

Abstract
Bifunctional alkyalating agent, Sulfur mustard (SM)-caused cutaneous injury is characterized by inflammation and delayed blistering. Our recent studies demonstrated that 2-chloroethyl ethyl sulfide (CEES), a monofunctional analog of SM that can be used in laboratory settings, induces oxidative stress. This could be the major cause of the activation of Akt/MAP kinase and AP1/NF-κB pathways that are linked to the inflammation and microvesication, and histopathological alterations in SKH-1 hairless mouse skin. To further establish a link between CEES-induced DNA damage and signaling pathways and inflammatory responses, skin samples from mice exposed to 2 or 4 mg CEES for 9–48 h were subjected to molecular analysis. Our results show a strong CEES-induced phosphorylation of H2A.X and an increase in COX-2, iNOS, and MMP-9 levels, indicating the involvement of DNA damage and inflammation in CEES-caused skin injury in male and female mice. Since, our recent studies showed reduction in CEES-induced inflammatory responses by glutathione (GSH), we further assessed the role of oxidative stress in CEES-caused DNA damage and the induction of inflammatory molecules. Oral GSH (300mg/kg) administration 1 h before CEES exposure attenuated the increase in both CEES-induced H2A.X phosphorylation (59%) as well as expression of COX-2 (68%), iNOS (53%) and MMP-9 (54%). Collectively, our results indicate that CEES-induced skin injuries involve DNA damage and an induction of inflammatory mediators, at least in part via oxidative stress. This study could help in identifying countermeasures that alone or in combination, can target the unveiled pathways for reducing skin injuries in humans by SM.
Keywords: Sulfur mustard, skin injury, inflammatory mediators, COX-2, iNOS, MMP-9, DNA damage, SKH-1 hairless mice, CEES, oxidative stress
1. Introduction
Sulfur mustard (bis (2-chloroethyl) sulfide, SM) is a powerful vesicating (blistering) and bifunctional alkylating warfare agent, which readily penetrates the skin causing histopathological changes, related to inflammation, and delayed blistering (Wormser, 1991; Dacre and Goldman, 1996; Pal et al., 2009). The first target of SM is the basal epidermal keratinocytes; it causes apoptotic cell death followed by protease digestion of anchoring filaments at epidermal-dermal junctions leading to a skin blistering response via epidermal-dermal separation (Kan et al., 2003; Greenberg et al., 2006; Hayden et al., 2009). The mechanism/s of SM-caused responses has been linked mainly to its alkylating property, and its effect on nucleophilic antioxidant, and glutathione (GSH) depletion. GSH depletion could lead to oxidative stress and macromolecular damage, including that of DNA that possibly induces the activation of signaling pathways (Rice, 2003; Kehe and Szinicz, 2005; Paromov et al., 2007; Brimfield et al., 2009; Jowsey et al., 2009; Laskin et al., 2010; Shakarjian et al., 2010; Tewari-Singh et al., 2010). The cellular GSH depletion could be due to its conjugation with SM, and protective effects of exogenous GSH and its precursor, NAC, in reducing SM or its analog-induced skin toxicity have been reported (Gross et al., 1993; Smith et al., 1997; Amir et al., 1998; Han et al., 2004; Arfsten et al., 2007; Paromov et al., 2007).
Several reported studies including a study in cylooxygenase-2 (COX-2) null mice suggest that COX-2, and COX-derived prostaglandins are important inflammatory mediators in SM-induced skin toxicity (Casillas et al., 2000; Nyska et al., 2001; Wormser et al., 2004). Nitric oxide synthases (NOSs), mainly endothelial NOS (eNOS) and inducible NOS (iNOS), predominantly found in mononuclear phagocytes and neutrophils, are the proposed important inflammatory mediators in SM-induced skin toxicity (Nyska et al., 2001; Gao et al., 2007). SM induced up-regulation of iNOS expression is shown to lead to the production of nitric oxide (NO), a potent oxidizing agent. However, at lower concentrations, NO acts as an antioxidant and plays a role in would healing (Gao et al., 2007). After SM exposure, matrix metalloproteinase (MMP) family proteases are reported to be up-regulated causing the degradation of extracellular proteins, which are the major component of basement membrane and separate epidermis from dermis causing vesication (Shakarjian et al., 2010). Constitutively expressed MMP-2 and MMP-9 (induced by cytokines, chemokines) are the most studied MMPs in SM-induced skin toxicity. Additionally, MMP-9 has been shown to play an important role in SM-induced skin toxicity using the mouse ear vesicant model (Johansson et al., 1997; Van den Steen et al., 2002; Shakarjian et al., 2006).
Because the molecular mechanism/s involved in SM-induced blistering and skin inflammation are not completely known, the identification of effective medical countermeasures for SM-caused skin injuries has not been possible. Consequently, to further understand the mechanism of SM-caused skin injury, we conducted studies in the SKH-1 hairless mouse using the monofunctional sulfur mustard analog, 2-chloroethyl ethyl sulfide (CEES). Due to the requirement of containment facilities for SM, CEES has been widely used as a valid experimental alternative to SM for studies in laboratory settings (Han et al., 2004; Jowsey et al., 2009; Tewari-Singh et al., 2009). This research demonstrated the induction of oxidative stress by CEES leading to the activation of transcription factors AP-1 and nuclear factor- kB (NF-kB) via upstream signaling pathways including mitogen activated protein kinases (MAPKs) and Akt, which were identified as possible mechanisms of the inflammatory responses observed in this skin injury model (Pal et al., 2009; Tewari-Singh et al., 2009). To further establish a link between CEES-induced signaling pathways and inflammatory responses, our objective in the present study was to analyze the involvement of DNA damage and inflammatory mediators in CEES-induced inflammatory and microvesication responses in SKH-1 hairless mice. Furthermore, because our earlier studies showed the involvement of GSH and oxidative stress in CEES-induced molecular and biological responses (Pal et al., 2009; Tewari-Singh et al., 2009; Tewari-Singh et al., 2011), we also assessed their impact on DNA damage and the expression of inflammatory mediators in the present study. This study is a valuable addition in understanding of the mechanism of action of SM analog. Additionally, the study outcomes could help in the selection of countermeasures, which alone or in combination can target the identified pathways to attenuate SM and other vesicating agents’ related skin injuries in humans.
2. Material and Methods
2.1. Materials
The sulfur mustard analog, CEES was from Sigma-Aldrich Chemicals Co (St. Louis, MO). MMP-2 antibody was from Calbiochem (San Diego, CA). MMP-9, H2A.X phospho ser139 antibodies and anti-rabbit IgG horseradish peroxidase (HRP) conjugated secondary antibody was from Cell Signaling Technology (Beverley, MA). COX-1 and COX-2 antibodies were from Cayman chemicals (Ann Arbor, MI), iNOS antibody was from Abcam (Cambridge, MA) and β-actin antibody was from Sigma-Aldrich Co. (St. Louis, MO). Protein assay kit was from Bio-Rad laboratories (Hercules, CA). Secondary anti-mouse IgG HRP linked antibody and enhanced chemiluminescence western blot detection reagents were from Amersham Biotech (Piscataway, NJ). Other chemicals used were from Sigma-Aldrich (St. Louis, MO) unless otherwise specified in the protocol.
2.2. Animals
Male and female SKH-1 hairless mice (5 animals per treatment group; 4–5 weeks of age) were purchased from Charles River Laboratories and housed under standard conditions at the Centre of Laboratory Animal Care, University of Colorado Denver, CO. The animals were acclimatized for one week before their use in our experimental work, which was carried out according to a specified protocol approved by the Institutional Animal Care and Use Committee (IACUC) of the University of Colorado Denver, CO, USA.
