

Background: SERCA1 is a membrane transporter responsive to various inhibitors.
Results: A novel synthesized compound (NF1058) interferes with calcium binding and ATP utilization, whereas phosphorylation with Pi is not inhibited.
Conclusion: NF1058 inhibits SERCA1 stabilizing an E2 state that can still be phosphorylated with Pi.
Significance: NF1058 differs in its inhibition mechanism from thapsigargin, which prevents both calcium binding and enzyme phosphorylation with Pi.
Keywords: Calcium ATPase, Calcium Transport, Drug Action, Enzyme Inhibitors, Membrane Proteins, Calcium Binding, Inhibition Mechanism, Phosphoenzyme
Abstract
Several clotrimazole (CLT) and 4-aminoquinoline derivatives were synthesized and found to exhibit in vitro antiplasmodial activity with IC50 ranging from nm to μm values. We report here that some of these compounds produce inhibition of rabbit sarcoplasmic reticulum Ca2+-ATPase (SERCA1) with IC50 values in the μm range. The highest affinity for the Ca2+-ATPase was observed with NF1442 (N-((3-chlorophenyl)(4-((4-(7-chloroquinolin-4-yl)piperazin-1-yl)methyl)phenyl)methyl)-7-chloro-4-aminoquinoline) and NF1058 (N-((3-chlorophenyl)(4-(pyrrolidin-1-ylmethyl)phenyl)methyl)-7-chloro-4-aminoquinoline),yielding IC50 values of 1.3 and 8.0 μm as demonstrated by measurements of steady state ATPase activity as well as single cycle charge transfer. Characterization of sequential reactions comprising the ATPase catalytic and transport cycle then demonstrated that NF1058, and similarly CLT, interferes with the mechanism of Ca2+ binding and Ca2+-dependent enzyme activation (E2 to E1·Ca2 transition) required for formation of phosphorylated intermediate by ATP utilization. On the other hand, Ca2+ independent phosphoenzyme formation by utilization of Pi (i.e. reverse of the hydrolytic reaction in the absence of Ca2+) was not inhibited by NF1058 or CLT. Comparative experiments showed that the high affinity inhibitor thapsigargin interferes not only with Ca2+ binding and phosphoenzyme formation with ATP but also with phosphoenzyme formation by utilization of Pi even though this reaction does not require Ca2+. It is concluded that NF1058 and CLT inhibit SERCA by stabilization of an E2 state that, as opposed to that obtained with thapsigargin, retains the functional ability to form E2-P by reacting with Pi.
Introduction
The sarco-endoplasmic reticulum Ca2+-ATPase (SERCA)2 is a member of the P-type ATPase family (1) that includes enzymes differentiated for transport of various cations such as Na+/K+, H+/K+, and Ca2+/H+. The P-type ATPases are membrane-bound and utilize the free energy of ATP for active transport, thereby playing a fundamental role in cellular signaling (2, 3). The SERCA catalytic and transport cycle includes initial enzyme activation (E2 to E1 transition) triggered by Ca2+ binding followed by ATP utilization to form a phosphoenzyme intermediate (E1P). The free energy derived from ATP is then utilized by the phosphoenzyme for a conformational transition (E1P to E2P) that favors vectorial displacement and release of bound Ca2+. The cycle is completed by hydrolytic cleavage of E2P (4) as shown in Fig. 1, where binding and dissociation of H+ in exchange for Ca2+ as well as the E2 to E1 conformational transition are also shown.
FIGURE 1.

Enzymatic cycle. Simplified enzymatic cycle of SERCA1 (see Introduction for details).
The Ca2+-ATPase (SERCA) is responsive to various compounds demonstrating different degrees of potency and inhibition (1, 5). These compounds include the highly specific and potent inhibitor thapsigargin (TG, IC50 ∼ 0.1 nm (6, 7)) as well as cyclopiazonic acid (IC50 ∼ 10–20 nm (8)) and 2,5-di-(t-butyl)-dihydroxybenzene (IC50 ∼ 1 μm, (9)), which have been used as aids for protein crystallization and structural studies (10). Medium affinity inhibitors such as clotrimazole (CLT, IC50 ∼ 35 μm (11)), curcumin (IC50 ∼ 15 μm (12)), and 1,3-dibromo-2,4,6-tri(methylisothiouronium)benzene (IC50 ∼ 15 μm (13)) are also known. As a point of pharmacological interest, TG derivatives have been considered for treatment of prostate cancer (14, 15) and cyclopiazonic acid for myocardial ischemia (16).
