and to stabilize the complex by essential hydrophobic interactions and electrostatic interactions with the p97 backbone. This interaction mode is quite similar to the observed interaction of p97 with the VIM binding motif of gp78 (Fig. 7, B and C). The UBX FPR motif, which is situated in the S3/S4 loop, aligns with the VIM α-helix in the region of Arg636. Both Phe619 of UBX and Arg636 of gp78 are located in the center of the hydrophobic binding groove in the p97N interdomain cleft. In addition, in each case, two arginines insert into this pocket. Strikingly, Arg625 of gp78, which is within hydrogen bonding distance of the carbonyl oxygen of Val108, overlaps with Arg579 of UBX, which is also deeply buried in p97N and is 3.8 Å from the carbonyl oxygen of Val108. ANKZF1, which lacks an Arg625 homolog but contains another arginine two amino acids later, would also make favorable electrostatic and hydrophobic contacts, in line with the high affinity interaction we observe. Tyr110 of p97, which is not involved in the recognition of VIM proteins with the exception of SVIP, as well as the non-essential Phe52 are significantly involved in interactions of p97 with the UBX domain (18). Asp35, which is one of the key residues in p97N-gp78 interaction by stabilizing the conformation of the gp78 Arg636 by hydrogen bonding, is not involved in UBX interaction. These observations demonstrate a convergence of interacting residue types in UBX proteins and VIM ligands despite the fact that these cofactors are not related at all in their architecture.
p97 Specificity for VIM Proteins
Based on bioinformatics as well as structural/functional studies presented in the accompanying paper of Stapf et al. (46) and here, VIM proteins can be divided into two subfamilies: (i) proteins with the signature motif RX5AAX2R (RAAR), including gp78, ANKZF1, and SVIP, and (ii) proteins with the signature motif AAX2R (AAR), such as UBXD1. The RAAR proteins are characterized by an additional conserved arginine N-terminal to the conserved AAX2R. This arginine (Arg626 of gp78, Arg658 of ANKZF1, Arg22 of SVIP) and further non-conserved positively charged residues (in gp78, Arg625; in ANKZF1, Lys660 and Arg661; in SVIP, Lys21 and Lys24) significantly enhance the interaction by electrostatic as well as hydrophobic interactions. The conserved Arg626 is largely solvent-accessible and contacts Arg53 and Phe52 of p97, which, however, are less important for p97-gp78 interaction. In contrast, Arg625 and Arg636 are 42 and 72% buried in the protein interface and apparently contribute more strongly to the stabilization of the p97-gp78 complex. Nevertheless, Arg658 of ANKZF1 (Arg626 of gp78) is important for the in vivo interaction with p97, as analyzed by Stapf et al. (46) in yeast two-hybrid interaction assays as well as co-immunoprecipitation assays in HEK293T cells. This binding mode, which involves positively charged residues separated by hydrophobic residues results in a high affinity interaction.
In contrast, proteins of the AAR subfamily, which lack the first positively charged residue, bind with much lower affinity, and the interaction is mainly driven by hydrophobic contacts. This binding mode where the helical VIM is anchored only by one arginine would presumably allow a higher degree of rotational flexibility. UBXD1 compensates for its rather low affinity mediated by its VIM, at least in part, by its bipartite binding mechanism. Although it could be shown recently that the p97-UBXD1 complex is involved in sorting of ubiquitylated cargo in the endocytic pathway (42), it is still unclear why UBXD1 associates with both the N-terminal domain and the C terminus of p97.
Although VIM proteins bind to the same p97 surface, they feature specific molecular details for recognition by p97 that depend on the amino acid composition and probably result in slight variations in the way the helix is oriented when bound to p97. However, the RX5AAX2R signature motif of RAAR proteins is not always sufficient for high affinity binding, as shown in this study, where we could not detect an interaction of the SVIP VIM with p97 using a peptide similar to gp78. The VIM of SVIP requires additional N-terminal as well as C-terminal α-helical residues; on the one hand, this probably stabilizes the VIM α-helix, and on the other hand, these residues interact with the ND1 linker and the adjacent D1 domain.
In vivo additional interaction partners also might contribute to specificity and the targeting of these complexes to different cellular functions. Although it has been known for years that p97-UFD1-NPL4 assemble into a heterodimeric complex and function together, among other pathways in the retrotranslocation step of ERAD, gp78 as well as ANKZF1 have been shown to co-exist with only NPL4, and the individual ternary complexes are involved in either ERAD or mitochondrial protein degradation (24, 44). In contrast to gp78 and ANKZF1, SVIP does not exist in any of these complexes (23). Clearly, further functional and structural studies, including studies of full-length proteins, are required to fully understand the p97-VIM interactome.
Supplementary Material
Acknowledgments
We thank Alexander Buchberger (University of Würzburg) for providing full-length p97 and full-length UBXD1 expression constructs and for critical reading of the manuscript and Christopher Lima (Sloan-Kettering Institute, New York) for the SenP2 plasmid.