2.3. CEES exposure
CEES was dissolved in acetone, and a dose of 2 mg (80 mg/kg) CEES in 200 μl acetone/mouse or acetone alone was applied topically on the dorsal skin of male and female SKH-1 hairless mice for 9, 12, 24 and 48 h as reported previously (Tewari-Singh et al., 2009). Similarly, 4 mg (160 mg/kg) CEES in 200 μl acetone/mouse was applied topically on the dorsal skin of male SKH-1 hairless mice for the above time points. The use of 2 or 4 mg CEES dose was based on the earlier reported doses of CEES used in animals, toxic doses of SM that cause microvesication, and taking into consideration the lesser toxic effect of CEES (Kehe et al., 2008). We have used 2 mg (80 mg/kg) CEES dose and established skin injury model as published earlier (Tewari-Singh et al., 2009), and a higher dose of 4 mg (160 mg/kg) CEES was employed to induce microvesication not observed with the lower dose in these mice (Jain et al., 2011). Comparison in the toxic responses related to 2 mg CEES exposure was carried out in both male and female mice that showed comparable inflammatory responses in our earlier published studies (Tewari-Singh et al., 2009; Jain et al., 2011). However, due to the possibility that males have more chances of exposure to this warfare agent in the battlefield as well as first responders in case of a terrorist attack, male mice were chosen for further studies using a higher dose of 4 mg CEES that induced microvesication in these mice. Acetone, a well established vehicle for skin topical treatments, was used as a vehicle for CEES application due to its ability to enhance the skin permeability to hydrophilic and amphipathic compounds (Tsai et al., 2001; Tewari-Singh et al., 2009). An untreated mice group was included as a control. For both male and female mice, five animals per control and CEES exposed groups were utilized. At the end of each experiment, mice were euthanized and dorsal skin was collected as described earlier (Tewari-Singh et al., 2009) and either snap frozen using liquid nitrogen or fixed in formalin for immunohistochemical (IHC) analysis.
2.4. GSH treatment
GSH was given at a dose of 300 mg/kg of body weight in 200μl saline by oral gavage, and five female mice were then either untreated, exposed to 2 mg CEES topically on the dorsal skin or exposed to acetone as vehicle control. At the end of the experiment, mice were euthanized and dorsal skin was collected and snap frozen using liquid nitrogen or fixed in formalin for immunohistochemistry (IHC).
2.5. Preparation of tissue lysates and western blot analysis
Approximately 100 mg of tissue was taken from the skin samples of two animals per control or CEES exposed group that were chosen randomly to avoid any bias. Subcutaneous fatty tissue and blood capillaries were removed. After washing with cold PBS, skin tissues were homogenized in lysis buffer (10mM Tris-HCL, 150 mM NaCl, 1% Triton X-100, 1mM EDTA, 1mM EGTA, 0.1 mM DTT, 0.5mM PMSF, 0.2 mM Sodium Orthovandate, 5μg/ml Aprotinin, 1.25 μg/ml Benzamidine and 0.5% NP-40) using a biospec homogenizer to prepare total cytosolic and nuclear lysates according to a previously published protocol (Gu et al., 2005; Gu et al., 2007). After preparation of aliquots, protein content was measured using Lowry’s method (Lowry et al., 1951). For Western blotting, 50 μg of protein per sample was denatured in 2x SDS–PAGE sample buffers and subjected to SDS-PAGE on 8, 12 or 16% polyacrylamide tris-glycine gels followed by immunoblotting as described earlier (Gu et al., 2005; Gu et al., 2007).. After blocking in 5% milk in TBST for 1 h, membranes were probed with primary antibodies against H2A.X at S139, COX-1, COX-2, iNOS, MMP-2 and MMP-9 for over night at 4°C, followed by peroxidase-conjugated appropriate secondary antibodies for 1 h at room temperature. Protein bands were visualized by an enhanced chemiluminescence detection system (GE Healthcare Life Sciences, Pittsburgh, PA). In each case, protein loading was monitored by stripping and re-probing the blots with β-actin. The autoradiograms/bands were scanned and where mentioned, mean density of bands was determined using Adobe Photoshop 6.0 (Adobe Systems, San Jose, CA). The densitometry analysis of the respective protein bands was performed using a Scion image program (NIH, Bethesda, MD). Change in mean band intensity is reported in terms of fold change to vehicle control and the values of band intensities were adjusted with β-actin.
2.6. Immunohistochemistry
The paraffin embedded sections were deparaffinized, rehydrated and antigen retrieval performed on them using 10 mM sodium citrate (pH 6.0) in a decloaking chamber for 30 min at 70°C. The endogenous peroxide activity was blocked using 3% hydrogen peroxide in methanol (v/v) and the sections were incubated with rabbit polyclonal phospho-H2A.X ser139 antibody at 1:100 dilution in PBS overnight at 4°C in a humidity chamber. The N-Universal negative control rabbit IgG antibody (DAKO) was used as a negative control. After washings in PBS, the sections were incubated with the anti-rabbit biotinylated secondary antibody for 1 h. This was followed by incubation with HRP conjugated streptavidin (DAKO) in PBS for 30 min at room temperature (RT) in a humidity chamber. The sections were then incubated in DAB (3,3′-diaminobenzidine) working solution for 5 min at RT and counterstained with diluted hematoxylin for 2 min followed by dehydration and mounting for microscopic observation. The brown colored DAB positive nuclei were counted in 10 randomly selected fields (400X magnification). The DNA damage nuclei index was determined by counting number of H2A.X ser 139 stained positive cells X 100/total number of cells (Black et al., 2010).
2.7. Statistical analysis
All the IHC stained slides were observed under a Zeiss Axioscop 2 microscope (Carl Zeiss, Inc., Germany), and image analysis was done using Carl Zeiss Axiovision Rel 4.5 software. The data were analyzed and all statistics calculations done using SigmaStat (software version 2.03, Jandal Scientific Corp., San Raphael, CA). Data are expressed as mean ± SEM (Standard Error Mean) and analyzed via one way ANOVA, followed by the Bonferroni t-test for multiple comparisons. P< 0.05 was considered to be statistically significant.
3. Results
3.1. Topical application of CEES causes an increase in H2A.X phosphorylation at ser139
SM is a highly reactive alkylating agent and its skin exposure is reported to lead to DNA damage, eventually inducing cell death, inflammation and cytotoxicity (Rogakou et al., 1998; Jowsey et al., 2009; Black et al., 2010). The DNA damage, especially the double strand breaks, triggers the phosphorylation of histone variant H2A.X at serine 139 and is considered to be a reporter of DNA double strand breaks required for cell cycle checkpoint arrest and DNA repair (Rogakou et al., 1998; Huang and Darzynkiewicz, 2006). Herein, we analyzed if the CEES-induced inflammatory and blistering response observed in our earlier study (Pal et al., 2009) is associated with DNA damage in SKH-1 hairless mouse skin. Phosphorylation of H2A.X at ser139 was evaluated by western blot analysis and IHC in the skin tissue exposed to CEES for 9–48 h. As shown in Fig. 1A, 2 mg CEES exposure increased H2A.X phosphorylation to 1.7 fold at 9 h of exposure as compared to vehicle control and was maxed out at 12 h and 48 h (3 or 3.1 fold), then decreasing at 24 h (2.1 fold) after exposure in female mice. The male mice study with 2 mg CEES exposure showed maximum phosphorylated protein expression of H2A.X at 12 h (7.4 fold) and at 24 h (5.5 fold) of exposure in comparison to vehicle control (Fig. 1B). Compared to the vehicle control group, 4 mg CEES exposure also resulted in an increase in H2A.X phosphorylation, which was 1.7, 1.5 and 4.7 fold at 12, 24 and 48 h exposure, respectively (Fig. 1C). Similar results were observed in IHC analysis for H2A.X ser139 phosphorylation in the skin tissue of male mice exposed to 4 mg CEES (Fig. 1D and E). An increase in the H2A.X ser139 phosphorylated epidermal cells (red arrows; Fig. 1D) at all time-points was observed following CEES exposure in comparison with the control group (Fig 1D and E).
