

Abstract
Glycosylation of proteins is a fundamental process that influences protein function. The Haemophilus influenzae HMW1 adhesin is an N-linked glycoprotein that mediates adherence to respiratory epithelium, an essential early step in the pathogenesis of H. influenzae disease. HMW1 is glycosylated by HMW1C, a novel glycosyltransferase in the GT41 family that creates N-glycosidic linkages with glucose and galactose at asparagine residues and di-glucose linkages at sites of glucose modification. Here we report the crystal structure of Actinobacillus pleuropneumoniae HMW1C (ApHMW1C), a functional homolog of HMW1C. The structure of ApHMW1C contains an N-terminal all α-domain (AAD) fold and a C-terminal GT-B fold with two Rossmann-like domains and lacks the tetratricopeptide repeat fold characteristic of the GT41 family. The GT-B fold harbors the binding site for UDP-hexose, and the interface of the AAD fold and the GT-B fold forms a unique groove with potential to accommodate the acceptor protein. Structure-based functional analyses demonstrated that the HMW1C protein shares the same structure as ApHMW1C and provided insights into the unique bi-functional activity of HMW1C and ApHMW1C, suggesting an explanation for the similarities and differences of the HMW1C-like proteins compared with other GT41 family members.
Keywords: Bacteria, Crystal Structure, Enzyme Structure, Glycoprotein, Glycosylation, Glycosyltransferase, HMW1 Adhesin, HMW1C, Haemophilus influenzae, Two-Partner Secretion
Introduction
Glycosylation of proteins is an essential process that plays an important role in protein structure and function. In recent years, glycoproteins have been identified increasingly in prokaryotes, including pathogenic bacteria. Some bacterial species contain complex O- and N-glycosylation pathways encoded by multiple gene clusters (1, 2), and others utilize a glycosyltransferase alone to modify a single target protein. Most bacterial glycoproteins are surface exposed, suggesting that glycosylation may influence interactions with the host (2–5).
Non-encapsulated Haemophilus influenzae is a common cause of localized respiratory tract disease in humans and initiates infection by colonizing the upper respiratory tract (6). Approximately 80% of non-encapsulated H. influenzae clinical isolates express two related high molecular weight proteins called HMW1 and HMW2 that mediate high level adherence to respiratory epithelial cells, a critical step in the pathogenesis of H. influenzae disease (7). HMW1 and HMW2 are encoded by homologous genes designated hmw1A and hmw2A, respectively. The hmw1A gene is flanked by the hmw1B and hmw1C accessory genes, and the hmw2A gene is flanked by the hmw2B and hmw2C accessory genes. The hmw1B and hmw2B genes and the hmw1C and hmw2C genes are highly homologous (8, 9).
The HMW1 adhesin is synthesized as a pre-pro-protein that contains an atypical signal peptide (amino acids 1–68), an adjacent pro-piece (amino acids 69–441), and a large exoprotein domain with adhesive activity (amino acids 442–1536) (10–12). HMW1 undergoes an elaborate maturation process and is ultimately presented on the bacterial surface via the two-partner secretion (TPS)2 pathway, a common secretion pathway in Gram-negative bacteria (12–13). In general, TPS systems consist of a large exoprotein (TpsA) and a cognate outer membrane channel-forming translocator protein (TpsB). In the HMW1 system, HMW1 is the TpsA protein, and HMW1B is the cognate TpsB protein. The HMW1 secretion system is characteristic of a subset of TPS systems and requires an additional protein for secretion called HMW1C (14–16).
Recent work established that the HMW1 adhesin is an N-linked glycoprotein and is modified at over 30 asparagine residues, in all except one case in the conventional consensus sequon of N-glycosylation, Asn-X-Ser/Thr (17). Glycosylation plays an essential role in preventing premature degradation of HMW1 during the process of secretion and in promoting tethering of HMW1 to the cell surface, a prerequisite for HMW1-mediated adherence (4). The glycan structures that modify HMW1 are simple mono-hexose or di-hexose sugars containing glucose or galactose rather than the characteristic N-acetylated sugars of N-glycosylation (supplemental Fig. S1), suggesting the presence of a novel glycosyltransferase (17).
The HMW1C protein is located in the cytoplasm and is the enzyme responsible for glycosylating HMW1 (4, 18). In recent work, we demonstrated that an Actinobacillus pleuropneumoniae HMW1C ortholog called ApHMW1C is an N-glycosyltransferase capable of transferring glucose and galactose to known asparagine glycosylation sites in HMW1, analogous to HMW1C (19). In addition, both ApHMW1C and HMW1C are capable of creating glucose-glucose linkages in in vitro reactions to account for the di-hexose modification of HMW1 (18, 19). It is not known whether the di-hexose is formed prior to modification of the acceptor asparagine residue or whether instead a single glucose is linked to the target asparagine and then a second glucose is linked to the first glucose, although the conventional interpretation is that the hexose is added to the protein and then the chain is extended (18). These observations indicate that HMW1C-like proteins are uniquely versatile, harboring N-glycosyltransferase activity that mediates N-linkage to the acceptor protein and O-glycosyltransferase activity that creates di-glucose structures on the acceptor protein. The CAZy (Carbohydrate Active Enzymes database at www.cazy.org) database currently classifies HMW1C-like proteins as members of the GT41 family, a family that otherwise exclusively contains O-GlcNAc transferases (OGT) with characteristic tetratricopeptide repeats (TPR) at the N terminus (20–23).
In the current study, we set out to elucidate the structure of HMW1C-like proteins and to define the structural and functional differences between HMW1C-like proteins and the OGT members of the GT41 family. Recombinant HMW1C was insoluble in high concentrations and was thus refractory to crystallization. As an alternative, we turned to ApHMW1C, the closest homolog of HMW1C. Here we report the crystal structure of ApHMW1C and structure-function studies of HMW1C, providing fundamental insights into HMW1C-like proteins and expanding our understanding of the GT41 family of glycosyltransferases.
EXPERIMENTAL PROCEDURES
General Materials
Restriction enzymes, Pfu DNA polymerase, and T4 DNA ligase were purchased from New England Biolabs, Stratagene, and Promega, respectively. Primers used for PCR were synthesized by IDT. The peptides (>95% purity) were synthesized by Genscript (Piscataway, NJ). Unless indicated otherwise, chemicals were purchased from Sigma.
Cloning, Expression, and Purification
Methods used for the cloning, expression, and purification of ApHMW1C and HMW1C have been described previously (18–19). Mutations in ApHMW1C and HMW1C were generated using the QuikChange II site-directed mutagenesis kit (Stratagene) and a mutagenic primer set according to the manufacturer's instruction. The plasmid pHMW1–15 encoding HMW1, HMW1B, and HMW1C served as the template for mutations in HMW1C (11).