We have used the CLT scaffold to synthesize novel compounds that turned out to be very active against several Plasmodium strains in vitro and in vivo (17–20). We report here a series of experiments conducted to evaluate the effects of several CLT analogues and related 4-aminoquinolines on the Ca2+-ATPase (SERCA1), revealing various degrees of inhibitory activity that were produced at relatively low concentrations by NF1442 (N-((3-chlorophenyl)(4-((4-(7-chloroquinolin-4-yl)piperazin-1-yl)methyl)phenyl)methyl)-7-chloro-4-aminoquinoline) and NF1058 (N-((3-chlorophenyl)(4-(pyrrolidin-1-ylmethyl) phenyl)methyl)-7-chloro-4-aminoquinoline). We then used SERCA1 to test drug effects on the sequential steps of the Ca2+-ATPase catalytic and transport cycle and to clarify in detail the inhibitory mechanism.
EXPERIMENTAL PROCEDURES
Chemicals
Analytical grade calcium, magnesium, potassium chlorides, and dithiothreitol (DTT ≥ 99%) were obtained from Merck. Adenosine-5′-triphosphate disodium salt (ATP, ∼97%) was purchased from Fluka (St. Louis, MO). Choline chloride, EGTA, MOPS, and calcimycin (calcium ionophore A23187) were obtained from Sigma. [γ-32P]ATP and [γ-32P]Pi were obtained from PerkinElmer Life Sciences. The chemical compounds tested, whose structures are reported in Table 1, were synthesized as previously described (17–20, 45, 46). The purity of all compounds was >98% as estimated by combustion analysis.
TABLE 1.
Structure of the screened compounds and values of IC50 determined from ATPase activity measurements
n.a., not active. n.p., not published.

ATPase Preparation
Sarcoplasmic reticulum vesicles were obtained with the microsomal fraction of homogenized fast-twitch rabbit hind leg muscle (21). The vesicles so obtained contain the Ca2+-ATPase SERCA1 isoform, accounting for ∼50% of the microsomal protein. Protein concentration was determined with the Folin phenol reagent using bovine serum albumin as a standard (22).
Functional Measurement
ATPase activity was determined by measuring production of Pi by the malachite green colorimetric method (23). The reaction mixture included 20 mm MOPS, pH 6.8, 80 mm KCl, 3 mm MgCl2, 2 μm A23187, 5 mm sodium azide, and 2 mm EGTA, or 0.2 mm EGTA plus 0.2 mm CaCl2 (≈8 μm free Ca2+ ions (24)). Microsomal vesicles containing the Ca2+-ATPase were added to yield protein concentrations varying between 0.3 and 30 μg/ml. The temperature was maintained at 37 °C, and the reaction was started by the addition of 1 mm ATP. Samples were then taken at serial times for Pi determination. Background enzyme activity was determined in the presence of 1 μm TG.
Enzyme phosphorylation by ATP was measured in a reaction mixture containing 50 mm MOPS, pH 7.0, 80 mm KCl, 2 mm MgCl2, 1 mm EGTA, 5 μm A23187, and 10–30 μg of microsomal protein/ml. The reaction was started by the addition of 10 μm [γ-32P]ATP and 0.95 mm CaCl2 (7 μm free Ca2+) and allowed to proceed for 90 s at 30 °C. Quenching was obtained with 5% trichloroacetic acid (TCA), and the quenched samples were passed through 0.45-μm Millipore filters. The protein adsorbed on the filter was washed with 15 ml of 0.1 m perchloric acid and 5 ml of cold water. The filters were then processed for determination of radioactivity by scintillation counting.
Phosphoenzyme formation with Pi in the reverse reaction of the cycle was obtained in a reaction mixture containing 50 mm MES-Tris, pH 6.0, 10 mm MgCl2, 2 mm EGTA, 20% Me2SO4, 0.5 mm [γ-32P]Pi, and 10 μg microsomal protein/ml. After 5 min of equilibration at 30 °C, the reaction was quenched with 5% TCA. The quenched reaction mixture was loaded onto a 0.45-μm Millipore filter, which was then washed with 15 ml of 0.125 m perchloric acid and 5 ml of water. The filter was then processed for determination of radioactivity by scintillation counting.
Ca2+ binding in the absence of ATP was measured at 30 °C temperature in a reaction mixture containing 50 mm MOPS, pH 7.0, 80 mm KCl, 2 mm MgCl2, 2–3 μm (45Ca)Ca2+, 5 μm A23187, and 10 μg of microsomal protein/ml. Total calcium was measured by atomic absorption spectroscopy or titration with EGTA in the presence of murexide. After equilibration for 10 min, the samples were loaded onto a 0.45-μm Millipore Filter, which were then blotted and processed for determination of radioactivity by scintillation counting. Control samples were obtained by the addition of 5 mm EGTA for chelation of free Ca2+.
Electrical Measurements
Presteady state measurements of charge displacement were performed after adsorption of vesicles containing the protein onto a surface-modified gold electrode (the solid supported membrane (SSM)) and activation by a fast concentration jump of an appropriate substrate (25, 26). If at least one electrogenic step is involved in the relaxation process after protein activation, a current transient can be observed. It should be pointed out that the SSM technique detects presteady state charge movements within the first transport cycle and is not sensitive to stationary currents after the first cycle. Further experimental details were published previously (27).