Figure 1.

Effect of topical CEES exposure on H2A.X phosphorylation, a marker of DNA damage, on female and male SKH-1 hairless mouse skin. Female mice were treated topically with 2 mg of CEES (A) and male with 2 mg (B) or 4 mg (C–E) of CEES or acetone alone. Dorsal skin was collected at 9, 12, 24 and 48 h time points and either skin tissue lysates were prepared for western immunoblotting of H2A.X ser139 phosphorylation (A–C) or skin samples were processed for IHC staining of H2A.X ser139 (D and E) as described in the Material and Methods. Data are presented as fold increase in comparison with their respective vehicle controls and values of band intensities were adjusted with β-actin loading control (A–C). Data are representative of the results from randomly chosen two CEES exposed animals taken at each time point of the study for both male and female mice (A–C). Following IHC analysis (panel D) in the skin tissue samples of male mice exposed to 4 mg CEES, H2A.X ser 139 phosphorylation positive cells were counted in five randomly selected fields per tissue section (400X magnification); data presented are mean ± SEM of five animals in each group (E). UC, untreated control; VC, vehicle (acetone) control; red arrows, H2A.X ser139 phosphorylation-positive cells.
3.2. Topical application of CEES induces COX-2 and iNOS expression
Cyclooxygenase enzymes, which play an important role in skin inflammation, exist in two isoforms; COX-1 and COX-2 (Smith et al., 2000). COX-1 is expressed constitutively while COX-2 is an inducible enzyme and is expressed in mononuclear phagocytes and neutrophils (Gilroy et al., 1999). COX-2, involved in prostaglandin biosynthesis, plays an important role in SM-induced skin inflammation by increasing the capillary permeability which lead to influx of inflammatory cells and other inflammatory mediator at the site of the injury (Shakarjian et al., 2010). To investigate the role of these enzymes in mediating the CEES-caused inflammatory and microvesicating responses and their link with the activation of signaling cascades observed in our earlier studies, we analyzed the expression of COX-1 and COX-2 in a dose (2 and 4 mg) and a time (9–48 h) response study in SKH-1 hairless mice. Western immunoblot analysis of skin samples showed that COX-2 expression increased following 2 mg of CEES exposure to the dorsal skin of both female and male mice, and following 4 mg of CEES exposure in male mice (Fig. 2). Mean band intensity measurements revealed that COX-2 expression was maximum at 24 h (3.0 fold) and 48 h (3.3 fold) in female mice exposed to 2 mg CEES (Fig. 2A). In male mice, 2 mg CEES exposure caused 1.8–2.7-fold increase in COX-2 levels at 9–48 h of exposure in comparison with the vehicle control group (Fig. 2B). Exposure to the 4 mg CEES dose also showed a strong induction in COX-2 expression, with a maximum at 24 h (4.3 fold) of exposure (Fig. 2C). On the other hand, a CEES-induced increase of 1.5 fold or more in COX-1 expression was not observed in both male and female mice (Fig. 2A–C).
Figure 2.

Effect of topical CEES exposure on the expression of COX-2, COX-1 and iNOS in female and male SKH-1 hairless mouse skin. Female mice were treated topically with 2 mg (A) of CEES or acetone alone and male mice were treated with 2 mg (B) or 4 mg (C) of CEES or acetone alone. Dorsal skin tissue was collected at 9, 12, 24 and 48 h time points following CEES exposure and lysates were prepared. Western blot analysis was carried out to assess the expression of COX-1, COX-2 and iNOS using specific antibodies as described under Material and Methods. The membranes were then stripped and reprobed for β-actin as a loading control. Data are presented as fold increase in comparison with their respective vehicle controls and values of band intensities were adjusted with β-actin loading control (A–C). Data are representative of the results from randomly chosen two CEES exposed animals taken at each time point of the study for both male and female mice. UC, untreated control; VC, vehicle (acetone) control.
It has been reported that iNOS and associated NO play an important role in SM-induced skin injury (Shimizu et al., 1997; Korkmaz et al., 2006; Ishida et al., 2008). Similar to COX immunoblot analysis, iNOS was also analyzed in the dorsal skin tissue from the mice exposed to 2 or 4 mg CEES. Results showed enhanced iNOS expression following 2 or 4 mg CEES exposure in both male and female mice (Fig. 2). In female mice, there was more than a two-fold increase in the expression of iNOS following 2 mg CEES exposure for 9–24 h in comparison to the vehicle control group (Fig 1A). In the male mouse study, 2 mg CEES exposure caused more than a 4-fold increase in the iNOS expression at 9, 12 and 24 h then declined at 48 h (Fig. 2B). On exposure to a higher dose of 4 mg CEES for 24 h, a two-fold increase in the iNOS expression as compared to vehicle control group was observed in male mice (Fig. 2C).
3.3. Topical application of CEES induces MMP expression
SM exposure causes an activation of proteolytic enzymes that degrade components of the extracellular matrix and basement membrane causing epidermal-dermal separation followed by blister formation on the skin (Cowan et al., 2003). Gelatinases (MMP-2 and MMP-9) are reported to be major proteases in SM-induced blistering and skin injury (Johansson et al., 1997; Kahari and Saarialho-Kere, 1997; Ravanti and Kahari, 2000; Ries et al., 2009). Our previous studies have shown microvesication and an inflammatory response with CEES (Jain et al., 2011). Therefore, we assessed the involvement of MMP-2 and MMP-9 in CEES-induced skin injury in SKH-1 hairless mice in this study. CEES exposure at both the 2 and 4 mg doses resulted in a strong induction of MMP-9 protein; however, a much smaller increase in MMP-2 was observed in response to CEES as compared to controls (Fig. 3). In female SKH-1 hairless mice, exposure to 2 mg CEES resulted in a 6-fold increase in MMP-9 expression at 9 h. This progressively decreased at 12 h (5.5 fold), 24 h (5.4 fold) and minimum at 48 h (3.5 fold) in comparison with the vehicle control (Fig. 3A). In the male mouse study, 2 mg CEES exposure caused an increase in the MMP-9 expression at 9, 12, 24 and 48 h, which was 1.9, 3.7, 2.9 and 3.2.-fold greater than respective vehicle control, (Fig. 3B). Exposure to 4 mg CEES for 24 h induced a maximum MMP-9 expression of 3.5 fold (Fig. 3C). With regard to MMP-2, the 2 mg CEES dose in male mice resulted in a maximum increase in MMP-2 expression at 12 h (2.0 fold), which did not increase further with the larger CEES dose of 4 mg (Fig. 3).
Figure 3.

