

Background: Nucleoprotein (NP) is an essential component of the virus replication complex.
Results: The crystal and quaternary structures of Lassa virus NP were determined.
Conclusion: Lassa virus NP assembles into a symmetric trimer in solution.
Significance: The trimeric complex may have a biological function in the virus life cycle, and its assembly could be a target for antivirals.
Keywords: Crystal Structure, Crystallography, Electron Microscopy (EM), Mutagenesis Site-specific, Negative-strand RNA Viruses, Viral Protein, Viral Transcription, Virology, Arenavirus, Lassa Virus
Abstract
The nucleoprotein (NP) of Lassa virus (LASV) strain AV was expressed in a recombinant baculovirus system. The crystal structure of full-length NP was solved at a resolution of 2.45 Å. The overall fold corresponds to that of NP of LASV strain Josiah (Qi, X., Lan, S., Wang, W., Schelde, L. M., Dong, H., Wallat, G. D., Ly, H., Liang, Y., and Dong, C. (2010) Nature 468, 779–783) with a root mean square deviation of 0.67 Å for all atoms (6.3% difference in primary sequence). As the packing in the crystal offers two different trimer architectures for the biological assembly, the quaternary structure of NP in solution was determined by small-angle x-ray scattering and EM. After classification and averaging of >6000 EM raw images, trimeric centrosymmetric structures were obtained, which correspond in size and shape to one trimer in the crystal structure formed around a crystallographic 3-fold rotation axis (symmetric trimer). The symmetric trimer is also a good model for the small-angle x-ray scattering data and could be well embedded into the ab initio model. The N-terminal domain of NP contains a deep nucleotide-binding cavity that has been proposed to bind cellular cap structures for priming viral mRNA synthesis. All residues implicated in m7GpppN binding were exchanged, and the transcription/replication phenotype of the NP mutant was tested using a LASV replicon system. None of the mutants showed a specific defect in mRNA expression; most were globally defective in RNA synthesis. In conclusion, we describe the full-length crystal structure and the quaternary structure in solution of LASV NP. The nucleotide-binding pocket of NP could not be assigned a specific role in viral mRNA synthesis.
Introduction
Lassa virus (LASV)5 causes hemorrhagic Lassa fever in West Africa (1). It belongs to the family Arenaviridae, which includes other important human pathogens such as Junin, Guanarito, and Machupo viruses. The prototype arenavirus is lymphocytic choriomeningitis virus. The arenavirus genome consists of two negative-strand RNA segments containing four genes. The L segment encodes the small matrix Z protein (2, 3) and the 220-kDa L protein (4, 5). L protein is a multidomain protein (5, 6) that harbors the viral RNA-dependent RNA polymerase (7) and an endonuclease required for viral mRNA synthesis (8, 9). The latter is most likely involved in snatching cap structures from cellular mRNA, in analogy to the endonuclease in the influenza virus polymerase PA subunit (10, 11). The S segment encodes the glycoprotein precursor and the nucleoprotein (NP). The virus genome, NP, and L protein represent the minimum components required for replication and transcription (12–14). NP is the major component of the viral ribonucleoprotein complex. It physically associates with itself as well as with L and Z proteins (15–18). The association with Z protein and ALIX/AIP1, an ESCRT-associated host protein, is important for incorporation of NP into virus-like particles (15, 17–19). In addition, NP acts as an interferon antagonist (20, 21). Recently published x-ray crystallographic data for full-length NP (22) and its C-terminal domain (23) of LASV strain Josiah revealed a nucleotide-binding site in the N terminus and a 3′–5′-exoribonuclease in the C terminus; the latter is critical for NP to function as an interferon antagonist. Here, we report the crystal structure of full-length NP of LASV strain AV expressed in a recombinant baculovirus system. The crystallographic data were complemented by small-angle x-ray scattering (SAXS) and EM studies to determine the quaternary structure of NP in solution. In addition, structure-guided functional studies using a LASV replicon system were conducted to substantiate the hypothesis that the nucleotide-binding site in the N terminus of NP represents a cap-binding site involved in cap snatching and viral mRNA synthesis (22).
EXPERIMENTAL PROCEDURES
Generation of Recombinant Baculovirus
The NP gene of LASV strain AV (GenBankTM accession number AAG41803) (24) was fused to a C-terminal FLAG-His6 sequence and cloned into baculovirus transfer vector pAcSG2 (BD Biosciences). Recombinant baculovirus expressing LASV NP-FLAG-His was generated using the flashBAC system (Oxford Expression Technologies). The transfer vector and baculovirus genome were cotransfected into Spodoptera frugiperda Sf9 cells (Invitrogen) using GeneJuice transfection reagent (Novagen). Cells were monitored for NP expression by immunoblotting using anti-FLAG antibody (Invitrogen) 5 days post-transfection. Supernatants of NP-expressing cultures were passaged twice to generate a high titer virus stock.