For ATP concentration jumps, the non-activating solution was composed of 150 mm choline chloride, 25 mm MOPS, pH 7.0, 0.25 mm EGTA, 1 mm MgCl2, 0.2 mm DTT, and 0.25 mm CaCl2 (10 μm free Ca2+). The activating solution also contained 100 μm ATP. Moreover, 1 μm A23187 was used to prevent Ca2+ accumulation into the sarcoplasmic reticulum vesicles.
The temperature was maintained at 22–23 °C for all the experiments. All measurements were carried out using the SURFE2ROne device by Scientific Devices Heidelberg (Heidelberg, Germany).
Data Analysis
To determine the half-inhibition constants of the compounds reported in Table 1, ATPase activity data were fitted by a sigmoid equation (Hill-type function),
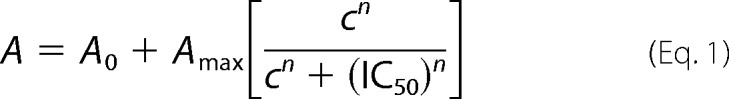 |
where A0 is the initial activity measured in the absence of the drug, Amax is the maximum variation of ATPase activity at highest drug concentration, c represents the drug concentration, and n is the Hill coefficient. IC50 represents the drug concentration producing half-maximal inhibition of steady state ATPase activity. Where indicated, the activity or charge were normalized with respect to the corresponding value measured in the absence of the drug.
RESULTS
Inhibition of Steady State Ca2+-ATPase Activity
Initial screening of CLT and of the newly synthesized 4-aminoquinoline derivatives was obtained by measuring steady state Pi production by the Ca2+-ATPase, with ATP as a substrate at 37 °C and in the presence of increasing drug concentrations. These measurements yielded the drug concentration producing 50% inhibition, which is here referred to as IC50.
Observed IC50 values ranged from 1.3 to 150 μm, as reported in Table 1, indicating that most of these compounds retain inhibitory activity with medium affinity as previously observed with CLT (11). On the other hand, NF1442 and NF1058 have higher apparent affinity for SERCA, with 1.3 ± 0.2 and 8 ± 2 μm IC50, respectively. These compounds are structurally related, although NF1442 presents a 4-piperazino-7-chloroquinoline moiety in place of the pyrrolidine ring of NF1058. Interestingly, even though NF1442 produced inhibition of ATPase hydrolytic activity at a lower concentration than NF1058 (Fig. 2), the 4-piperazino-7-chloroquinoline moiety alone (NF1610) produced no inhibition of steady state ATPase hydrolytic activity (Table 1 and Fig. 2). This suggests that the 4-piperazino-7-chloroquinoline moiety contributes to binding of NF1442 to the ATPase protein but is not directly involved in the inhibitory mechanism. Due to limited aqueous solubility of NF1442 with respect to NF1058, more detailed experiments were performed using NF1058. Nevertheless, concentrated (50 mm) stock solutions of NF1058 were best obtained with ethanol as a solvent.
FIGURE 2.

Hydrolytic activity measurements. Normalized hydrolytic activity of the SERCA pump in the presence of NF1442 (filled circles), NF1610 (empty circles), and NF1058 (empty triangles). Solid and dashed lines represent best fittings to the experimental data obtained for NF1442 and NF1058, respectively. The kinetic constants are IC50 = 1.3 ± 0.2 μm, n = 1.1 ± 0.2 (solid line) and IC50 = 8 ± 2 μm, n = 0.9 ± 0.2 (dashed line). Error bars are the S.D.
It is worth noting that steady state ATPase activity sustained by our preparation derived from skeletal muscle includes a main component that is inhibited by TG, accounts for ∼90% of the total activity, and is attributed specifically to SERCA. The remaining 10% is referred to as “TG-insensitive.” We found that this low level of TG-insensitive ATPase was not inhibited by NF1058 at 20 and 50 μm concentrations but was inhibited ∼35% by 100 μm NF1058 (not shown).
Experiments conducted by preincubation of protein with NF1058 followed by a 10-fold dilution with medium containing or not containing NF1058 demonstrated reversibility of the inhibitory effect within the minute time scale, consistent with kinetic constants of drug association and dissociation to yield IC50 in the μm range (not shown).
Measurements of Charge Transfer
Further characterization of the effect of NF1058 was obtained by presteady state charge measurements with microsomes (containing SERCA) adsorbed onto a SSM and subjected to ATP jumps in the presence of saturating Ca2+. In these experiments a current transient was observed after the addition of 100 μm ATP in the presence of 10 μm free Ca2+ (Fig. 3A), and the charge obtained by numerical integration of the transient is attributed to an electrogenic event corresponding to vectorial displacement of bound Ca2+ at the end of the first cycle after utilization of ATP (26). Although mixed orientation of microsomal membranes may underestimate the charge movement, this technique provides a unique method for direct measurements of charge transfer within the first catalytic cycle.