Effect of topical CEES exposure on the expression of MMP-9 and MMP-2 in female and male SKH-1 hairless mouse skin. Female mice were treated topically with 2 mg (A) of CEES or acetone alone and male mice with 2 mg (B) or 4 mg (C) of CEES or acetone alone. Dorsal skin tissue was collected at 9, 12, 24 and 48 h time points following CEES exposure and lysates were prepared. Western blot analysis was carried out to assess the expression of MMP-9 and MMP-2 using specific antibodies as detailed under material and methods. The membranes were then stripped and reprobed for β-actin as a loading control. Data are presented as fold increase in comparison with their respective vehicle control and values of band intensities were adjusted with β-actin loading control (A–C). Data are representative of the results from randomly chosen two CEES exposed animals taken at each time point of the study for both male and female mice. UC, untreated control; VC, vehicle (acetone) control. Values of band intensities were adjusted with β-actin and data presented are representative of the results from two animals at each time point of the study.
3.4. GSH treatment reduces the CEES-caused increase in H2A.X ser139 phosphorylation and COX-2, iNOS and MMP9 levels
GSH is an important antioxidant found in tissues or cells, which protects cells from oxidative stress (Meister and Anderson, 1983; Meister, 1994). SM and CEES exposures are known to deplete GSH leading to oxidative stress that causes DNA damage and triggers signaling pathways, which result in cell death and skin tissue injury (Paromov et al., 2007; Shakarjian et al., 2010; Tewari-Singh et al., 2010). Our in vivo studies in female SKH-1 hairless mice have shown the involvement of oxidative stress in CEES-caused inflammatory responses and that treatment of female mice with GSH via oral gavage 1 h prior to CEES exposure was found to significantly protect against CEES-caused inflammatory responses (Pal et al., 2009; Tewari-Singh et al., 2011). To further study the involvement of oxidative stress in CEES-caused DNA damage, skin tissues from the current study were analyzed for H2A.X ser139 phosphorylation with GSH pre-treatment. Results from the IHC analysis in terms of the H2A.X ser139 stained nuclei (red arrows; Fig. 4A and B) and protein expression (Fig. 4C) showed that GSH pre-treatment caused a 59% and 58% decrease, respectively, in the CEES-caused increase in H2A.X ser139 phosphorylation (Fig. 4). To assess the role of oxidative stress in CEES-induced expression of inflammatory mediators, skin tissues from female mice were also analyzed for COX-2, iNOS, and MMP-9 expression by western immunoblotting following GSH pre-treatment. GSH pre-treatment also attenuated the CEES-induced increase in the expression of inflammatory mediators COX-2 iNOS and MMP-9 by 68%, 53% and 54%, respectively (Fig. 5).
Figure 4.

Effect of GSH treatment 1 h before topical CEES exposure on H2A.X ser139 phosphorylation in female SKH-1 hairless mouse skin. Mice were treated either with 300 mg/kg/mouse GSH by oral gavage 1 h before 2 mg CEES exposure, GSH alone, CEES alone, acetone alone as CEES vehicle control or left untreated. Mice were sacrificed after 24 h of CEES exposure, dorsal skin was collected, and western immunoblotting or IHC analysis for H2A.X ser 139 phosphorylation was carried out as detailed in the Material and Methods. Representative pictures of H2A.X ser139 IHC staining are shown in panel A. In IHC stained skin tissue, the H2A.X ser139 positive cells were counted in five randomly selected fields per tissue section (400X magnification); data presented are mean ± SEM of five animals in each group (B). After western immunoblotting for H2A.X ser139 phosphorylation, membranes were reprobed with β-actin as a loading control (C). Data are presented as fold increase in comparison with their respective vehicle control and values of band intensities were adjusted with β-actin loading control. Data are representative of the results from randomly chosen two animals taken at each time point of the study for CEES exposed and GSH treated groups (C). UC, untreated control; VC, vehicle (acetone) control; G, Glutathione alone; GC, Glutathione + CEES; red arrows, H2A.X ser139 positive cells.
Figure 5.

Effect of GSH treatment 1 h before topical CEES exposure on the protein expression of COX-2, iNOS, MMP-9, MMP-2 in female SKH-1 hairless mouse skin. Mice were treated with either 300 mg/kg/mouse GSH by oral gavage 1 h before 2 mg CEES exposure, GSH alone, CEES alone, acetone alone as CEES vehicle control or left untreated. Mice were sacrificed after 24 h of CEES exposure, dorsal skin was collected, and western blot analysis was carried out to assess the expression of COX-2, iNOS, MMP-2 and MMP-9 using specific antibodies as detailed under Material and Methods. The membranes were then stripped and reprobed for β-actin as a loading control. Data are presented as fold increase in comparison with their respective vehicle control and values of band intensities were adjusted with β-actin loading control. Data are representative of the results from randomly chosen two animals taken at each time point of the study for CEES exposed and GSH treated groups. UC, untreated control; VC, vehicle (acetone) control; GSH, Glutathione alone; GSH+CEES, Glutathione + CEES.
4. Discussion
Outcomes of this study suggest that both DNA damage and inflammatory mediators are an important link between our earlier reported CEES-induced signaling pathways and skin injury responses involving oxidative stress (Pal et al., 2009; Tewari-Singh et al., 2009; Jain et al., 2011). Our studies employed SKH-1 hairless mouse model as reported earlier (Pal et al., 2009; Tewari-Singh et al., 2009; Jain et al., 2011) because it is the most widely used mouse model in skin research due to being unpigmented and immunocompetent. These mice are equipped for skin manipulation, application of topical drugs, UVR, etc. as well as cutaneous response can be easily analyzed in these mice (Kligman and Kligman, 1998; Benavides et al., 2009). Mice were exposed to a monofunctional analog of SM, CEES, that alkylates biological molecules similar to SM including the formation DNA monoadducts, and lead to similar pathological lesions (Han et al., 2004; Jowsey et al., 2009; Tewari-Singh et al., 2009).
DNA damage is reported as a key consequence of SM exposure and can occur either via direct DNA alkylation by SM or via oxidative or nitrosative stress (Ruff and Dillman, 2007; Jowsey et al., 2009). The signaling cascades activated by DNA damage phosphorylate downstream target proteins, mainly p53, histone H2A.X and cell cycle checkpoint kinases (Jowsey et al., 2009). Both 2 and 4 mg CEES-caused DNA damage was observed in this study in the strong phosphorylation of H2A.X at ser139 at 12–24 h of exposure that decreased by 48 h in both male and female SKH-1 hairless mice. These results indicate a decrease in DNA damage by CEES by 48 h of exposure suggesting a repair response by this time following CEES exposure. These findings further support previous studies reporting DNA damage as an important early consequence of SM exposure and related skin injury (Jowsey et al., 2009; Kehe et al., 2009; Black et al., 2010; Shakarjian et al., 2010). Furthermore, the results of this in vivo study also corroborate our earlier study in skin epidermal cells showing that DNA damaging effects of CEES activates ataxia telangiectasia mutated (ATM)/ataxia telangiectasia-Rad3-related (ATR) cell cycle signaling as well as PARP pathways leading to cell cycle arrest and apoptotic cell death (Tewari-Singh et al., 2010). Overall, the results of this and our other recent study suggest that DNA damage-related signaling could be one of the major mechanisms of CEES-caused skin inflammation and vesication, together with MAPKs/Akt activated NF-κB/AP-1 related signaling cascades involving COX-2, iNOS and MMP-9 as important mediators (Fig. 6).
Figure 6.