This work was supported by the Deutsche Forschungsgemeinschaft (Rudolf Virchow Center for Experimental Biomedicine, FZ 82) (to H. S.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1 and S2.
The atomic coordinates and structure factors (code 3TIW) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
- ERAD
- endoplasmic reticulum-associated degradation
- ITC
- isothermal titration calorimetry
- PUB
- peptide:N-glycanase/UBA or UBX
- UFD
- ubiquitin fusion degradation
- UBX
- ubiquitin regulatory X
- ULD
- ubiquitin-like domain
- VCP
- valosin-containing protein
- VIM
- VCP-interacting motif
- IBMPFD
- inclusion body myopathy, Paget disease of the bone, and frontotemporal dementia
- N domain
- N-terminal domain
- p97N
- p97 N-terminal domain
- PUL
- PLAP, Ufd3p, Lub1p
- SVIP
- small valosin-interacting protein
- aa
- amino acids
- ATPγS
- adenosine 5′-O-(thiotriphosphate).
REFERENCES
- 1. Halawani D., Latterich M. (2006) Mol. Cell 22, 713–717 [DOI] [PubMed] [Google Scholar]
- 2. Ye Y. (2006) J. Struct. Biol. 156, 29–40 [DOI] [PubMed] [Google Scholar]
- 3. Vij N. (2008) J. Cell Mol. Med. 12, 2511–2518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Schuberth C., Buchberger A. (2008) Cell Mol. Life Sci. 65, 2360–2371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Haines D. S. (2010) Genes Cancer 1, 753–763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ju J. S., Weihl C. C. (2010) Hum. Mol. Genet. 19, R38–R45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Johnson J. O., Mandrioli J., Benatar M., Abramzon Y., Van Deerlin V. M., Trojanowski J. Q., Gibbs J. R., Brunetti M., Gronka S., Wuu J., Ding J., McCluskey L., Martinez-Lage M., Falcone D., Hernandez D. G., Arepalli S., Chong S., Schymick J. C., Rothstein J., Landi F., Wang Y. D., Calvo A., Mora G., Sabatelli M., Monsurrò M. R., Battistini S., Salvi F., Spataro R., Sola P., Borghero G., Galassi G., Scholz S. W., Taylor J. P., Restagno G., Chiò A., Traynor B. J. (2010) Neuron 68, 857–864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. DeLaBarre B., Brunger A. T. (2003) Nat. Struct. Biol. 10, 856–863 [DOI] [PubMed] [Google Scholar]
- 9. Zhang X., Shaw A., Bates P. A., Newman R. H., Gowen B., Orlova E., Gorman M. A., Kondo H., Dokurno P., Lally J., Leonard G., Meyer H., van Heel M., Freemont P. S. (2000) Mol. Cell 6, 1473–1484 [DOI] [PubMed] [Google Scholar]
- 10. Yeung H. O., Kloppsteck P., Niwa H., Isaacson R. L., Matthews S., Zhang X., Freemont P. S. (2008) Biochem. Soc. Trans. 36, 62–67 [DOI] [PubMed] [Google Scholar]
- 11. Jentsch S., Rumpf S. (2007) Trends Biochem. Sci. 32, 6–11 [DOI] [PubMed] [Google Scholar]
- 12. Madsen L., Seeger M., Semple C. A., Hartmann-Petersen R. (2009) Int. J. Biochem. Cell Biol. 41, 2380–2388 [DOI] [PubMed] [Google Scholar]
- 13. Zhao G., Zhou X., Wang L., Li G., Schindelin H., Lennarz W. J. (2007) Proc. Natl. Acad. Sci. U.S.A. 104, 8785–8790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhao G., Li G., Schindelin H., Lennarz W. J. (2009) Proc. Natl. Acad. Sci. U.S.A. 106, 16197–16202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kern M., Fernandez-Sáiz V., Schäfer Z., Buchberger A. (2009) Biochem. Biophys. Res. Commun. 380, 303–307 [DOI] [PubMed] [Google Scholar]
- 16. Isaacson R. L., Pye V. E., Simpson P., Meyer H. H., Zhang X., Freemont P. S., Matthews S. (2007) J. Biol. Chem. 282, 21361–21369 [DOI] [PubMed] [Google Scholar]
- 17. Dreveny I., Kondo H., Uchiyama K., Shaw A., Zhang X., Freemont P. S. (2004) EMBO J. 23, 1030–1039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hänzelmann P., Buchberger A., Schindelin H. (2011) Structure 19, 833–843 [DOI] [PubMed] [Google Scholar]
- 19. Morreale G., Conforti L., Coadwell J., Wilbrey A. L., Coleman M. P. (2009) FEBS J. 276, 1208–1220 [DOI] [PubMed] [Google Scholar]
- 20. Ballar P., Shen Y., Yang H., Fang S. (2006) J. Biol. Chem. 281, 35359–35368 [DOI] [PubMed] [Google Scholar]