FIGURE 3.

Charge measurements in the presence of NF1058. Panel A, current transients after ATP jumps in the presence of Ca2+ ions were recorded in the absence (solid line) and in the presence of 100 μm NF1058 (dashed line). Before ATP was added, the sarcoplasmic reticulum vesicles were incubated with NF1058 for 3 min. The ATP concentration jump occurs at time zero. Panel B, normalized charges related to ATP jumps in the presence of Ca2+ ions (full squares) as a function of NF1058 concentration. The solid line represents the fitting curve to the ATP-induced charges (IC50 = 52 ± 9 μm; n = 1.1 ± 0.2). The error bars represent S.E. of four independent measurements.
We found that NF1058, added to the same SSM prepared for control measurements, decreases the peak current obtained after the ATP jump (Fig. 3A). The concentration dependence of this effect (Fig. 3B) is slightly higher than observed for inhibition of steady state ATPase activity. This is likely due to partitioning of the drug in the lipid phase of the SSM and adsorbed vesicles, thereby decreasing the effective drug concentration interacting directly with the ATPase protein. On the other hand, the decay time constant of the residual signal in the presence of NF1058 concentrations producing partial inhibition was identical (25 ± 3 ms) to that observed in the absence of the drug (24 ± 2 ms). Therefore, the charge displacement measurements reflect a concentration-dependent inhibition by NF1058 whereby the observed charge transfer corresponds to single cycles sustained by the ATPase molecules not yet affected by the drug.
Characterization of Partial Reactions
Calcium Binding
Activation of SERCA requires initial binding of two calcium ions per ATPase molecule (28) that can be measured after equilibration of ATPase protein with Ca2+ in the absence of ATP. Under these conditions we observed a significant reduction of the equilibrium level of calcium binding in the presence of NF1058 concentrations that also produced inhibition of steady state ATPase activity (Fig. 4). It is noteworthy that the same NF1058 concentration produced stronger inhibition of steady state ATPase activity than of calcium binding (Fig. 4). Similar effects were obtained with CLT.
FIGURE 4.

Calcium binding and ATPase activity. Calcium binding (panel A) was measured under equilibrium conditions in the absence of ATP. ATPase activity (panel B) was measured under steady state conditions in the presence of 1 mm ATP and 8 μm free Ca2+ and reported in the graph as calcium-dependent activity. The temperature was 30 °C. See “Experimental Procedures” for details. Black columns indicate control measurements in the absence of drugs. White columns were obtained in the presence of 20, 50, and 100 μm NF1058, gray columns were obtained in the presence of 20 and 50 μm CLT, and dark gray columns were obtained in the presence of 0.2 and 1.0 μm TG. Error bars represent the S.D. of four independent measurements.
The equilibrium binding measurements were performed in the presence of 2–3 μm free Ca2+, a concentration that allows full saturation of the high affinity Ca2+ sites. In fact, the average level of Ca2+ binding obtained in the absence of drug was 8.2 nmol/mg of microsomal protein, corresponding to 2 mol of Ca2+/mol of ATPase (based on the ATPase stoichiometry present in the microsomes used). Because catalytic activation is produced by the second calcium (28), our experiments suggest that inhibition by NF1058 interferes preferentially with binding of the second calcium, which is an absolute requirement for catalytic activation. In this case, a fraction of ATPase molecules may still bind the first calcium even though their catalytic activation is interfered with by defective binding of the second calcium.
In comparative experiments (Fig. 4, A and B) we found that very low concentrations of the highly potent SERCA inhibitor TG produced total inhibition of ATPase activity and interfered with binding of both (i.e. first and second) calcium ions that are expected to bind to each ATPase molecule (Fig. 4, A and B).
Phosphoenzyme Formed by Utilization of ATP
The first intermediate step of the Ca2+-ATPase cycle after the addition of calcium and ATP to the enzyme is formation of a phosphorylated enzyme intermediate by transfer of the ATP terminal phosphate to an aspartyl residue conserved in all cation transport P-type ATPases (2, 3). When we added calcium and ATP to sarcoplasmic reticulum vesicles preincubated with EGTA (to chelate contaminant Ca2+), we obtained a good time resolution of phosphoenzyme formation (Fig. 5), reaching a steady state level determined by its rates of formation and subsequent hydrolytic cleavage. We then found that Ca2+-dependent formation of phosphoenzyme intermediate was strongly inhibited by NF1058 (Fig. 5).
FIGURE 5.