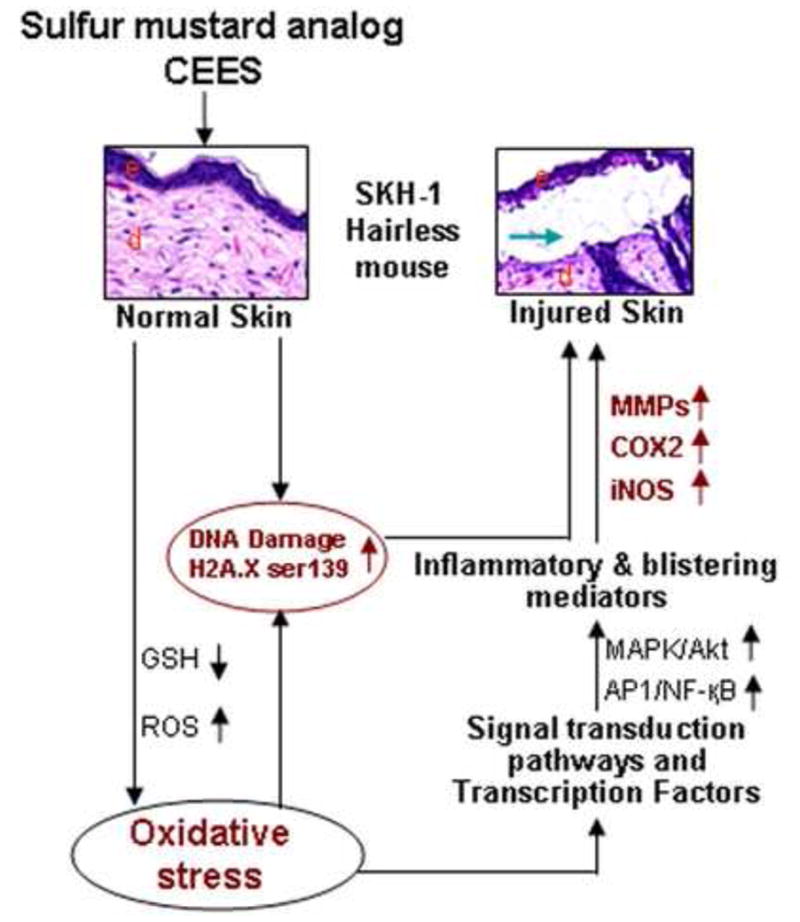
Schematic representation of the possible mechanism of CEES-caused cutaneous inflammation and micro-vesication in SKH-1 hairless mouse skin. CEES could damage DNA directly, or via an induction of oxidative stress due to a decrease in GSH and an increase in reactive oxygen species (ROS) leading to oxidative DNA damage. This would further trigger a signal transduction pathway leading to cell death and tissue injury. CEES-induced oxidative stress could also cause the activation of transcription factors such as AP-1 and NF-κB via MAPK and Akt pathways, inducing the level of inflammatory mediators like COX-2, iNOS and MMPs leading to inflammation and micro-vesication and skin injury. e, epidermis; d, dermis; green arrow, microvesication.
The main pathogenic effect of SM on the skin is inflammation, which includes erythema, edema and infiltration of inflammatory cells including mast cells, macrophages and neutrophils at the site of injury (Smith et al., 1995b; Millard et al., 1997; Valent et al., 1998; Rice, 2003). Our previous studies have shown CEES-induced histopathological changes to correlate with a high infiltration of mast cells, macrophages and neutrophils in SKH-1 hairless mice (Tewari-Singh et al., 2009). COX-derived prostaglandins play an important role in inflammation and are reported in SM-induced skin injury, including a study where SM exposure in COX-2 null mice or in mice treated with COX-2 inhibitor resulted in reduced inflammatory responses (Yourick et al., 1995; Nyska et al., 2001; Wormser et al., 2004). Our results here show an induction of COX-2 expression in both CEES dose- and time- response studies in male and female SKH-1 hairless mouse skin, whereas only a slight CEES-associated induction of COX-1 expression was observed. This is in accord with earlier reports that COX-2 is an important inflammatory mediator in CEES-caused skin injury (Rikimaru et al., 1991; Shakarjian et al., 2010). Our findings also suggest that CEES exposure induces COX-2 expression, which increases vascular permeability and facilitates the influx of inflammatory cells.
iNOS is an inducible form of nitric oxide synthase which is expressed by epithelial cells as well as activated neutrophils and macrophages that infiltrate during inflammation (Bloch et al., 2007; Gao et al., 2007). CEES-induced infiltration of neutrophils and macrophages, which is linked to the activation of MAPK/Akt-NF-κB/AP-1 pathways, was observed in our earlier studies in SKH-1 hairless mice (Pal et al., 2009; Tewari-Singh et al., 2009), indicating that iNOS is another important inflammatory mediator in the skin injury response observed in the CEES-skin injury model. Indeed, our results in the present study showed an induction of iNOS in both female and male mice at both the 2 and 4 mg CEES doses which have been previously reported to cause skin injury including microvesication (Tewari-Singh et al., 2009; Jain et al., 2011). NO is synthesized from L-argenine mainly by iNOS proteins. NO could affect the wound healing process either positively or negatively depending upon the production of peroxynitrite (ONOO−) following reaction with superoxide (O2−) radicals. This process can cause subsequent oxidation of cellular macromolecules (Ishida et al., 2008). The present study supports earlier reports that iNOS-related ONOO−, alone or together with other free radicals, can also cause increased DNA strand breakage. This event triggers activation of DNA repair enzymes including PARP, leading to necrotic cell death and related pathogenesis by SM (Korkmaz et al., 2006; Debiak et al., 2009). Because calcium and calmodulin have been identified to play an important role in formation of NO (Kehe et al., 2009), further studies are required to analyze the role of these molecules, as well as the positive and/or negative action of NO produced by iNOS in wound healing as well as in DNA damage induced by SM.
Proteases play an important role in blister formation at epidermal-dermal junctions in SM- induced skin injury (Ray et al., 2002; Cowan et al., 2003). SM directly reacts with anchoring filament laminin-5 and or activates different kinds of proteases, mainly MMPs, which degrade collagen and other extracellular matrix protein (ECM) (Smith et al., 1995a; Sabourin et al., 2002; Chang et al., 2009; Mol et al., 2009). Our results show CEES-induced a strong expression of MMP-9 as compared to MMP-2 suggesting MMP-9 as an important mediator in CEES-caused skin microvesication and inflammation in both male and female SKH-1 hairless mice. These results are in accordance with other reports on differential expression of MMP-2 and MMP-9 in SM exposure studies in different skin injury models (Nova et al., 2003; Shakarjian et al., 2006; Ries et al., 2009). In addition, our study suggests an important mediatory role of MMP-9 in the CEES-caused activation of MAPK/Akt-NF-κB/AP-1 pathways and related inflammatory and microvesication responses observed in our earlier studies in SKH-1 hairless mice (Pal et al., 2009; Tewari-Singh et al., 2009).
Oxidative stress is one of the most studied mechanisms in SM-induced skin injury (Han et al., 2004; Pal et al., 2009; Pohanka and Stetina, 2009; Laskin et al., 2010). Our previous studies have shown that oxidative stress plays an important role in the activation of complex signaling pathways that lead to CEES-caused skin injury (Pal et al., 2009; Tewari-Singh et al., 2010). It has also been reported that SM or its analog CEES exposure decreases the intracellular GSH levels leading to elevated production of ROS and oxidation of biomolecules including DNA (Han et al., 2004; Laskin et al., 2010). Several studies have shown that pre-treatment with antioxidants including GSH, N-acetyl cysteine (NAC), superoxide dismutase (SOD) and catalytic antioxidant AEOL 10150 can reduce SM-induced skin toxicity (Paromov et al., 2007; Gould et al., 2009; Tewari-Singh et al., 2010). Our earlier study in female SKH-1 hairless mice showed a protective effect of oral GSH in ameliorating CEES-caused inflammatory responses, which supports the involvement of GSH and oxidative stress in CEES-caused activation of signaling cascades and skin inflammation (Tewari-Singh et al., 2010). Our previous study supported the oral administration of GSH because oral route is possibly a more practical and fast treatment route in case of a chemical emergency. In addition, following oral uptake, an increase in the GSH levels in the blood and other tissues including skin have been reported (Campbell and Griffith, 1989; Aw et al., 1991; Stabler et al., 2000; Kariya et al., 2007). The present study further confirms the impact of GSH and oxidative stress in the CEES-induced expression of COX-2, iNOS and MMP-9. CEES-caused oxidative DNA damage was observed in our previous study in SKH-1 hairless mice (Pal et al., 2009). In the present study, CEES-caused increases in H2A.X ser139 phosphorylation were significantly decreased in GSH-treated mice asserting a role of GSH and oxidative stress in CEES-induced DNA damage. The involvement of these processes in etiology of blistering and microvesication caused by CEES exposure is supported by this study.