Protein Crystallization
The purified ApHMW1C protein was concentrated to 9 mg/ml in buffer containing 50 mm HEPES pH 7.0, 0.2 m NaCl, 5% glycerol and 0.1 m EDTA. The solution of peptide pN1131 (NVTVNNNITSHK, corresponding to residues 1131 to 1142 of HMW1) was prepared to a concentration of 10 mm peptide in sterilized water. To obtain ApHMW1C-peptide complex crystals, ApHMW1C and pN1131 solutions (10:1 (v/v)) were mixed and incubated at room temperature for 2 h. The protein-peptide sample was screened against commercial screen solutions (Hampton Research Inc). Small three-dimensional crystals were observed in drops containing PEG8000. Crystallization was optimized by employing microseeding methods. Sizable crystals (in space group P212121) of protein-peptide complex were grown in a mixture of 1.5 μl of protein-peptide solution, 0.3 μl of seed solution, and 1.5 μl of reservoir solution (0.1 m MES pH 6.5, 0.12–0.16 m (NH4)2SO4, and 22–30% PEG 8000) using the hanging drop vapor diffusion method at 17 °C for a week.
To obtain native apo crystals, microseeding methods were also performed, since the ApHMW1C solution alone (i.e. in the absence of pN1131) only produced P1 crystals. The initial apo crystals were obtained in similar crystallization conditions to protein-peptide complex crystals using a fresh seed stock prepared from the pN1131::ApHMW1C complex crystals. Subsequently, sizable apo crystals used for data collection were grown in the same conditions, but the seed stock was made from the apo crystals. To obtain UDP-Glc::ApHMW1C complex crystals, the 0.5 μl of 10 mm UDP-Glc solution was directly added into the crystallization drops of apo ApHMW1C crystals and then incubated at 17 °C for 2 h.
Heavy Atom Labeling and Data Collection
For phasing, five different heavy atom compounds, 0.5 and 1 mm HgCl2, 1, 5, and 10 mm EMTS (ethyl mercury thiosalicylate)/Thimerosal, 1, 5, and 10 mm K2PtCl4, 1, 5, and 10 mm KAu(CN)2 and 1 mm (CH3)Pb(CH3COO)2, were prepared in mother liquor solution (0.1 m MES pH 6.5, 0.12 m (NH4)2SO4 and 23% PEG 8000). Heavy atom derivatives were prepared by soaking apo ApHMW1C crystals in each of these solutions at 17 °C for 3, 6, and 24 h. After each time point, crystals were washed by “back-soaking” in mother liquor solution lacking heavy atom compounds. Crystals were cryo-protected with mother liquor solution containing 25% (v/v) glycerol and cooled in liquid N2. Similarly, native apo ApHMW1C crystals, UDP-Glc::ApHMW1C complex crystals, and pN1131::ApHMW1C peptide complex crystals were treated with cryoprotectants and flash cooled in liquid N2. Diffraction data were collected at 1.008 Å for the Hg SAD datasets and at 0.9794 Å for crystals of native apo ApHMW1C, UDP-Glc::ApHMW1C complex, and pN1131::ApHMW1C peptide complex on beamline 19ID at the Advanced Photon Source, Argonne National Laboratory, using an ADSC Quantum 315 CCD detector. All data sets were indexed and integrated with HKL2000 or HKL3000 and scaled with SCALEPACK (24). General handling of the scaled data were carried out with programs from the CCP4 suite.
Structure Determination
The structure was solved by SAD phasing from EMTS-derivatized crystals of apo ApHMW1C using Shelx, MLphare, and dm on HKL3000 (25). SHELXC/D was used to obtain eight mercury sites and the initial phases were determined by MLphare. After the density modification, the starting model was built using buccaneer on CCP4 suite. Subsequently, one of two molecules in the asymmetric unit was manually built using COOT (26). When ∼90% main chain and ∼70% side chain was traced, the structure model was used as a template for Molecular Replacement (MOLREP) against native apo crystal dataset. MOLREP found two molecules in the asymmetric unit with the Cc values 0.465 and 0.590 for the 1st protomer and the 2nd protomer, respectively. The final model of the apo-ApHMW1C structure was obtained after iterative cycles of manual model building with COOT and refinement with REFMAC5 (27). To solve ligand-complexed structures, one of two protomers from the final model of the apo structure was used as a template against UDP-Glc::ApHMW1C complex dataset and pN1131::ApHMW1C complex dataset. For each structure, modeling and refinement were performed with similar procedures used for the apo structure. In pN1131::ApHMW1C complex refinement, individual coordinates and B-factors refinement and simulated annealing were simultaneously applied using PHENIX (28). In each structure, solvent molecules were assigned at positions, where the electron density peaks were found above 1.3 σ in the 2Fo-Fc map (above 3.0σ in the Fo-Fc map) and hydrogen bonds were stereochemically reasonable. The final model of the apo structure contained residues 1–619 of chain A and residues 4–118, 134–413, and 419–619 of chain B. The final model of UDP-Glc::ApHMW1C complex contained residues 1–619 of chain A and residues 4–118, 134–412, and 419–619 of chain B. The final model of pN1131::ApHMW1C complex contained residues 1–619 of chain A and residues 6–119, 134–412, and 420–619 of chain B. Validation of all three final models was carried out using the Protein Data Bank validation server. Except for Val-106 (both chain A and B), almost all residues are in the most favored regions on the Ramachandran plot. A summary of the data collection and refinement statistics is given in Table 1. Coordinates and structure factors have been deposited at the Protein Data Bank with PDB ID 3Q3E (apo ApHMW1C), 3Q3H (UDP-Glc::ApHMW1C complex), and 3Q3I (pN1131::ApHMW1C complex).
TABLE 1.
Statistics from crystallographic analysis
| Data Collection | EMTS (Hg) | apo ApHMW1C | UDP-Glc::ApHMW1C | Peptide::ApHMW1C |
| Wavelength (Å) | 1.008 | 0.9794 | 0.9794 | 0.9794 |
| Space group | P212121 | |||
| Cell parameters (Å) | a = 79.739 | a = 80.368 | a = 80.245 | a = 79.701 |
| b = 93.888 | b = 94.677 | b = 94.896 | b = 93.262 | |
| c = 177.482 | c = 176.911 | c = 176.791 | c = 176.705 | |
| Resolution (Å) | 50.00–3.00 (3.05–3.00) | 50.00–2.10 (2.18–2.10) | 50.00–2.25 (2.33–2.25) | 50.00–2.45 (2.54–2.45) |
| Reflections (Total / Unique) | 260058 / 27518 | 262452 / 73875 | 209323 / 61133 | 183950 / 46099 |
| Completeness (%)a | 96.4 (91.5) | 93.3 (83.3) | 92.8 (74.2) | 94.1 (83.1) |
| Rmerge (%)a, c | 12.8 (61.2) | 8.0 (29.1) | 8.4 (27.1) | 8.0 (30.3) |
| Mean <I/σ(I)> | 28.6 | 12.5 | 16.5 | 13.4 |
| Redundancya | 9.5 (9.6) | 3.7 (2.4) | 3.6 (1.9) | 4.1 (2.5) |
| Sigma cutoff | 0.5 | 0.5 | 0.3 | |
| Phasing | ||||
| Overall FOMb (before / after DM) | 0.167 / 0.836 | |||
| Overall Phasing Power | 1.09 | |||
| Structure Refinement | ||||
| Resolution (Å) | 2.10 | 2.25 | 2.45 | |
| No. reflections (working/test) | 70123 / 3720 | 57402 / 3065 | 43748 / 2329 | |
| R (%)a, d/Rfreee (%) | 18.4 / 22.6 | 18.6/24.4 | 17.8 / 24.3 | |
| Number of atoms (protein/water/GOL/UDP) | 9742 / 484 / 18 / - | 9736 / 280 / 12 / 50 | 9721 / 163 / 24 / - | |
| Average B factor (protein/water/GOL/UDP, Å2) | 25.2 / 28.2 / 43.1 / - | 35.7 / 34.7 / 52.4 / 52.1 | 37.6 / 33.0 / 47.2 / - | |
| rms deviation | ||||
| Bonds (Å) | 0.016 | 0.019 | 0.021 | |
| Angles (°) | 1.607 | 1.812 | 1.933 | |
| Ramachandran plot (%) | ||||
| Most favored regions | 92.1 | 91.6 | 91.7 | |
| Additional allowed regions | 7.8 | 7.8 | 7.8 | |
| Generously allowed regions | 0.0 | 0.4 | 0.4 | |
| Disallowed regions | 0.2 | 0.2 | 0.2 | |
a Completeness and Rmerge are given for overall data and for the highest resolution shell.
