

Abstract
Callophycin A was originally isolated from the red algae Callophycus oppositifolius and shown to mediate anticancer and cytotoxic effects. In our collaborative effort to identify potential chemopreventive and anticancer agents with enhanced potency and selectivity, we employed a tetrahydro-β-carboline-based template inspired by callophycin A for production of a chemical library. Utilizing a parallel synthetic approach, 50 various functionalized tetrahydro-β-carboline derivatives were prepared and assessed for activities related to cancer chemoprevention and cancer treatment: induction of quinone reductase 1 (QR1) and inhibition of aromatase, nitric oxide (NO) production, tumor necrosis factor (TNF)-α-induced NFκB activity, and MCF7 breast cancer cell proliferation. Biological results showed that the n-pentyl urea S-isomer 6a was the strongest inducer of QR1 with an induction ratio (IR) value of 4.9 at 50 μM [the concentration to double the activity (CD) = 3.8 μM] and its corresponding R-isomer 6f had an IR value of 4.3 (CD = 0.2 μM). The isobutyl carbamate derivative 3d with R stereochemistry demonstrated the most potent inhibitory activity of NFκB, with the half maximal inhibitory concentration (IC50) value of 4.8 μM, and also showed over 60% inhibition at 50 μM of NO production (IC50 = 2.8 μM). The R-isomer urea derivative 6j, having an appended adamantyl group, exhibited the most potent MCF7 cell proliferation inhibitory activity (IC50 = 14.7 μM). The S-isomer 12a of callophycin A showed the most potent activity in aromatase inhibition (IC50 = 10.5 μM).
Keywords: Callophycin A, Tetrahydro-β-carboline, Anticancer agent, Chemoprevention, Aromatase, NFκB, Quinone reductase 1, iNOS
1. Introduction
Tetrahydro-β-carbolines have been featured as important templates for many naturally occurring and synthetic compounds, which mediate a variety of biological activities.1-3 They have been characterized as a new class of orally bioavailable compounds for the potential treatment of malaria.4, 5 Some were reported as potential anti-thrombotic agents6, 7 as well as human papillomavirus antiinfective agents.8 More importantly for our research, several tetrahydro-β-carboline derivatives have also been investigated as anticancer agents.2,9
Callophycin A (Figure 1), a marine natural product displaying a tetrahydro-β-carboline scaffold, was first isolated from the red algae Callophycus oppositifolius, and shown to have antiproliferative effects on various human cancer cell lines at low micromolar concentrations.10 Although no selectivity between tumor and normal mammalian cells was noted, due to facile chemistry accessibility and a variety of potential sites for chemical derivatization, callophycin A offers a valuable chemical starting point for further investigation in an attempt to discover more selective and potent anticancer and chemopreventive agents. To that end, we have undertaken synthesis and biological evaluation of a chemical library of callophycin A derivatives.11 In this paper, the synthetic efforts that focused on the modifications of the 2- and 3-positions of callophycin A are described. Subsequently, a series of N2-substituted tetrahydro-β-carboline derivatives were synthesized and assessed in chemopreventive and anticancer target-based bioassays: induction of quinone reductase 1 (QR1) and inhibition of aromatase, nitric oxide (NO) production by inducible nitric oxide synthases (iNOS) in lipopolysaccharide (LPS)-stimulated RAW 264.7 murine macrophage cells, tumor necrosis factor (TNF)-α-induced NFκB activity, and MCF7 cell proliferation.
Figure 1.
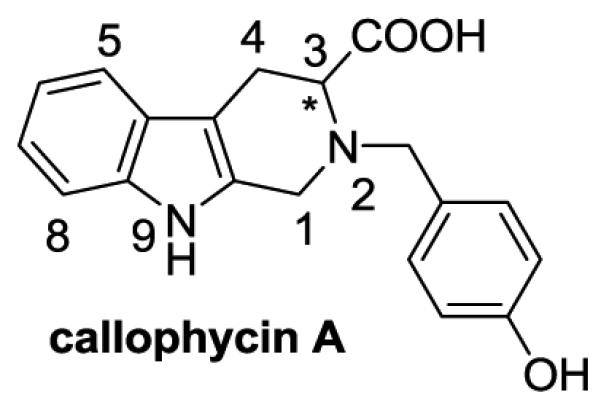
Chemical structure of callophycin A(MW: 322.36, calculated log P : 2.31).
More specifically, QR1 is categorized as a detoxifying enzyme which is responsible for the reduction of electrophilic quinones to their non-toxic hydroquinone forms.12 It is believed that QR1 elevation provides a reasonable biomarker for cancer chemoprevention.13 Aromatase catalyzes the conversion of androgen to estrogen in the late stage of female sex hormone biosynthesis, and it is found to be over expressed in breast cancer tissues.14, 15 Clinically, non-steroidal aromatase inhibitors such as letrozole and anastrozole represent an important class of therapeutic agents for the treatment of breast cancer and have shown their potential as chemopreventive agents.16-18 NO elevation has been shown to be associated with the early stages of cancer development, and inhibitors of NO production have been considered as cancer chemopreventive agents.19 In contrast, the role of NFκB in many cellular processes is well studied; it has been shown that NFκB is involved in inflammation, cell-cycle regulation, and apoptosis.20 Blocking of NFκB can stop cell proliferation and ultimately lead to apoptosis, thus, inhibitors of NFκB have been investigated as promising anticancer and cancer chemoprevention agents.20
Herein, we report our effort toward synthesis and biological evaluation of callophycin A and analogues as potential chemopreventive and anticancer agents; preliminary structure-activity relationships (SARs) are also discussed.
2. Results and Discussion
2.1. Chemistry
Design and synthesis of callophycin A-inspired tetrahydro-β-carboline derivatives 2-12, including callophycin A itself, are shown in Scheme 1. The design rationale of this focused carboline library was to explore the chemical diversity at the 2 and 3 positions of the tetrahydro-β-carboline template and to improve the poor solubility associated with 1a and 1b. Esterification of chiral isomer 1a or 1b21 with methanol in the presence of SOCl2 yielded the enantiomeric methyl ester 2a or 2b in excellent yields, respectively.22 The esters 2a-b were then subject to reaction with allyl or isopropyl chloroformates to afford a series of carbamate derivatives 3a-d in high yields (79-100%).
Scheme 1.

Synthesis of callophycin A analogues 2-12. Reagents and conditions: (i) SOCl2, MeOH, rt; (ii) R1OCOCl, Et3N, CH2Cl2, rt; (iii) R2SO2Cl, Et3N, CH2Cl2, rt; (iv) RM3Br or 4-BnOBnBr, DIPEA, CH3CN, reflux; (v) R4NCO, CH2Cl2, rt; (vi) LiOH, THF/H2O (1:1), rt; (vii) 10% Pd/C, H2, MeOH, rt, 4.5h.
Carboline sulfonamides 4a-f were obtained by reacting 2a or 2b with various sulfonyl chlorides in the presence of triethylamine at room temperature. The reactions of 2a or 2b with 4-bromobenzenesulfonyl chloride or p-toluenesulfonyl chloride proceeded smoothly to completion in 40 h, providing 4a-b and 4d-e in good yields (78-100%). The reactions with methanesulfonyl chloride occurred much faster and went to completion in 0.5 h to give 4c and 4f in 100% and 93% yield, respectively. The N-alkylation was carried out by the method reported by Cook’s group.23 A reaction mixture of secondary amine 2a or 2b and various bromides in acetonitrile was heated at reflux for 2-5 h in the presence of N,N-diisopropylethylamine (DIPEA, Hunig’s base). Subsequent purification by flash column chromatography on silica gel gave the alkylation products 5a-f in 62-84% yields. The urea derivatives 6a-j were prepared by reaction of 2a or 2b with an array of substituted isocyanates in dichloromethane at room temperature in > 88% yields.
Compounds 3-5 were further hydrolyzed to the corresponding free acids 7-9 in the presence of LiOH in aqueous THF at room temperature. These hydrolysis reactions were quite clean based on HPLC monitoring and products 7-9 were subsequently isolated in 39-84% yields. However, hydrolysis of carboline urea derivatives 6 in basic conditions led to the facile formation of intramolecular cyclized hydantoin byproduct.24,25
Finally, the first total synthesis of callophycin A started with the N-alkylation of 2a, b with 4-(benzyloxy)benzyl bromide26. The experiments were conducted under the same alkylation conditions as those of 5a-f, to give 10a, b in 58% and 61% yields, respectively, followed by O-debenzylation (H2, Pd/C) to afford 11a, b.27 Enantiomeric callophycin A (12a, b) were then obtained after ester hydrolysis under the same reaction conditions as in the preparation of 7-9. The mass spectrometric and NMR (1H, 13C NMR, HSQC, HMBC, and COSY) spectroscopic data of 12a and 12b were in good agreement with those of the reported natural callophycin A.10
All compounds were characterized by 1H, 13C NMR, and mass spectrometry.28 Purity was determined by reverse-phase C18 HPLC with UV monitoring at 254 and 220 nm. Interestingly, the coupling and splitting patterns of the 1H NMR spectra of carbamates 3 differred from those of sulfonamides 4, alkylation products 5, and urea derivatives 6 in that they were more complex and displayed two sets of signals for most of the protons at room temperature (24 °C). This phenomenon indicates that 3 exists as a mixture of rotamers due to restricted rotation of the carbamate side chain.29 To confirm this deduction, temperature-dependent NMR studies of 3d were undertaken. The 1H NMR spectra of 3d in DMSO-d6 were recorded at 24, 40, and 90 °C. For clarity, only four sets of proton signals of 3d are shown in Figure 2 to demonstrate the influence of temperature on the four sets of resonances shown. These signals include: i) 5.30-5.26 (Ha), ii) 4.86-4.76 (Hb), iii) 4.52-4.36 (Hc), and iv) 3.99-3.82 (Hd). As illustrated in Figure 2, all these signals were doubled at 24 °C. When the sample temperature was increased to 40 °C, the two sets of signals for Ha, Hb, and Hd started to merge. At 90 °C, all signals coalesced. As rotamers were only observed for the carbamates, it was concluded that the phenomenon is possibly caused by an interaction, probably a weak hydrogen bond, between the carbamate oxygen and Ha that resulted in restricted rotation around the N-CO(O) bond, giving rise to two distinct sets of NMR signals at normal room temperatures.
Figure 2.

1H NMR spectra of 3d at 24, 40, and 90 °C.
To evaluate the possible racemization in the final step of basic hydrolysis, we performed chiral HPLC analysis for the targeted compounds to determine their enantiomeric purity. For comparison, the ester derivatives were also evaluated. Ten pairs of enantiomers were randomly selected for analysis, and the chiral HPLC analysis data are given in Table 1. In all cases, good to excellent separations were achieved. As expected, no racemization occurred for all the evaluated ester compounds (entries 1-8). Among the free carboxylic acid derivatives analyzed, no racemization took place and clean chiral HPLC profiles were obtained for the alkylation hydrolysis products (entries 17-20, Table 1). However, for the sulfonamide series (entries 11-15), approximately 20-40% of partial racemization ocurred in the hydrolysis process due to increased acidity originating from the electron withdrawing effect of the sulfonamide group. The representative chiral HPLC chromatographs are illustrated in Figure 3.
Table 1.
Chiral HPLC analysis of selected ester and targeted compoundsa
| Entry | Compd | Config. | Mobile phases (hexanes : IPA)b |
Enantiomeric purity (%) |
tR (min) |
|---|---|---|---|---|---|
| 1 | 2a | S- | 80 : 20 | 100.0 | 13.72 |
| 2 | 2b | R- | 80 : 20 | 100.0 | 13.06 |
| 3 | 4b | S- | 60 : 40 | 100.0 | 9.94 |
| 4 | 4e | R- | 60 : 40 | 100.0 | 22.03 |
| 5 | 6a | S- | 80 : 20 | 96.0 | 15.84 |
| 6 | 6f | R- | 80 : 20 | 89.3 | 11.19 |
| 7 | 6d | S- | 70 : 30 | 99.3 | 18.46 |
| 8 | 6i | R- | 70 : 30 | 97.8 | 9.11 |
| 9 | 7b | S- | 80 : 20 | 92.4 | 7.67 |
| 10 | 7d | R- | 80 : 20 | 98.9 | 8.36 |
| 11 | 8a | S- | 60 : 40 | 72.4c | 11.22 |
| 12 | 8d | R- | 60 : 40 | 79.3c | 12.68 |
| 13 | 8b | S- | 50 : 50 | 78.5c | 11.22 |
| 14 | 8e | R- | 50 : 50 | 82.5c | 13.61 |
| 15 | 8c | S- | 60 : 40 | 62.7c | 13.61 |
| 16 | 8f | R- | 60 : 40 | 100.0 | 14.47 |
| 17 | 9a | S- | 60 : 40 | 100.0 | 10.79 |
| 18 | 9d | R- | 60 : 40 | 100.0 | 12.25 |
| 19 | 12a | S- | 40 : 60 | 100.0 | 11.21 |
| 20 | 12b | R- | 40 : 60 | 100.0 | 11.19 |
Chiral HPLC analysis was performed on a Lux 5 μm Amylose-2 analytical column (250 × 4.6 mm, Phenomenex) at ambient temperature using isopropanol (IPA) in hexanes as mobile phase at a flow rate of 1.0 mL/min. For entries 1-2, 6 μL of 0.5 mg/mL of sample in IPA was injected. For entries 3-20, 3 μL of 1 mg/mL of sample in IPA was injected. UV absorbance was measured at 220 nm.
0.1% of formic acid was added in mobile phases except entries 1-2.
Approximately 20-40% of partial racemization in the sulfonamide series 8 was observed.
Figure 3.

Representative chiral HPLC chromatographs: (A) enantiomeric urea derivatives 6d, i; (B) enantiomeric alkylation derivatives 9a, d.
2.2. Biological activities
Biological evaluation of tetrahydro-β-carboline derivatives 1-12 was undertaken in a series of bioassays related to cancer chemoprevention and cancer treatment. An initial screen for activity was performed at 50 μM. If a compound exhibited more than 50% inhibition (or 2.0 IR, for QR1) at this concentration, further testing was then performed for the determination of an IC50 (or CD) value. In the case of the evaluation of antiproliferative effects using a sulforhodamine B (SRB) assay, compounds which exhibited less than 50% survival are considered active, and IC50 values were determined. Results are summarized in Table 2.
Table 2.