Phosphoenzyme formation by utilization of ATP. The reaction was started by the addition of 10 μm [γ-32P]ATP and 0.95 mm CaCl2 to protein preincubated with 1 mm EGTA to yield 7 μm free Ca2+. The temperature was 30 °C. See the “Experimental Procedures” for details. Filled circles are controls in the absence of drug. Empty circles were obtained in the presence of 50 μm NF1058.
Phosphoenzyme Formed by Utilization of Pi
The final hydrolytic step of the ATPase cycle can be demonstrated in reverse direction (E2 + Pi ↔ E2-P) by reacting the ATPase with [γ-32P]Pi in the absence of Ca2+ (29, 30) whereby equilibrium levels of phosphoenzyme are obtained after a few minutes of incubation at 30 °C temperature. It is shown in Fig. 6 that NF1058, even at concentrations producing total inhibition of steady state ATPase activity, produced no significant reduction of the phosphoenzyme level obtained by utilization of Pi in the absence of Ca2+. A similar behavior was observed with CLT (Fig. 6). On the contrary, TG produced strong inhibition of ATPase phosphorylation with Pi (Fig. 6) even though this reaction did not require Ca2+.
FIGURE 6.

Phosphoenzyme formation by utilization of Pi. [γ-32P]Phosphoenzyme was obtained by 2 min of incubation with 0.5 mm [γ-32P]Pi, 2 mm EGTA (to chelate contaminant Ca2+), and other medium components as specified under “Experimental Procedures.” The temperature 30 °C. The black column is a control in the absence of drugs. The values for the white columns were obtained in the presence of NF1058, for the gray columns were obtained in the presence of CLT, and for the dark gray columns were obtained in the presence of TG.
DISCUSSION
We screened several CLT and 4-aminoquinoline derivatives (Table 1) for their effects on the Ca2+-ATPase (SERCA1) activity of rabbit skeletal muscle microsomes. The initial screening revealed that the compounds more closely related to CLT, namely NF990 (17), NF1023 (17), and NF1041 (19), displayed SERCA inhibitory activity comparable to that of CLT (11), with IC50 values within the μm range. This is a lower potency when compared with the in vitro antiplasmodial activity of these compounds, which is in the nm range (with the exception of NF1023, which is in the μm range) (17, 19). The carbinol derivative NF1067 (19), lacking the 5-membered heterocycle, showed an even lower affinity for SERCA with respect to the previously mentioned compounds. On the other hand, NF1022 (18), a heterobivalent molecule that bears the pharmacophoric elements of both NF990 and the 4-aminoquinoline type antimalarials (such as chloroquine), displayed a SERCA inhibitory activity similar to that of other CLT-related structures (Table 1).
A significant increase of affinity for SERCA was observed for NF1058 (20) and even more for NF1442, which is the most active compound of the whole series (Table 1). Although compound NF1058 bears a pyrrolidine ring at the 4-position of the benzhydril system, this latter heterocycle is replaced in NF1442 by a 4-piperazino-7-chloroquinoline system that has no effect on SERCA hydrolytic activity if used alone (NF1610, Table 1 and Fig. 2). It is, therefore, clear that this moiety favors binding of the drug but has no inhibitory effect.
A possible relationship between calcium transport ATPases and drugs effective against plasmodium has been raised by a reported interference by P-type cation transport ATPase (PfATP4) mutations with the plasmodium toxicity of spiroindolones (31, 32). On the other hand, a possible effect of the antiplasmodium drug artemisinin on the Plasmodium falciparum Ca2+-ATPase PfATP6 has been disproved (33). We find that CLT, NF1058, and NF1442 inhibit SERCA with IC50 values of 35, 8, and 1.3 μm (Table 1), therefore, with much lower affinity as compare with the IC50 values for antiplasmodial activity (250, 65, and 10 nm, respectively; Refs. 24 and 25). The different potencies observed in our experiments indicate that a relation between the antiplasmodium and the SERCA inhibitory activities of CLT and 4-aminoquinoline derivatives is unlikely. It should also be noted that CLT, widely known as an antifungal drug, is endowed with several biochemical and pharmacological activities. It is a general inhibitor of P-type ATPases including SERCA (11), Na+,K+-ATPase (34), and H+,K+-ATPase (35), interacts with the Gardos channels (36), inhibits cytochromes (both fungal and human) and has also moderate antiplasmodial activity (37, 38). Therefore, CLT and its derivatives cannot be considered as specific SERCA inhibitors for a possible therapeutic use.
The most interesting observations resulting from our experiments are related to the specific and unique mechanism of SERCA inhibition by CLT and 4-aminoquinoline derivatives, which we characterized in greatest detail with NF1058. It is clear that SERCA inhibition is primarily due to interference with Ca2+ binding and the related catalytic activation. In fact, we find that formation of phosphorylated enzyme intermediate is strongly reduced (Fig. 5) by NF1058 when ATP and Ca2+ are added simultaneously to the enzyme preincubated with a Ca2+ chelator. On the contrary, Ca2+ independent phosphorylation of the same residue (Asp-351) by Pi is not inhibited by NF1058 (Fig. 6).