Expression and Purification of NP
Sf9 cells in several T75 cell culture flasks were inoculated with 0.5 ml of high titer virus stock/flask. 5 days post-infection, cells were lysed in 50 mm NaH2PO4 (pH 8), 500 mm NaCl, 10 mm imidazole, 0.5% Nonidet P-40, 1× Complete protease inhibitor mixture (Roche Applied Science), and 25 units/ml Benzonase (Novagen) and sonicated. Cell debris was removed by centrifugation, and the supernatant was incubated with nickel-nitrilotriacetic acid-agarose (Invitrogen) overnight at 4 °C. The agarose was washed five times with 50 mm NaH2PO4 (pH 8), 500 mm NaCl, and 30 mm imidazole, and NP was eluted in 50 mm NaH2PO4 (pH 8), 500 mm NaCl, and 500 mm imidazole. NP was further purified by gel filtration chromatography using a HiLoad 16/60 Superdex 200 column (GE Healthcare) with 50 mm Tris-HCl (pH 7.2) and 150 mm NaCl as a running buffer. Fractions containing NP were concentrated using an Amicon Ultra-4 centrifugal filter unit with a 10-kDa cutoff (Millipore). NP was diluted 1:10 in 50 mm Tris-HCl (pH 7.2), loaded onto a 1-ml HiTrap heparin HP column (GE Healthcare), and eluted with 1 m NaCl. Fractions containing NP were concentrated to ∼5 mg/ml using an Amicon Ultra-0.5 centrifugal filter unit with a 10-kDa cutoff (Millipore). Purity of the protein solution was verified by PAGE.
Crystallization
The NP solution was monitored by dynamic light scattering with a Spectroscatter 201 apparatus (Molecular Dimensions) over a suitable period of time, and a stable particle radius (hydrodynamic radius) of ∼6 nm was observed. Several hundred crystallization conditions were screened using the Honeybee 961 dispensing robot (Genomic Solutions, Ann Arbor, MI) at 20 °C in 96-well crystallization plates (NeXtal QIA1 μplates, Qiagen) using the sitting-drop vapor-diffusion method based on various commercially available screens. A 300-nl droplet of the NP solution was mixed with the same volume of reservoir solution and equilibrated against 35 μl of reservoir solution. Promising conditions were optimized by applying the hanging-drop technique and up-scaling in 24-well Linbro plates (ICN Biomedicals). Crystals were obtained reproducibly using a reservoir solution containing 7% PEG 300, 10% glycerol, and 0.5 m KCl. A 1-μl droplet of a 4.7 mg/ml protein solution was mixed with the same volume of reservoir solution and equilibrated against 1 ml of reservoir solution at 20 °C.
X-ray Crystallography
Diffraction data were collected to a resolution of 2.45 Å from a flash-frozen crystal at −173 °C at Advanced Light Source beamline 5.0.1. The initial crystal characterization and space group assignment were performed using the software Denzo (25) and scaled using Scalepack (26). NP of LASV strain AV was determined by molecular replacement using the program MolRep (27) and the native structure of NP of LASV strain Josiah (Protein Data Bank code 3MWP) as a search model. The solution was further refined by iterative model building using the COOT graphics package (28) combined with REFMAC (29). The crystals showed heavy twining with a twin fraction of 0.34. However, after the inclusion of intensity-based twin refinement in REFMAC, the R and Rfree values dropped dramatically. The final model was well refined with good quality electron density and crystallographic R and Rfree values of 18.3 and 22.1%, respectively.
SAXS
The SAXS measurements for the NP sample were performed at EMBL BioSAXS beamline X33 (30, 31) at the DORIS storage ring, DESY (Hamburg, Germany). A photon-counting PILATUS detector of 1 million pixels was used to record the scattered x-rays at a wavelength of 1.54 Å. The sample-to-detector distance was 2.7 m, yielding a maximum recordable momentum transfer (s = 4πsinθ/λ, where 2θ is the scattering angle and λ is the wavelength) of 0.6 Å−1. Bovine serum albumin solution at 4.5 mg/ml in 50 mm HEPES (pH 7.5) was used for calibrating the molecular mass. A series of concentrations (0.5, 0.9, 1.9, 3.8, and 6.7 mg/ml) was measured for the NP sample at 20 °C. The standard SAXS data reduction and analysis were done using the program package ATSAS (32). With the increasing concentration, a systematic change in the Guinier region of the SAXS pattern was observed, which indicated the presence of concentration-driven oligomerization or aggregation. To eliminate the effects of aggregation, the low angle part of the scattering pattern corresponding to the protein at 0.5 mg/ml and the high angle part of the curve for the protein at 6.7 mg/ml were merged using PRIMUS (33), and the merged curve was used for further analysis and modeling. The maximum size of the protein molecule (Dmax) and the pair distance distribution function (p(r)) was calculated with the program GNOM (34). Ab initio models were obtained with DAMMIN (35) by enforcing P3 symmetry and an oblate anisometry. The crystal structure was docked into the model using the program SUPCOMB (36). Rigid body modeling was performed with SASREF (37) by applying P3 symmetry to the monomeric subunit.
EM
NP was prepared for EM by the GraFix (gradient centrifugation in combination with fixation) method (38) with gradients ranging from 5 to 20% sucrose and 0 to 0.15% glutaraldehyde and standard buffer conditions. Gradient fractions were negatively stained with 2% uranyl acetate. Low dose images were recorded on a 4k × 4k CCD camera on a Philips CM200 FEG transmission electron microscope using an acceleration voltage of 160 kV at magnification ×161,000. 6644 single particle images were extracted from 32 micrographs using SIGNATURE (39). Single particles were analyzed by reference-free alignment and classification (40) using IMAGIC-5 software (41).