Similar to our previous study where comparable CEES-induced inflammatory responses were observed in male and female SKH-1 mice (Tewari-Singh et al., 2009; Jain et al., 2011), this study also shows comparable CEES-caused increase in DNA damage and expression of inflammatory mediators in male and female mice. Based on the known mechanism of SM-caused skin injury, efficacy studies have been conducted using COX-2, iNOS, protease inhibitors as well as antioxidants to reduce SM-related inflammation and vesication (Han et al., 2004; Wormser et al., 2004; Gould et al., 2009; Mol et al., 2009; Tewari-Singh et al., 2011). Findings in the present study suggest that oxidative stress, in part, mediates a simultaneous mechanism of CEES-caused skin injury involving both DNA damage and inflammatory signaling cascades (Fig. 6). CEES-induced GSH depletion and oxidative stress or direct alkylating effect can cause DNA damage leading to skin injury via activation of DNA damaging pathways (Fig. 6). In addition, CEES-induced GSH depletion and oxidative stress can lead to the activation of signal transduction pathways and transcription factors (Pal et al., 2009) that can cause skin inflammation and vesication via inflammatory mediators like COX-2, iNOS and MMP-9 (Fig. 6). The outcomes of the present study and the SKH-1 hairless mouse model could be useful in designing efficacy studies to identify and develop effective countermeasures alone or in combination to overcome the SM and other vesicating agents’ related skin inflammation and vesication in humans.
Highlights.
CEES causes phosphorylation of H2A.X and increases COX-2, iNOS and MMP-9 levels in SKH1 hairless mouse skin.
GSH reverses CEES-caused DNA damage and induction of inflammatory regulators signifying oxidative stress as a major player.
Our results indicate a two-hit mechanism of CEES-caused skin injury in SKH-1 hairless mouse skin.
Acknowledgments
This research was supported by the Countermeasures Against Chemical Threats (CounterACT) Program, National Institutes of Health (NIH) Office of the Director, and National Institute of Environmental Health Sciences, Grant Number (U54 ES-015678). The study sponsor (NIH) had no involvement in the study design; collection, analysis and interpretation of data; the writing of the manuscript; nor the decision to submit the manuscript for publication.
Abbreviations
- ATM
ataxia telangiectasia
- ATR
ataxia telangiectasia-Rad3-related
- CEES
2-chloroethyl ethyl sulfide
- COX-1
Cyclooxygenase-1
- COX-2
Cyclooxygenase-2
- ECM
extra cellular matrix protein
- eNOS
endothelial NOS
- GSH
glutathione
- iNOS
inducible NOS
- MAPKs
mitogen activated protein kinases
- MMPs
Matrix metalloproteinases
- MMP-2
Matrix metalloproteinase-2
- MMP-9
Matrix metalloproteinase-9
- NF-kB
nuclear factor- kB
- NOS
Nitric oxide synthase
- (O2−
superoxide
- ONOO−
peroxinitrite
- ROS
reactive oxygen species
- SM
bis (2-chloroethyl) sulfide
Footnotes
Conflict of Interest Statement
The authors declare that there are no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Anil K. Jain, Email: Anil.Jain@ucdenver.edu.
Neera Tewari-Singh, Email: Neera.Tewari-Singh@ucdenver.edu.
Mallikarjuna Gu, Email: Mallikarjuna.Gu@ucdenver.edu.
Swetha Inturi, Email: Swetha.Inturi@ucdenver.edu.
Carl W. White, Email: whitec@njc.org.
References
- Amir A, Chapman S, Gozes Y, Sahar R, Allon N. Protection by extracellular glutathione against sulfur mustard induced toxicity in vitro. Hum Exp Toxicol. 1998;17:652–660. doi: 10.1177/096032719801701202. [DOI] [PubMed] [Google Scholar]
- Arfsten DP, Johnson EW, Wilfong ER, Jung AE, Bobb AJ. Distribution of radio-labeled N-Acetyl-L-Cysteine in Sprague-Dawley rats and its effect on glutathione metabolism following single and repeat dosing by oral gavage. Cutan Ocul Toxicol. 2007;26:113–134. doi: 10.1080/15569520701212233. [DOI] [PubMed] [Google Scholar]
- Aw TY, Wierzbicka G, Jones DP. Oral glutathione increases tissue glutathione in vivo. Chem Biol Interact. 1991;80:89–97. doi: 10.1016/0009-2797(91)90033-4. [DOI] [PubMed] [Google Scholar]
- Benavides F, Oberyszyn TM, VanBuskirk AM, Reeve VE, Kusewitt DF. The hairless mouse in skin research. J Dermatol Sci. 2009;53:10–18. doi: 10.1016/j.jdermsci.2008.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black AT, Hayden PJ, Casillas RP, Heck DE, Gerecke DR, Sinko PJ, Laskin DL, Laskin JD. Expression of proliferative and inflammatory markers in a full-thickness human skin equivalent following exposure to the model sulfur mustard vesicant, 2-chloroethyl ethyl sulfide. Toxicol Appl Pharmacol. 2010 doi: 10.1016/j.taap.2010.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloch W, Elischer A, Schriek M, Böhm K, Moghbeli F, Kehe K, Szinicz L, Steinritz D. Comparison of sulfur mustard induced mechanism of cell damage in dependency of time course and cell type. Toxicology. 2007;233:223–223. [Google Scholar]
- Brimfield AA, Mancebo AM, Mason RP, Jiang JJ, Siraki AG, Novak MJ. Free radical production from the interaction of 2-chloroethyl vesicants (mustard gas) with pyridine nucleotide-driven flavoprotein electron transport systems. Toxicol Appl Pharmacol. 2009;234:128–134. doi: 10.1016/j.taap.2008.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell EB, Griffith OW. Glutathione monoethyl ester: high-performance liquid chromatographic analysis and direct preparation of the free base form. Anal Biochem. 1989;183:21–25. doi: 10.1016/0003-2697(89)90165-6. [DOI] [PubMed] [Google Scholar]
- Casillas RP, Kiser RC, Truxall JA, Singer AW, Shumaker SM, Niemuth NA, Ricketts KM, Mitcheltree LW, Castrejon LR, Blank JA. Therapeutic approaches to dermatotoxicity by sulfur mustard. I. Modulaton of sulfur mustard-induced cutaneous injury in the mouse ear vesicant model. J Appl Toxicol. (20) 2000;(Suppl 1):S145–151. doi: 10.1002/1099-1263(200012)20:1+<::aid-jat665>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- Chang YC, Sabourin CL, Lu SE, Sasaki T, Svoboda KK, Gordon MK, Riley DJ, Casillas RP, Gerecke DR. Upregulation of gamma-2 laminin-332 in the mouse ear vesicant wound model. J Biochem Mol Toxicol. 2009;23:172–184. doi: 10.1002/jbt.20275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowan FM, Broomfield CA, Lenz DE, Smith WJ. Putative role of proteolysis and inflammatory response in the toxicity of nerve and blister chemical warfare agents: implications for multi-threat medical countermeasures. J Appl Toxicol. 2003;23:177–186. doi: 10.1002/jat.901. [DOI] [PubMed] [Google Scholar]