It is interesting that NF1058 reduces Ca2+ binding to a lesser extent than ATPase activity (Fig. 4). This may be explained considering that each ATPase molecule binds two Ca2+ ions, but catalytic activation follows binding of the second Ca2+, as demonstrated by the effects of site directed mutations and then shown by structural representation of the two sites (Fig. 7). In fact, single mutations of residues (Glu-309 or Asn-796) that are involved in binding the second calcium result in total ATPase inhibition (39, 40) even though calcium binding is only reduced by 50%.
FIGURE 7.

Functional effects of single residue mutations (left panel), related to structural representation (right panel) of their involvement in calcium binding. All mutations listed produce total ATPase inhibition. Mutations of residues involved in binding the first calcium interfere with binding of the second calcium and produce total ATPase inhibition. On the other hand, mutations of residues involved in binding of the second calcium allow binding of the first calcium but still produce total ATPase inhibition. This demonstrates that binding of the second calcium is required for enzyme activation (39, 40). The side chain positions of the binding residues are shown in the absence (yellow) and in the presence (blue) of calcium, as previously described (41, 42).
From the functional standpoint, these effects are clarified by the detailed reaction scheme used for the kinetic simulation shown in Fig. 8. The sequence of partial reactions included in the simulation is derived from the SERCA cycle shown in Fig. 1 but includes more detailed steps such as binding of the first Ca2+ (reaction A) and the consequent isomeric transition (reaction B) producing cooperative binding of the second Ca2+ (reaction C). A comparison of the simulations shown in the upper and lower panels of Fig. 8 shows that lowering the forward rate constants of reaction A from 107 to 106 m−1 s−1 and of reaction B from 120 to 1.2 s−1 reduces the ATPase velocity from 30.11 to 1.74 nmol/(mg of protein·s), whereas reduction of calcium binding involves mostly the second (E′CaCa) rather than the first calcium (ECa). Note also that the steady state phosphoenzyme level is reduced from 3.27 to 0.19 nmol/mg of protein even though the rates of phosphoenzyme formation (reaction E) and cleavage (reactions F to N) are not lowered. The simulations clearly demonstrate that reduction of the forward rate constants for the isomeric transition (reaction B) after binding of the first Ca2+ (reaction A), without changing any phosphorylation or dephosphorylation constant, is sufficient to obtain the results observed in our experiments.
FIGURE 8.

Steady state kinetic simulations. Shown is the reaction scheme used for computations of steady state velocities (nmol of Pi s−1) and intermediate states levels (nmol/mg of protein). Bidirectional rate constants (s−1) of each sequential step and concentrations (μm) of substrates and products used for the computations are given in the diagram. The number of ATPase sites is assumed to be 5 nmol/mg of microsomal protein, corresponding to the high concentration of ATPase protein in the microsomes obtained from skeletal muscle. The upper panel shows a control simulation with normal constants. For the simulation shown in the lower panel, the constants for reactions A and B were modified, resulting in reduced binding of the second calcium, a reduced phosphoenzyme intermediate level, and a reduced steady state ATPase velocity. A detailed description of this analysis and computational method was published previously (43, 44).
Further demonstration of the specific inhibitory mechanism targeting the Ca2+ activation mechanism is provided by the experiments on ATPase phosphorylation with Pi through reverse of the hydrolytic reaction. In fact, ATPase phosphorylation by Pi involving the same residue (Asp-351) phosphorylated by ATP does not require Ca2+ and is not affected at all by NF1058 and only slightly by CLT (Fig. 6). It is of interest, on the other hand, that the Pi reaction is inhibited by TG (Fig. 6) at the very low concentrations, which also inhibit Ca2+ binding and overall ATPase activity (Fig. 4, A and B). These experiments indicate that the TG-ATPase complex is inactive in all respects and does not correspond to a functional E2 state even though it may retain an E2-like conformation (41).
In conclusion, we demonstrate that CLT and 4-aminoquinoline derivatives inhibit SERCA1 with IC50 values in the μm range by an inhibitory mechanism that interferes specifically with ATPase activation by Ca2+ and results in stabilization of a functional E2 state with respect to its phosphorylation by Pi in the absence of Ca2+.
This work was supported, in whole or in part, by National Institutes of Health Grant RO301-69830 (NHBLI). This work was also supported by the Ministero dell'Istruzione, dell'Università e della Ricerca (PRIN2008), the Ente Cassa di Risparmio di Firenze, Monte dei Paschi di Siena and NatSynDrugs.
- SERCA
- sarco-endoplasmic reticulum Ca2+-ATPase
- SSM
- solid supported membrane
- CLT
- clotrimazole
- TG
- thapsigargin.