LASV Replicon Assay
Constructs pCITE-Las-NP, pCITE-Las-L, and pLAS-MG for T7 RNA polymerase-driven expression in mammalian cells of NP, L protein, and minigenome, respectively, of LASV strain AV have been described (12, 42). For transfection, the functional cassette of pCITE-Las-L and pLAS-MG was PCR-amplified with Phusion DNA polymerase (Finnzymes). NP mutants were generated via a two-step PCR mutagenesis approach (7) using pCITE-Las-NP as a template. The presence of artificial mutations was ascertained by sequencing. PCR-amplified DNA for expression of NP, L protein, and minigenome was purified using a PCR purification kit (Macherey-Nagel), quantified spectrophotometrically, and used directly for transfection.
BSR-T7/5 cells stably expressing T7 RNA polymerase (43) were grown in Glasgow's minimal essential medium (Invitrogen) supplemented with 5% fetal calf serum. Cells were seeded at a density of 1 × 105 cells/well in a 24-well plate 1 day prior to transfection. Cells in a well were transfected with DNA for expression of minigenome (250 ng), L protein (250 ng), and NP (250 ng) as well as with 10 ng of pCITE-FF-luc for expression of firefly luciferase (transfection control) using 3 μl of Lipofectamine 2000 (Invitrogen)/μg of DNA. 1 day after transfection, total RNA was prepared for Northern blotting, or cells were lysed in passive lysis buffer (Promega), and the lysate was assayed for firefly and Renilla luciferase activities using the Dual-Luciferase reporter assay system (Promega). Renilla luciferase levels were corrected with the firefly luciferase levels, resulting in standardized relative light units, to compensate for differences in transfection efficiency or cell density.
Northern Blot Analysis
Total RNA was purified using RNeasy columns (Qiagen) with on-column DNase treatment. RNA (0.5 μg) was separated on a 1.5% agarose-formaldehyde gel and transferred onto a Hybond N+ membrane (Amersham Biosciences). Blots were hybridized with an antisense 32P-labeled riboprobe targeting the Renilla luciferase gene. Autoradiography signals were recorded and quantified using an FLA-7000 PhosphorImager (Fujifilm) and TINA software (Raytest, Straubenhardt, Germany).
Immunoblot Analysis
HA tags were fused to the C terminus of NP genes integrated in pCITE-Las-NP via PCR mutagenesis. HeLa cells were inoculated with modified vaccinia virus Ankara expressing T7 RNA polymerase (44) and transfected with PCR-amplified DNA for expression of HA-tagged NP. Cytoplasmic lysate was prepared 1 day after transfection, separated by 4–12% BisTris-PAGE, and transferred to nitrocellulose membrane (Schleicher & Schüll). NP-HA was detected using anti-HA antibody (1:10,000; Sigma-Aldrich H6908) and peroxidase-conjugated secondary antibodies. Protein bands were visualized by chemiluminescence using SuperSignal West Pico substrate (Pierce) and x-ray film (Kodak).
RESULTS
Crystal Structure of LASV NP
As NP of LASV could not be expressed in soluble form in Escherichia coli, a recombinant baculovirus expressing LASV NP was constructed. Full-length NP of strain AV was expressed in Sf9 cells and purified successively by nickel chelate affinity, gel filtration, and heparin affinity chromatography. Calibration of the gel filtration column indicated that NP formed complexes with an estimated molecular mass of 250 kDa corresponding to trimers or tetramers; higher order complexes or a monomeric fraction was not observed (64.5 kDa for the NP monomer including FLAG and His6 tags) (Fig. 1, A and B). Dynamic light scattering analysis showed monodispersity of NP in solution with a hydrodynamic radius of 6 nm (Fig. 1C). Crystals were grown by the sitting-drop vapor-diffusion method. Diffraction data were collected to a resolution of 2.45 Å. Data collection and refinement statistics are summarized in Table 1. The final model included 513 of the 569 residues (excluding FLAG and His tags) of LASV NP. The regions comprising residues 1–7, 147–157, 339–363, 517–521, and 562–569 were disordered and could not be identified in the electron density. The NP monomer features a mainly α-helical N-terminal domain and a C-terminal domain composed of α-helices and β-strands (Fig. 2A). The fold corresponds to that of the recently solved structure of LASV NP of strain Josiah (22) with a root mean square deviation of 0.67 Å for all atoms. The 6.3% difference in amino acid sequence between both NPs has no remarkable effect on the structure. The architecture of functionally important substructures, such as exonuclease in the C-terminal domain (22, 23) and a deep nucleotide-binding cavity in the N-terminal domain, is conserved (22). The C-terminal domain contains electron densities compatible with the presence of Zn2+ and Mn2+ ions as found in the NP structure of strain Josiah (Fig. 2A) (22).
FIGURE 1.

Purification of NP expressed by recombinant baculovirus in Sf9 cells. A, gel filtration peak of NP (filled curve) overlaid with a molecular mass marker (HMW gel filtration calibration kit, GE Healthcare). B, PAGE of NP after nickel chelate affinity and gel filtration chromatography (GF), heparin affinity chromatography (Hep), and a final concentration step using an Amicon Ultra-0.5 centrifugal filter (Conc). C, dynamic light scattering analysis of NP before crystallization.
TABLE 1.