Biological activities of compounds 1-12 evaluated in cancer chemopreventive and anticancer assaysa
| Comp. | R | Conf ig. |
QR1 | Aromatase | Nitrite assay | NFκB | SRB assay | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| IRb | % surv.c |
CDd (μM) |
% inhib.e |
IC50 (μM) |
% inhib.e |
% surv.c |
IC50 (μM) |
% inhib.e |
% surv.c |
IC50 (μM) |
% surv.c |
IC50 (μM) |
|||
| 1a | - | S- | 1.1 | 99.2 | 20.7 | 2.0 | 100.1 | 4.6 | 103.0 | ||||||
| 1b | - | R- | 0.8 | 90.7 | 34.9 | 3.5 | 95.9 | −72.5 | 95.4 | ||||||
| 2a | - | S- | 1.1 | 124.9 | 18.3 | 12.5 | 104.1 | 58.3 | 38.6 | 38.9 | |||||
| 2b | - | R- | 1.1 | 115.0 | 14.3 | 7.2 | 116.5 | 58.4 | 60.3 | 22.1 | |||||
| 3a | Allyl | S- | 1.9 | 83.4 | 54.6 | 44.8 | 8.5 | 94.2 | 33.3 | 191.2 | 70.4 | ||||
| 3b | Isobutyl | S- | 1.7 | 88.3 | 41.5 | 64.5 | 11.1 | 104.8 | 59.8 | 95.6 | 16.2 | 76.7 | |||
| 3c | Allyl | R- | 2.8 | 77.3 | 15.9 | 35.1 | 57.1 | 91.6 | 27.7 | 50.7 | 97.6 | 69.5 | |||
| 3d | Isobutyl | R- | 1.1 | 78.8 | 31.7 | 60.1 | 92.6 | 2.8 | 91.6 | 112.3 | 4.8 | 72.0 | |||
| 4a | 4-Bromophenyl | S- | 0.2 | 36.4 | 36.7 | 70.6 | 89.2 | 23.1 | 32.8 | 186.0 | −2.1 | 22.0 | |||
| 4b | 4-Methylphenyl | S- | 1.3 | 61.0 | 41.7 | 63.2 | 73.8 | 116.1 | 22.6 | 6.0 | 171.1 | 19.0 | 37.0 | ||
| 4c | Methyl | S- | 1.6 | 85.8 | 30.5 | 10.6 | 91.9 | 38.6 | 153.0 | 72.5 | |||||
| 4d | 4-Bromophenyl | R- | 2.1 | 83.7 | 29.7 | 35.5 | 3.8 | 89.7 | 43.7 | 154.3 | 30.6 | 42.1 | |||
| 4e | 4-Methylphenyl | R- | 1.3 | 85.8 | 28.2 | 7.4 | 91.4 | 25.2 | 114.0 | 86.5 | |||||
| 4f | Methyl | R- | 1.6 | 86.3 | 40.2 | 68.1 | 12.9 | 79.9 | 42.2 | 158.5 | 90.9 | ||||
| 5a | Benzyl | S- | 1.6 | 100.5 | 31.5 | 68.2 | 85.9 | 32.0 | 60.4 | 28.8 | 30.7 | ||||
| 5b | Cyclopropylmethyl | S- | 2.2 | 68.6 | 34.0 | 47.7 | 58.6 | 23.5 | 99.8 | 54.1 | 152.2 | 85.6 | |||
| 5c | Propargyl | S- | 1.7 | 89.6 | 24.4 | 22.3 | 84.4 | 21.1 | 116.8 | 81.1 | |||||
| 5d | Benzyl | R- | 1.2 | 96.8 | 38.4 | 74.7 | 81.2 | 32.9 | 54.4 | 77.1 | 44.2 | ||||
| 5e | Cyclopropylmethyl | R- | 1.8 | 81.8 | 37.9 | 5.6 | 124.5 | 29.9 | 253.9 | 101.9 | |||||
| 5f | Propargyl | R- | 2.0 | 77.2 | 50.0 | 31.7 | 41.1 | 114.3 | 43.6 | 226.9 | 77.8 | ||||
| 6a | n-Pentyl | S- | 4.9 | 7.8 | 3.8 | −0.04 | 8.7 | 94.9 | 35.5 | 109.5 | 40.3 | 38.4 | |||
| 6b | Isopropyl | S- | 2.9 | 61.2 | 6.4 | 54.0 | 47.3 | 37.8 | 94.3 | 69.0 | 116.8 | 26.5 | 71.0 | ||
| 6c | Phenethyl | S- | 0.5 | 59.6 | 31.8 | 48.2 | 68.9 | 87.7 | 94.6 | 16.9 | 1.3 | 25.4 | |||
| 6d | Cyclohexyl | S- | 1.1 | 71.5 | 33.0 | 53.8 | 79.0 | 41.1 | 44.7 | 111.2 | 26.3 | 35.8 | |||
| 6e | 1-Adamantyl | S- | 0.9 | 78.1 | 30.3 | 64.1 | 66.7 | 13.4 | 60.7 | 139.0 | 10.2 | 37.6 | 36.9 | ||
| 6f | n-Pentyl | R- | 4.3 | 12.7 | 0.2 | 11.1 | 36.2 | 90.5 | 62.1 | 140.2 | 15.9 | 50.5 | |||
| 6g | Isopropyl | R- | 2.7 | 77.0 | 8.6 | 32.6 | 16.2 | 89.4 | 73.7 | 142.8 | 21.2 | 66.1 | |||
| 6h | Phenethyl | R- | 1.5 | 51.7 | 24.6 | 66.2 | 66.1 | 11.0 | 81.7 | 108.2 | 13.3 | 4.3 | 27.7 | ||
| 6i | Cyclohexyl | R- | 1.0 | 71.8 | 24.4 | 55.0 | 83.8 | 41.6 | 20.9 | 100.6 | 5.3 | 18.4 | |||
| 6j | 1-Adamantyl | R- | 0.4 | 30.6 | 39.9 | 64.7 | 63.2 | 11.7 | 36.5 | 110.3 | 9.9 | 14.7 | |||
| 7a | Allyl | S- | 2.4 | 82.9 | 26.5 | 38.4 | −6.3 | 134.9 | 35.6 | 109.1 | 107.9 | ||||
| 7b | Isobutyl | S- | 1.8 | 94.2 | 40.0 | 72.0 | −3.4 | 137.3 | 42.6 | 137.8 | 104.6 | ||||
| 7c | Allyl | R- | 1.9 | 93.8 | 32.9 | −4.0 | 134.5 | 49.4 | 203.7 | 101.5 | |||||
| 7d | Isobutyl | R- | 1.5 | 87.5 | 36.3 | 1.5 | 134.4 | 46.1 | 264.3 | 97.4 | |||||
| 8a | 4-Bromophenyl | S- | 1.3 | 97.8 | 23.2 | 50.0 | 80.6 | 49.7 | 55.4 | 246.8 | 77.0 | ||||
| 8b | 4-Methylphenyl | S- | 1.4 | 88.8 | 16.9 | 7.0 | 85.5 | 42.5 | 271.9 | 96.5 | |||||
| 8c | Methyl | S- | 1.1 | 96.7 | 26.5 | 6.4 | 94.2 | 51.1 | 255.6 | 96.2 | |||||
| 8d | 4-Bromophenyl | R- | 1.3 | 88.3 | 32.3 | 56.2 | 102.6 | 46.1 | 50.9 | 251.9 | 91.2 | ||||
| 8e | 4-Methylphenyl | R- | 1.2 | 93.8 | 23.0 | 2.9 | 102.3 | 26.0 | 194.8 | 99.8 | |||||
| 8f | Methyl | R- | 1.9 | 80.6 | 30.7 | 5.0 | 93.1 | −2.3 | 178.4 | 88.7 | |||||
| 9a | Benzyl | S- | 2.4 | 105.2 | 8.1 | 28.0 | 6.9 | 119.4 | 59.5 | 49.1 | 21.4 | ||||
| 9b | Cyclopropylmethyl | S- | 2.4 | 90.7 | 27.6 | 30.2 | −7.0 | 134.6 | 33.7 | 234.1 | 108.4 | ||||
| 9c | Propargyl | S- | 2.2 | 91.9 | 18.7 | 30.5 | 7.0 | 126.5 | 50.2 | 228.1 | 111.6 | ||||
| 9d | Benzyl | R- | 1.8 | 107.5 | 19.2 | 9.9 | 108.9 | 41.8 | 89.5 | ||||||
| 9e | Cyclopropylmethyl | R- | 1.8 | 101.6 | 31.5 | −10.3 | 129.8 | 48.4 | 168.9 | 113.4 | |||||
| 9f | Propargyl | R- | 1.6 | 94.6 | 23.5 | 16.4 | 87.9 | −7.7 | 72.7 | 106.6 | |||||
| 10a | - | S- | 1.8 | 72.8 | 28.0 | 58.0 | 93.9 | 39.2 | 24.7 | 99.7 | 99.4 | ||||
| 10b | - | R- | 1.4 | 85.4 | 27.3 | 75.9 | 86.8 | 29.4 | 40.5 | 91.0 | 96.7 | ||||
| 11a | - | S- | 1.3 | 91.7 | 46.1 | 57.4 | 62.1 | 75.5 | 38.8 | 47.8 | 96.0 | 94.7 | |||
| 11b | - | R- | 1.8 | 98.6 | 38.7 | 63.7 | 87.3 | 37.5 | 33.5 | 122.0 | 100.3 | ||||
|
12a (callophycin A) |
- | S- | 1.7 | 88.3 | 57.9 | 10.5 | 16.0 | 90.3 | 55.0 | 102.4 | 85.2 | ||||
|
12b (callophycin A) |
- | R- | 1.5 | 91.2 | 38.4 | 27.8 | 94.7 | 54.6 | 132.9 | 71.1 | |||||
|
| |||||||||||||||
| Standard Controlf |
0.01 | 0.23 | 22.1 | 4.9 | 0.056 | ||||||||||
Concentration for testing: 50 μM.
IR: induction ratio.
Percentage of cell survival in comparison with vehicle-treated controls.
CD is the concentration that doubles the activity. CD values were determined for compounds with IR > 2.
Percentage of inhibition in comparison with vehicle-treated controls.
IC50 or CD values of known bioactive compounds for the each assay; 4′-bromoflavone for QR1, naringenin for aromatase, N-tosyl-L-phenylalaninechloromethyl ketone for NFκB assay, L-NG-monomethyl arginine for nitrite assay, and camptothecin for SRB assay.
QR1 induction activity
The effect of compounds 1-12 on QR1 induction was tested as described in the experimental section. As shown in Table 2, the two isomers of N2-unsubstituted β-carboline 2a and 2b showed the same QR1 induction activity with an IR value of 1.1. Among the carbamate derivatives 3a-d, allyl substituted R-isomer 3c demonstrated higher activity (IR = 2.8, CD = 15.9 μM). There were no differences observed between the two chiral isomers of 4-methylphenyl and methyl substituted sulfonamide derivatives (4b and 4e, IR = 1.3; 4c and 4f, IR = 1.6). In contrast, the 4-bromophenyl substituted R-isomer 4d (IR = 2.1, CD = 29.7 μM, 83.7% survival at 50 μM) was much more active and less cytotoxic than its corresponding S-isomer (4a, IR = 0.2, 36.4% survival at 50 μM). For the alkylation products 5a-f and 10-11, the S-isomers of benzyl 5a (IR = 1.6), cyclopropylmethyl substituted compound 5b (IR = 2.2, CD = 34.0 μM), and compound 10a (IR = 1.8) showed higher activity than their respective R-isomers 5d (IR = 1.2), 5e (IR = 1.8), and 10b (IR = 1.4). However, for the propargyl alkylation R-isomer product 5f and compound 11b, higher IR values (2.0 for 5f and 1.8 for 11b) were observed compared with their corresponding S-isomers 5c (IR = 1.7) and 11a (IR = 1.3). It was further noted that higher activities were observed for urea derivatives with an alkyl chain. For example, compounds 6b and 6g with the isopropyl chain showed almost equal activity with IR values of 2.9 and 2.7, respectively. More potent activity was observed for the urea derivative with a longer alkyl chain, and the compounds with an n-pentyl group proved to be the best inducers of QR1: the R-isomer 6f had an IR value of 4.3 and a CD value of 0.2 μM and its corresponding S-isomer 6a had an IR value of 4.9 (CD = 3.8 μM). Unfortunately, both compounds also demonstrated significant cytotoxicity (12.7% and 7.8% survival at 50 μM for 6f and 6a, respectively). On the basis of these data, there appears to be a linear correlation between QR1 induction and the length of alkyl side chains. In general, most compounds with a free carboxylic acid group showed lower QR1 induction activity than the corresponding ester compounds. Higher activity was observed for the allyl substituted carbamate carboxylic acid 7a, with an IR value of 2.4, than other derivatives 7b-d in this carbamate acid series. Overall, the alkylation free acid products 9a-f, 12a, b were more active, with IR values in the 1.5-2.4 range, compared with other acid compounds 8a-f.
Nitrite inhibition
The ability of carboline compounds 1-12 to inhibit NO production was assessed, results are given in Table 2. Stereoisomers of carbamate derivatives 3a-d and sulfonamide derivatives 4a-f showed different activities in the applied assay. In the case of carbamate derivatives, both allyl and isobutyl substituted R-isomers (3c and 3d) were more active than their corresponding S-isomers (3a and 3b). For instance, compound 3d with an isobutyl substituent showed much lower micromolar inhibition of NO production (IC50 = 2.8 μM) than the corresponding S-isomer 3b, 11.1% at 50 μM. For sulfonamide derivatives, S-isomers with a 4-bromophenyl substituent (4a, IC50 = 23.1 μM) and a 4-methylphenyl group (4b, IC50 = 22.6 μM) showed more potent activity than their corresponding R-isomers (4d and 4e, both less than 10% inhibition at 50 μM). Both methyl substituted isomers 4c (S) and 4f (R) exhibited low inhibition. These data demonstrated that substituted phenylsulfonamides with S stereochemistry were more effective inhibitors of NO production than their R and less bulky methylsulfonamide analogues. In the alkylation series, clearly, the pair of chiral isomers 5a (S) and 5d (R) with N-benzyl substitution showed more potent inhibitory activities with IC50 = 32.0 and 32.9 μM, respectively. The activity was maintained with further substitution on the benzyl group (10-11, IC50 = 29.4-39.2 μM). Nevertheless, compounds 5b-c and 5e-f with alkyl and less bulky substituents, such as cyclopropylmethyl and propargyl groups, showed less than 50% inhibition at 50 μM. In the cases of urea derivatives 6a-j, compounds with a larger substituent showed enhanced activities. For example, compounds 6d and 6i, each with a cyclohexyl group, showed better inhibition of NO production (IC50 = 41.1 and 41.6 μM, respectively) than n-pentyl and isopropyl derivatives (6a-b, 6f-g, inhibition < 50% at 50 μM). Further improved activity was observed for adamantyl urea derivatives 6e and 6j, which exhibited IC50 values of 13.4 and 11.7 μM, respectively. Most compounds with free carboxylic acid functionality displayed negligible inhibition, except that the 4-bromophenyl sulfonamide derivatives 8a and 8d demonstrated moderate inhibitions with IC50 values of 49.7 and 46.1 μM, respectively.
NFκB inhibitory activity
Most of the R-isomers were slightly more active than their corresponding S-isomers in the NFκB assay. The carbamate derivatives 3b and 3d with an isobutyl substituent showed significant inhibitory activity in this assay. The R-isomer 3d demonstrated the most potent inhibitory activity with an IC50 of 4.8 μM. Low activities were observed for the sulfonamide derivatives 4a-f (inhibition < 50% at 50 μM). For the alkylation products, compounds with a benzyl substituent (5a with IC50 = 30.7 μM and 5d with IC50 = 44.2 μM) exhibited higher inhibition than compounds with cyclopropylmethyl and propargyl groups (5b-c, 5e-f, 21.1-54.1% inhibition at 50 μM). Further substitution on the benzene ring resulted in lower activities (10, 11, inhibition < 50% at 50 μM). Most urea derivatives (6b-c, 6e-h) exhibited good activities in the NFκB inhibition assay. However, both isomers with a cyclohexyl group (6d with 44.7% and 6i with 20.9% inhibition at 50 μM) were significantly less potent than other urea derivatives (6b-c, 6e-h with IC50 values in the 10.2-26.5 μM range). All the synthesized carboline derivatives with a free acid, except for alkylation products 9a and callophycin A, demonstrated weak NFκB activity at 50 μM. The benzyl substituted alkylation S-isomer 9a showed inhibition of NFκB activity with an IC50 of 21.4 μM.
Inhibition of MCF7 cell proliferation
All carbamate derivatives (3a-d) and alkylation products (5a-f, 10-12) were inactive at 50 μM in this assay. For sulfonamide derivatives, S-isomers were more active than their corresponding R-isomers. Compound 4a with a 4-bromophenyl substituent exhibited good inhibition of MCF7 cell proliferation with an IC50 of 22.0 μM, while the R-isomer (4d) demonstrated an IC50 of 42.1 μM. The difference between the two isomers of the 4-methylphenyl substituted compound 4b and 4e was larger: the S-isomer inhibited the growth of MCF7 cells with an IC50 of 37.0 μM, while the R-isomer was inactive at 50 μM. Most of the urea derivatives, except 6b and 6g containing an isopropyl group, showed activities with IC50 values in the 14.7-38.4 μM range. The R-isomer 6j having an adamantyl group being the most potently active (IC50 = 14.7 μM). Compounds 7-9 and 12, all displaying a carboxylic acid group, showed no inhibition of MCF7 cell growth at 50 μM presumably due to poor cell membrane penetration. Notably, compared with the previously reported antiproliferative activity of natural callophycin A,10 our synthetic sample didn’t show the inhibitory activity in our MCF7 cell proliferation assay.
Aromatase inhibitory activity
For all compounds tested, the carbamate methyl ester 3a, urea methyl ester 6b, and callophycin A (S) 12a showed more than 50% inhibition at 50 μM in an aromatase inhibitory assay, with 12a being the most potent inhibitor (IC50 = 10.5 μM); all the other compounds were shown to be ineffective inhibitors of aromatase at 50 μM (Table 2).
3. Conclusion
In summary, a focused chemical library of 50 tetrahydro-β-carboline analogues inspired by callophycin A was designed, synthesized, and tested in a series of bioassays related to cancer chemoprevention and cancer treatment. This library yielded five compounds considered to have significant activity in one or more of the applied assays. The S-isomer of the carboline urea derivative 6a bearing an n-pentyl group showed significant QR1 induction with an IR of 4.9 at 50 μM (CD = 3.8 μM), the corresponding R-isomer 6f has an IR value of 4.3 (CD = 0.2 μM). The R-isomer of the carbamate derivative 3d having an isobutyl group was the most potent inhibitor of NFκB, with an IC50 = 4.8 μM. Good inhibition of NO production was also observed for 3d (IC50 = 2.8 μM). The most potent inhibitor of MCF7 cell proliferation was the R-isomer of the urea derivative containing an adamantyl group, 6j (IC50 = 14.7 μM). The S-isomer 12a of callophycin A demonstrated the most inhibitory activity of aromatase (IC50 = 10.5 μM). Based on these results, it is evident that these emerging callophycin A analogues will serve as important leads for further synthesis and optimization. Future synthetic efforts will focus on optimization of the methyl ester group at the C2 position to improve metabolic stability, and production of a series of N9 derivatives.