- Dacre JC, Goldman M. Toxicology and pharmacology of the chemical warfare agent sulfur mustard. Pharmacol Rev. 1996;48:289–326. [PubMed] [Google Scholar]
- Debiak M, Kehe K, Burkle A. Role of poly(ADP-ribose) polymerase in sulfur mustard toxicity. Toxicology. 2009;263:20–25. doi: 10.1016/j.tox.2008.06.002. [DOI] [PubMed] [Google Scholar]
- Gao X, Ray R, Xiao Y, Barker PE, Ray P. Inhibition of sulfur mustard-induced cytotoxicity and inflammation by the macrolide antibiotic roxithromycin in human respiratory epithelial cells. BMC Cell Biol. 2007;8:17. doi: 10.1186/1471-2121-8-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilroy DW, Colville-Nash PR, Willis D, Chivers J, Paul-Clark MJ, Willoughby DA. Inducible cyclooxygenase may have anti-inflammatory properties. Nat Med. 1999;5:698–701. doi: 10.1038/9550. [DOI] [PubMed] [Google Scholar]
- Gould NS, White CW, Day BJ. A role for mitochondrial oxidative stress in sulfur mustard analog 2-chloroethyl ethyl sulfide-induced lung cell injury and antioxidant protection. J Pharmacol Exp Ther. 2009;328:732–739. doi: 10.1124/jpet.108.145037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenberg S, Kamath P, Petrali J, Hamilton T, Garfield J, Garlick JA. Characterization of the initial response of engineered human skin to sulfur mustard. Toxicol Sci. 2006;90:549–557. doi: 10.1093/toxsci/kfi306. [DOI] [PubMed] [Google Scholar]
- Gross CL, Innace JK, Hovatter RC, Meier HL, Smith WJ. Biochemical manipulation of intracellular glutathione levels influences cytotoxicity to isolated human lymphocytes by sulfur mustard. Cell Biol Toxicol. 1993;9:259–267. doi: 10.1007/BF00755604. [DOI] [PubMed] [Google Scholar]
- Gu M, Dhanalakshmi S, Singh RP, Agarwal R. Dietary feeding of silibinin prevents early biomarkers of UVB radiation-induced carcinogenesis in SKH-1 hairless mouse epidermis. Cancer Epidemiol Biomarkers Prev. 2005;14:1344–1349. doi: 10.1158/1055-9965.EPI-04-0664. [DOI] [PubMed] [Google Scholar]
- Gu M, Singh RP, Dhanalakshmi S, Agarwal C, Agarwal R. Silibinin inhibits inflammatory and angiogenic attributes in photocarcinogenesis in SKH-1 hairless mice. Cancer Res. 2007;67:3483–3491. doi: 10.1158/0008-5472.CAN-06-3955. [DOI] [PubMed] [Google Scholar]
- Han S, Espinoza LA, Liao H, Boulares AH, Smulson ME. Protection by antioxidants against toxicity and apoptosis induced by the sulphur mustard analog 2-chloroethylethyl sulphide (CEES) in Jurkat T cells and normal human lymphocytes. Br J Pharmacol. 2004;141:795–802. doi: 10.1038/sj.bjp.0705591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayden PJ, Petrali JP, Stolper G, Hamilton TA, Jackson GR, Jr, Wertz PW, Ito S, Smith WJ, Klausner M. Microvesicating effects of sulfur mustard on an in vitro human skin model. Toxicol In Vitro. 2009;23:1396–1405. doi: 10.1016/j.tiv.2009.07.021. [DOI] [PubMed] [Google Scholar]
- Huang X, Darzynkiewicz Z. Cytometric assessment of histone H2AX phosphorylation: a reporter of DNA damage. Methods Mol Biol. 2006;314:73–80. doi: 10.1385/1-59259-973-7:073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishida H, Ray R, Ray P. Sulfur mustard downregulates iNOS expression to inhibit wound healing in a human keratinocyte model. J Dermatol Sci. 2008;49:207–216. doi: 10.1016/j.jdermsci.2007.09.002. [DOI] [PubMed] [Google Scholar]
- Jain AK, Tewari-Singh N, Orlicky DJ, White CW, Agarwal R. 2-Chloroethyl ethyl sulfide causes microvesication and inflammation-related histopathological changes in male hairless mouse skin. Toxicology. 2011;282:129–138. doi: 10.1016/j.tox.2011.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson N, Westermarck J, Leppa S, Hakkinen L, Koivisto L, Lopez-Otin C, Peltonen J, Heino J, Kahari VM. Collagenase 3 (matrix metalloproteinase 13) gene expression by HaCaT keratinocytes is enhanced by tumor necrosis factor alpha and transforming growth factor beta. Cell Growth Differ. 1997;8:243–250. [PubMed] [Google Scholar]
- Jowsey PA, Williams FM, Blain PG. DNA damage, signalling and repair after exposure of cells to the sulphur mustard analogue 2-chloroethyl ethyl sulphide. Toxicology. 2009;257:105–112. doi: 10.1016/j.tox.2008.12.001. [DOI] [PubMed] [Google Scholar]
- Kahari VM, Saarialho-Kere U. Matrix metalloproteinases in skin. Exp Dermatol. 1997;6:199–213. doi: 10.1111/j.1600-0625.1997.tb00164.x. [DOI] [PubMed] [Google Scholar]
- Kan RK, Pleva CM, Hamilton TA, Anderson DR, Petrali JP. Sulfur mustard-induced apoptosis in hairless guinea pig skin. Toxicol Pathol. 2003;31:185–190. doi: 10.1080/01926230390183661. [DOI] [PubMed] [Google Scholar]
- Kariya C, Leitner H, Min E, van Heeckeren C, van Heeckeren A, Day BJ. A role for CFTR in the elevation of glutathione levels in the lung by oral glutathione administration. Am J Physiol Lung Cell Mol Physiol. 2007;292:L1590–1597. doi: 10.1152/ajplung.00365.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kehe K, Balszuweit F, Emmler J, Kreppel H, Jochum M, Thiermann H. Sulfur mustard research--strategies for the development of improved medical therapy. Eplasty. 2008;8:e32. [PMC free article] [PubMed] [Google Scholar]
- Kehe K, Balszuweit F, Steinritz D, Thiermann H. Molecular toxicology of sulfur mustard-induced cutaneous inflammation and blistering. Toxicology. 2009;263:12–19. doi: 10.1016/j.tox.2009.01.019. [DOI] [PubMed] [Google Scholar]
- Kehe K, Szinicz L. Medical aspects of sulphur mustard poisoning. Toxicology. 2005;214:198–209. doi: 10.1016/j.tox.2005.06.014. [DOI] [PubMed] [Google Scholar]
- Kligman AM, Kligman LH. A hairless mouse model for assessing the chronic toxicity of topically applied chemicals. Food Chem Toxicol. 1998;36:867–878. doi: 10.1016/s0278-6915(98)00045-3. [DOI] [PubMed] [Google Scholar]
- Korkmaz A, Yaren H, Topal T, Oter S. Molecular targets against mustard toxicity: implication of cell surface receptors, peroxynitrite production, and PARP activation. Arch Toxicol. 2006;80:662–670. doi: 10.1007/s00204-006-0089-x. [DOI] [PubMed] [Google Scholar]