Data collection and refinement statistics
| X-ray source | Advanced Light Source |
| Wavelength (Å) | 0.977 |
| Space group | P3 |
| Unit cell dimensions (Å) | a = 176.32, b = 176.32, c = 56.58 |
| VM (Å3/Da) | 2.68 (3 molecules/AU)a |
| Solvent content (%) | 54.14 |
| Completeness of data (%) | 99.9 |
| No. of total reflections | 627,570 |
| No. of unique reflections | 37,576 |
| Average I/σ intensity | 23.8 (2.7) |
| Resolution (Å) | 80–2.45 |
| Rmerge (%)b | 14.8 |
| No. of reflections used in refinement | 35,333 |
| Rcrystal (%)c | 18.3 (22.1) |
| No. of reflections used in Rfree | 1832 |
| Rfree (%)d | 21.9 (26.1) |
| r.m.s.d. bonds (Å) | 0.006 |
| r.m.s.d. bond angles | 0.960 |
a AU, asymmetric unit; r.m.s.d., root mean square deviation.
b Rmerge = 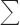 hkli|Ii(hkl) − (I(hkl))|/hkliIi(hkl), where Ii(hkl) and (I(hkl)) are the observed individual and mean intensities of a reflection with indices hkl, respectively; i is the sum over the individual measurements of a reflection with indices hkl; and hkl is the sum over all reflections.
hkli|Ii(hkl) − (I(hkl))|/hkliIi(hkl), where Ii(hkl) and (I(hkl)) are the observed individual and mean intensities of a reflection with indices hkl, respectively; i is the sum over the individual measurements of a reflection with indices hkl; and hkl is the sum over all reflections.
c Rcrystal = ‖Fobs − Fcalc‖/|Fobs|, where Fobs and Fcal are the observed and calculated structure factors, respectively.
d Rfree was calculated using a randomly selected 5% of the reflections.
FIGURE 2.

Crystal structure of NP of LASV strain AV. A, ribbon diagram of the NP monomer. NP is composed of N- and C-terminal domains. The N-terminal domain contains a deep cavity (dashed blue oval) previously shown to bind pyrimidine nucleotides (22). The C-terminal domain contains two metal ions: Mn2+ (red sphere), coordinated by Asp-389, Glu-391, and Asp-533; and Zn2+ (green sphere), coordinated by Glu-399, Cys-506, His-509, and Cys-529. The dotted black line depicts the chain in loop regions that could not be solved. B, asymmetric trimer representing the asymmetric unit composed of chains A, B, and C (left, ribbon diagram; right, molecular surface). C, symmetric trimer composed of C chains from three asymmetric units viewed along a crystallographic 3-fold rotation axis (left, ribbon diagram; right, molecular surface).
In the crystal, the monomers are arranged to form two types of trimers, an asymmetric and a symmetric trimer. The asymmetric trimer (corresponding to the content of the asymmetric unit) is composed of chains A and B, which form a symmetric dimer via a tightly packed N-terminal/N-terminal interface, complemented by the loosely connected chain C (Fig. 2B). The symmetric trimer is composed of monomers (chain C) assembled around a crystallographic 3-fold axis. The three monomers are packed in a head-to-tail arrangement with tight C-terminal/N-terminal interfaces (Fig. 2C).
EM Analysis of NP Complexes in Solution
Because the packing of NP monomers in the crystal is ambiguous or may be artificial, the overall shape of NP complexes in solution was determined by EM and SAXS analyses. For EM, NP was fixed with the GraFix method (38) and negatively stained. Most of the structures visible in EM were ring-shaped (Fig. 3D). More than 6000 raw images of ring-shaped structures were recorded and processed. After reference-free alignment and multivariate statistical analysis of the NP complexes, the eigenimages (Fig. 3, A--C) revealed circular components of varying diameter. The third eigenimage (Fig. 3C) clearly reveals a 3-fold centrosymmetric structure of the NP complexes. After classification and averaging of intact ring-shaped complexes, the trimeric centrosymmetric shape was confirmed (Fig. 3E). When applying 3-fold symmetry to the class averages, their appearance is faithfully conserved (Fig. 3F), supporting the notion that NP assembles into a 3-fold symmetric complex. The size of the NP complex in EM corresponds to that of the symmetric trimer in the crystal (Fig. 3G). The latter projects reasonably well onto the two-dimensional shape of the EM structure (Fig. 3H). The deep intersections in the EM structure probably correspond to the interfaces between the monomers, whereas the intersections within the monomers map to the nucleotide-binding cavity (Fig. 3G, arrows). In conclusion, the EM data suggest that NP in solution adopts a conformation that is similar or even identical to the symmetric trimer found in the crystal.
FIGURE 3.

Analysis of LASV NP complexes by EM. A–C, eigenimages obtained after alignment and multivariate statistical analysis of negatively stained raw images shown in D. D, raw electron micrograph. Circular views are circled in white. Another particle view is circled in gray. E, class averages obtained after alignment and multivariate statistical analysis. Scale bar = 5 nm. F, views corresponding to the class averages shown in E but with 3-fold symmetry applied. Scale bar = 5 nm. G, larger magnification of the class average shown in F (middle panel). The large and small arrows mark the presumed monomer interface and nucleotide-binding cavity in the N-terminal domain, respectively. H, overlay of the symmetric trimer from the crystal structure onto the class average view shown in G.