4. Experimental
4.1. Chemistry
All reagents and solvents obtained from commercial sources were used without further purification. Reactions were monitored either by thin-layer chromatography (TLC) or by analytical HPLC employing a Shimadzu LC-20A series high performance liquid chromatography (HPLC) system. TLC was performed using glass plates pre-coated with silica gel (0.25 mm, 60-Å pore size, 230-400 mesh, Sorbent Technologies, GA) impregnated with a fluorescent indicator (254 nm). TLC plates were visualized by exposure to ultraviolet light (UV). Hydrogenation reactions were done using domnick hunter NITROX UHP-60H hydrogen generator, USA. Flash column chromatography on silica gel was performed using a Biotage Isolera One system and a Biotage SNAP cartridge. Proton and carbon nuclear magnetic resonance (1H and 13C NMR) spectra were recorded employing a Bruker Avance DRX-400 spectrometer. Chemical shifts were expressed in ppm, J values were in Hz. ESI mass spectra in either positive or negative mode were recorded on a Varian 500-MS IT Mass Spectrometer. Compounds’ purity was determined by analytical HPLC using a Gemini, 3 μm, C18, 110Å column (50 mm × 4.6 mm, Phenomenex) and a flow rate of 1.0 mL/min. Gradient conditions: solvent A (0.1% trifluoroacetic acid in water) and solvent B (acetonitrile): 0-2.00 min 100% A, 2.00-7.00 min 0-100% B (linear gradient), 7.00-8.00 min 100% B, UV detection at 254 and 220 nm.
4.1.1. General procedure for compounds 2a-b 22
At 0 °C, thionyl chloride (2.1 mL, 24.1 mmol) was added dropwise to a solution of 1a or 1b (1 g, 98%, 4.6 mmol) in methanol (80 mL). The reaction mixture was stirred at room temperature for 3 days and completion of the reaction was monitored by HPLC. The excess methanol and thionyl chloride were removed by evaporation. The residue was dissolved in dichloromethane and washed successively with saturated Na2CO3 and NaCl solutions. The dried dichloromethane layer was evaporated and purified by flash column chromatography on silica gel to provide 2a or 2b.
(S)-Methyl 2,3,4,9-tetrahydro-1H-pyrido[3,4-b]indole-3-carboxylate (2a)
white powder. Yield: 91%. 1H NMR (DMSO-d6, 400 MHz): δ = 10.73 (1H, s, 9-NH), 7.38 (1H, d, J = 7.7 Hz, 5-H), 7.28 (1H, d, J = 7.9 Hz, 8-H), 7.02 (1H, t, J = 7.5 Hz, 7-H), 6.94 (1H, t, J = 7.4 Hz, 6-H), 3.97 (2H, ABq, J = 15.9 Hz, 1-H), 3.74 (1H, dd, J = 8.8 and 4.8 Hz, 3-H), 3.69 (3H, s, -COOCH3), 3.33 (1H, br s, 2-NH), 2.94 (1H, dd, J = 14.9 and 4.6 Hz, 4β-H), 2.75 (1H, dd, J = 15.0 and 8.8 Hz, 4α-H) ppm. 13C NMR (DMSO-d6, 100 MHz): δ = 174.1, 136.2, 133.9, 127.4, 120.9, 118.7, 117.7, 111.3, 106.0, 55.7, 52.1, 41.9, 25.5 ppm. ESI-MS: calc. for C13H15N2O2 [M+H]+: 231.1, found: 231.2. HPLC purity: 99.5% (254 nm), tR: 5.22 min; 99.5% (220 nm), tR: 5.22 min.
(R)-Methyl 2,3,4,9-tetrahydro-1H-pyrido[3,4-b]indole-3-carboxylate (2b)
white powder. Yield: 90%. 1H NMR (DMSO-d6, 400 MHz): δ = 10.73 (1H, s, 9-NH), 7.38 (1H, d, J = 7.7 Hz, 5-H), 7.27 (1H, d, J = 8.0 Hz, 8-H), 7.02 (1H, t, J = 7.5 Hz, 7-H), 6.94 (1H, t, J = 7.4 Hz, 6-H), 3.97 (2H, ABq, J = 15.9 Hz, 1-H), 3.74 (1H, dd, J = 8.7 and 4.8 Hz, 3-H), 3.68 (3H, s, -COOCH3), 3.34 (1H, br s, 2-NH), 2.94 (1H, dd, J = 15.0 and 4.5 Hz, 4β-H), 2.75 (1H, dd, J = 15.0 and 8.8 Hz, 4α-H) ppm. 13C NMR (DMSO-d6, 100 MHz): δ = 174.1, 136.2, 133.9, 127.4, 120.9, 118.7, 117.7, 111.3, 106.0, 55.7, 52.1, 41.9, 25.4 ppm. ESI-MS: calc. for C13H15N2O2 [M+H]+ : 231.1, found: 231.2. HPLC purity: 100% (254 nm), tR: 5.21 min; 99.2% (220 nm), tR: 5.21 min.
4.1.2. General procedure for compounds 3a-d
Triethylamine (29 μL, 0.21 mmol) was added to a solution of 2a or 2b (0.17 mmol) in dichloromethane (2 mL), followed by the addition of various chloroformates (0.21 mmol). The reaction mixture was stirred at room temperature for 0.5-1.5 h. After the reaction was complete (monitored by HPLC), the reaction mixture was evaporated. The residue was purified by flash column chromatography on silica gel to give products 3a-d.
(S)-2-Allyl 3-methyl 3,4-dihydro-1H-pyrido[3,4-b]indole-2,3(9H)-dicarboxylate (3a)
yellow viscous oil. Yield: 100%. 1H NMR (DMSO-d6, 400 MHz): a mixture of rotamers, δ = 10.92 and 10.87 (1H, s, 9-NH), 7.44 (1H, d, J = 7.8 Hz, 5-H), 7.31 (1H, d, J = 8.0 Hz, 8-H), 7.09-7.05 (1H, m, 7-H), 7.00-6.96 (1H, m, 6-H), 6.05-5.91 (1H, m, -CH2CH=CH2), 5.39-5.21 (3H, m, 3-H and -CH2CH=CH2), 4.86 and 4.81 (1H, d, J = 16.4 and 17.4 Hz, 1α-H), 4.71-4.63 (2H, m, -CH2CH=CH2), 4.51 and 4.39 (1H, d, J = 16.2 and 16.6 Hz, 1β-H), 3.58 (3H, s, -COOCH3), 3.33 (1H, d, J = 15.6 Hz, 4β-H), 3.09-3.01 (1H, m, 4α-H) ppm. 13C NMR (DMSO-d6, 100 MHz): δ = 172.0, 171.9, 156.1, 155.6, 136.7, 136.6, 133.6, 130.1, 130.0, 126.6, 121.6 (2C), 119.1, 118.2, 117.7, 117.6, 111.6, 104.7, 104.4, 66.4, 66.2, 53.6, 53.3, 52.9 (2C), 40.9, 40.7, 23.5, 23.2 ppm. ESI-MS: calc. for C17H17N2O4[M-H]-: 313.1, found: 313.3. HPLC purity: 99.8% (254 nm), tR: 6.82 min; 99.7% (220 nm), tR: 6.82 min.
(S)-2-Isobutyl 3-methyl 3,4-dihydro-1H-pyrido[3,4-b]indole-2,3(9H)-dicarboxylate (3b)
yellow viscous oil. Yield: 100%. 1H NMR (DMSO-d6, 400 MHz): a mixture of rotamers, δ = 10.91 and 10.87 (1H, s, 9-NH), 7.44 (1H, d, J = 7.7 Hz, 5-H), 7.30 (1H, d, J = 8.1 Hz, 8-H), 7.08-7.04 (1H, m, 7-H), 7.00-6.96 (1H, m, 6-H), 5.30 and 5.27 (1H, d, J = 5.3 and 5.4 Hz, 3-H), 4.84 and 4.78 (1H, d, J = 16.1 and 16.7Hz, 1α-H), 4.50 and 4.38 (1H, d, J = 16.2 and 16.6 Hz, 1β-H), 3.99-3.83 (2H, m, -CH2CH(CH3)2), 3.58 and 3.57 (3H, s, -COOCH3), 3.32 (1H, d, J = 16.0 Hz, 4β-H), 3.09-3.01 (1H, m, 4α-H), 2.01-1.86 (1H, m, -CH2CH(CH3)2), 0.96 and 0.90 (6H, d, J = 6.7 Hz; dd, J = 6.7 and 1.9 Hz, -CH2CH(CH3)2) ppm. 13C NMR (DMSO-d6, 100 MHz): δ = 172.1, 156.5, 156.1, 136.6 (2C), 130.2, 130.1, 126.6, 121.6 (2C), 119.1, 118.1, 111.6, 104.7, 104.3, 71.8 (2C), 53.5, 53.2, 52.9 (2C), 40.9, 28.0 (2C), 23.4, 23.2, 19.3(2C), 19.2(2C) ppm. ESI-MS: calc. for C18H21N2O4 [M-H]-: 329.2, found: 329.4. HPLC purity: 98.4% (254 nm), tR: 7.20 min; 98.4% (220 nm), tR: 7.19 min.
(R)-2-Allyl 3-methyl 3,4-dihydro-1H-pyrido[3,4-b]indole-2,3(9H)-dicarboxylate (3c)
yellow viscous oil. Yield: 79%. 1H NMR (DMSO-d6, 400 MHz): a mixture of rotamers, δ = 10.92 and 10.87 (1H, s, 9-NH), 7.44 (1H, d, J = 7.7 Hz, 5-H), 7.31 (1H, d, J = 8.0 Hz, 8-H), 7.08-7.05 (1H, m, 7-H), 7.00-6.96 (1H, m, 6-H), 6.05-5.88 (1H, m, -CH2CH=CH2), 5.39-5.21 (3H, m, 3-H and -CH2CH=CH2), 4.86 and 4.81 (1H, d, J = 16.4 and 17.3 Hz, 1α-H), 4.68-4.60 (2H, m, -CH2CH=CH2), 4.51 and 4.39 (1H, d, J = 16.2 and 16.2 Hz, 1β-H), 3.58 (3H, s, -COOCH3), 3.33 (1H, d, J = 15.6 Hz, 4β-H), 3.11-2.98 (1H, m, 4α-H) ppm. 13C NMR (DMSO-d6, 100 MHz): δ = 172.0, 156.1, 155.6, 136.7, 136.6, 133.6, 130.1, 130.0, 126.6, 121.6, 119.1, 118.2, 117.7, 117.6, 111.6, 104.7, 104.4, 66.4, 66.2, 53.6, 53.3, 52.9 (2C), 40.6, 40.4, 23.5, 23.2 ppm. ESI-MS: calc. for C17H17N2O4 [M-H]-: 313.1, found: 313.3. HPLC purity: 98.9% (254 nm), tR: 6.83 min; 98.7% (220 nm), tR: 6.84 min.
(R)-2-Isobutyl 3-methyl 3,4-dihydro-1H-pyrido[3,4-b]indole-2,3(9H)-dicarboxylate (3d)
yellow powder. Yield: 89%. 1H NMR (DMSO-d6, 400 MHz): a mixture of rotamers, δ = 10.91 and 10.87 (1H, s, 9-NH), 7.44 (1H, d, J = 7.8 Hz, 5-H), 7.30 (1H, d, J = 8.0 Hz, 8-H), 7.08-7.04 (1H, m, 7-H), 7.00-6.96 (1H, m, 6-H), 5.30 and 5.27 (1H, d, J = 5.3 and 5.4 Hz, 3-H), 4.84 and 4.78 (1H, d, J = 16.2 and 16.6 Hz, 1α-H), 4.50 and 4.38 (1H, d, J = 16.2 and 16.5 Hz, 1β-H), 3.99-3.82 (2H, m, -CH2CH(CH3)2), 3.58 and 3.57 (3H, s, -COOCH3), 3.32 (1H, d, J = 16.0 Hz, 4β-H), 3.09-3.01 (1H, m, 4α-H), 2.01-1.86 (1H, m, -CH2CH(CH3)2), 0.95 and 0.90 (6H, d, J = 6.7 Hz; dd, J = 6.7 and 1.9 Hz, -CH2CH(CH3)2) ppm. 13C NMR (DMSO-d6, 100 MHz): δ = 172.0, 156.5, 156.1, 136.7 (2C), 130.3, 130.1, 126.6, 121.6 (2C), 119.1, 118.1, 111.6, 104.7, 104.3, 71.8 (2C), 53.5, 53.2, 52.9, 52.8, 40.9, 28.1, 28.0, 23.4, 23.2, 19.3(2C), 19.2(2C) ppm. ESI-MS: calc. for C18H21N2O4 [M-H]- : 329.2, found: 329.3. HPLC purity: 97.4% (254 nm), tR: 7.15 min; 97.9% (220 nm), tR: 7.15 min
4.1.3. General procedure for compound s 4a-f
Triethylamine (25.4 μL, 0.18 mmol) was added to a solution of compound 2a or 2b (0.15 mmol) in dichloromethane (2 mL), followed by the addition of substituted sulfonyl chlorides (0.15 mmol). The reaction mixture was stirred at room temperature for 40-60 h. After the reaction was complete (monitored by HPLC), the reaction mixture was evaporated. The residue was purified by flash column chromatography on silica gel to give products 4a-f.
For 4c and 4f: 1.5 equiv of methanesulfonyl chloride and triethylamine were used and the reaction was complete in 30 min based on HPLC monitoring.
(S)-Methyl 2-(4-bromophenylsulfonyl)-2,3,4,9-tetrahydro-1H-pyrido[3,4-b]indole-3-carboxylate (4a)
white floc. Yield: 83%. 1H NMR (DMSO-d6, 400 MHz): δ = 10.86 (1H, s, 9-NH), 7.84-7.79 (4H, m, BrPh-H), 7.39 (1H, d, J = 7.8 Hz, 5-H), 7.31-7.29 (1H, m, 8-H), 7.08-7.04 (1H, m, 7-H), 6.98-6.94 (1H, m, 6-H), 5.21 (1H, dd, J = 6.5 and 1.2 Hz, 3-H), 4.74 (1H, d, J = 15.4 Hz, 1α-H), 4.46 (1H, d, J = 15.6 Hz, 1β-H), 3.42 (3H, s, -COOCH3), 3.24 (1H, d, J = 15.8 Hz, 4β-H), 2.97 (1H, dd, J = 15.9 and 6.5 Hz, 4α-H) ppm. 13C NMR (DMSO-d6, 100 MHz): δ = 170.8, 138.7, 136.5, 132.9, 129.4, 129.0, 127.5, 126.5, 121.8, 119.2, 118.2, 111.7, 104.4, 54.4, 52.8, 41.2, 24.3 ppm. ESI-MS: calc. for C19H16BrN2O4S [M-H]-: 447.0, found: 447.2. HPLC purity: 99.5% (254 nm), tR: 7.24 min; 100% (220 nm), tR: 7.24 min.
(S)-Methyl 2-tosyl-2,3,4,9-tetrahydro-1H-pyrido[3,4-b]indole-3-carboxylate (4b)
white powder. Yield: 93%. 1H NMR (DMSO-d6, 400 MHz): δ = 10.84 (1H, s, 9-NH), 7.74 (2H, d, J = 8.3 Hz, MePh-H), 7.39 (2H, d, J = 8.1 Hz, MePh-H), 7.38 (1H, d, J = 7.2 Hz, 5-H), 7.29 (1H, d, J = 8.0 Hz, 8-H), 7.07-7.03 (1H, m, 7-H), 6.97-6.93 (1H, m, 6-H), 5.18 (1H, dd, J = 6.4 and 1.2 Hz, 3-H), 4.70 (1H, d, J = 15.4 Hz, 1α-H), 4.47 (1H, d, J = 15.6 Hz, 1β-H), 3.42 (3H, s, -COOCH3), 3.21 (1H, d, J = 15.9 Hz, 4β-H), 2.94 (1H, dd, J = 15.8 and 6.5 Hz, 4α-H), 2.36 (3H, s, -PhCH3) ppm. 13C NMR (DMSO-d6, 100 MHz): δ = 171.0, 143.9, 136.7, 136.6, 130.2, 129.2, 127.3, 126.7, 121.7, 119.2, 118.1, 111.6, 104.4, 54.2, 52.6, 41.1, 24.3, 21.4 ppm. ESI-MS: calc. for C20H19N2O4S [M-H]-: 383.1, found: 383.3. HPLC purity: 99.4% (254 nm), tR: 7.05 min; 99.9% (220 nm), tR: 7.05 min.