b The figure of merit (FOM) = |Fbest| − |F|.
c Rmerge = 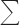 |Ii − I〉/|/sigma]|Ii|, where Ii is the intensity of an observation, I〉 is the mean value for that reflection, and the summations are overall equivalents.
|Ii − I〉/|/sigma]|Ii|, where Ii is the intensity of an observation, I〉 is the mean value for that reflection, and the summations are overall equivalents.
d R-factor = h||Fo(h)| − Fc(h)||/hFo(h), where Fo and Fc are the observed and calculated structure factor amplitudes, respectively.
e Rfree was calculated with 5% of the data excluded from the refinement.
Glycosyltransferase Assay of ApHMW1C and Data Analysis
Glycosyltransferase activity of ApHMW1C was assessed as previously described (19, 29). Experimental details are given in the supplemental information.
Glycosyltransferase Assay of HMW1C
In vitro glycosyltransferase assays were performed as described previously (18). Briefly, 1.5 μg of purified HMW1C or mutant HMW1C, 1.5 μg of purified HMW1802–1406, and 20 μl of 50 mm UDP-α-d-glucose (Calbiochem) were combined in a final volume of 150 μl in 25 mm Tris pH 7.2, 150 mm NaCl. Samples were incubated for 60 min at room temperature, then further incubated at 4 °C overnight, and then resolved on an SDS-PAGE gel. Protein was transferred to a nitrocellulose membrane, and glycosylation was detected using DIG Glycan reagents (Roche).
Adherence Assays
Adherence assays were performed with Chang epithelial cells (human conjunctiva; ATCC CCL 20.2) (Wong-Kilbourne derivative clone 1–5c-4) as described previously (30). Escherichia coli expressing HMW1, HMW1B, and either wild type or mutant HMW1C was prepared by inoculating LB broth containing ampicillin to select for pHMW1–15 or the relevant plasmid derivative and incubating overnight. Percent adherence was calculated by dividing the number of adherent colony-forming units by the number of inoculated colony-forming units. All strains were examined in triplicate, and assays were repeated three times.
Protein Analysis
Whole cell lysates were prepared by suspending bacterial pellets in 10 mm HEPES, pH 7.4 and sonicating to clarity. Proteins were resolved by SDS-PAGE using 7.5% polyacrylamide gels. Western blots were performed using guinea pig antiserum GP96 against the HMW1 protein or guinea pig antiserum 64 against HMW1C.
RESULTS
Structure Determination of ApHMW1C
In initial experiments we purified recombinant H. influenzae HMW1C for crystallography. However, purified HMW1C precipitated at high concentrations, precluding crystallization. As an alternative, we turned to A. pleuropneumoniae HMW1C (ApHMW1C), which is 65% identical and 85% similar to HMW1C and shares the same ability to glycosylate the H. influenzae HMW1 adhesin in vivo and in vitro (19). We focused first on native and selenomethionine-derivatized ApHMW1C proteins but generated only thin plate-shaped triclinic (P1) crystals with high mosaicity. As an alternative approach, we exploited knowledge of the carbohydrate modification sites of HMW1 (17) and synthesized peptides that are known to be glycopeptides. Co-crystallization of ApHMW1C with the NVTVNNNITSHK peptide (referred to as pN1131) resulted in crystals in the space group P212121 diffracting to 2.45 Å resolution. Using these crystals and seeding techniques, we obtained apo crystals and UDP-Glc containing crystals, which diffracted to 2.1 Å and 2.25 Å resolution, respectively. The ApHMW1C structure was solved by SAD phasing with an EMTS derivative of the apo crystals and refined for three different crystal systems: apo form, UDP-Glc::ApHMW1C (the glucose moiety was not observed in the electron density map), and pN1131::ApHMW1C (although the peptide was critical for crystallization, no electron density was observed for the peptide) (Table 1). The two protomers in the asymmetric unit did not have significant molecular contacts, indicating that the functional enzyme is a monomer, consistent with previous biochemical studies (19).
Overall Structure of ApHMW1C
The ApHMW1C structure consists of three discrete domains, including an all α-helical domain (referred to as AAD) at the N terminus (residues 1 to 257) and two Rossmann-like domains that create a GT-B fold at the C terminus (residues 258 to 620) (Fig. 1). Together the AAD and the two Rossmann-like domains form a quasi equilateral (∼70 Å) triangle face. The AAD fold contains 13 consecutive α-helical motifs and appears to be conserved among HMW1C-like proteins, as demonstrated by the structure-based alignment of ApHMW1C and 3 other HMW1C-like proteins (Fig. 2A). Although the AAD of ApHMW1C and TPR repeats adopt all α-structures, the fold of AAD differs from the fold of TPR repeats (Fig. 2, B and C), providing strong structural evidence that HMW1C-like proteins are distinct from other members of the GT41 family (20–23).
FIGURE 1.

Structure of ApHMW1C. A, discrete domains of ApHMW1C are determined in this study. The corresponding domains of HMW1C are indicated. B, cartoon representation of ApHMW1C in complex with UDP. The UDP molecule is in stick representation. C, ApHMW1C structure is shown as a semi-transparent surface. The N-terminal all α-helical domain (AAD), GT-1 and GT-2 of the C-terminal GT-B fold, and the inter-domain connecting GT-1 and GT-2 are rendered in blue, green, red, and orange hues, respectively. D, superimposition of six protomers from the three crystal systems (apo form, red and green; UDP-complex, blue and yellow; and peptide complex, magenta and cyan). High flexibility regions, where electron density was not observed in the chain B of each crystal system, are indicated with circles.
FIGURE 2.

Structural features of the AAD. A, structure-based sequence alignment of the N-terminal domain (AAD) of representative HMW1C-like proteins highlights the conserved feature of the AAD among HMW1C-like proteins. The identical and conserved residues are highlighted in blue and light blue, respectively. The helices observed in the ApHMW1C structure are indicated above the sequence: Ap, A. pleuropneumoniae HMW1C; Hi, H. influenzae HMW1C; Ec, Escherichia coli EtpC; and Ye, Yersinia enterocolitica RcsC. B, topology of the AAD in ApHMW1C. Shown are α-helices of the AAD in cylinders. The α1 to α5 helices are highlighted in green, cyan, magenta, yellow, and blue, and the α9 to α13 helices are colored in gold. In the ribbon representations, α1 to α5 of ApHMW1C and the corresponding C-terminal domain of GST-2 (PDB 3ERF) are colored as in the topology diagram. C, topology of TPRs in XcOGT. The TPR-like (TRL) helices of XcOGT are highlighted in gold. The superimposition of ApHMW1C (gray) and XcOGT (pale blue) shows the dissimilarity of N-terminal helices and the similarity of TRL helices in the two structures.