SAXS Analysis of NP Complexes in Solution
The molecular mass of the NP complex estimated from the SAXS analysis is 160 ± 10 kDa, and the excluded volume of the scattering solute is (340 ± 20) × 103 Å3, corresponding to a molecular mass of 170 ± 10 kDa. These values are about three times the molecular mass of 64.5 kDa calculated from the sequence of a monomer, indicating that the particle is a trimer in solution. The linear Guinier plot of the SAXS profile yields a radius of gyration (Rg) of 42.7 ± 0.6 Å, which is close to the Rg of 41.7 Å evaluated from the atomic structure of the doughnut-shaped symmetric trimer using the program CRYSOL (45). In contrast, the calculated Rg for the structure corresponding to the asymmetric unit in the crystal is 46.2 Å. The maximum particle size (Dmax) derived from the SAXS data is 120 ± 10 Å, which agrees with the value of 120 Å for the symmetric trimer, whereas the Dmax for the asymmetric trimer is ∼160 Å. A direct comparison of the experimental scattering profile with that calculated from the atomic structure for the symmetric and asymmetric trimers is given in Fig. 4A (upper curves). The asymmetric trimer yields a worse fit with discrepancy χ = 4.68 in contrast to χ = 2.30 for the symmetric trimer. (The discrepancy χ should equal 1 for a perfect fit.) In summary, the SAXS results suggest that the symmetric trimer is a better model in aqueous solution than the asymmetric trimer. However, as shown in Fig. 4A, the calculated scattering of the symmetric structure shows some misfit in the s range between 0.05 and 0.15 Å−1. This indicates that the structure in solution might be slightly different from the structure in the crystal. To investigate this hypothesis, a rigid body model was obtained using the monomer subunit and P3 symmetry. The fit to experimental data improved significantly with χ = 1.30 (Fig. 4A, lower curves). The root mean square deviation between the rigid body model and the crystal trimer is 6 Å for the Cα atoms and 6.8 Å when all atoms are used in the alignment (Fig. 4C). Nevertheless, considering the low resolution of the SAXS technique (nominal resolution of 18 Å for these data), we conclude that the trimeric crystal structure is well preserved in solution. The ab initio model obtained with DAMMIN (35) using P3 symmetry and the constraint of an oblate shape yields a good fit to the experimental data (Fig. 4A, lower curves). The symmetric trimer can be well embedded into the ab initio model, confirming that there are no dramatic conformational changes between the structures in solution and in the crystal (Fig. 4B).
FIGURE 4.

SAXS data and models. A, upper curves, comparison of the experimental SAXS pattern with the calculated profile of the symmetric trimer and the trimer from the asymmetric unit of the crystal; lower curves, fits of the rigid body model and the ab initio model to the experimental data. (The SAXS pattern of the upper curve was duplicated and shifted by −1 log I unit to facilitate presentation of multiple curves in one plot.) s, momentum transfer; I, scattered intensity. B, symmetric trimer from the crystal structure embedded into the three-dimensional profile of the ab initio model (gray beads) derived from the experimental data. C, symmetric trimer from the crystal structure (red) superimposed onto the rigid body model (blue) derived from the SAXS data using the monomeric subunit and P3 symmetry.
Functional Relevance of Residues Forming the Nucleotide-binding Cavity
The N-terminal domain of LASV contains a deep cavity of ∼20 Å in length (Fig. 2A). The architecture of this cavity, including the positions of side chains within the cavity, is conserved between the NP structures of LASV strains AV and Josiah. Previous co-crystallization studies have shown that this cavity is able to bind UTP and dTTP (22). It has been proposed that the biological function of this cavity is to bind the cap structure of cellular mRNA, thus facilitating cap snatching and priming of viral mRNA synthesis by L protein (22). To provide further evidence for this hypothesis, we mutated all residues in the cavity implicated in binding the m7GpppN1 cap structure (Fig. 5A) and tested the ability of the NP mutant to support transcription (synthesis of mRNA) and replication (synthesis of the antigenome) using a mammalian cell-based LASV replicon system (Fig. 5B) (9, 12). The two RNA species were separated and detected by Northern blotting. In addition, expression of functional mRNA was measured by Renilla luciferase reporter gene assay. The exchange of side chains critically involved in cap binding or snatching will reduce the mRNA and Renilla luciferase levels but is not expected to affect synthesis of the de novo primed antigenome.
FIGURE 5.

Mutational analysis of the nucleotide-binding site of NP. A, view of the residues implicated in cap binding (22) in the NP structure of strain AV. Main chain atoms are shown in black, and side chain atoms are shown in gray. B, analysis of NP mutants in the LASV replicon system. Transcriptional activity was measured via Renilla luciferase (Ren-Luc) reporter gene expression. The Renilla luciferase activity is shown in the bar graph (mean ± range of standardized relative light units as a percentage of the wild type in two independent transfection experiments). Synthesis of the antigenome and Renilla luciferase mRNA was evaluated by Northern blotting. The methylene blue-stained 18 S rRNA is shown below the blot as a marker for gel loading and RNA transfer. WT, wild-type NP; Neg, defective NP with a deletion of residues 294–339. C, quantification of Northern blot signals and ratio between the signal intensities for mRNA and the antigenome. The mRNA/antigenome ratio of wild-type NP was set at 100%. The mean ± range of the intensity data of two Northern blots is shown. D, immunoblot of NP mutants. NP was expressed with a C-terminal HA tag to facilitate detection by anti-HA antibody. Cells, control cells infected with modified vaccinia virus Ankara expressing T7 RNA polymerase but not transfected.