(S)-Methyl 2-(methylsulfonyl)-2,3,4,9-tetrahydro-1H-pyrido[3,4-b]indole-3-carboxylate (4c)
light yellow powder. Yield: 100%. 1H NMR (DMSO-d6, 400 MHz): δ = 10.94 (1H, s, 9-NH), 7.44 (1H, d, J = 7.8 Hz, 5-H), 7.32 (1H, d, J = 8.0 Hz, 8-H), 7.09-7.05 (1H, m, 7-H), 7.01-6.97 (1H, m, 6-H), 5.07 (1H, dd, J = 6.3 and 1.4 Hz, 3-H), 4.70 (1H, d, J = 15.7 Hz, 1α-H), 4.56 (1H, d, J = 15.5 Hz, 1β-H), 3.60 (3H, s, -COOCH3), 3.30 (1H, d, J = 15.8 Hz, 4β-H), 3.12-3.06 (1H, m, 4α-H), 3.09 (3H, s, -SO2CH3) ppm. 13 C NMR (DMSO-d6, 100 MHz): δ = 171.6, 136.6, 129.5, 126.7, 121.7, 119.2, 118.2, 111.6, 104.6, 54.4, 52.9, 40.8, 24.5 ppm. ESI-MS: calc. for C14H15N2O4S [M-H]-: 307.1, found: 307.3. HPLC purity: 99.3% (254 nm), tR: 6.45 min; 99.3% (220 nm), tR: 6.45 min.
(R)-Methyl 2-(4-bromophenylsulfonyl)-2,3,4,9-tetrahydro-1H-pyrido[3,4-b]indole-3-carboxylate (4d)
white powder. Yield: 78%. 1H NMR (DMSO-d6, 400 MHz): δ = 10.86 (1H, s, 9-NH), 7.84-7.79 (4H, m, Ph-H), 7.39 (1H, d, J = 7.8 Hz, 5-H), 7.31-7.29 (1H, m, 8-H), 7.08-7.04 (1H, m, 7-H), 6.98-6.94 (1H, m, 6-H), 5.22 (1H, dd, J = 6.3 and 1.1 Hz, 3-H), 4.74 (1H, d, J = 15.5 Hz, 1α-H), 4.46 (1H, d, J = 15.6 Hz, 1β-H), 3.42 (3H, s, -COOCH3), 3.24 (1H, d, J = 15.9 Hz, 4β-H), 2.97 (1H, dd, J = 15.8 and 6.5 Hz, 4α-H) ppm. 13C NMR (DMSO-d6, 100 MHz): δ = 170.8, 138.7, 136.5, 132.9, 129.4, 129.0, 127.5, 126.5, 121.8, 119.2, 118.2, 111.7, 104.4, 54.4, 52.8, 41.2, 24.3 ppm. ESI-MS: calc. for C19H16BrN2O4S [M-H]-: 447.0, found: 447.2. HPLC purity: 100% (254 nm), tR: 7.19 min; 100% (220 nm), tR: 7.19 min.
(R)-Methyl 2-tosyl-2,3,4,9-tetrahydro-1H-pyrido[3,4-b]indole-3-carboxylate (4e)
white powder. Yield: 99%. 1H NMR (DMSO-d6, 400 MHz): δ = 10.84 (1H, s, 9-NH), 7.74 (2H, d, J = 8.3 Hz, MePh-H), 7.39 (2H, d, J = 8.1 Hz, MePh-H), 7.38 (1H, d, J = 7.1 Hz, 5-H), 7.29 (1H, d, J = 8.0 Hz, 8-H), 7.07-7.03 (1H, m, 7-H), 6.97-6.93 (1H, m, 6-H), 5.18 (1H, dd, J = 6.4 and 1.0 Hz, 3-H), 4.70 (1H, d, J = 15.1 Hz, 1α-H), 4.47 (1H, d, J = 15.7 Hz, 1β-H), 3.42 (3H, s, -COOCH3), 3.21 (1H, d, J = 15.9 Hz, 4β-H), 2.94 (1H, dd, J = 15.8 and 6.5 Hz, 4α-H), 2.36 (3H, s, -PhCH3) ppm. 13C NMR (DMSO-d6, 100 MHz): δ = 171.0, 144.0, 136.6, 136.5, 130.2, 129.2, 127.3, 126.6, 121.7, 119.2, 118.2, 111.6, 104.3, 54.2, 52.7, 41.0, 24.3, 21.4 ppm. ESI-MS: calc. for C20H19N2O4S [M-H]-: 383.1, found: 383.3. HPLC purity: 97.6% (254 nm), tR: 7.11 min; 99.5% (220 nm), tR: 7.11 min.
(R)-Methyl 2-(methylsulfonyl)-2,3,4,9-tetrahydro-1H-pyrido[3,4-b]indole-3-carboxylate (4f)
pale yellow powder. Yield: 93%. 1H NMR (DMSO-d6, 400 MHz): δ = 10.94 (1H, s, 9-NH), 7.44 (1H, d, J = 7.8 Hz, 5-H), 7.32 (1H, d, J = 8.0 Hz, 8-H), 7.09-7.05 (1H, m, 7-H), 7.01-6.97 (1H, m, 6-H), 5.06 (1H, dd, J = 6.4 and 1.4 Hz, 3-H), 4.70 (1H, d, J = 15.4 Hz, 1α-H), 4.56 (1H, d, J = 15.5 Hz, 1β-H), 3.60 (3H, s, -COOCH3), 3.30 (1H, d, J = 15.8 Hz, 4β-H), 3.14-3.06 (1H, m, 4α-H), 3.09 (3H, s, -SO2CH3) ppm. 13 C NMR (DMSO-d6, 100 MHz): δ = 171.6, 136.6, 129.5, 126.7, 121.7, 119.2, 118.2, 111.6, 104.6, 54.4, 52.9, 40.8, 24.5 ppm. ESI-MS: calc. for C14H15N2O4S [M-H]-: 307.1, found: 307.2. HPLC purity: 95.4% (254 nm), tR: 6.44 min; 97.7% (220 nm), tR: 6.44 min.
4.1.4. General procedure for compound s 5a-f 23
Substituted bromides (0.12 mmol) and N,N-diisopropylethylamine (DIPEA, 0.15 mmol) were added to the solution of 2 (0.1 mmol) in acetonitrile (5 mL). This mixture was heated under reflux for 2-5 h. After the reaction was complete (monitored by HPLC), the solvent was removed by evaporation. The residue was dissolved in cold EtOAc (10 mL) and the mixture was then filtered to remove the precipitate. The filtrate was concentrated in vacuo to yield the yellow crude residue, which was further purified by flash column chromatography on silica gel to give products 5a-f.
(S)-Methyl 2-benzyl-2,3,4,9-tetrahydro-1H-pyrido[3,4-b]indole-3-carboxylate (5a)
pale yellow powder. Yield: 62%. 1H NMR (DMSO-d6, 400 MHz): δ = 10.66 (1H, s, 9-NH), 7.40-7.24 (7H, m, -CH2PhH, 5-H, and 8-H), 7.03-6.99 (1H, m, 7-H), 6.97-6.93 (1H, m, 6-H), 4.02-3.92 (4H, m, 3-H, -CH2Ph, 1α-H), 3.74 (1H, d, J = 15.3 Hz, 1β-H), 3.60 (3H, s, -COOCH3), 3.10-3.00 (2H, m, 4-H) ppm. 13C NMR (DMSO-d6, 100 MHz): δ = 173.3, 139.4, 136.4, 132.6, 129.0, 128.8, 127.6, 127.1, 121.0, 118.8, 117.8, 111.3, 104.8, 59.4, 58.7, 51.8, 45.8, 24.3 ppm. 13C NMR (DMSO-d6, 100 MHz): δ = 173.3, 139.4, 136.4, 132.6, 129.0, 128.8, 127.6, 127.1, 121.0, 118.8, 117.8, 111.3, 104.8, 59.4, 58.7, 51.8, 45.8, 24.3 ppm. ESI-MS: calc. for C20H19N2O2 [M-H]-: 319.1, found: 319.3. HPLC purity: 100% (254 nm), tR: 5.74 min; 100% (220 nm), tR: 5.74 min.
(S)-Methyl 2-(cyclopropylmethyl)-2,3,4,9-tetrahydro-1H-pyrido[3,4-b]indole-3-carboxylate (5b)
brown viscous oil. Yield: 72%. 1H NMR (DMSO-d6, 400 MHz): δ = 10.75 (1H, s, 9-NH), 7.37 (1H, d, J = 7.7 Hz, 5-H), 7.26 (1H, d, J = 8.0 Hz, 8-H), 7.03-6.99 (1H, m, 7-H), 6.95-6.92 (1H, m, 6-H), 4.09-3.99 (3H, m, 3-H, 1-H), 3.54 (3H, s, -COOCH3), 3.05-2.96 (2H, m, -CH2CH(CH2)2), 2.72 (1H, dd, J = 12.6 and 6.3 Hz, 4β-H), 2.62 (1H, dd, J = 12.6 and 6.9 Hz, 4α-H), 0.96-0.86 (1H, m, -CH2CH(CH2)2), 0.57-0.46 (2H, m, -CH2CH(CH2)2), 0.19-0.07 (2H, m, -CH2CH(CH2)2) ppm. 13C NMR (CDCl3, 100 MHz): δ = 173.3, 136.2, 131.5, 127.1, 121.4, 119.3, 117.9, 110.7, 106.1, 59.4, 59.2, 51.6, 46.5, 23.8, 9.4, 4.2, 3.6 ppm. ESI-MS: calc. for C17H19N2O2 [M-H]-: 283.1, found: 283.3. HPLC purity: 96.3% (254 nm), tR: 5.50 min; 95.1% (220 nm), tR: 5.50 min.
(S)-Methyl 2-(prop-2-ynyl)-2,3,4,9-tetrahydro-1H-pyrido[3,4-b]indole-3-carboxylate (5c)
light yellow powder. Yield: 73%. 1H NMR (DMSO-d6, 400 MHz): δ = 10.79 (1H, s, 9-NH), 7.38 (1H, d, J = 7.7 Hz, 5-H), 7.28 (1H, d, J = 8.0 Hz, 8-H), 7.05-7.01 (1H, m, 7-H), 6.97-6.93 (1H, m, 6-H), 4.05-3.96 (3H, m, 3-H, 1-H), 3.72 (1H, dd, J = 16.7 and 2.4 Hz, -CH2C≡CH), 3.66 (1H, dd, J = 16.7 and 2.4 Hz, -CH2C≡CH), 3.58 (3H, s, -COOCH3), 3.26 (1H, t, J = 2.4 Hz, -CH2C≡CH), 3.01 (2H, d, J = 4.6 Hz, 4-H) ppm. 13C NMR (DMSO-d6, 100 MHz): δ = 173.0, 136.4, 132.2, 126.9, 121.0, 118.8, 117.9, 111.4, 104.5, 80.6, 76.1, 58.6, 51.8, 45.9, 43.7, 24.7 ppm. ESI-MS: calc. for C16H15N2O2 [M-H]-: 267.1, found: 267.2. HPLC purity: 93.8% (254 nm), tR: 5.59 min; 92.9% (220 nm), tR: 5.59 min.
(R)-Methyl 2-benzyl-2,3,4,9-tetrahydro-1H-pyrido[3,4-b]indole-3-carboxylate (5d)
pale yellow powder. Yield: 84%. 1H NMR (DMSO-d6, 400 MHz): δ = 10.66 (1H, s, 9-NH), 7.40-7.24 (7H, m, -CH2PhH, 5-H, and 8-H), 7.03-6.99 (1H, m, 7-H), 6.97-6.93 (1H, m, 6-H), 4.02-3.92 (4H, m, 3-H, -CH2Ph, 1α-H), 3.74 (1H, d, J = 15.2 Hz, 1β-H), 3.60 (3H, s, -COOCH3), 3.10-3.00 (2H, m, 4-H) ppm. 13C NMR (DMSO-d6, 100 MHz): δ = 173.3, 139.4, 136.4, 132.6, 129.0, 128.8, 127.6, 127.1, 121.0, 118.8, 117.8, 111.3, 104.8, 59.4, 58.7, 51.8, 45.8, 24.3 ppm. ESI-MS: calc. for C20H19N2O2 [M-H]-: 319.1, found: 319.3. HPLC purity: 100% (254 nm), tR: 5.75 min; 99.5% (220 nm), tR: 5.74 min
(R)-Methyl 2-(cyclopropylmethyl)-2,3,4,9-tetrahydro-1H-pyrido[3,4-b]indole-3-carboxylate (5e)
light brown solid. Yield: 69%. 1H NMR (DMSO-d6, 400 MHz): δ = 10.76 (1H, s, 9-NH), 7.37 (1H, d, J = 7.7 Hz, 5-H), 7.26 (1H, d, J = 7.9 Hz, 8-H), 7.03-6.99 (1H, m, 7-H), 6.96-6.92 (1H, m, 6-H), 4.10-3.99 (3H, m, 3-H, 1-H), 3.54 (3H, s, -COOCH3), 3.05-2.96 (2H, m, -CH2CH(CH2)2), 2.72 (1H, dd, J = 12.6 and 6.3 Hz, 4β-H), 2.62 (1H, dd, J = 12.6 and 6.8 Hz, 4α-H), 0.96-0.85 (1H, m, -CH2CH(CH2)2), 0.57-0.46 (2H, m, -CH2CH(CH2)2), 0.19-0.07 (2H, m, -CH2CH(CH2)2) ppm. 13C NMR (DMSO-d6, 100 MHz): δ = 173.5, 136.4, 133.0, 127.1, 120.9, 118.7, 117.8, 111.3, 104.6, 59.7, 59.1, 51.6, 46.2, 24.5, 9.8, 4.5, 3.7 ppm. ESI-MS: calc. for C17H19N2O2 [M-H]-: 283.1, found: 283.3. HPLC purity: 98.8% (254 nm), tR: 5.44 min; 99.6% (220 nm), tR: 5.44 min.
(R)-Methyl 2-(prop-2-ynyl)-2,3,4,9-tetrahydro-1H-pyrido[3,4-b]indole-3-carboxylate (5f)
yellow powder. Yield: 84%. 1H NMR (DMSO-d6, 400 MHz): δ = 10.79 (1H, s, 9-NH), 7.38 (1H, d, J = 7.7 Hz, 5-H), 7.28 (1H, d, J = 8.0 Hz, 8-H), 7.05-7.00 (1H, m, 7-H), 6.97-6.93 (1H, m, 6-H), 4.05-3.96 (3H, m, 3-H, 1-H), 3.72 (1H, dd, J = 16.7 and 2.4 Hz, -CH2C≡CH), 3.66 (1H, dd, J = 16.7 and 2.5 Hz, -CH2C≡CH), 3.58 (3H, s, -COOCH3), 3.26 (1H, t, J = 2.4 Hz, -CH2C≡CH), 3.01 (2H, d, J = 4.3 Hz, 4-H) ppm. 13C NMR (DMSO-d6, 100 MHz): δ = 173.0, 136.4, 132.2, 126.9, 121.0, 118.8, 117.9, 111.4, 104.5, 80.6, 76.1, 58.6, 51.8, 45.9, 43.7, 24.6 ppm. ESI-MS: calc. for C16H15N2O2 [M-H]-: 267.1, found: 267.2. HPLC purity: 96.5% (254 nm), tR: 5.59 min; 95.1% (220 nm), tR: 5.58 min.
4.1.5. General procedure for compounds 6a-j
The solutions of 2 (0.25 mmol) and a series of isocyanates in dichloromethane (2 mL) were stirred at room temperature overnight. After the reaction was complete (monitored by HPLC), the reaction mixture was evaporated. The residue was purified by flash column chromatography on silica gel to give products 6a-j.
(S)-Methyl 2-(pentylcarbamoyl)-2,3,4,9-tetrahydro-1H-pyrido[3,4-b]indole-3-carboxylate (6a)
light yellow powder. Yield: 88%. 1H NMR (DMSO-d6, 400 MHz): δ = 10.89 (1H, s, 9-NH), 7.42 (1H, d, J = 7.8 Hz, 5-H), 7.30 (1H, d, J = 8.0 Hz, 8-H), 7.07-7.03 (1H, m, 7-H), 6.99-6.95 (1H, m, 6-H), 6.76 (1H, t, J = 5.4 Hz, -CONH), 5.36 (1H, d, J = 5.0 Hz, 3-H), 4.75 (1H, d, J = 15.6 Hz, 1α-H), 4.38 (1H, d, J = 15.7 Hz, 1β-H), 3.52 (3H, s, -COOCH3), 3.27 (1H, d, J = 15.6 Hz, 4β-H), 3.12-3.07 (2H, m, -CONHCH2), 2.96 (1H, dd, J = 15.6 and 6.3 Hz, 4α-H), 1.50-1.43 (2H, m, -CONHCH2CH2), 1.34-1.22 (4H, m, -CONHCH2CH2CH2CH2), 0.88 (3H, t, J = 7.0 Hz, -CONH(CH2)4CH3) ppm. 13C NMR (DMSO-d6, 100 MHz): δ = 172.8, 158.5, 136.8, 130.9, 126.7, 121.4, 119.0, 118.1, 111.5, 105.0, 52.4(2C), 40.8, 29.9, 29.1, 23.7, 22.4, 14.4 ppm. ESI-MS: calc. for C19H24N3O3 [M-H]-: 342.2, found: 342.3. HPLC purity: 97.2% (254 nm), tR: 6.85 min; 97.5% (220 nm), tR: 6.85 min.