- 21. Chen B., Mariano J., Tsai Y. C., Chan A. H., Cohen M., Weissman A. M. (2006) Proc. Natl. Acad. Sci. U.S.A. 103, 341–346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Nagahama M., Suzuki M., Hamada Y., Hatsuzawa K., Tani K., Yamamoto A., Tagaya M. (2003) Mol. Biol. Cell 14, 262–273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ballar P., Zhong Y., Nagahama M., Tagaya M., Shen Y., Fang S. (2007) J. Biol. Chem. 282, 33908–33914 [DOI] [PubMed] [Google Scholar]
- 24. Heo J. M., Livnat-Levanon N., Taylor E. B., Jones K. T., Dephoure N., Ring J., Xie J., Brodsky J. L., Madeo F., Gygi S. P., Ashrafi K., Glickman M. H., Rutter J. (2010) Mol. Cell 40, 465–480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Reverter D., Lima C. D. (2004) Structure 12, 1519–1531 [DOI] [PubMed] [Google Scholar]
- 26. Battye T. G., Kontogiannis L., Johnson O., Powell H. R., Leslie A. G. (2011) Acta Crystallogr. D Biol. Crystallogr. 67, 271–281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Evans P. (2006) Acta Crystallogr. D Biol. Crystallogr. 62, 72–82 [DOI] [PubMed] [Google Scholar]
- 28. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H. (2010) Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bailey S. (1994) Acta Crystallogr. D Biol. Crystallogr. 50, 760–763 [DOI] [PubMed] [Google Scholar]
- 30. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J. (2007) J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Rich R. L., Myszka D. G. (2007) Anal. Biochem. 361, 1–6 [DOI] [PubMed] [Google Scholar]
- 32. Tang W. K., Li D., Li C. C., Esser L., Dai R., Guo L., Xia D. (2010) EMBO J. 29, 2217–2229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Fernández-Sáiz V., Buchberger A. (2010) EMBO Rep. 11, 479–485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Vagenende V., Yap M. G., Trout B. L. (2009) Biochemistry 48, 11084–11096 [DOI] [PubMed] [Google Scholar]
- 35. Bruderer R. M., Brasseur C., Meyer H. H. (2004) J. Biol. Chem. 279, 49609–49616 [DOI] [PubMed] [Google Scholar]
- 36. Meyer H. H., Shorter J. G., Seemann J., Pappin D., Warren G. (2000) EMBO J. 19, 2181–2192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Madsen L., Andersen K. M., Prag S., Moos T., Semple C. A., Seeger M., Hartmann-Petersen R. (2008) Int. J. Biochem. Cell Biol. 40, 2927–2942 [DOI] [PubMed] [Google Scholar]
- 38. Beuron F., Dreveny I., Yuan X., Pye V. E., McKeown C., Briggs L. C., Cliff M. J., Kaneko Y., Wallis R., Isaacson R. L., Ladbury J. E., Matthews S. J., Kondo H., Zhang X., Freemont P. S. (2006) EMBO J. 25, 1967–1976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Pye V. E., Beuron F., Keetch C. A., McKeown C., Robinson C. V., Meyer H. H., Zhang X., Freemont P. S. (2007) Proc. Natl. Acad. Sci. U.S.A. 104, 467–472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Rouiller I., Butel V. M., Latterich M., Milligan R. A., Wilson-Kubalek E. M. (2000) Mol. Cell 6, 1485–1490 [DOI] [PubMed] [Google Scholar]
- 41. Manno A., Noguchi M., Fukushi J., Motohashi Y., Kakizuka A. (2010) Genes Cells 15, 911–922 [DOI] [PubMed] [Google Scholar]
- 42. Ritz D., Vuk M., Kirchner P., Bug M., Schutz S., Hayer A., Bremer S., Lusk C., Baloh R. H., Lee H., Glatter T., Gstaiger M., Aebersold R., Weihl C. C., Meyer H. (2011) Nat. Cell Biol. 13, 1116–1123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Alexandru G., Graumann J., Smith G. T., Kolawa N. J., Fang R., Deshaies R. J. (2008) Cell 134, 804–816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ballar P., Pabuccuoglu A., Kose F. A. (2011) Int. J. Biochem. Cell Biol. 43, 613–621 [DOI] [PubMed] [Google Scholar]
- 45. Davis I. W., Leaver-Fay A., Chen V. B., Block J. N., Kapral G. J., Wang X., Murray L. W., Arendall W. B., 3rd, Snoeyink J., Richardson J. S., Richardson D. C. (2007) Nucleic Acids Res. 35, W375–W383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Stapf C., Cartwright E., Bycroft M., Hofmann K., Buchberger A. (2011) J. Biol. Chem. 286, 38670–38678 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.