REFERENCES
- 1. Yatime L., Buch-Pedersen M. J., Musgaard M., Morth J. P., Lund Winther A. M., Pedersen B. P., Olesen C., Andersen J. P., Vilsen B., Schiøtt B., Palmgren M. G., Møller J. V., Nissen P., Fedosova N. (2009) Biochim. Biophys. Acta 1787, 207–220 [DOI] [PubMed] [Google Scholar]
- 2. Andersen J. P., Vilsen B. (1995) FEBS Lett. 359, 101–106 [DOI] [PubMed] [Google Scholar]
- 3. Inesi G., Toyoshima C. (2004) in Handbook of ATPases: Biochemistry, Cell Biology, Pathophysiology (Futai M., Wada Y., Kaplan J. H. eds) pp. 63–87, Wiley-VCH, Weinheim, Germany [Google Scholar]
- 4. de Meis L., Vianna A. L. (1979) Annu. Rev. Biochem. 48, 275–292 [DOI] [PubMed] [Google Scholar]
- 5. Michelangeli F., East J. M. (2011) Biochem. Soc. Trans. 39, 789–797 [DOI] [PubMed] [Google Scholar]
- 6. Lytton J., Westlin M., Hanley M. R. (1991) J. Biol. Chem. 266, 17067–17071 [PubMed] [Google Scholar]
- 7. Sagara Y., Inesi G. (1991) J. Biol. Chem. 266, 13503–13506 [PubMed] [Google Scholar]
- 8. Seidler N. W., Jona I., Vegh M., Martonosi A. (1989) J. Biol. Chem. 264, 17816–17823 [PubMed] [Google Scholar]
- 9. Moore G. A., McConkey D. J., Kass G. E., O'Brien P. J., Orrenius S. (1987) FEBS Lett. 224, 331–336 [DOI] [PubMed] [Google Scholar]
- 10. Takahashi M., Kondou Y., Toyoshima C. (2007) Proc. Natl. Acad. Sci. U.S.A. 104, 5800–5805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bartolommei G., Tadini-Buoninsegni F., Hua S., Moncelli M. R., Inesi G., Guidelli R. (2006) J. Biol. Chem. 281, 9547–9551 [DOI] [PubMed] [Google Scholar]
- 12. Bilmen J. G., Khan S. Z., Javed M. H., Michelangeli F. (2001) Eur. J. Biochem. 268, 6318–6327 [DOI] [PubMed] [Google Scholar]
- 13. Berman M. C., Karlish S. J. (2003) Biochemistry 42, 3556–3566 [DOI] [PubMed] [Google Scholar]
- 14. Denmeade S. R., Isaacs J. T. (2005) Cancer Biol. Ther 4, 14–22 [DOI] [PubMed] [Google Scholar]
- 15. Søhoel H., Jensen A. M., Møller J. V., Nissen P., Denmeade S. R., Isaacs J. T., Olsen C. E., Christensen S. B. (2006) Bioorg. Med. Chem. 14, 2810–2815 [DOI] [PubMed] [Google Scholar]
- 16. Avellanal M., Rodriguez P., Barrigon S. (1998) J. Cardiovasc. Pharmacol. 32, 845–851 [DOI] [PubMed] [Google Scholar]
- 17. Gemma S., Campiani G., Butini S., Kukreja G., Joshi B. P., Persico M., Catalanotti B., Novellino E., Fattorusso E., Nacci V., Savini L., Taramelli D., Basilico N., Morace G., Yardley V., Fattorusso C. (2007) J. Med. Chem. 50, 595–598 [DOI] [PubMed] [Google Scholar]
- 18. Gemma S., Kukreja G., Campiani G., Butini S., Bernetti M., Joshi B. P., Savini L., Basilico N., Taramelli D., Yardley V., Bertamino A., Novellino E., Persico M., Catalanotti B., Fattorusso C. (2007) Bioorg. Med. Chem. Lett. 17, 3535–3539 [DOI] [PubMed] [Google Scholar]
- 19. Gemma S., Campiani G., Butini S., Kukreja G., Coccone S. S., Joshi B. P., Persico M., Nacci V., Fiorini I., Novellino E., Fattorusso E., Taglialatela-Scafati O., Savini L., Taramelli D., Basilico N., Parapini S., Morace G., Yardley V., Croft S., Coletta M., Marini S., Fattorusso C. (2008) J. Med. Chem. 51, 1278–1294 [DOI] [PubMed] [Google Scholar]
- 20. Gemma S., Campiani G., Butini S., Joshi B. P., Kukreja G., Coccone S. S., Bernetti M., Persico M., Nacci V., Fiorini I., Novellino E., Taramelli D., Basilico N., Parapini S., Yardley V., Croft S., Keller-Maerki S., Rottmann M., Brun R., Coletta M., Marini S., Guiso G., Caccia S., Fattorusso C. (2009) J. Med. Chem. 52, 502–513 [DOI] [PubMed] [Google Scholar]