(S)-Methyl 2-(isopropylcarbamoyl)-2,3,4,9-tetrahydro-1H-pyrido[3,4-b]indole-3-carboxylate (6b)
yellow powder. Yield: 99%. 1H NMR (DMSO-d6, 400 MHz): δ = 10.87 (1H, s, 9-NH), 7.42 (1H, d, J = 7.8 Hz, 5-H), 7.30 (1H, d, J = 8.0 Hz, 8-H), 7.07-7.03 (1H, m, 7-H), 6.99-6.95 (1H, m, 6-H), 6.48 (1H, d, J = 7.5 Hz, -CONH), 5.35 (1H, d, J = 5.0 Hz, 3-H), 4.80 (1H, d, J = 15.7 Hz, 1α-H), 4.36 (1H, d, J = 15.8 Hz, 1β-H), 3.87-3.78 (1H, m, -CONHCH), 3.53 (3H, s, -COOCH3), 3.26 (1H, d, J = 15.6 Hz, 4β-H), 2.96 (1H, dd, J = 15.6 and 6.3 Hz, 4α-H), 1.13 (3H, d, J = 6.6 Hz, -CONHCH(CH3)2), 1.10 (3H, d, J = 6.5 Hz, -CONHCH(CH3)2) ppm. 13C NMR (DMSO-d6, 100 MHz): δ = 172.8, 158.0, 136.8, 131.0, 126.7, 121.4, 119.0, 118.1, 111.5, 104.9, 52.5, 52.4, 42.6, 23.7, 23.4, 23.3 ppm. ESI-MS: calc. for C17H20N3O3 [M-H]-: 314.2, found: 314.2. HPLC purity: 94.8% (254 nm), tR: 6.40 min; 97.1% (220 nm), tR: 6.40 min.
(S)-Methyl 2-(phenethylcarbamoyl)-2,3,4,9-tetrahydro-1H-pyrido[3,4-b]indole-3-carboxylate (6c)
yellow powder. Yield: 100%. 1H NMR (DMSO-d6, 400 MHz): δ = 10.92 (1H, s, 9-NH), 7.43 (1H, d, J = 7.7 Hz, 5-H), 7.32-7.19 (6H, m, -CH2PhH, 8-H), 7.07-7.03 (1H, m, 7-H), 6.99-6.95 (1H, m, 6-H), 6.93 (1H, t, J = 5.2 Hz, -CONH), 5.37 (1H, d, J = 5.8 Hz, 3-H), 4.73 (1H, d, J = 15.5 Hz, 1α-H), 4.38 (1H, d, J = 15.8 Hz, 1β-H), 3.54 (3H, s, -COOCH3), 3.32-3.26 (3H, m, -CONHCH2, 4β-H), 2.96 (1H, dd, J = 15.5 and 6.3 Hz, 4α-H), 2.83-2.72 (2H, m, -CONHCH2CH2Ph) ppm. 13C NMR (DMSO-d6, 100 MHz): δ = 172.8, 158.4, 140.3, 136.8, 130.8, 129.2, 128.8, 126.7, 126.5, 121.4, 119.0, 118.1, 111.5, 105.0, 52.5, 52.4 (2C), 42.6, 36.3, 23.7 ppm. ESI-MS: calc. for C22H22N3O3 [M-H]-: 376.2, found: 376.3. HPLC purity: 96.5% (254 nm), tR: 6.88 min; 96.9% (220 nm), tR: 6.88 min.
(S)-Methyl 2-(cyclohexylcarbamoyl)-2,3,4,9-tetrahydro-1H-pyrido[3,4-b]indole-3-carboxylate (6d)
white powder. Yield: 100%. 1H NMR (DMSO-d6, 400 MHz): δ = 10.86 (1H, s, 9-NH), 7.42 (1H, d, J = 7.8 Hz, 5-H), 7.30 (1H, d, J = 8.0 Hz, 8-H), 7.07-7.03 (1H, m, 7-H), 6.98-6.95 (1H, m, 6-H), 6.47 (1H, d, J = 7.7 Hz, -CONH), 5.34 (1H, dd, J = 6.0 and 1.1 Hz, 3-H), 4.80 (1H, d, J = 15.7 Hz, 1α-H), 4.36 (1H, d, J = 15.9 Hz, 1β-H), 3.52 (3H, s, -COOCH3), 3.50-3.44 (1H, m, -CONHCH), 3.26 (1H, d, J = 15.6 Hz, 4β-H), 2.95 (1H, dd, J = 15.6 and 6.3 Hz, 4α-H), 1.81-1.58 (5H, m, -CONHCH(CH2)5), 1.31-1.05 (5H, m, -CONHCH(CH2)5) ppm. 13C NMR (DMSO-d6, 100 MHz): δ = 172.8, 157.9, 136.8, 131.1, 126.7, 121.4, 119.0, 118.1, 111.5, 104.9, 52.4, 49.9, 33.5 (2C), 25.9, 25.6, 25.5, 23.7 ppm. ESI-MS: calc. for C20H24N3O3 [M-H]-: 354.2, found: 354.3. HPLC purity: 98.4% (254 nm), tR: 6.84 min; 98.9% (220 nm), tR: 6.84 min.
(S)-Methyl 2-(1-adamantylcarbamoyl)-2,3,4,9-tetrahydro-1H-pyrido[3,4-b]indole-3-carboxylate (6e)
yellow powder. Yield: 93%. 1H NMR (DMSO-d6, 400 MHz): δ = 10.85 (1H, s, 9-NH), 7.42 (1H, d, J = 7.8 Hz, 5-H), 7.29 (1H, d, J = 8.0 Hz, 8-H), 7.06-7.02 (1H, m, 7-H), 6.98-6.94 (1H, m, 6-H), 5.94 (1H, s, -CONH), 5.31 (1H, dd, J = 6.3 and 1.5 Hz, 3-H), 4.81 (1H, d, J = 15.6 Hz, 1α-H), 4.37 (1H, d, J = 15.7 Hz, 1β-H), 3.53 (3H, s, -COOCH3), 3.25 (1H, d, J = 15.5 Hz, 4β-H), 2.95 (1H, dd, J = 15.6 and 6.3 Hz, 4α-H), 2.03-1.98 (9H, m, adamantyl-H), 1.66-1.60 (6H, m, adamantyl-H) ppm. 13C NMR (DMSO-d6, 100 MHz): δ = 172.8, 157.6, 136.7, 131.3, 126.7, 121.3, 119.0, 118.0, 111.5, 104.9, 52.4, 51.3, 42.0, 40.9, 40.6, 36.7, 29.5, 23.7 ppm. ESI-MS: calc. for C24H28N3O3 [M-H]-: 406.2, found: 406.3. HPLC purity: 96.5% (254 nm), tR: 7.35 min; 97.9% (220 nm), tR: 7.35 min.
(R)-Methyl 2-(pentylcarbamoyl)-2,3,4,9-tetrahydro-1H-pyrido[3,4-b]indole-3-carboxylate (6f)
white powder. Yield: 89%. 1H NMR (DMSO-d6, 400 MHz): δ = 10.90 (1H, s, 9-NH), 7.42 (1H, d, J = 7.7 Hz, 5-H), 7.30 (1H, d, J = 8.0 Hz, 8-H), 7.07-7.03 (1H, m, 7-H), 6.99-6.95 (1H, m, 6-H), 6.77 (1H, t, J = 5.4 Hz, -CONH), 5.35 (1H, d, J = 5.2 Hz, 3-H), 4.75 (1H, d, J = 15.7 Hz, 1α-H), 4.37 (1H, d, J = 15.7 Hz, 1β-H), 3.52 (3H, s, -COOCH3), 3.27 (1H, d, J = 15.6 Hz, 4β-H), 3.15-3.03 (2H, m, -CONHCH2), 2.96 (1H, dd, J = 15.7 and 6.2 Hz, 4α-H), 1.49-1.42 (2H, m, -CONHCH2CH2), 1.36-1.20 (4H, m, -CONHCH2CH2CH2CH2), 0.88 (3H, t, J = 6.9 Hz, -CONH(CH2)4CH3) ppm. 13C NMR (DMSO-d6, 100 MHz): δ = 172.8, 158.5, 136.8, 130.9, 126.7, 121.4, 119.0, 118.1, 111.5, 105.0, 52.5, 52.4, 40.7, 29.9, 29.1, 23.7, 22.4, 14.5 ppm. ESI-MS: calc. for C19H24N3O3 [M-H]-: 342.2, found: 342.4. HPLC purity: 98.1% (254 nm), tR: 6.85 min; 98.4% (220 nm), tR: 6.85 min.
(R)-Methyl 2-(isopropylcarbamoyl)-2,3,4,9-tetrahydro-1H-pyrido[3,4-b]indole-3-carboxylate (6g)
yellow powder. Yield: 100%. 1H NMR (DMSO-d6, 400 MHz): δ = 10.87 (1H, s, 9-NH), 7.42 (1H, d, J = 7.8 Hz, 5-H), 7.30 (1H, d, J = 8.0 Hz, 8-H), 7.07-7.03 (1H, m, 7-H), 6.99-6.95 (1H, m, 6-H), 6.48 (1H, d, J = 7.5 Hz, -CONH), 5.35 (1H, d, J = 5.2 Hz, 3-H), 4.79 (1H, d, J = 15.7 Hz, 1α-H), 4.36 (1H, d, J = 15.7 Hz, 1β-H), 3.89-3.77 (1H, m, -CONHCH), 3.53 (3H, s, -COOCH3), 3.26 (1H, d, J = 15.6 Hz, 4β-H), 2.96 (1H, dd, J = 15.6 and 6.3 Hz, 4α-H), 1.13 (3H, d, J = 6.6 Hz, -CONHCH(CH3)2), 1.10 (3H, d, J = 6.5 Hz, -CONHCH(CH3)2) ppm. 13C NMR (DMSO-d6, 100 MHz): δ = 172.8, 158.0, 136.8, 131.0, 126.7, 121.4, 119.0, 118.1, 111.5, 104.9, 52.4 (2C), 42.6, 23.7, 23.4, 23.3 ppm. ESI-MS: calc. for C17H20N3O3 [M-H]-: 314.2, found: 314.3. HPLC purity: 95.4% (254 nm), tR: 6.42 min; 97.1% (220 nm), tR: 6.42 min.
(R)-Methyl 2-(phenethylcarbamoyl)-2,3,4,9-tetrahydro-1H-pyrido[3,4-b]indole-3-carboxylate (6h)
yellow powder. Yield: 100%. 1H NMR (DMSO-d6, 400 MHz): δ = 10.92 (1H, s, 9-NH), 7.43 (1H, d, J = 7.7 Hz, 5-H), 7.32-7.19 (6H, m, -CH2PhH, 8-H), 7.07-7.04 (1H, m, 7-H), 6.99-6.95 (1H, m, 6-H), 6.94 (1H, t, J = 5.2 Hz, -CONH), 5.37 (1H, d, J = 5.5 Hz, 3-H), 4.73 (1H, d, J = 15.6 Hz, 1α-H), 4.38 (1H, d, J = 15.7 Hz, 1β-H), 3.54 (3H, s, -COOCH3), 3.34-3.26 (3H, m, -CONHCH2, 4β-H), 2.96 (1H, dd, J = 15.6 and 6.2 Hz, 4α-H), 2.83-2.72 (2H, m, -CONHCH2CH2Ph) ppm. 13C NMR (DMSO-d6, 100 MHz): δ = 172.8, 158.5, 140.3, 136.8, 130.8, 129.2, 128.8, 126.7, 126.5, 121.4, 119.0, 118.1, 111.5, 105.0, 52.5, 52.4 (2C), 42.6, 36.3, 23.7 ppm. ESI-MS: calc. for C22H22N3O3 [M-H]-: 376.2, found: 376.4. HPLC purity: 96.5% (254 nm), tR: 6.86 min; 98.1% (220 nm), tR: 6.86 min.
(R)-Methyl 2-(cyclohexylcarbamoyl)-2,3,4,9-tetrahydro-1H-pyrido[3,4-b]indole-3-carboxylate (6i)
white powder. Yield: 97%. 1H NMR (DMSO-d6, 400 MHz): δ = 10.86 (1H, s, 9-NH), 7.42 (1H, d, J = 7.7 Hz, 5-H), 7.30 (1H, d, J = 8.0 Hz, 8-H), 7.07-7.03 (1H, m, 7-H), 6.98-6.95 (1H, m, 6-H), 6.47 (1H, d, J = 7.6 Hz, -CONH), 5.35 (1H, dd, J = 6.2 and 1.4 Hz, 3-H), 4.80 (1H, d, J = 15.8 Hz, 1α-H), 4.36 (1H, d, J = 15.8 Hz, 1β-H), 3.52 (3H, s, -COOCH3), 3.51-3.42 (1H, m, -CONHCH), 3.26 (1H, d, J = 15.6 Hz, 4β-H), 2.95 (1H, dd, J = 15.6 and 6.2 Hz, 4α-H), 1.84-1.58 (5H, m, -CONHCH(CH2)5), 1.31-1.05 (5H, m, -CONHCH(CH2)5) ppm. 13C NMR (DMSO-d6, 100 MHz): δ = 172.8, 158.0, 136.8, 131.1, 126.8, 121.4, 119.0, 118.0, 111.5, 104.9, 52.5, 52.4, 50.0, 33.5 (2C), 25.9, 25.6, 25.5, 23.7 ppm. ESI-MS: calc. for C20H24N3O3 [M-H]-: 354.2, found: 354.3. HPLC purity: 99.2% (254 nm), tR: 6.85 min; 99.8% (220 nm), tR: 6.84 min.
(R)-Methyl 2-(1-adamantylcarbamoyl)-2,3,4,9-tetrahydro-1H-pyrido[3,4-b]indole-3-carboxylate (6j)
yellow powder. Yield: 100%. 1H NMR (DMSO-d6, 400 MHz): δ = 10.84 (1H, s, 9-NH), 7.41 (1H, d, J = 7.7 Hz, 5-H), 7.28 (1H, d, J = 8.0 Hz, 8-H), 7.06-7.02 (1H, m, 7-H), 6.98-6.94 (1H, m, 6-H), 5.92 (1H, s, -CONH), 5.31 (1H, dd, J = 6.0 and 1.3 Hz, 3-H), 4.80 (1H, d, J = 15.8 Hz, 1α-H), 4.37 (1H, d, J = 15.7 Hz, 1β-H), 3.53 (3H, s, -COOCH3), 3.25 (1H, d, J = 15.5 Hz, 4β-H), 2.95 (1H, dd, J = 15.6 and 6.3 Hz, 4α-H), 2.03-1.98 (9H, m, adamantyl-H), 1.66-1.60 (6H, m, adamantyl-H) ppm. 13C NMR (DMSO-d6, 100 MHz): δ = 172.9, 157.6, 136.7, 131.3, 126.7, 121.3, 119.0, 118.0, 111.5, 104.9, 52.4, 51.3, 42.0, 40.9, 40.6, 36.7, 29.5, 23.7 ppm. ESI-MS: calc. for C24H28N3O3 [M-H]-: 406.2, found: 406.2. HPLC purity: 96.9% (254 nm), tR: 7.35 min; 98.4% (220 nm), tR: 7.36 min.
4.1.6. General procedure for compounds 7-9
A solution of an appropriate methyl ester 3, 4, or 5 (0.1 mmol) and LiOH (0.3 mmol) in THF/H2O (1:1, 4 mL) was stirred at room temperature until the completion of the reaction (monitored by HPLC). THF was removed in vacuo and then the solution was carefully neutralized with glacial acetic acid, whereupon a heavy precipitate was formed. The product was collected by filtration, washed with cold water, and dried.
(S)-2-(Allyloxycarbonyl)-2,3,4,9-tetrahydro-1H-pyrido[3,4-b]indole-3-carboxylic acid (7a)
light brown powder. Yield: 55%. 1H NMR (DMSO-d6, 400 MHz): a mixture of rotamers, δ = 12.90 (1H, br, -COOH), 10.90 and 10.85 (1H, s, 9-NH), 7.43 (1H, d, J = 7.7 Hz, 5-H), 7.31 (1H, d, J = 8.0 Hz, 8-H), 7.06 (1H, t, J = 7.2 Hz, 7-H), 6.98 (1H, t, J = 7.3 Hz, 6-H), 6.05-5.92 (1H, m, -CH2CH=CH2), 5.39-5.17 (3H, m, 3-H and -CH2CH=CH2), 4.83 and 4.77 (1H, d, J = 16.2 and 16.3 Hz, 1α-H), 4.70-4.59 (2H, m, -CH2CH=CH2), 4.52 and 4.41 (1H, d, J = 16.1 and 16.5 Hz, 1β-H), 3.30-3.17 (1H, m, 4β-H), 3.04-2.96 (1H, m, 4α-H) ppm. 13C NMR (DMSO-d6, 100 MHz): δ = 173.1 (2C), 156.2, 155.8, 136.7, 136.6, 133.7, 130.4, 130.2, 126.7, 121.5 (2C), 119.1, 118.1, 117.7, 117.4, 111.5, 105.0, 104.7, 66.2, 66.1, 53.6, 53.3, 41.0, 40.8, 23.6, 23.3 ppm. ESI-MS: calc. for C16H15N2O4 [M-H]-: 299.1, found: 299.2. HPLC purity: 96.0% (254 nm), tR: 6.45 min; 97.9% (220 nm), tR: 6.45 min.