The GT-B fold of ApHMW1C contains the GT-1 domain (residues 258 to 403), the GT-2 domain (residues 427 to 620), and an inter-domain region that connects GT-1 and GT-2 (residues 404 to 426) (Fig. 1, A–C). The GT-1 and GT-2 domains have a similar core structure (β/α/β folds) and form the UDP-sugar binding site at their interface. The long helical tail that includes C-terminal helices α24, α25, and α26 (residues 578–618) extends from the GT-2 domain back to the GT-1 domain and secures interdomain (GT-1 versus GT-2) contacts, similar to observations for most GT-B fold glycosyltransferases (reviewed in Refs. 31, 32). The sequence alignment and secondary structure assignment of ApHMW1C and HMW1C (Fig. 1, A–C, and supplemental Figs. S2 and S3) highlights that these proteins are virtually identical structurally except for a disordered 30 residue N-terminal tail in HMW1C (DISORDER2 Disorder Prediction server (33)).
The two protomers of each crystal system superimposed with an rmsd of 0.56 Å in apo-ApHMW1C, 0.45 Å in UDP-Glc::ApHMW1C, and 0.51 Å in pN1131::ApHMW1C. Pair-wise comparison of the six protomers from these three individual crystal systems ranged from 0.23 Å (apo chain A versus UDP complex chain A) to 0.61 Å (apo chain B versus peptide complex chain A). Overall, the six protomers from these three crystal systems superimposed well, except for three highly flexible peptide segments (Fig. 1D). Two of these segments (residues 1–3 and residues 119–133, connecting the loop between α6 and α7) correspond to the most variable sequence regions of the AAD (Fig. 2A) and protrude from the triangle plane in the opposite direction. The third segment (residues 414–418) belongs to the inter-domain, which does not contain secondary structure and appears to be highly flexible.
UDP-sugar Binding Pocket
The difference Fourier electron density maps calculated using data collected from apo and UDP-Glc soaked crystals revealed a clear density for the UDP moiety, which is almost completely buried in the interdomain cleft between GT-1 and GT-2 and makes extensive contact with residues of the GT-2 domain (Figs. 1B and 3A). Interestingly, UDP occupied slightly different conformations in the two molecules of the asymmetric unit (referred to as UDP-A and UDP-B), suggesting a possible mechanistic snapshot of the enzyme. No electron density was observed for the glucose moiety on either protomer, suggesting release from the UDP molecule. The UDP binding pocket is defined by the C-terminal ends of β9 (residues 437–441) and β10 (residues 468–471), the loop between β11 and α20 (residues 495–497), and the N-terminal ends of α20 (residues 498–501) and α21 (residues 519–522) (Figs. 3A and 5A).
FIGURE 3.

Active site, interactions of UDP, and functional implications. A, two different conformations of UDP molecules from two protomers in the asymmetric unit are shown in stick representation with electron density (2Fo-Fc contoured at 1.3σ). Key residues involved in the interaction between ApHMW1C and UDP are indicated with side chains (stick presentation) and black letters. The corresponding residues of HMW1C are labeled in magenta letters. The protein backbone is shown with a scheme, and hydrogen-bonding interactions are indicated with black dashed lines. Water molecules mediating hydrogen bonds are shown in blue spheres. B, in vitro transferase assays examining HMW1C mutants. Assays were performed with purified HMW1802–1406, UPD-Glc, and either wild type HMW1C (lane 1), HMW1C-T464A (lane 2), HMW1C-K467A (lane 3), HMW1C-N547A (lane 4), HMW1C-D551A (lane 5), or no HMW1C (lane 6). Glycosylation was detected using DIG-Glycan reagents. C, Western immunoblots of whole cell sonicates of E. coli DH5α harboring vector only (lane 1), HMW1, HMW1B, and wild type HMW1C (lane 2), HMW1, HMW1B, and HMW1C-T464A (lane 3), HMW1, HMW1B, and HMW1C-K467A (lane 4), HMW1, HMW1B, and HMW1C-N547A (lane 5), and HMW1, HMW1B, and HMW1C-D551A (lane 6). The upper blot was performed with guinea pig antiserum GP85 against HMW1, and the lower blot was performed with guinea pig antiserum GP64 against HMW1C. D, in vitro adherence assay showing HMW1-mediated adherence by E. coli DH5α expressing HMW1 and HMW1B along with wild type H. influenzae HMW1C or an HMW1C mutant. The HWM1C residues Thr-464, Lys-467, Asn-547, and Asp-551 correspond to the ApHMW1C residues Thr-438, Lys-441, Asn-521, and Asp-525, respectively, of the binding pocket shown in A.
FIGURE 5.

Comparison of the GT-B domain of representative HMW1C-like proteins and OGT-like proteins, two subfamilies of GT41. A, structure-based sequence alignment of the GT-B domains. Residues involved in UDP binding and proposed to be involved in hexose moiety binding are marked with blue asterisks and pink diamonds, respectively. It is noticed that there is a large insertion in HsOGT and DmOGT between GT-1 and GT-2, as indicated in the blue box. Examples of N-glycosyltransferases include A. pleuropneumoniae HMW1C (Ap), H. influenzae HMW1C (Hi), E. coli EtpC (Ec), and Yersinia enterocolitica RcsC (Ye). Examples of O-glycosyltransferases include Homo sapiens OGT (Hs), Drosophila melanogaster OGT (Dm), Arabidopsis thaliana OGT (At), and Xanthomonas campestris OGT (Xc). B, structural comparison of ApHMW1C with GT-B adopting proteins, XcOGT (PDB 2JLB) and E. coli glycogen synthase (PDB 2R4T). The structures are shown in cartoon representation with color spectrum from blue (N terminus) to red (C terminus) for AAD and TPR, and GT-1 and GT-2 domains are in green and pink hues, respectively.
The uracil bases on both UDP conformations are stabilized by a stacking interaction with the side chain of Tyr-501 and further van der Waals contacts with Gly-468. The N3 and O4 atoms on both uracil bases make hydrogen bonds with Ser496N,O and with Pro494O and Ser496OG via a water molecule (Fig. 3A). Both ribose rings make hydrogen bonds with Asp-525 and make additional van der Waals contacts with Tyr-521. The O2′ and O3′ atoms of UDP-A make further water molecule-mediated hydrogen bonds with Asp-525. The phosphate groups on UDP-A make hydrogen bond interactions with Lys441NZ, Gln469OE1 (via a water molecule), and Asn521N, ND2. The phosphate groups on UDP-B interact with Lys441NZ, Asn519OD1 (via a water molecule), Thr520N, Asn521N, and Gly522N. While the binding of UDP yielded only subtle changes in the overall ApHMW1C structure, binding appears to foster localized conformational changes. The side chain of Gln-469 sways into the UDP binding pocket, placing its amido group nearby the phosphate group of UDP. The side chain of Asn-521 swings away from UDP, allowing the main chain N and the side chain NH2 to form critical hydrogen bonds with β-phosphate oxygen O1B and O3B and to thus resolve electrostatic clashes. Additionally, localized shifts are observed for the main chains of the loops between β10 and α19, β11 and α20, and β12 and α21, resulting in a fine-tuning effect of the binding pocket in the presence or absence of the UDP-sugar donor.