Ten of the 18 mutants (W164A, L172A, F176A, Y209A, K253A, L265A, E266A, K309A, Y319A, and R323A) were completely defective in expression of functional mRNA as measured by Northern blotting and Renilla luciferase assay. However, they were also defective in de novo primed antigenome synthesis. Five mutants (M54A, L120A, I241A, S242A, and R300A) showed reduced expression of mRNA and Renilla luciferase with a concomitant decrease in the antigenome level. Mutation of three residues (Y213A, K236A, and L239A) had no remarkable influence on the competence of NP in supporting transcription and replication (Fig. 5B and Table 2). A minor decrease in the mRNA/antigenome ratio was seen for mutants Y213A, I241A, and R300A (Fig. 5C). However, none of the tested mutants showed the typical mRNA-defective phenotype characterized by low or lacking mRNA synthesis as measured via reporter gene expression in combination with a wild type-like antigenome signal in Northern blotting and a mRNA/antigenome ratio of ∼10% (7–9). All mutants were expressed at a level comparable with wild-type NP (Fig. 5D). In conclusion, exchange of side chains in the cavity of the N terminus of NP in most cases results in a global defect in viral RNA synthesis rather than a specific defect in mRNA synthesis.
TABLE 2.
Summary of transcription and replication phenotypes of NP mutants with changes in the putative cap-binding site
| Presumed function of residue in m7GpppN1 binding (22) | Phenotype of alanine mutant |
||
|---|---|---|---|
| Ren-Luca activity | Ren-Luc mRNA | Antigenomic RNA | |
| Pocket for m7G | |||
| Met-54 | + | ± | ± |
| Leu-120 | + | + | + |
| Trp-164 | − | − | − |
| Leu-172 | − | − | − |
| Phe-176 | − | − | − |
| Leu-239 | ++ | ++ | ++ |
| Ile-241 | ± | ± | ± |
| Bonds to triphosphate | |||
| Lys-253 | − | − | − |
| Arg-300 | + | + | + |
| Lys-309 | − | − | − |
| Arg-323 | − | − | − |
| Pocket for base N1 | |||
| Tyr-209 | − | − | − |
| Tyr-213 | ++ | ++ | ++ |
| Leu-265 | − | − | − |
| Glu266 | − | − | − |
| Tyr-319 | − | − | − |
| Gate to binding cavity | |||
| Lys-236 | ++ | ++ | ++ |
| Ser-242 | + | + | + |
a Ren-Luc, Renilla luciferase.
DISCUSSION
In this study, we solved the crystal structure of NP of LASV strain AV using protein expressed in a baculovirus system. In solution, NP assembles into a symmetric trimer as determined by EM and SAXS analyses. Functional analysis of residues forming the deep cavity in the N-terminal domain of NP suggested their relevance in global viral RNA synthesis rather than a specific role in mRNA synthesis.
The crystal structure of full-length NP of LASV strain AV is largely identical to that of strain Josiah. The main structural features, including exoribonuclease and Mn2+ and Zn2+ binding in the C terminus and the nucleotide-binding cavity in the N terminus, are conserved (22, 23). In the crystal, two forms of trimers are observed, an asymmetric trimer in the asymmetric unit and a symmetric trimer formed around a crystallographic 3-fold rotation axis. The crystallographic packing suggests two tight dimerization interfaces, a tail-to-tail (N-terminal/N-terminal) interface in the asymmetric unit and a head-to-tail (C-terminal/N-terminal) interface in the symmetric trimer. Insight into the biological unit and the interface regions was obtained by determining the quaternary structure of NP in solution. Both EM and SAXS analyses revealed that NP assembles into a 3-fold symmetric complex in solution. The size and shape of these complexes faithfully reflect the size and shape of the trimeric structure formed by the C chains of three neighboring asymmetric units in the crystal. It is therefore likely that the atomic interactions at the C-terminal/N-terminal interface in the crystal reflect true interactions between NP monomers in solution. This predicts the involvement in NP/NP interaction of Asp-434, Asp-437, Asn-496, Asp-500, Thr-510, and Lys-522 in the C-terminal domain of one NP molecule and Arg-52, Lys-56, Lys-189, Gln-190, and Asn-230 in the N-terminal domain of the other molecule as determined by LIGPLOT (Fig. 6) (46). Most of these residues are conserved among the Old World arenaviruses. Some conformational changes may occur at the interface in solution, as suggested by the rigid body model obtained for the SAXS data. However, this model shall not be overestimated considering the relatively low resolution of the SAXS analysis. Neither the crystallographic studies nor the EM and SAXS analyses revealed the presence of RNA in the complex. In the gel filtration chromatography, only the trimeric complex was observed, and dynamic light scattering analysis showed monodispersity of the trimeric NP complex in solution. Thus, RNA-free NP seems to exist exclusively as a trimer, which presumably has biological relevance. Besides being the building block of ribonucleoproteins, NP has additional functions during the life cycle of the virus, such as interaction with L and Z proteins and cellular proteins, budding of particles, suppression of the interferon response, exoribonuclease activity, and nucleotide binding (15–23). Some of these or other so far unknown functions of NP might be associated specifically with the trimeric configuration. Upon binding of RNA or by another trigger such as phosphorylation, the NP complex may reorganize, forming higher order ribonucleoprotein structures that function in genome replication, transcription, and virion assembly. It is conceivable that a structural adjustment in LASV NP switches the protein to an interaction mode that facilitates multimerization and RNA encapsidation. Recent studies demonstrated two different conformations of Rift Valley fever virus NP, one of which would allow the formation of ribonucleoprotein complexes (47, 48).
FIGURE 6.

Presumed noncovalent interactions at the head-to-tail interface in the symmetric NP trimer. The plot was created using the atomic coordinates from the crystal structure and the program LIGPLOT (46). The residues in the C-terminal domain of one partner are in brown; those in the N-terminal domain of the interacting partner are shown in blue. Arcs with radiating spokes indicate residues involved in hydrophobic interactions. Residues forming salt bridges or hydrogen bonds are shown in ball-and-stick representation; dashed red lines depict the bonds. Black, main chain carbon atoms; gray, side chain carbon atoms; red, oxygen atoms; blue, nitrogen atoms.