(S)-2-(Isobutoxycarbonyl)-2,3,4,9-tetrahydro-1H-pyrido[3,4-b]indole-3-carboxylic acid (7b)
pale orange powder. Yield: 58%. 1H NMR (DMSO-d6, 400 MHz): a mixture of rotamers, δ = 12.87 (1H, br, -COOH), 10.90 and 10.86 (1H, s, 9-NH), 7.43 (1H, d, J = 7.7 Hz, 5-H), 7.30 (1H, dd, J = 7.9 and 2.7 Hz, 8-H), 7.08-7.04 (1H, m, 7-H), 6.98 (1H, t, J = 7.2 Hz, 6-H), 5.17 and 5.14 (1H, d, J = 5.3 and 5.4 Hz, 3-H), 4.81 and 4.75 (1H, d, J = 16.2 and 16.5 Hz, 1α-H), 4.51 and 4.40 (1H, d, J = 16.1 and 16.5Hz, 1β-H), 3.98-3.82 (2H, m, -CH2CH(CH3)2), 3.32 (1H, d, J = 16.0 Hz, 4β-H), 3.04-2.96 (1H, m, 4α-H), 1.99-1.89 (1H, m, -CH2CH(CH3)2), 0.95 and 0.91 (6H, d, J = 6.7 and dd, J = 6.7 and 1.3 Hz, -CH2CH(CH3)2) ppm. 13C NMR (DMSO-d6, 100 MHz): δ = 173.2, 156.5, 156.2, 136.6 (2C), 130.5, 130.3, 126.7, 121.5, 119.1, 118.1, 111.5, 105.0, 104.6, 71.7, 71.6, 53.4, 53.1, 41.0, 28.1, 28.0, 23.5, 23.3, 19.4, 19.3, 19.2 ppm. ESI-MS: calc. for C17H19N2O4 [M-H]-: 315.1, found: 315.3. HPLC purity: 98.1% (254 nm), tR: 6.74 min; 99.1% (220 nm), tR: 6.74 min.
(R)-2-(Allyloxycarbonyl)-2,3,4,9-tetrahydro-1H-pyrido[3,4-b]indole-3-carboxylic acid (7c)
yellow viscous oil. Yield: 58%. 1H NMR (DMSO-d6, 400 MHz): a mixture of rotamers, δ = 12.91 (1H, br, -COOH), 10.90 and 10.85 (1H, s, 9-NH), 7.43 (1H, d, J = 7.8 Hz, 5-H), 7.31 (1H, d, J = 8.0 Hz, 8-H), 7.08-7.04 (1H, m, 7-H), 7.00-6.96 (1H, m, 6-H), 6.05-5.92 (1H, m, -CH2CH=CH2), 5.39-5.17 (3H, m, 3-H and -CH2CH=CH2), 4.83 and 4.77 (1H, d, J = 15.9 and 16.3 Hz, 1α-H), 4.70-4.59 (2H, m, -CH2CH=CH2), 4.52 and 4.41 (1H, d, J = 16.2 and 16.5 Hz, 1β-H), 3.35-3.30 (1H, m, 4β-H), 3.04-2.96 (1H, m, 4α-H) ppm. 13C NMR (DMSO-d6, 100 MHz): δ = 173.1, 156.2, 155.8, 136.7, 136.6, 133.7, 130.3, 130.2, 126.7, 121.5 (2C), 119.1, 118.1, 117.7, 117.4, 111.5, 105.0, 66.2, 66.1, 53.6, 53.3, 41.0, 40.8, 23.6, 23.2 ppm. ESI-MS: calc. for C16H15N2O4 [M-H]-: 299.1, found: 299.2. HPLC purity: 96.3% (254 nm), tR: 6.49 min; 99.6% (220 nm), tR: 6.49 min.
(R)-2-(Isobutoxycarbonyl)-2,3,4,9-tetrahydro-1H-pyrido[3,4-b]indole-3-carboxylic acid (7d)
white powder. Yield: 62%. 1H NMR (DMSO-d6, 400 MHz): a mixture of rotamers, δ = 12.89 (1H, br, -COOH), 10.90 and 10.86 (1H, s, 9-NH), 7.43 (1H, d, J = 7.6 Hz, 5-H), 7.30 (1H, dd, J = 7.7 and 2.5 Hz, 8-H), 7.08-7.04 (1H, m, 7-H), 7.00-6.96 (1H, m, 6-H), 5.17 and 5.14 (1H, d, J = 5.2 and 5.2 Hz, 3-H), 4.81 and 4.75 (1H, d, J = 16.2 and 16.5 Hz, 1α-H), 4.51 and 4.40 (1H, d, J = 16.1 and 16.6 Hz, 1β-H), 3.98-3.82 (2H, m, -CH2CH(CH3)2), 3.35-3.30 (1H, m, 4β-H), 3.04-2.95 (1H, m, 4α-H), 1.98-1.88 (1H, m, -CH2CH(CH3)2), 0.95 and 0.91 (6H, d, J = 6.7 and dd, J = 6.7 and 1.4 Hz, -CH2CH(CH3)2) ppm. 13C NMR (DMSO-d6, 100 MHz): δ = 173.2, 156.5, 156.2, 136.6, 130.5, 130.3, 126.7, 121.5(2C), 119.1, 118.1, 111.5, 105.0, 104.6, 71.7, 71.6, 53.5, 53.1, 41.0, 40.7, 28.1, 28.0, 23.5, 23.3, 19.4, 19.3, 19.2 ppm. ESI-MS: calc. for C17H19N2O4 [M-H]-: 315.1, found: 315.3. HPLC purity: 98.9% (254 nm), tR: 6.76 min; 99.6% (220 nm), tR: 6.76 min.
(S)-2-(4-Bromophenylsulfonyl)-2,3,4,9-tetrahydro-1H-pyrido[3,4-b]indole-3-carboxylic acid (8a)
white powder. Yield: 61%. 1H NMR (DMSO-d6, 400 MHz): δ = 12.94 (1H, br, -COOH), 10.85 (1H, s, 9-NH), 7.80 (4H, s, BrPh-H), 7.39 (1H, d, J = 7.8 Hz, 5-H), 7.30 (1H, d, J = 8.1 Hz, 8-H), 7.07-7.03 (1H, m, 7-H), 6.98-6.94 (1H, m, 6-H), 5.04 (1H, d, J = 5.5 Hz, 3-H), 4.72 (1H, d, J = 15.2 Hz, 1α-H), 4.53 (1H, d, J = 15.5 Hz, 1β-H), 3.24 (1H, d, J = 15.7 Hz, 4β-H), 2.93 (1H, dd, J = 15.7 and 6.5 Hz, 4α-H) ppm. 13C NMR (DMSO-d6, 100 MHz): δ = 171.9, 138.9, 136.5, 132.8, 129.4, 129.2, 127.3, 126.6, 121.7, 119.2, 118.1, 111.6, 104.6, 54.3, 41.2, 24.5 ppm. ESI-MS: calc. for C18H14BrN2O4S [M-H]-: 433.0, found: 433.2. HPLC purity: 99.8% (254 nm), tR: 6.85 min; 99.3% (220 nm), tR: 6.85 min.
(S)-2-Tosyl-2,3,4,9-tetrahydro-1H-pyrido[3,4-b]indole-3-carboxylic acid (8b)
white powder. Yield: 84%. 1H NMR (DMSO-d6, 400 MHz): δ = 12.90 (1H, br, -COOH), 10.83 (1H, s, 9-NH), 7.74 (2H, d, J = 8.3 Hz, MePh-H), 7.37 (3H, d, J = 8.0 Hz, MePh-H and 5-H), 7.29 (1H, d, J = 8.0 Hz, 8-H), 7.06-7.02 (1H, m, 7-H), 6.95 (1H, t, J = 7.4 Hz, 6-H), 5.03 (1H, d, J = 5.6 Hz, 3-H), 4.67 (1H, d, J = 15.5 Hz, 1α-H), 4.55 (1H, d, J = 15.6 Hz, 1β-H), 3.21 (1H, d, J = 15.6 Hz, 4β-H), 2.89 (1H, dd, J = 15.6 and 6.5 Hz, 4α-H), 2.35 (3H, s, -PhCH3) ppm. 13C NMR (DMSO-d6, 100 MHz): δ = 172.1, 143.7, 137.0, 136.5, 130.2, 129.4, 127.3, 126.7, 121.7, 119.1, 118.1, 111.6, 104.6, 54.1, 41.0, 24.4, 21.4 ppm. ESI-MS: calc. for C19H17N2O4S [M-H]-: 369.1, found: 369.3. HPLC purity: 99.6% (254 nm), tR: 6.68 min; 100% (220 nm), tR: 6.68 min.
(S)-2-(Methylsulfonyl)-2,3,4,9-tetrahydro-1H-pyrido[3,4-b]indole-3-carboxylic acid (8c)
white powder. Yield: 70%. 1H NMR (DMSO-d6, 400 MHz): δ = 13.04 (1H, br, -COOH), 10.93 (1H, s, 9-NH), 7.44 (1H, d, J = 7.7 Hz, 5-H), 7.32 (1H, d, J = 8.0 Hz, 8-H), 7.09-7.05 (1H, m, 7-H), 6.99 (1H, t, J = 7.2 Hz,, 6-H), 4.93 (1H, dd, J = 6.4 and 1.2 Hz, 3-H), 4.67 (1H, d, J = 15.2 Hz, 1α-H), 4.58 (1H, d, J = 15.4 Hz, 1β-H), 3.30-3.29 (1H, m, 4β-H), 3.08 (3H, s, -SO2CH3), 3.03 (1H, dd, J = 15.8 and 6.5 Hz, 4α-H) ppm. 13C NMR (DMSO-d6, 100 MHz): δ = 172.7, 136.6, 129.7, 126.8, 121.6, 119.1, 118.1, 111.6, 104.9, 54.4, 40.8, 39.3, 24.7 ppm. ESI-MS: calc. for C13H13N2O4S [M-H]-: 293.0, found: 293.2. HPLC purity: 99.3% (254 nm), tR: 6.02 min; 99.5% (220 nm), tR: 6.02 min.
(R)-2-(4-Bromophenylsulfonyl)-2,3,4,9-tetrahydro-1H-pyrido[3,4-b]indole-3-carboxylic acid (8d)
white powder. Yield: 40%. 1H NMR (DMSO-d6, 400 MHz): δ = 10.85 (1H, s, 9-NH), 7.80 (4H, s, BrPh-H), 7.39 (1H, d, J = 7.8 Hz, 5-H), 7.30 (1H, d, J = 8.0 Hz, 8-H), 7.07-7.03 (1H, m, 7-H), 6.98-6.94 (1H, m, 6-H), 5.04 (1H, d, J = 5.4 Hz, 3-H), 4.72 (1H, d, J = 14.9 Hz, 1α-H), 4.53 (1H, d, J = 15.3 Hz, 1β-H), 3.24 (1H, d, J = 15.7 Hz, 4β-H), 2.93 (1H, dd, J = 15.9 and 6.5 Hz, 4α-H) ppm. 13C NMR (DMSO-d6, 100 MHz): δ = 171.9, 138.9, 136.5, 132.8, 129.3, 129.2, 127.3, 126.6, 121.7, 119.2, 118.1, 111.6, 104.6, 54.3, 41.2, 24.5 ppm. ESI-MS: calc. for C18H14BrN2O4S [M-H]-: 433.0, found: 433.2. HPLC purity: 98.5% (254 nm), tR: 6.88 min; 100% (220 nm), tR: 6.88 min.
(R)-2-Tosyl-2,3,4,9-tetrahydro-1H-pyrido[3,4-b]indole-3-carboxylic acid (8e)
white powder. Yield: 81%. 1H NMR (DMSO-d6, 400 MHz): δ = 12.89 (1H, br, -COOH), 10.83 (1H, s, 9-NH), 7.74 (2H, d, J = 8.3 Hz, MePh-H), 7.37 (3H, d, J = 8.2 Hz, MePh-H and 5-H), 7.29 (1H, d, J = 8.0 Hz, 8-H), 7.06-7.02 (1H, m, 7-H), 6.97-6.93 (1H, m, 6-H), 5.03 (1H, dd, J = 6.2 and 0.9 Hz, 3-H), 4.67 (1H, d, J = 15.5 Hz, 1α-H), 4.54 (1H, d, J = 15.6 Hz, 1β-H), 3.21 (1H, d, J = 15.6 Hz, 4β-H), 2.88 (1H, dd, J = 15.6 and 6.5 Hz, 4α-H), 2.35 (3H, s, -PhCH3) ppm. 13C NMR (DMSO-d6, 100 MHz): δ = 172.1, 143.7, 137.0, 136.5, 130.2, 129.4, 127.3, 126.7, 121.6, 119.1, 118.1, 111.6, 104.6, 54.1, 41.0, 24.4, 21.4 ppm. ESI-MS: calc. for C19H17N2O4S [M-H]-: 369.1, found: 369.3. HPLC purity: 99.1% (254 nm), tR: 6.66 min; 99.1% (220 nm), tR: 6.66 min.
(R)-2-(Methylsulfonyl)-2,3,4,9-tetrahydro-1H-pyrido[3,4-b]indole-3-carboxylic acid (8f)
white powder. Yield: 58%. 1H NMR (DMSO-d6, 400 MHz): δ = 13.04 (1H, br, -COOH), 10.93 (1H, s, 9-NH), 7.44 (1H, d, J = 7.8 Hz, 5-H), 7.32 (1H, d, J = 8.0 Hz, 8-H), 7.09-7.05 (1H, m, 7-H), 7.00-6.96 (1H, m, 6-H), 4.93 (1H, dd, J = 6.3 and 1.3 Hz, 3-H), 4.67 (1H, d, J = 14.9 Hz, 1α-H), 4.57 (1H, d, J = 15.4 Hz, 1β-H), 3.34-3.28 (1H, m, 4β-H), 3.07 (3H, s, -SO2CH3), 3.04 (1H, dd, J = 15.8 and 6.5 Hz, 4α-H) ppm. 13C NMR (DMSO-d6, 100 MHz): δ = 172.7, 136.6, 129.7, 126.8, 121.6, 119.1, 118.1, 111.6, 104.9, 54.4, 40.8, 39.3, 24.6 ppm. ESI-MS: calc. for C13H13N2O4S [M-H]-: 293.0, found: 293.2. HPLC purity: 97.4% (254 nm), tR: 6.02 min; 99.4% (220 nm), tR: 6.02 min.
(S)-2-Benzyl-2,3,4,9-tetrahydro-1H-pyrido[3,4-b]indole-3-carboxylic acid (9a)
light yellow powder. Yield: 39%. 1H NMR (DMSO-d6, 400 MHz): δ = 10.64 (1H, s, 9-NH), 7.40-7.27 (6H, m, -CH2Ph-H and 5-H), 7.25 (1H, d, J = 7.8 Hz, 8-H), 7.03-6.99 (1H, m, 7-H), 6.96-6.92 (1H, m, 6-H), 4.04-3.94 (3H, m, 1α-H and -CH2Ph), 3.86 (1H, dd, J = 6.1 and 3.4 Hz, 3-H), 3.72 (1H, d, J = 15.2 Hz, 1β-H), 3.08 (1H, dd, J = 15.6 and 2.5 Hz, 4β-H), 2.99 (1H, dd, J = 15.2 and 5.9 Hz, 4α-H) ppm. 13C NMR (DMSO-d6, 100 MHz): δ = 174.7, 139.6, 136.4, 132.8, 129.0, 128.8, 127.5, 127.2, 120.9, 118.7, 117.8, 111.3, 105.2, 59.6, 58.7, 45.9, 24.4 ppm. ESI-MS: calc. for C19H17N2O2 [M-H]-: 305.1, found: 305.2. HPLC purity: 99.6% (254 nm), tR: 5.55 min; 99.1% (220 nm), tR: 5.56 min.