To test the mechanism of UDP-sugar binding, we generated a number of ApHMW1C variants with point mutations and examined enzymatic activity using a continuous spectroscopy assay (19). Lys-441, Asn-521, and Asp-525 form critical interactions with the ribose and phosphate moieties, and mutation of these residues eliminated enzymatic activity. Similarly, mutation of Tyr-498 resulted in a marked decrease in enzymatic activity (∼9% of wild type) (Table 2). In contrast, mutation of Thr-438 had little effect on enzymatic activity, consistent with the fact that this residue does not make specific contact with UDP in the ApHMW1C structure (Fig. 3A).
TABLE 2.
Kinetic parameters of ApHMWC wild type and mutants
These are apparent values, determined by varying the concentration of one substrate (sugar donor substrate) at a fixed concentration of the second (protein acceptor).
| Proteins | Kmapp for UDP-Glc | Vmax | kcat | kcat/Km |
|---|---|---|---|---|
| μm | nmol min−1mg−1 | s−1 | m−1s−1 | |
| ApHMW1C | 39 ± 6 | 109 ± 4 | 0.130 ∼ 0.135 | (3.00∼3.34) × 103 |
| ApHMW1C-F39A | 85 ± 20 | 60 ± 4 | 0.067 ∼ 0.077 | (0.79∼0.90) × 103 |
| ApHMW1C-H214A | 59 ± 7 | 99 ± 3 | 0.115 ∼ 0.122 | (1.95∼2.07) × 103 |
| ApHMW1C-D215A | ND | ND | ND | ND |
| ApHMW1C-H219A | 65 ± 12 | 56 ± 3 | 0.063 ∼ 0.071 | (0.98∼1.09) × 103 |
| ApHMW1C-H272A | 62 ± 8 | 84 ± 3 | 0.097 ∼ 0.104 | (1.56∼1.68) × 103 |
| ApHMW1C-H277A | 82 ± 15 | 12 ± 1 | 0.014 ∼ 0.016 | (0.16∼0.18) × 103 |
| ApHMW1C-H277D | ND | ND | ND | ND |
| ApHMW1C-T438A | 31 ± 6 | 90 ± 4 | 0.108 ∼ 0.112 | (3.04∼3.47) × 103 |
| ApHMW1C-K441A | ND | ND | ND | ND |
| ApHMW1C-Y498A | 101 ± 13 | 24 ± 1 | 0.029 ∼ 0.030 | (0.26∼0.28) × 103 |
| ApHMW1C-N521A | ND | ND | ND | ND |
| ApHMW1C-D525A | ND | ND | ND | ND |
The Interface between the AAD and GT-B Domains and the Acceptor Protein Binding Groove
The crystal structure of ApHMW1C revealed extensive contacts between the AAD and the GT-B domain, creating a unique groove adjacent to the UDP-sugar binding pocket (Figs. 1C, 2A, and 4A). The narrow end of the groove is ∼7 Å, and the wide side of the groove measures ∼18 Å. The hydrophobic core residues in the AAD and the GT-B domain are absolutely conserved between ApHMW1C and HMW1C and are highly conserved among representative HMW1C-like proteins. The surface of the groove is also remarkably conserved between ApHMW1C and HMW1C, including His214/His241, Asp215/Asp242, Met218/Met245, His219/His246, Tyr222/Tyr249 (from α12 HDVYMHCSY), His272/His298, Met349/Met375, and Asp350/Aps376 (supplemental Fig. S3 and Fig. 4A). The close association of the AAD and the GT-B domain renders the overall enzyme structure original and rather rigid, perhaps providing an explanation for the lack of significant conformational changes among different crystal systems (apo versus complex structures).
FIGURE 4.

Putative sugar/peptide-binding sites and functional implications. A, surface representation of ApHMW1C-UDP complex (left) and XcOGT-UDPGlcNAc analog (UDM) complex (right). For comparison of the putative peptide binding clefts, the ApHMW1C and XcOGT (PDB 2JLB) structures were first superimposed. Thus, the two views are from the same orientation. B, close-up view of the groove area, as indicated with boxes in A: ApHMW1C (left) and XcOGT (right). ApHMW1C and HMW1C residues were tested for activity by mutagenesis and functional assays. For clarity, representative residues are colored and labeled: ApHMW1 residues (top) and HMW1C (bottom). The corresponding groove region of XcOGT (right panel) differs from the groove in HMW1C-like proteins. C, Western immunoblots of whole cell sonicates of E. coli DH5α expressing HMW1, HMW1B, and wild type HMW1C (lane 1), HMW1 and HMW1B (lane 2), HMW1, HMW1B, and HMW1C-D242A/H246A/Y249A (lane 3), and HMW1, HMW1B, and HMW1C-H303R (lane 4). The upper blot was performed with guinea pig antiserum GP85 against HMW1, and the lower blot was performed with guinea pig antiserum GP64 against HMW1C. D, in vitro adherence assay showing HMW1-mediated adherence by E. coli DH5α expressing HMW1 and HMW1B along with wild type H. influenzae HMW1C, no HMW1C, or an HMW1C mutant.
The structure-based sequence alignment revealed that the absolutely conserved residue His-277 corresponds to a putative catalytic base proposed in XcOGT homologs and is adjacent to the UDP binding pocket (Figs. 3A, 4, A and B, and 5A). As the first step to delineate the acceptor protein binding site and the catalytic mechanism, we examined a number of ApHMW1C mutants with point mutations affecting the region adjacent to the UDP-binding site and the groove (Tables 2 and 3). While most of the mutants showed decreased enzyme activity, mutation of Asp-215 resulted in null enzymatic activity. Interestingly, while the H277D mutant showed no apparent activity, the H277A mutant showed low specific activity (∼5% of wild type).
TABLE 3.