Another important feature of LASV NP is the presence of a deep cavity in the N-terminal domain that has been shown to bind pyrimidine triphosphate (22). The appealing hypothesis has been put forward that this cavity represents the cap-binding site of the LASV replication complex and thus plays a crucial role in cap snatching and viral mRNA synthesis (22). In an attempt to substantiate this hypothesis, we mutated all side chains proposed to contribute to m7GpppN1 binding. Evidence for the involvement of these residues in cap binding would be a phenotype characterized by reduced or defective mRNA synthesis but intact de novo primed synthesis of the virus genome, as has been observed by mutation of critical residues in the cap-binding site of influenza virus polymerase subunit PB2 (49, 50). This phenotype has also been generated by mutation of important residues in the endonuclease of LASV and lymphocytic choriomeningitis virus L protein, which is assumed to function in cap snatching (8, 9). However, an analogous phenotype was not observed with the NP mutants. This means that there is no functional evidence for NP being involved in cap binding, cap snatching, and priming of viral mRNA synthesis, although the lack of phenotype is no direct evidence against this hypothesis. An alternative explanation for the outcome of the experiment is a relevance of the nucleotide-binding pocket in global RNA synthesis. It can also not be excluded that modifications of the binding pocket induce structural changes rendering NP generally nonfunctional. In conclusion, further experiments are needed to assign the nucleotide-binding capacity of NP a function in the virus life cycle.
Acknowledgments
We thank Katja Kleinsteuber, Nina Schmalstieg, Lars Redecke, Christel Schmetz, and Silke Retzlaff for providing technical assistance and Sankaran Banumathi (Berkeley Center for Structural Biology) for diffraction data collection.
This work was supported by Deutsche Forschungsgemeinschaft (DFG) Grant GU 883/1-1 and Bundesministerium für Bildung und Forschung (BMBF) Sync-Life Contract 05K10YEA.
The atomic coordinates and structure factors (code 3R3L) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
- LASV
- Lassa virus
- NP
- nucleoprotein
- SAXS
- small-angle X-ray scattering
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol.
REFERENCES
- 1. Günther S., Lenz O. (2004) Crit. Rev. Clin. Lab. Sci. 41, 339–390 [DOI] [PubMed] [Google Scholar]
- 2. Perez M., Craven R. C., de la Torre J. C. (2003) Proc. Natl. Acad. Sci. U.S.A. 100, 12978–12983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Strecker T., Eichler R., Meulen J., Weissenhorn W., Dieter Klenk H., Garten W., Lenz O. (2003) J. Virol. 77, 10700–10705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lukashevich I. S., Djavani M., Shapiro K., Sanchez A., Ravkov E., Nichol S. T., Salvato M. S. (1997) J. Gen. Virol. 78, 547–551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kranzusch P. J., Schenk A. D., Rahmeh A. A., Radoshitzky S. R., Bavari S., Walz T., Whelan S. P. (2010) Proc. Natl. Acad. Sci. U.S.A. 107, 20069–20074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Brunotte L., Lelke M., Hass M., Kleinsteuber K., Becker-Ziaja B., Günther S. (2011) J. Virol. 85, 324–333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hass M., Lelke M., Busch C., Becker-Ziaja B., Günther S. (2008) J. Virol. 82, 10207–10217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Morin B., Coutard B., Lelke M., Ferron F., Kerber R., Jamal S., Frangeul A., Baronti C., Charrel R., de Lamballerie X., Vonrhein C., Lescar J., Bricogne G., Günther S., Canard B. (2010) PLoS Pathog. 6, e1001038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lelke M., Brunotte L., Busch C., Günther S. (2010) J. Virol. 84, 1934–1944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dias A., Bouvier D., Crépin T., McCarthy A. A., Hart D. J., Baudin F., Cusack S., Ruigrok R. W. (2009) Nature 458, 914–918 [DOI] [PubMed] [Google Scholar]
- 11. Yuan P., Bartlam M., Lou Z., Chen S., Zhou J., He X., Lv Z., Ge R., Li X., Deng T., Fodor E., Rao Z., Liu Y. (2009) Nature 458, 909–913 [DOI] [PubMed] [Google Scholar]
- 12. Hass M., Gölnitz U., Müller S., Becker-Ziaja B., Günther S. (2004) J. Virol. 78, 13793–13803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lee K. J., Novella I. S., Teng M. N., Oldstone M. B., de La Torre J. C. (2000) J. Virol. 74, 3470–3477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. López N., Jácamo R., Franze-Fernández M. T. (2001) J. Virol. 75, 12241–12251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Casabona J. C., Levingston Macleod J. M., Loureiro M. E., Gomez G. A., Lopez N. (2009) J. Virol. 83, 7029–7039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jácamo R., López N., Wilda M., Franze-Fernández M. T. (2003) J. Virol. 77, 10383–10393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Levingston Macleod J. M., D'Antuono A., Loureiro M. E., Casabona J. C., Gomez G. A., Lopez N. (2011) J. Virol. 