- 21. Eletr S., Inesi G. (1972) Biochim. Biophys. Acta 290, 178–185 [DOI] [PubMed] [Google Scholar]
- 22. Lowry O. H., Rosebrough N. J., Farr A. L., Randall R. J. (1951) J. Biol. Chem. 193, 265–275 [PubMed] [Google Scholar]
- 23. Lanzetta P. A., Alvarez L. J., Reinach P. S., Candia O. A. (1979) Anal. Biochem. 100, 95–97 [DOI] [PubMed] [Google Scholar]
- 24. Patton C., Thompson S., Epel D. (2004) Cell Calcium 35, 427–431 [DOI] [PubMed] [Google Scholar]
- 25. Pintschovius J., Fendler K. (1999) Biophys. J. 76, 814–826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tadini-Buoninsegni F., Bartolommei G., Moncelli M. R., Guidelli R., Inesi G. (2006) J. Biol. Chem. 281, 37720–37727 [DOI] [PubMed] [Google Scholar]
- 27. Tadini-Buoninsegni F., Bartolommei G., Moncelli M. R., Fendler K. (2008) Arch. Biochem. Biophys. 476, 75–86 [DOI] [PubMed] [Google Scholar]
- 28. Inesi G., Kurzmack M., Coan C., Lewis D. E. (1980) J. Biol. Chem. 255, 3025–3031 [PubMed] [Google Scholar]
- 29. de Meis L., Masuda H. (1974) Biochemistry 13, 2057–2062 [DOI] [PubMed] [Google Scholar]
- 30. Masuda H., de Meis L. (1973) Biochemistry 12, 4581–4585 [DOI] [PubMed] [Google Scholar]
- 31. Krishna S., Woodrow C., Webb R., Penny J., Takeyasu K., Kimura M., East J. M. (2001) J. Biol. Chem. 276, 10782–10787 [DOI] [PubMed] [Google Scholar]
- 32. Rottmann M., McNamara C., Yeung B. K., Lee M. C., Zou B., Russell B., Seitz P., Plouffe D. M., Dharia N. V., Tan J., Cohen S. B., Spencer K. R., González-Páez G. E., Lakshminarayana S. B., Goh A., Suwanarusk R., Jegla T., Schmitt E. K., Beck H. P., Brun R., Nosten F., Renia L., Dartois V., Keller T. H., Fidock D. A., Winzeler E. A., Diagana T. T. (2010) Science 329, 1175–1180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Arnou B., Montigny C., Morth J. P., Nissen P., Jaxel C., Møller J. V., Maire M. (2011) Biochem. Soc. Trans. 39, 823–831 [DOI] [PubMed] [Google Scholar]
- 34. Bartolommei G., Devaux N., Tadini-Buoninsegni F., Moncelli M., Apell H. J. (2008) Biophys. J. 95, 1813–1825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Witzke A., Lindner K., Munson K., Apell H. J. (2010) Biochemistry 49, 4524–4532 [DOI] [PubMed] [Google Scholar]
- 36. Brugnara C., Gee B., Armsby C. C., Kurth S., Sakamoto M., Rifai N., Alper S. L., Platt O. S. (1996) J. Clin. Investig. 97, 1227–1234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Saliba K. J., Kirk K. (1998) Trans. R. Soc. Trop. Med. Hyg. 92, 666–667 [DOI] [PubMed] [Google Scholar]
- 38. Tiffert T., Ginsburg H., Krugliak M., Elford B. C., Lew V. L. (2000) Proc. Natl. Acad. Sci. U.S.A. 97, 331–336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Inesi G., Ma H., Lewis D., Xu C. (2004) J. Biol. Chem. 279, 31629–31637 [DOI] [PubMed] [Google Scholar]
- 40. Strock C., Cavagna M., Peiffer W. E., Sumbilla C., Lewis D., Inesi G. (1998) J. Biol. Chem. 273, 15104–15109 [DOI] [PubMed] [Google Scholar]
- 41. Toyoshima C., Nomura H. (2002) Nature 418, 605–611 [DOI] [PubMed] [Google Scholar]
- 42. Toyoshima C., Nakasako M., Nomura H., Ogawa H. (2000) Nature 405, 647–655 [DOI] [PubMed] [Google Scholar]
- 43. Inesi G., Kurzmack M., Lewis D. (1988) Methods Enzymol. 157, 154–190 [DOI] [PubMed] [Google Scholar]
- 44. Yu X., Inesi G. (1995) J. Biol. Chem. 270, 4361–4367 [DOI] [PubMed] [Google Scholar]
- 45. Andrews S., Burgess S. J., Skaalrud D., Kelly J. X., Peyton D. H. (2010) J. Med. Chem. 53, 916–919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Campiani G. (August 28th, 2008), Italy patent WO2008/101891