Kinetic parameters of ApHMWC wild type and mutants
These are apparent values, determined by varying the concentration of one substrate (protein acceptor) at a fixed concentration of the second (sugar donor substrate).
| Proteins | Kmapp for HMW1ct | Vmax | kcat | kcat/Km |
|---|---|---|---|---|
| μm | nmol min−1mg−1 | s−1 | m−1s−1 | |
| ApHMW1C | 5 ± 0.2 | 415 ± 6 | 0.489 ∼ 0.504 | (9.79∼10.1) × 104 |
| ApHMW1C-F39A | 5 ± 1 | 222 ± 23 | 0.238 ∼ 0.293 | (4.76∼5.86) × 104 |
| ApHMW1C-H214A | 6 ± 1 | 419 ± 29 | 0.467 ∼ 0.536 | (7.78∼8.94) × 104 |
| ApHMW1C-D215A | ND | ND | ND | ND |
| ApHMW1C-H219A | 3 ± 0.5 | 151 ± 6 | 0.174 ∼ 0.188 | (5.78∼6.26) × 104 |
| ApHMW1C-H272A | 6 ± 0.7 | 297 ± 14 | 0.339 ∼ 0.372 | (5.64∼6.20) × 104 |
Structure-Function Analysis of HMW1C
Based on the ApHMW1C-UDP complex structure and the HMW1C model, we predicted that Lys-467, Asn-547, and Asp-551 are key residues for UDP binding in HMW1C (Fig. 3A). To test these predictions, we generated a series of HMW1C point mutations and then examined the effect of the mutant proteins on HMW1 glycosylation, HMW1 tethering to the bacterial surface, and HMW1-mediated adherence to human epithelial cells. As controls, we used wild type HMW1C and an HMW1C variant with a mutation of Thr-464 (Thr-438 in ApHMW1C), a residue that is predicted to be unrelated to UDP-sugar binding. As shown in Fig. 3B, mutation of Lys-467, Asn-547, or Asp-551 abrogated glycosylation as assessed by in vitro glycosylation assays using the purified HMW1C derivatives, a purified fragment of HMW1, and UDP-glucose and assessing glycosylation using DIG-Glycan reagents. As shown in Fig. 3C, examination of whole cell sonicates of bacteria expressing HMW1 and HMW1B with either wild type HMW1C or HMW1C-T464A revealed 160 kDa and 125 kDa bands corresponding to the glycosylated HMW1 pre-pro-protein and the glycosylated HMW1 mature protein, respectively. In contrast, in bacteria expressing HMW1 and HMW1B with HMW1C-K467A, HMW1C-N547A, or HMW1C-D551A, the HMW1 pre-pro-protein and the HMW1 mature protein were less abundant and migrated at lower apparent molecular masses, consistent with a lack of glycosylation. Analysis of adherence demonstrated that bacteria expressing HMW1C-K467A, HMW1C-N547A, or HMW1C-D551A were nonadherent in assays with Chang epithelial cells (Fig. 3D).
To assess whether residues along the groove at the interface of the AAD and the GT-1 domain are critical for HMW1C binding of the HMW1 acceptor protein or sugar moiety (Fig. 4A), we generated a point mutant involving His-303 (His-277 in ApHMW1C) and a triple mutant involving Asp-242, His-246, and Tyr-249 and then examined these derivatives in whole bacteria expressing HMW1 and HMW1B for an effect on HMW1 glycosylation as assessed by Western analysis and adherence assays. As shown in Fig. 4, C and D, mutation of His-303 by itself and of Asp-242, His-246, and Tyr-249 together resulted in a change in molecular mass of the HMW1 pre-pro-protein and the HMW1 mature protein and a loss of HMW1-mediated adherence, consistent with elimination of HMW1 glycosylation.
DISCUSSION
HMW1C-like Proteins Define a Novel Subfamily of the GT41 Family
In this study we report the first structure of an HMW1C-like protein, defining a new subfamily of the GT41 family characterized by versatile catalytic activities that include N-glycosylation of protein acceptor sites and O-glycosylation of sugar acceptor sites (18, 19). The unique architecture of ApHMW1C consists of an N-terminal all α-domain (AAD) fold and a C-terminal GT-B fold. The AAD fold differs from the TPR fold that is characteristic of the GT41 family based on other members of the family. The GT-B fold contains the GT-1 and GT-2 domains and harbors the binding site for UDP-hexose. The interface of the AAD and the GT-B domain creates a unique groove with potential to accommodate the acceptor protein. Based on kinetic analyses of active site mutants of ApHMW1C, we validated the critical role of key residues of the UDP-hexose binding pocket in glycosylation of HMW1. Using the structure-guided HMW1C model, site-directed mutagenesis, and glycosylation assays, we demonstrated that the H. influenzae HMW1C protein adopts the same structure as ApHMW1C, with critical residues for binding UDP-hexose including Lys-467, Asn-547, and Asp-551 in the GT-2 domain. In addition, we delineated the binding region for the HMW1 acceptor protein at the interface groove between AAD fold and the GT-1 domain.
Structural Comparison with XcOGT, an OGT-like GT41 Member
The GT41 family contains both bacterial and eukaryotic enzymes and was previously believed to include just O-GlcNAc transferases (OGT), with a characteristic TPR domain at the N terminus. Limited structural information is available for the GT41 family, namely the structure of a bacterial OGT homolog called XcOGT and the structure of human OGT (20–23). The TPR domain of XcOGT contains three complete TPR repeats that form the standard TPR superhelix and are followed by two extra pairs of antiparallel α-helices called TPR-like repeats (TLRs). Although the ApHMW1C AAD and the XcOGT TPR regions (sharing ∼11% sequence identity) have two different folds according to DALI (34) searches, we were able to manually superimpose the last four helices of the AAD with the corresponding helices of XcOGT (Fig. 2, B and C). Both the ApHMW1C AAD and the XcOGT TPR regions are closely associated with the GT-B domain. However, the resulting molecular surfaces of the two proteins are very distinct in shapes and chemical properties (Fig. 4, A and B), suggesting an explanation for the different donor and acceptor specificities between HMW1C-like proteins and other OGT members of the GT41 family.
While the closest structural homolog of the ApHMW1C GT-B domain was the XcOGT GT-B domain (DALI Z-score, 30; rmsd, 3.1 Å; and 17% sequence identity over 332 residues of the GT-B domain), the structure-based sequence alignment of GT-B domains clearly visualizes two distinct subfamilies of GT41 members, namely HMW1C-like proteins and OGT-like proteins (Fig. 5, A and B). Consistent with the different mechanistic strategies utilized by these subfamilies, GT41 members show appreciable resemblance only in the UDP-binding pocket, the common structural moiety for substrates, with conservation of critical amino acids interacting with UDP (Fig. 6, A and B). At the same time, there are important dissimilarities between the UDP-binding pockets in HMW1C-like proteins and OGT-like proteins. In particular, Asn-385 in XcOGT (mapping to Gln-839 in hOGT) makes a hydrogen bond with the α-phosphate group of UDP, whereas the corresponding residue in ApHMW1C (Thr-438 in ApHMW1C, mapping to Thr-464 in HMW1C) makes no direct contact with UDP. Our efforts to obtain the structure of the complete UDP-Glc substrate bound to ApHMW1C have been unsuccessful so far, probably reflecting the cleavage activity of ApHMW1C in the absence of the HMW1 protein acceptor, similar to reports of T4 phage β-glucosyltransferase (35, 36). Nevertheless, on the basis of the structural comparison with other UDP-sugar complex structures, we predicted the plausible position of the glucose moiety for ApHMW1C (Fig. 6, A and B, and supplemental Fig. S4). As expected, no clearly conserved residues of the sugar moiety sites were detected, reflecting the fact that the HMW1C-like proteins are specific for UDP-hexoses while the OGTs are specific for UDP-GlcNAc.
FIGURE 6.