(S)-2-(Cyclopropylmethyl)-2,3,4,9-tetrahydro-1H-pyrido[3,4-b]indole-3-carboxylic acid (9b)
pale yellow powder. Yield: 41%. 1H NMR (DMSO-d6, 400 MHz): δ = 10.76 (1H, s, 9-NH), 7.38 (1H, d, J = 7.7 Hz, 5-H), 7.27 (1H, d, J = 8.0 Hz, 8-H), 7.04-7.00 (1H, m, 7-H), 6.96-6.92 (1H, m, 6-H), 4.18 (1H, d, J = 14.9 Hz, 1α-H), 4.04 (1H, d, J = 15.2 Hz, 1β-H), 3.93 (1H, dd, J = 5.9 and 3.7 Hz, 3-H), 3.08 (1H, dd, J = 15.6 and 2.8 Hz, 4β-H), 2.97 (1H, dd, J = 15.5 and 6.2 Hz, 4α-H), 2.81-2.68 (2H, m, -CH2CH(CH2)2), 0.98-0.91 (1H, m, -CH2CH(CH2)2), 0.58-0.49 (2H, m, -CH2CH(CH2)2), 0.20-0.11 (2H, m, -CH2CH(CH2)2) ppm. 13C NMR (DMSO-d6, 100 MHz): δ = 173.9, 136.5, 132.2, 127.1, 120.9, 118.7, 117.8, 111.3, 105.1, 59.7, 59.2, 46.3, 24.2, 9.5, 4.5, 3.8 ppm. ESI-MS: calc. for C16H17N2O2 [M-H]-: 269.1, found: 269.3. HPLC purity: 96.8% (254 nm), tR: 5.29 min; 97.5% (220 nm), tR: 5.29 min.
(S)-2-(Prop-2-ynyl)-2,3,4,9-tetrahydro-1H-pyrido[3,4-b]indole-3-carboxylic acid (9c)
brown viscous oil. Yield: 44%. 1H NMR (DMSO-d6, 400 MHz): δ = 10.74 (1H, s, 9-NH), 7.35 (1H, d, J = 7.7 Hz, 5-H), 7.26 (1H, d, J = 7.9 Hz, 8-H), 7.02-6.98 (1H, m, 7-H), 6.95-6.91 (1H, m, 6-H), 4.08 (1H, d, J = 14.8 Hz, 1α-H), 3.88 (1H, d, J = 14.8 Hz, 1β-H), 3.76 (1H, dd, J = 16.5 and 2.5 Hz, -CH2C≡CH), 3.67-3.62 (2H, m, 3-H and -CH2C≡CH), 3.17 (1H, t, J = 2.4 Hz, -CH2C≡CH), 3.02 (1H, dd, J = 15.0 and 4.5 Hz, 4β-H), 2.88 (1H, dd, J = 15.1 and 5.8 Hz, 4α-H) ppm. 13C NMR (DMSO-d6, 100 MHz): δ = 175.1, 136.4, 132.7, 127.2, 120.7, 118.6, 117.8, 111.3, 105.6, 81.5, 75.4, 60.2, 46.4, 43.6, 25.0 ppm. ESI-MS: calc. for C15H13N2O2 [M-H]-: 253.1, found: 253.2. HPLC purity: 94.2% (254 nm), tR: 5.18 min; 92.1% (220 nm), tR: 5.18 min.
(R)-2-Benzyl-2,3,4,9-tetrahydro-1H-pyrido[3,4-b]indole-3-carboxylic acid (9d)
yellow powder. Yield: 52%. 1H NMR (DMSO-d6, 400 MHz): δ = 10.66 (1H, s, 9-NH), 7.40-7.27 (6H, m, -CH2Ph-H and 5-H), 7.25 (1H, d, J = 7.8 Hz, 8-H), 7.03-6.99 (1H, m, 7-H), 6.96-6.93 (1H, m, 6-H), 4.04-3.94 (3H, m, 1α-H and -CH2Ph), 3.86 (1H, dd, J = 6.0 and 3.3 Hz, 3-H), 3.71 (1H, d, J = 15.1 Hz, 1β-H), 3.08 (1H, dd, J = 14.9 and 2.3 Hz, 4β-H), 2.99 (1H, dd, J = 15.3 and 6.0 Hz, 4α-H) ppm. 13C NMR (DMSO-d6, 100 MHz): δ = 174.7, 139.6, 136.4, 132.8, 129.0, 128.8, 127.5, 127.2, 120.9, 118.7, 117.8, 111.3, 105.2, 59.6, 58.7, 45.9, 24.4 ppm. 13C NMR (DMSO-d6, 100 MHz): δ = 173.9, 136.5, 132.2, 127.1, 120.9, 118.7, 117.8, 111.3, 105.1, 59.7, 59.2, 46.3, 24.2, 9.5, 4.5, 3.8 ppm. ESI-MS: calc. for C19H17N2O2 [M-H]-: 305.1, found: 305.3. HPLC purity: 100% (254 nm), tR: 5.53 min; 99.4% (220 nm), tR: 5.53 min.
(R)-2-(Cyclopropylmethyl)-2,3,4,9-tetrahydro-1H-pyrido[3,4-b]indole-3-carboxylic acid (9e)
yellow semisolid. Yield: 42%. 1H NMR (DMSO-d6, 400 MHz): δ = 10.68 (1H, s, 9-NH), 7.34 (1H, d, J = 7.6 Hz, 5-H), 7.24 (1H, d, J = 7.9 Hz, 8-H), 7.00-6.96 (1H, m, 7-H), 6.94-6.90 (1H, m, 6-H), 4.24 (1H, d, J = 15.1 Hz, 1α-H), 3.90 (1H, d, J = 15.1 Hz, 1β-H), 3.66-3.65 (1H, m, 3-H), 3.04 (1H, dd, J = 15.1 and 3.7 Hz, 4β-H), 2.86 (1H, dd, J = 15.2 and 6.0 Hz, 4α-H), 2.70 (2H, d, J = 6.6 Hz, -CH2CH(CH2)2), 0.96-0.86 (1H, m, -CH2CH(CH2)2), 0.53-0.44 (2H, m, -CH2CH(CH2)2), 0.16-0.08 (2H, m, -CH2CH(CH2)2) ppm. 13C NMR (DMSO-d6, 100 MHz): δ = 173.9, 136.5, 132.2, 127.1, 120.9, 118.7, 117.8, 111.3, 105.1, 59.7, 59.2, 46.3, 24.2, 9.5, 4.5, 3.8 ppm. ESI-MS: calc. for C16H17N2O2 [M-H]-: 269.1, found: 269.2. HPLC purity: 99.1% (254 nm), tR: 5.35 min; 98.7% (220 nm), tR: 5.35 min.
(R)-2-(Prop-2-ynyl)-2,3,4,9-tetrahydro-1H-pyrido[3,4-b]indole-3-carboxylic acid (9f)
light brown solid. Yield: 43%. 1H NMR (DMSO-d6, 400 MHz): δ = 10.65 (1H, s, 9-NH), 7.32 (1H, d, J = 7.6 Hz, 5-H), 7.24 (1H, d, J = 7.9 Hz, 8-H), 6.98 (1H, t, J = 7.5 Hz,, 7-H), 6.91 (1H, t, J = 7.2 Hz,, 6-H), 4.12 (1H, d, J = 14.7 Hz, 1α-H), 3.80-3.75 (2H, m, 1β-H and -CH2C≡CH), 3.65 (1H, dd, J = 16.4 and 2.4 Hz, -CH2C≡CH), 3.39 (1H, overlapped with water peak, 3-H), 3.09 (1H, t, J = 2.1 Hz, -CH2C≡CH), 3.00 (1H, dd, J = 14.9 and 5.2 Hz, 4β-H), 2.80 (1H, dd, J = 14.9 and 5.6 Hz, 4α-H) ppm. 13C NMR (DMSO-d6, 100 MHz): δ = 175.3, 136.4, 133.0, 127.4, 120.5, 118.5, 117.7, 111.2, 106.4, 82.1, 75.1, 59.9, 46.7, 43.5, 25.3 ppm. ESI-MS: calc. for C15H13N2O2 [M-H]-: 253.1, found: 253.2. HPLC purity: 98.9% (254 nm), tR: 5.18 min; 98.8% (220 nm), tR: 5.18 min.
4.1.7. General procedure for compounds 10a, b
4-(Benzyloxy)benzyl bromide (1.48 mmol) and N,N-diisopropylethylamine (DIPEA, 1.48 mmol) were added to the solution of 2 (0.74 mmol) in acetonitrile (40 mL). This mixture was heated under reflux for 5 h. After the reaction was complete (monitored by HPLC), the solvent was removed by evaporation. The residue was dissolved in EtOAc and washed with water and saturated NaCl solution. The organic layer was dried over anhydrous Na2SO4 and then filtered. The filtrate was concentrated in vacuo to yield the yellow crude residue, which was further purified by flash column chromatography on silica gel to give products 10a and 10b.
(S)-Methyl 2-(4-(benzyloxy)benzyl)-2,3,4,9-tetrahydro-1H-pyrido[3,4-b]indole-3-carboxylate (10a)
yellow powder. Yield: 58%. 1H NMR (DMSO-d6, 400 MHz): δ = 10.66 (1H, s, 9-NH), 7.47-7.45 (2H, m, Ar-H), 7.42-7.37 (3H, m, Ar-H), 7.35-7.24 (4H, m, Ar-H), 7.03-6.92 (4H, m, Ar-H), 5.10 (2H, s, -OCH2Ph), 3.99-3.95 (2H, m, 3-H and 1α-H), 3.89 (2H, ABq, J = 13.2 Hz, -NCH2Ph), 3.72 (1H, d, J = 15.2 Hz, 1β-H), 3.59 (3H, s, -COOCH3), 3.09-2.98 (2H, m, 4-H) ppm. 13C NMR (DMSO-d6, 100 MHz): δ = 173.3, 158.0, 137.6, 136.4, 132.7, 131.4, 130.3, 128.9, 128.3, 128.1, 127.1, 120.9, 118.8, 117.8, 115.1, 111.3, 104.8, 69.7, 59.2, 58.1, 51.7, 45.7, 24.3 ppm. ESI-MS: calc. for C27H25N2O3 [M-H]-: 425.2, found: 425.4. HPLC purity: 99.0% (254 nm), tR: 6.15 min; 98.6% (220 nm), tR: 6.15 min.
(R)-Methyl 2-(4-(benzyloxy)benzyl)-2,3,4,9-tetrahydro-1H-pyrido[3,4-b]indole-3-carboxylate (10b)
yellow powder. Yield: 61%. 1H NMR (DMSO-d6, 400 MHz): δ = 10.67 (1H, s, 9-NH), 7.46-7.45 (2H, m, Ar-H), 7.42-7.37 (3H, m, Ar-H), 7.35-7.24 (4H, m, Ar-H), 7.03-6.92 (4H, m, Ar-H), 5.09 (2H, s, -OCH2Ph), 3.99-3.95 (2H, m, 3-H and 1α-H), 3.89 (2H, ABq, J = 13.2 Hz, -NCH2Ph), 3.72 (1H, d, J = 15.3 Hz, 1β-H), 3.59 (3H, s, -COOCH3), 3.09-2.98 (2H, m, 4-H) ppm. 13C NMR (DMSO-d6, 100 MHz): δ = 173.3, 158.0, 137.6, 136.4, 132.7, 131.4, 130.3, 128.9, 128.3, 128.1, 127.1, 120.9, 118.8, 117.8, 115.1, 111.3, 104.8, 69.6, 59.2, 58.1, 51.7, 45.7, 24.3 ppm. ESI-MS: calc. for C27H25N2O3 [M-H]- : 425.2, found: 425.5. HPLC purity: 98.6% (254 nm), tR: 6.14 min; 96.5% (220 nm), tR: 6.15 min.
4.1.8. General procedure for compounds 11a, b
A mixture of 10a or 10b (150 mg, 0.35 mmol) and 10% Pd/C (15 mg) in MeOH (10 mL) was stirred under an atmosphere of H2 (1.5 bar) at room temperature for 5 h. The reaction mixture was filtered through a pad of celite. Solvent was removed by evaporation to give the crude product, which was further purified by flash column chromatography on silica gel (hexanes/ethyl acetate = 80/30) to give 11a or 11b.
(S)-Methyl 2-(4-hydroxybenzyl)-2,3,4,9-tetrahydro-1H-pyrido[3,4-b]indole-3-carboxylate (11a)
yellow powder. Yield: 43%. 1H NMR (DMSO-d6, 400 MHz): δ = 10.66 (1H, s, 9-NH), 9.30 (1H, s, -OH), 7.38 (1H, d, J = 7.7 Hz, 5-H), 7.25 (1H, d, J = 7.9 Hz, 8-H), 7.15 (2H, d, J = 8.4 Hz, Ph-H), 7.01 (1H, t, J = 7.9 Hz, 7-H), 6.94 (1H, t, J = 7.2 Hz, 6-H), 6.73 (2H, d, J = 8.4 Hz, Ph-H), 3.97-3.94 (2H, m, 3-H and 1α-H), 3.83 (2H, ABq, J = 13.1 Hz, -NCH2Ph), 3.71 (1H, d, J = 15.6 Hz, 1β-H), 3.59 (3H, s, -COOCH3), 3.07-2.96 (2H, m, 4-H) ppm. 13C NMR (DMSO-d6, 100 MHz): δ = 173.4, 156.9, 136.4, 132.8, 130.3, 129.3, 127.1, 120.9, 118.8, 117.8, 115.5, 111.3, 104.8, 59.1, 58.2, 51.7, 45.6, 24.3 ppm. ESI-MS: calc. for C20H19N2O3 [M-H]-: 335.1, found: 335.3. HPLC purity: 98.1% (254 nm), tR: 5.50 min; 98.2% (220 nm), tR: 5.51 min.
(R)-Methyl 2-(4-hydroxybenzyl)-2,3,4,9-tetrahydro-1H-pyrido[3,4-b]indole-3-carboxylate (11b)
yellow semisolid. Yield: 43%. 1H NMR (DMSO-d6, 400 MHz): δ = 10.66 (1H, s, 9-NH), 9.31 (1H, s, -OH), 7.38 (1H, d, J = 7.7 Hz, 5-H), 7.25 (1H, d, J = 8.0 Hz, 8-H), 7.15 (2H, d, J = 8.4 Hz, Ph-H), 7.01 (1H, t, J = 7.9 Hz, 7-H), 6.94 (1H, t, J = 7.1 Hz, 6-H), 6.73 (2H, d, J = 8.4 Hz, Ph-H), 3.98-3.93 (2H, m, 3-H and 1α-H), 3.83 (2H, ABq, J = 13.0 Hz, -NCH2Ph), 3.71 (1H, d, J = 15.3 Hz, 1β-H), 3.59 (3H, s, -COOCH3), 3.07-2.97 (2H, m, 4-H) ppm. 13C NMR (DMSO-d6, 100 MHz): δ = 173.4, 156.9, 136.4, 132.8, 130.2, 129.3, 127.1, 120.9, 118.8, 117.8, 115.5, 111.3, 104.7, 59.1, 58.2, 51.7, 45.6, 24.3 ppm. ESI-MS: calc. for C20H19N2O3 [M-H]-: 335.1, found: 335.3. HPLC purity: 98.9% (254 nm), tR: 5.50 min; 99.1% (220 nm), tR: 5.50 min.
4.1.9. General procedure for compounds 12a, b
A solution of 11a, or 11b (0.1 mmol) and LiOH (0.5 mmol) in THF/H2O (1:1, 3 mL) was stirred at room temperature until completion of the reaction (monitored by HPLC). THF was removed in vacuo and then the solution was carefully neutralized with glacial acetic acid, whereupon a heavy precipitate was formed. The product was collected by filtration, washed with cold water, and dried.
(S)-Callophycin A (12a)
pale yellow powder. Yield: 66%. 1H NMR (DMSO-d6, 400 MHz): δ = 10.65 (1H, s, 9-NH), 9.33 (1H, br s, -OH), 7.38 (1H, d, J = 7.7 Hz, 5-H), 7.25 (1H, d, J = 7.8 Hz, 8-H), 7.16 (2H, d, J = 8.4 Hz, Ph-H), 7.01 (1H, t, J = 7.5 Hz, 7-H), 6.94 (1H, t, J = 7.3 Hz, 6-H), 6.73 (2H, d, J = 8.4 Hz, Ph-H), 4.00 (1H, d, J = 15.3 Hz, 1α-H), 3.86 (2H, ABq, J = 13.0 Hz, -NCH2Ph), 3.81 (1H, dd, J = 6.2 and 3.4 Hz, 3-H), 3.70 (1H, d, J = 15.1 Hz, 1β-H), 3.06 (1H, dd, J = 15.3 and 2.3 Hz, 4α-H), 2.95 (1H, dd, J = 15.5 and 6.2 Hz, 4β-H) ppm. 13C NMR (DMSO-d6, 100 MHz): δ = 174.5, 156.9, 136.4, 132.7, 130.3, 129.3, 127.1, 120.9, 118.7, 117.8, 115.5, 111.3, 105.1, 59.2, 58.1, 45.7, 24.3 ppm. ESI-MS: calc. for C19H17N2O3 [M-H]-: 321.1, found: 321.3. HPLC purity: 96.3% (254 nm), tR: 5.37 min; 99.0% (220 nm), tR: 5.37 min.