85, 2012–2023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Shtanko O., Imai M., Goto H., Lukashevich I. S., Neumann G., Watanabe T., Kawaoka Y. (2010) J. Virol. 84, 5415–5422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Shtanko O., Watanabe S., Jasenosky L. D., Watanabe T., Kawaoka Y. (2011) J. Virol. 85, 3631–3641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Martínez-Sobrido L., Emonet S., Giannakas P., Cubitt B., García-Sastre A., de la Torre J. C. (2009) J. Virol. 83, 11330–11340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Martínez-Sobrido L., Zúñiga E. I., Rosario D., García-Sastre A., de la Torre J. C. (2006) J. Virol. 80, 9192–9199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Qi X., Lan S., Wang W., Schelde L. M., Dong H., Wallat G. D., Ly H., Liang Y., Dong C. (2010) Nature 468, 779–783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hastie K. M., Kimberlin C. R., Zandonatti M. A., MacRae I. J., Saphire E. O. (2011) Proc. Natl. Acad. Sci. U.S.A. 108, 2396–2401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Günther S., Emmerich P., Laue T., Kühle O., Asper M., Jung A., Grewing T., ter Meulen J., Schmitz H. (2000) Emerg. Infect. Dis. 6, 466–476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Otwinowski Z. (1993) in Data Collection and Processing (Sawyer L., Isaacs N., Bailey S. eds) pp. 56–62, Science and Engineering Research Council, Daresbury Laboratory, Warrington, UK [Google Scholar]
- 26. Otwinowski Z., Minor W. (1997) Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 27. Vagin A., Teplyakov A. (2010) Acta Crystallogr. D 66, 22–25 [DOI] [PubMed] [Google Scholar]
- 28. Emsley P., Cowtan K. (2004) Acta Crystallogr. D 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 29. Murshudov G. N., Vagin A. A., Dodson E. J. (1997) Acta Crystallogr. D 53, 240–255 [DOI] [PubMed] [Google Scholar]
- 30. Roessle M. W., Klaering R., Ristau U., Robrahn B., Jahn D., Gehrmann T., Konarev P., Round A., Fiedler S., Hermes C., Svergun D. (2007) J. Appl. Crystallogr. 40, 190–194 [Google Scholar]
- 31. Round A. R., Franke D., Moritz S., Huchler R., Fritsche M., Malthan D., Klaering R., Svergun D. I., Roessle M. (2008) J. Appl. Crystallogr. 41, 913–917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Petoukhov M. V., Konarev P. V., Kikheny A. G., Svergun D. I. (2007) J. Appl. Crystallogr. 40, 223–228 [Google Scholar]
- 33. Konarev P. V., Volkov V. V., Sokolova A. V., Koch M. H., Svergun D. I. (2003) J. Appl. Crystallogr. 36, 1277–1282 [Google Scholar]
- 34. Semenyuk A. V., Svergun D. I. (1991) J. Appl. Crystallogr. 24, 537–540 [Google Scholar]
- 35. Svergun D. I. (1999) Biophys. J. 76, 2879–2886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kozin M. B., Svergun D. I. (2001) J. Appl. Crystallogr. 34, 33–41 [Google Scholar]
- 37. Petoukhov M. V., Svergun D. I. (2005) Biophys. J. 89, 1237–1250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kastner B., Fischer N., Golas M. M., Sander B., Dube P., Boehringer D., Hartmuth K., Deckert J., Hauer F., Wolf E., Uchtenhagen H., Urlaub H., Herzog F., Peters J. M., Poerschke D., Lührmann R., Stark H. (2008) Nat. Methods 5, 53–55 [DOI] [PubMed] [Google Scholar]
- 39. Chen J. Z., Grigorieff N. (2007) J. Struct. Biol. 157, 168–173 [DOI] [PubMed] [Google Scholar]
- 40. Dube P., Tavares P., Lurz R., van Heel M. (1993) EMBO J. 12, 1303–1309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. van Heel M., Harauz G., Orlova E. V., Schmidt R., Schatz M. (1996) J. Struct. Biol. 116, 17–24 [DOI] [PubMed] [Google Scholar]
- 42. Hass M., Westerkofsky M., Müller S., Becker-Ziaja B., Busch C., Günther S. (2006) J. Virol. 80, 12414–12419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Buchholz U. J., Finke S., Conzelmann K. K. (1999) J. Virol. 73, 251–259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sutter G., Ohlmann M., Erfle V. (1995) FEBS Lett. 371, 9–12 [DOI] [PubMed] [Google Scholar]
- 45. Svergun D. I., Barberato C., Koch M. H. (1995) J. Appl. Crystallogr. 28, 768–773 [Google Scholar]
- 46. Wallace A. C., Laskowski R. A., Thornton J. M. (1995) Protein Eng. 8, 127–134 [DOI] [PubMed] [Google Scholar]
- 47. Ferron F., Li Z., Danek E. I., Luo D., Wong Y., Coutard B., Lantez V., Charrel R., Canard B., Walz T., Lescar J. (2011) PLoS Pathog. 7, e1002030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Raymond D. D., Piper M. E., Gerrard S. R., Smith J. L. (2010) Proc. Natl. Acad. Sci. U.S.A. 107, 11769–11774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Fechter P., Mingay L., Sharps J., Chambers A., Fodor E., Brownlee G. G. (2003) J. Biol. Chem. 278, 20381–20388 [DOI] [PubMed] [Google Scholar]
- 50. Guilligay D., Tarendeau F., Resa-Infante P., Coloma R., Crepin T., Sehr P., Lewis J., Ruigrok R. W., Ortin J., Hart D. J., Cusack S. (2008) Nat. Struct. Mol. Biol. 15, 500–506 [DOI] [PubMed] [Google Scholar]