Comparative representation of the active sites of ApHMW1C, XcOGT, and EcGS. A, UDP-hexose binding pocket of ApHMW1C. Key residues involved in the interaction between ApHMW1C and the UDP moiety are in stick presentation (green) and labeled in black. Comparison with XcOGT and EcGS suggested putative residues for glucose binding (sticks in gray). Resides corresponding to residues involved in sugar binding in XcOGT and EcGS are labeled in blue and pink, respectively. B, UDP-GlcNAc binding pocket of XcOGT (PDB 2JLB, XcOGT complex with UDP-GlcNAc phosphonate). Key residues involved in the interaction between XcOGT and the UDP moiety are in stick presentation (blue) and labeled in black. Representative residues interacting with the sugar moiety (GlcNAc) are indicated with gray stick side chains and labeled in blue. C, ADP-glucose binding pocket of EcGS (PDB 2R4T, EcGS complex with ADP, glucose, and HEPPSO). Key residues involved in the interactions between EcGS and the ADP moiety are in stick presentation (pink) and labeled in black. Representative residues interacting with the sugar moiety (glucose and HEPPSO) are indicated with gray stick side chains and labeled in pink.
Comparison with Other Structural Homologs
Searches for structural homologs of the AAD of ApHMW1C (residues 1–257) using DALI revealed that the first 5 helices of the AAD have limited similarity to the C-terminal domain of glutathione S-transferase (GST), aligning with an rmsd of 2.7 Å and a Z-score of 5.3 for 79 Cα atoms (Fig. 2B). Thus, the AAD in ApHMW1C appears to have a composite structure, with a partial GST-like motif at the N terminus and TLRs at the C terminus, making the AAD distinct from other α-helical bundle structures.
Searches for structural homologs of the GT-B fold in ApHMW1C (residues 258 to 620) revealed a number of other GT-B proteins, including members of the glycogen synthase 1 family (Z-score, ∼19; rmsd, 4.2–4.8 Å; and 7–9% sequence identity on 308–316 residues) and the GT4 family (Z-score, ∼19; rmsd, 4.8–5.2 Å; and 6–8% sequence identity on 303–311 residues). Glycogen synthases are classified in two large GT families, namely the GT3 and GT5 families (31). Animal and yeast glycogen synthases belong to the GT3 family and use UDP-glucose as the glucose donor (37), while bacterial glycogen synthases are grouped in the GT5 family and use ADP-glucose exclusively as the glucose source (38). Currently, glycogen synthase structures are available from Agrobacterium tumefaciens, E. coli, and Pyrococcus abyssi, all from the GT5 family (39–41). Unlike HMW1C-like proteins and OGTs, these glycogen synthases contain the GT-B domain alone, without an extra N-terminal appendage (Fig. 5B). Although the nucleotide-sugar donor bound to available crystal structures of glycogen synthases is ADP rather than UDP, these enzymes transfer glucose, an important common feature with HMW1C-like proteins. These enzymes catalyze O-glycosidic bond formation between glucoses, another shared characteristic with HMW1C-like proteins. Comparison of ApHMW1C with structures of glycogen synthases containing glucose or the glucose polymer analog HEPPSO did not show clearly conserved residues in the glucose-binding site but suggested that ApHMW1C can accommodate a di-glucose in the reaction center (Fig. 6C and supplemental Fig. S4), consistent with the observation that ApHMW1C catalyzes O-glycosidic bond formation. In addition, the position of the sugar moiety in these structures is consistent with the position of the sugar analog in XcOGT (Fig. 6, B and C).
The GT4 family is perhaps the largest of all the GT families (32) and contains sucrose synthase, α-glucosyltransferase, and diglucosyl diacylglycerol synthase, among others. Based on the wide range of donor and acceptor substrates, the GT4 family appears to have both functional and sequence diversity (42–44). When we discovered that HMW1C and ApHMW1C are glycosyltransferases with structural homology with the GT41 family, it was surprising to consider that enzymes in the same family would be capable of generating peptide N-linked, peptide O-linked, and sugar O-linked glycosides, especially since these activities would be anticipated to employ different mechanistic strategies. The current study clearly illustrates the unique characteristics of HMW1C-like proteins that combine features from several GT families to accommodate versatile activities.
Molecular Insights into the H. influenzae HMW1C Structure and Function
Given the high sequence identity between ApHMW1C and HMW1C, the ApHMW1C structure provided a basis for probing the molecular mechanism of HMW1C glycosyltransferase activity. Site-directed mutagenesis demonstrated that the UDP-hexose binding pocket in HMW1C is absolutely conserved with the pocket in ApHMW1C. The structure also revealed a funnel shaped groove adjacent to the UDP-hexose binding site with an orientation and configuration that suggested a mechanism for accommodating the acceptor protein. Indeed, mutation of Asp-242, His-246, and Tyr-249 in this groove abolished glycosylation of HMW1, consistent with the conclusion that this groove is critical for binding the acceptor protein. Based on previous studies of XcOGT and the crystal structure of ApHMW1C, we predicted that His-303 (His-277 in ApHMW1C, His-218 in XcOGT, and His-558 in human OGT) is the catalytic base. In fact, this His residue is invariant in the GT41 family (Fig. 5A). However, mutant ApHMW1C-H277A retained low but appreciable activity, raising a question about the identity of the catalytic base. Considering the position of this His residue in the active site and the observation that mutants ApHMW1C-H277D and HMW1C-H303R lack activity, we suspect that this residue is important for binding the sugar moiety or the acceptor protein, but not as the catalytic base. Based on the observation that mutant ApHMW1C-D215A resulted in null activity, we speculate that this absolutely conserved residue in HMW1C-like proteins (Asp-242 in HMW1C) plays a critical role in the recognition of the acceptor substrate. The recent structure of human OGT indicated that the His-498 of human OGT is the probable catalytic base, not the previously proposed His-558 (23). This His-498 residue is not conserved in XcOGT, but corresponds to Phe-165 in XcOGT that is located in helix α9 of the structure (Fig. 2C). A structure overlay aligned Tyr-222 of helix α12 in ApHMW1C (Tyr-249 in HMW1C) with Phe-165 of XcOGT (Fig. 2, B and C, and 4B).
In conclusion, this study demonstrates the structural basis for the glycosyltransferase activity in HMW1C and ApHMW1C, members of a novel subfamily of the GT41 family of glycosyltransferases. The HMW1C-like proteins share features of glycogen synthases and OGTs, in part accounting for their dual function as glycosyltransferases that catalyze N-linkage to HMW1 and O-glycosidic bonds between glucose residues on HMW1.
Supplementary Material
Acknowledgments
Results shown in this report are derived from work performed at the Argonne National Laboratory, Structural Biology Center (19ID) at the Advanced Photon Source. Argonne is operated by the University of Chicago, Argonne, LLC, for the U.S. Dept. of Energy, Office of Biological and Environmental Research under Contract DE-AC02-06CH11357.
This work was supported, in whole or in part, by National Institutes of Health Grant AI068943 and Grant E-1616 from the Welch Foundation (to H.-J. Y.) and by National Institutes of Health Grant R01-DC02873 (to J. W. S.).
The atomic coordinates and structure factors (codes 3Q3E, 3Q3H, and 3Q3I) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S4.
- TPS
- two-partner secretion
- ApHMW1C
- Actinobacillus pleuropneumoniae HMW1C
- TPR
- tetratricopeptide repeats
- EMTS
- ethyl mercury thiosalicylate
- OGT
- O-GlcNAc transferases
- HEPPSO
- 4-(2-hydroxyethyl)piperazine-1-(2-hydroxypropane)sulfonic acid
- AAD
- all α-domain.