(R)-Callophycin A (12b)
pale yellow powder. Yield: 76%. 1H NMR (DMSO-d6, 400 MHz): δ = 10.65 (1H, s, 9-NH), 9.31 (1H, br s, -OH), 7.38 (1H, d, J = 7.7 Hz, 5-H), 7.25 (1H, d, J = 7.9 Hz, 8-H), 7.16 (2H, d, J = 8.4 Hz, Ph-H), 7.01 (1H, t, J = 7.5 Hz, 7-H), 6.94 (1H, t, J = 7.4 Hz, 6-H), 6.73 (2H, d, J = 8.4 Hz, Ph-H), 4.00 (1H, d, J = 15.1 Hz, 1α-H), 3.87 (2H, ABq, J = 13.1 Hz, -NCH2Ph), 3.82 (1H, dd, J = 6.0 and 3.3 Hz, 3-H), 3.71 (1H, d, J = 15.5 Hz, 1β-H), 3.06 (1H, dd, J = 15.4 and 2.5 Hz, 4α-H), 2.96 (1H, dd, J = 15.3 and 6.0 Hz, 4β-H) ppm. 13C NMR (DMSO-d6, 100 MHz): δ = 174.5, 156.9, 136.4, 132.7, 130.3, 129.3, 127.1, 120.9, 118.7, 117.8, 115.5, 111.3, 105.1, 59.2, 58.1, 45.7, 24.3 ppm. ESI-MS: calc. for C19H17N2O3 [M-H]-: 321.1, found: 321.3. HPLC purity: 98.3% (254 nm), tR: 5.37 min; 97.8% (220 nm), tR: 5.37 min.
4.2. Biology
NFκB luciferase assay
Human embryonic kidney cells 293 were used to monitor any changes occurring along the NFκB pathway. This cell line contains chromosomal integration of a luciferase reporter construct regulated by the NFκB response element. Transcription factors can bind to the response element when stimulated by certain agents, allowing transcription of the luciferase gene. Following an incubation period of 6 h with TNFα and test compounds, cells were analyzed for luciferase activity using the Luc assay system from Promega Corporation (Madison, WI). Results were expressed as a percentage, relative to control (TNFα-treated) samples, and dose-response curves were constructed for the determination of IC50 values, which were generated from the results of five serial dilutions of test compounds and were the mean of two different experiments.
Measurement of the production of NO in LPS-stimulated RAW 264.7 murine macrophage cells (nitrite assay)
The level of NO in the cultured media was estimated by measuring the level of nitrite due to the instability of NO and its subsequent conversion to nitrite. The nitrite assay was performed as previously described.30 Briefly, RAW 264.7 cells were incubated in 96-well culture plates at 37°C, 5% CO2 in a humidified air incubator for 24 h. Then cells were treated with serially diluted compounds for 15 min, followed by treatment with or without LPS (1 μg/mL) for an additional 20 h. After the incubation, nitrite released in the cultured media was measured using Griess reagent [1:1 mixture (v/v) of 1% sulfanilamide in 5% H3PO4 and 0.1% N-(1-naphthyl)ethylenediamine dihydrochloride solution], and absorbance was measured at 540 nm. The concentration of nitrite was calculated using a standard curve created with known concentrations of sodium nitrite. To evaluate the cytotoxic effects of compounds with RAW 264.7 cells under the same experimental condition, the SRB assay was performed.
Evaluation of antiproliferative effect of compounds in MCF7 breast cancer cells (sulforhodamine B assay)
The effect of compounds on MCF7 breast cancer cell proliferation was evaluated using the sulforhodamine B (SRB) cellular protein-staining method31. In brief, MCF7 cells (1 × 104 cells in 190 μl of the complete media) were plated in 96-well plates containing compounds and incubated at 37°C, 5% CO2 in humidified air for 72 h. After the incubation, cells were fixed with 10% trichloroacetic acid solution for 30 min and stained with 0.4% SRB in 1% acetic acid solution for 30 min. After washing with 1% acetic acid solution, protein-bound SRB was dissolved in 10 mM Tris buffer (pH 10.0) and the absorbance was measured at 515 nm. The effect of compounds on cell survival was demonstrated as % survival in comparison with vehicle (DMSO)-treated control cells.
Determination of QR1 activity in cell culture
QR1 was assessed using Hepa 1c1c7 murine hepatoma cells as previously reported.32 QR1 was measured as a function of the NADPH-dependent menadiol-mediated reduction of 3-(4,5-dimetylthiazo-2-yl)-2,5-diphenyltetrazolium bromide (MTT) to a blue formazan. Protein content was determined via crystal violet staining of identical plates. Specific activity is defined as nmol of formazan formed per mg protein per min. The induction ratio (IR) of QR activity represents the specific enzyme activity of agent-treated cells compared with a DMSO-treated control. The concentration to double activity (CD) was determined through a dose-response assay for active substances (IR >2).
Aromatase assay
Aromatase activity was assayed as previously reported33 with the necessary modifications to perform the test in 384-well plates. The test substance (3.5 μL at 20 μg/mL) was pre-incubated with 30 μL of NADPH regenerating system (2.6 mM NADP+, 7.6 mM glucose 6-phosphate, 0.8 U/mL glucose 6-phosphate dehydrogenase, 13.9 mM MgCl2, and 1 mg/mL albumin in 50 mM potassium phosphate buffer, pH 7.4) for 10 min. The enzyme and substrate mixture [33 μL of 1 μM enzyme (165 μU CYP19, BD Biosciences), 0.4 μM dibenzylfluorescein, and 4 mg/mL albumin in 50 mM potassium phosphate, pH 7.4] was added and the plate was incubated for 30 min at 37 °C before quenching with 25 μL of 2 N NaOH solution. After termination of the reaction and shaking for 5 min, the plate was further incubated for 2 h at 37 °C. This enhances the ratio of signal to background. Fluorescence was measured using a BioTek Synergy 2 plate reader at 485 nm (excitation) and 530 nm (emission). IC50 values and dose-response curves were based on three independent experiments performed in duplicate using five concentrations of tested substance.
Supplementary Material
Acknowledgments
We thank UH Hilo CoP for start-up funding. This work was also supported in part by NCI P01 CA48112 (JMP) and NCRR INBRE program P20 RR016467 (DS). We thank Annie Ma at Phenomenex for the assistance with the chiral HPLC method development. We also thank Dr. Cherie A. Motti of the Australian Institute of Marine Sciences (AIMS), Queensland, Australia, for providing NMR spectra of the isolated natural callophycin A.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supplementary data
Supplementary data (1H and 13C NMR spectra and HPLC chromatographs of compounds 2-12 and HSQC, HMBC, and COSY spectra of 12b) associated with this article can be found, in the online version, at doi:
References and notes
- 1.Düsman Tonin LT, Barbosa VA, Bocca CC, Ramos ÉRF, Nakamura CV, Ferreira da Costa W, Basso EA, Nakamura TU, Sarragiotto MH. Eur. J. Med. Chem. 2009;44:1745. doi: 10.1016/j.ejmech.2008.03.044. [DOI] [PubMed] [Google Scholar]
- 2.Kumar R, Gupta L, Pal P, Khan S, Singh N, Katiyar SB, Meena S, Sarkar J, Sinha S, Kanaujiya JK, Lochab S, Trivedi AK, Chauhan PMS. Eur. J. Med. Chem. 2010;45:2265. doi: 10.1016/j.ejmech.2010.02.001. [DOI] [PubMed] [Google Scholar]
- 3.Barsanti PA, Wang W, Ni Z-J, Duhl D, Brammeier N, Martin E, Bussiere D, Walter AO. Bioorg. Med. Chem. Lett. 2010;20:157. doi: 10.1016/j.bmcl.2009.11.012. [DOI] [PubMed] [Google Scholar]
- 4.Yeung BK, Zou B, Rottmann M, Lakshminarayana SB, Ang SH, Leong SY, Tan J, Wong J, Keller-Maerki S, Fischli C, Goh A, Schmitt EK, Krastel P, Francotte E, Kuhen K, Plouffe D, Henson K, Wagner T, Winzeler EA, Petersen F, Brun R, Dartois V, Diagana TT, Keller TH. J. Med. Chem. 2010;53:5155. doi: 10.1021/jm100410f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gupta L, Srivastava K, Singh S, Puri SK, Chauhan PMS. Bioorg. Med. Chem. Lett. 2008;18:3306. doi: 10.1016/j.bmcl.2008.04.030. [DOI] [PubMed] [Google Scholar]
- 6.Bi W, Bi L, Cai J, Liu S, Peng S, Fischer NO, Tok JBH, Wang G. Bioorg. Med. Chem. Lett. 2006;16:4523. doi: 10.1016/j.bmcl.2006.06.024. [DOI] [PubMed] [Google Scholar]
- 7.Bi W, Cai J, Liu S, Baudy-Floc’h M, Bi L. Bioorg. Med. Chem. 2007;15:6909. doi: 10.1016/j.bmc.2007.08.022. [DOI] [PubMed] [Google Scholar]
- 8.Miller JF, Turner EM, Sherrill RG, Gudmundsson K, Spaltenstein A, Sethna P, Brown KW, Harvey R, Romines KR, Golden P. Bioorg. Med. Chem. Lett. 2010;20:256. doi: 10.1016/j.bmcl.2009.10.123. [DOI] [PubMed] [Google Scholar]
- 9.Jenkins PR, Wilson J, Emmerson D, Garcia MD, Smith MR, Gray SJ, Britton RG, Mahale S, Chaudhuri B. Bioorg. Med. Chem. 2008;16:7728. doi: 10.1016/j.bmc.2008.07.002. [DOI] [PubMed] [Google Scholar]
- 10.Ovenden SPB, Nielson JL, Liptrot CH, Willis RH, Tapiolas DM, Wright AD, Motti CA. Phytochem. Lett. 2011;4:69. [Google Scholar]
- 11.Part of this work has been presented at the following meeting. Shen L, Park E-J, Kondratyuk TP, Guendisch D, Marler L, Pezzuto JM, Wright AD, Sun D. 241st American Chemical Society National Meeting; Anaheim, CA, USA. March 27-31, 2011; MEDI-40.
- 12.Foppoli C, Marco FD, Blarzino C, Perluigi M, Cini C, Coccia R. Int. J. Biochem. Cell Biol. 2005;37:852. doi: 10.1016/j.biocel.2004.11.002. [DOI] [PubMed] [Google Scholar]
- 13.Dinkova-Kostova AT, Talalay P. Free Radical Biol. Med. 2000;29:231. doi: 10.1016/s0891-5849(00)00300-2. [DOI] [PubMed] [Google Scholar]
- 14.Siiteri PK. Cancer Res. 1982;42:3269s. [PubMed] [Google Scholar]
- 15.James VH, McNeill JM, Lai LC, Newton CJ, Ghilchik MW, Reed M. J. Steroids. 1987;50:269. doi: 10.1016/0039-128x(83)90077-6. [DOI] [PubMed] [Google Scholar]
- 16.Brueggemeier RW, Hackett JC, Diaz-Cruz ES. Endocr. Rev. 2005;26:331. doi: 10.1210/er.2004-0015. [DOI] [PubMed] [Google Scholar]
- 17.Miller WR. Semin. Oncol. 2003;30:3. doi: 10.1016/s0093-7754(03)00302-6. [DOI] [PubMed] [Google Scholar]
- 18.Recanatini M, Cavalli A, Valenti P. Med. Res. Rev. 2002;22:282. doi: 10.1002/med.10010. [DOI] [PubMed] [Google Scholar]
- 19.Schupp PJ, Kohlert-Schupp C, Whitefield S, Engemann A, Rohde S, Hemscheidt T, Pezzuto JM, Kondratyuk TP, Park EJ, Marler L, Rostama B, Wright AD. Nat. Prod. Commun. 2009;4:1717. [PMC free article] [PubMed] [Google Scholar]
- 20.Luqman S, Pezzuto JM. Phytother. Res. 2010;24:949. doi: 10.1002/ptr.3171. [DOI] [PubMed] [Google Scholar]
- 21.S- and R-1,2,3,4-tetrahydro-β-carboline-3-carboxylic acids were purchased from Chem-Impex International, Inc., IL, USA.
- 22.Liu J, Jiang X, Zhao M, Zhang X, Zheng M, Peng L, Peng S. J. Med. Chem. 2010;53:3106. doi: 10.1021/jm901816j. [DOI] [PubMed] [Google Scholar]
- 23.Kumpaty HJ, Van Linn ML, Kabir MS, Forsterling FH, Deschamps JR, Cook JM. J. Org. Chem. 2009;74:2771. doi: 10.1021/jo8028168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lopez-Rodriguez ML, Morcillo MJ, Garrido M, Benhamu B, Perez V, Campa J. G. d. l. J. Org. Chem. 1994;59:1583. [Google Scholar]
-
25.Representative characterization data of cyclization hydantoin byproduct of 6c:
 1H NMR (DMSO-d6, 400 MHz): δ = 11.03 (1H, s, 9-NH), 7.47 (1H, d, J = 7.8 Hz, 5-H), 7.35 (1H, d, J = 8.1 Hz, 8-H), 7.30-7.27 (2H, m, -CH2CH2PhH), 7.22-7.19 (3H, m, -CH2CH2PhH), 7.11-7.07 (1H, m, 7-H), 7.00 (1H, t, J = 7.3 Hz, 6-H), 4.89 (1H, d, J = 16.2 Hz, 1α-H), 4.41-4.34 (2H, m, 1β-H and 3-H), 371-3.66 (2H, m, -NCH2CH2Ph), 3.19 (1H, dd, J = 14.9 and 5.2 Hz, 4β-H), 2.90 (2H, t, J = 7.3 Hz, -NCH2CH2Ph), 2.60-2.53 (1H, m, 4α-H) ppm. ESI-MS: calc. for C21H20N3O2 [M+H]+: 346.2, found: 346.4. HPLC purity: 96.5% (254 nm), tR: 6.88 min; 96.9% (220 nm), tR: 6.88 min.
1H NMR (DMSO-d6, 400 MHz): δ = 11.03 (1H, s, 9-NH), 7.47 (1H, d, J = 7.8 Hz, 5-H), 7.35 (1H, d, J = 8.1 Hz, 8-H), 7.30-7.27 (2H, m, -CH2CH2PhH), 7.22-7.19 (3H, m, -CH2CH2PhH), 7.11-7.07 (1H, m, 7-H), 7.00 (1H, t, J = 7.3 Hz, 6-H), 4.89 (1H, d, J = 16.2 Hz, 1α-H), 4.41-4.34 (2H, m, 1β-H and 3-H), 371-3.66 (2H, m, -NCH2CH2Ph), 3.19 (1H, dd, J = 14.9 and 5.2 Hz, 4β-H), 2.90 (2H, t, J = 7.3 Hz, -NCH2CH2Ph), 2.60-2.53 (1H, m, 4α-H) ppm. ESI-MS: calc. for C21H20N3O2 [M+H]+: 346.2, found: 346.4. HPLC purity: 96.5% (254 nm), tR: 6.88 min; 96.9% (220 nm), tR: 6.88 min.
- 26.Nieddu G, De Luca L, Giacomelli G. Synthesis 2008. 2008:3947. [Google Scholar]
- 27.According to the HPLC monitoring of the reaction mixture, about 30% of byproduct 2a or 2b was observed due to the further N-debenzylation of the corresponding product 11a or 11b. The byproduct can be easily removed by flash column chromatography on silica gel.
- 28.Identical 1H NMR spectra were obtained for all the pairs of enantiomers except for two pairs of alkylation products (9b and 9e, 9c and 9f) in which slight chemical shifts of 1-H, 3-H, 4-H, and protons from N2 substituted group were observed likely due to the interaction of carboxylate with N2-propargyl or cyclopropylmethyl group.
- 29.Saxena AK, Pandey SK, Tripathi RC, Raghubir R. Bioorg. Med. Chem. 2001;9:1559. doi: 10.1016/s0968-0896(01)00042-6. [DOI] [PubMed] [Google Scholar]
- 30.Hoshino J, Park EJ, Kondratyuk TP, Marler L, Pezzuto JM, van Breemen RB, Mo S, Li Y, Cushman M. J. Med. Chem. 2010;53:5033. doi: 10.1021/jm100274c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Park E-J, Min H-Y, Chung H-J, Hong J-Y, Kang Y-J, Hung TM, Youn UJ, Kim YS, Bae K, Kang SS, Lee SK. Cancer Lett. 2009;277:133. doi: 10.1016/j.canlet.2008.11.029. [DOI] [PubMed] [Google Scholar]
- 32.Song LL, II JWK, Lee SK, Gerhäuser C, Lantvit D, Moon RC, Moriarty RM, Pezzuto JM. Cancer Res. 1999;59:578. [PubMed] [Google Scholar]
- 33.Maiti A, Cuendet M, Croy VL, Endringer DC, Pezzuto JM, Cushman M. J. Med. Chem. 2007;50:2799. doi: 10.1021/jm070109i. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.