REFERENCES
- 1. Schmidt M. A., Riley L. W., Benz I. (2003) Trends Microbiol. 11, 554–561 [DOI] [PubMed] [Google Scholar]
- 2. Szymanski C. M., Wren B. W. (2005) Nat. Rev. Microbiol. 3, 225–237 [DOI] [PubMed] [Google Scholar]
- 3. Charbonneau M. E., Girard V., Nikolakakis A., Campos M., Berthiaume F., Dumas F., Lépine F., Mourez M. (2007) J. Bacteriol. 189, 8880–8889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Grass S., Buscher A. Z., Swords W. E., Apicella M. A., Barenkamp S. J., Ozchlewski N., St Geme J. W., 3rd (2003) Mol. Microbiol. 48, 737–751 [DOI] [PubMed] [Google Scholar]
- 5. Rudd P. M., Elliott T., Cresswell P., Wilson I. A., Dwek R. A. (2001) Science 291, 2370–2376 [DOI] [PubMed] [Google Scholar]
- 6. St Geme J. W., 3rd (1993) Infect. Agents Dis. 2, 1–16 [PubMed] [Google Scholar]
- 7. St Geme J. W., 3rd, Falkow S., Barenkamp S. J. (1993) Proc. Natl. Acad. Sci. U.S.A. 90, 2875–2879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Barenkamp S. J., Leininger E. (1992) Infect. Immun. 60, 1302–1313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Barenkamp S. J., St Geme J. W., 3rd (1994) Infect. Immun. 62, 3320–3328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Buscher A. Z., Grass S., Heuser J., Roth R., St Geme J. W., 3rd (2006) Mol. Microbiol. 61, 470–483 [DOI] [PubMed] [Google Scholar]
- 11. Grass S., St Geme J. W., 3rd (2000) Mol. Microbiol. 36, 55–67 [DOI] [PubMed] [Google Scholar]
- 12. St Geme J. W., 3rd, Yeo H. J. (2009) Trends Microbiol. 17, 355–360 [DOI] [PubMed] [Google Scholar]
- 13. Jacob-Dubuisson F., Locht C., Antoine R. (2001) Mol. Microbiol. 40, 306–313 [DOI] [PubMed] [Google Scholar]
- 14. St Geme J. W., 3rd, Grass S. (1998) Mol. Microbiol. 27, 617–630 [DOI] [PubMed] [Google Scholar]
- 15. Fleckenstein J. M., Roy K., Fischer J. F., Burkitt M. (2006) Infect. Immun. 74, 2245–2258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Nelson K. M., Young G. M., Miller V. L. (2001) Infect. Immun. 69, 6201–6208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gross J., Grass S., Davis A. E., Gilmore-Erdmann P., Townsend R. R., St Geme J. W., 3rd (2008) J. Biol. Chem. 283, 26010–26015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Grass S., Lichti C. F., Townsend R. R., Gross J., St Geme J. W., 3rd (2010) PLoS Pathog. 6, e1000919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Choi K. J., Grass S., Paek S., St Geme J. W., 3rd, Yeo H. J. (2010) PLoS ONE 5, e15888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Clarke A. J., Hurtado-Guerrero R., Pathak S., Schüttelkopf A. W., Borodkin V., Shepherd S. M., Ibrahim A. F., van Aalten D. M. (2008) EMBO J. 27, 2780–2788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jinek M., Rehwinkel J., Lazarus B. D., Izaurralde E., Hanover J. A., Conti E. (2004) Nat. Struct. Mol. Biol. 11, 1001–1007 [DOI] [PubMed] [Google Scholar]
- 22. Martinez-Fleites C., Macauley M. S., He Y., Shen D. L., Vocadlo D. J., Davies G. J. (2008) Nat. Struct. Mol. Biol. 15, 764–765 [DOI] [PubMed] [Google Scholar]
- 23. Lazarus M. B., Nam Y., Jiang J., Sliz P., Walker S. (2011) Nature 469, 564–567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Otwinowski Z., Minor W. (1997) Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 25. Minor W., Cymborowski M., Otwinowski Z., Chruszcz M. (2006) Acta Crystallogr. D Biol. Crystallogr. 62, 859–866 [DOI] [PubMed] [Google Scholar]
- 26. Emsley P., Cowtan K. (2004) Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 27. Murshudov G. N., Vagin A. A., Lebedev A., Wilson K. S., Dodson E. J. (1999) Acta Crystallogr. D Biol. Crystallogr. 55, 247–255 [DOI] [PubMed] [Google Scholar]
- 28. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H. (2010) Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Liu F., Tanner M. E. (2006) J. Biol. Chem. 281, 20902–20909 [DOI] [PubMed] [Google Scholar]
- 30. Buscher A. Z., Burmeister K., Barenkamp S. J., St. Geme J. W., 3rd (2004) J. Bacteriol. 186, 4209–4217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Coutinho P. M., Deleury E., Davies G. J., Henrissat B. (2003) J. Mol. Biol. 328, 307–317 [DOI] [PubMed] [Google Scholar]
- 32. Lairson L. L., Henrissat B., Davies G. J., Withers S. G. (2008) Annu. Rev. Biochem. 77, 521–555 [DOI] [PubMed] [Google Scholar]
- 33. Ward J. J., McGuffin L. J., Bryson K., Buxton B. F., Jones D. T. (2004) Bioinformatics 20, 2138–2139 [DOI] [PubMed] [Google Scholar]
- 34. Holm L., Sander C. (1998) Nucleic Acids Res. 26, 316–319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Moréra S., Imberty A., Aschke-Sonnenborn U., Rüger W., Freemont P. S. (1999) J. Mol. Biol. 292, 717–730 [DOI] [PubMed] [Google Scholar]
- 36. Murray B. W., Wittmann V., Burkart M. D., Hung S. C., Wong C. H. (1997) Biochemistry 36, 823–831 [DOI] [PubMed] [Google Scholar]
- 37. Roach P. J. (2002) Curr. Mol. Med. 2, 101–120 [DOI] [PubMed] [Google Scholar]
- 38. Ball S. G., Morell M. K. (2003) Annu. Rev. Plant Biol. 54, 207–233 [DOI] [PubMed] [Google Scholar]
- 39. Buschiazzo A., Ugalde J. E., Guerin M. E., Shepard W., Ugalde R. A., Alzari P. M. (2004) EMBO J. 23, 3196–3205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Horcajada C., Guinovart J. J., Fita I., Ferrer J. C. (2006) J. Biol. Chem. 281, 2923–2931 [DOI] [PubMed] [Google Scholar]
- 41. Sheng F., Jia X., Yep A., Preiss J., Geiger J. H. (2009) J. Biol. Chem. 284, 17796–17807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Martinez-Fleites C., Proctor M., Roberts S., Bolam D. N., Gilbert H. J., Davies G. J. (2006) Chem. Biol. 13, 1143–1152 [DOI] [PubMed] [Google Scholar]
- 43. Ruane K. M., Davies G. J., Martinez-Fleites C. (2008) Proteins 73, 784–787 [DOI] [PubMed] [Google Scholar]
- 44. Vetting M. W., Frantom P. A., Blanchard J. S. (2008) J. Biol. Chem. 283, 15834–15844 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.