- Laskin JD, Black AT, Jan YH, Sinko PJ, Heindel ND, Sunil V, Heck DE, Laskin DL. Oxidants and antioxidants in sulfur mustard-induced injury. Ann N Y Acad Sci. 2010;1203:92–100. doi: 10.1111/j.1749-6632.2010.05605.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- Meister A. Glutathione, ascorbate, and cellular protection. Cancer Res. 1994;54:1969s–1975s. [PubMed] [Google Scholar]
- Meister A, Anderson ME. Glutathione. Annu Rev Biochem. 1983;52:711–760. doi: 10.1146/annurev.bi.52.070183.003431. [DOI] [PubMed] [Google Scholar]
- Millard CB, Bongiovanni R, Broomfield CA. Cutaneous exposure to bis-(2-chloroethyl)sulfide results in neutrophil infiltration and increased solubility of 180,000 Mr subepidermal collagens. Biochem Pharmacol. 1997;53:1405–1412. doi: 10.1016/s0006-2952(97)00008-7. [DOI] [PubMed] [Google Scholar]
- Mol MA, van den Berg RM, Benschop HP. Involvement of caspases and transmembrane metalloproteases in sulphur mustard-induced microvesication in adult human skin in organ culture: directions for therapy. Toxicology. 2009;258:39–46. doi: 10.1016/j.tox.2009.01.004. [DOI] [PubMed] [Google Scholar]
- Nova D, Le Griel C, Holvoet S, Gentilhomme E, Vincent C, Staquet MJ, Schmitt D, Serres M. Comparative studies on the secretion and activation of MMPs in two reconstructed human skin models using HaCaT- and HaCaT-ras-transfected cell lines. Clin Exp Metastasis. 2003;20:675–683. doi: 10.1023/b:clin.0000006816.09548.31. [DOI] [PubMed] [Google Scholar]
- Nyska A, Lomnitski L, Maronpot R, Moomaw C, Brodsky B, Sintov A, Wormser U. Effects of iodine on inducible nitric oxide synthase and cyclooxygenase-2 expression in sulfur mustard-induced skin. Arch Toxicol. 2001;74:768–774. doi: 10.1007/s002040000199. [DOI] [PubMed] [Google Scholar]
- Pal A, Tewari-Singh N, Gu M, Agarwal C, Huang J, Day BJ, White CW, Agarwal R. Sulfur mustard analog induces oxidative stress and activates signaling cascades in the skin of SKH-1 hairless mice. Free Radic Biol Med. 2009;47:1640–1651. doi: 10.1016/j.freeradbiomed.2009.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paromov V, Suntres Z, Smith M, Stone WL. Sulfur mustard toxicity following dermal exposure: role of oxidative stress, and antioxidant therapy. J Burns Wounds. 2007;7:e7. [PMC free article] [PubMed] [Google Scholar]
- Pohanka M, Stetina R. Shift of oxidants and antioxidants levels in rats as a reaction to exposure to sulfur mustard. J Appl Toxicol. 2009;29:643–647. doi: 10.1002/jat.1451. [DOI] [PubMed] [Google Scholar]
- Ravanti L, Kahari VM. Matrix metalloproteinases in wound repair (review) Int J Mol Med. 2000;6:391–407. [PubMed] [Google Scholar]
- Ray P, Chakrabarti AK, Broomfield CA, Ray R. Sulfur mustard-stimulated protease: a target for antivesicant drugs. J Appl Toxicol. 2002;22:139–140. doi: 10.1002/jat.829. [DOI] [PubMed] [Google Scholar]
- Rice P. Sulphur mustard injuries of the skin. Pathophysiology and management. Toxicol Rev. 2003;22:111–118. doi: 10.2165/00139709-200322020-00006. [DOI] [PubMed] [Google Scholar]
- Ries C, Popp T, Egea V, Kehe K, Jochum M. Matrix metalloproteinase-9 expression and release from skin fibroblasts interacting with keratinocytes: Upregulation in response to sulphur mustard. Toxicology. 2009;263:26–31. doi: 10.1016/j.tox.2008.08.011. [DOI] [PubMed] [Google Scholar]
- Rikimaru T, Nakamura M, Yano T, Beck G, Habicht GS, Rennie LL, Widra M, Hirshman CA, Boulay MG, Spannhake EW, et al. Mediators, initiating the inflammatory response, released in organ culture by full-thickness human skin explants exposed to the irritant, sulfur mustard. J Invest Dermatol. 1991;96:888–897. doi: 10.1111/1523-1747.ep12475292. [DOI] [PubMed] [Google Scholar]
- Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem. 1998;273:5858–5868. doi: 10.1074/jbc.273.10.5858. [DOI] [PubMed] [Google Scholar]
- Ruff AL, Dillman JF. Signaling molecules in sulfur mustard-induced cutaneous injury. Eplasty. 2007;8:e2. [PMC free article] [PubMed] [Google Scholar]
- Sabourin CL, Danne MM, Buxton KL, Casillas RP, Schlager JJ. Cytokine, chemokine, and matrix metalloproteinase response after sulfur mustard injury to weanling pig skin. J Biochem Mol Toxicol. 2002;16:263–272. doi: 10.1002/jbt.10050. [DOI] [PubMed] [Google Scholar]
- Shakarjian MP, Bhatt P, Gordon MK, Chang YC, Casbohm SL, Rudge TL, Kiser RC, Sabourin CL, Casillas RP, Ohman-Strickland P, Riley DJ, Gerecke DR. Preferential expression of matrix metalloproteinase-9 in mouse skin after sulfur mustard exposure. J Appl Toxicol. 2006;26:239–246. doi: 10.1002/jat.1134. [DOI] [PubMed] [Google Scholar]
- Shakarjian MP, Heck DE, Gray JP, Sinko PJ, Gordon MK, Casillas RP, Heindel ND, Gerecke DR, Laskin DL, Laskin JD. Mechanisms mediating the vesicant actions of sulfur mustard after cutaneous exposure. Toxicol Sci. 2010;114:5–19. doi: 10.1093/toxsci/kfp253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu Y, Sakai M, Umemura Y, Ueda H. Immunohistochemical localization of nitric oxide synthase in normal human skin: expression of endothelial-type and inducible-type nitric oxide synthase in keratinocytes. J Dermatol. 1997;24:80–87. doi: 10.1111/j.1346-8138.1997.tb02748.x. [DOI] [PubMed] [Google Scholar]
- Smith CN, Lindsay CD, Upshall DG. Presence of methenamine/glutathione mixtures reduces the cytotoxic effect of sulphur mustard on cultured SVK-14 human keratinocytes in vitro. Hum Exp Toxicol. 1997;16:247–253. doi: 10.1177/096032719701600502. [DOI] [PubMed] [Google Scholar]
- Smith KJ, Graham JS, Moeller RB, Okerberg CV, Skelton H, Hurst CG. Histopathologic features seen in sulfur mustard induced cutaneous lesions in hairless guinea pigs. J Cutan Pathol. 1995a;22:260–268. doi: 10.1111/j.1600-0560.1995.tb00748.x. [DOI] [PubMed] [Google Scholar]
- Smith KJ, Hurst CG, Moeller RB, Skelton HG, Sidell FR. Sulfur mustard: its continuing threat as a chemical warfare agent, the cutaneous lesions induced, progress in understanding its mechanism of action, its long-term health effects, and new developments for protection and therapy. J Am Acad Dermatol. 1995b;32:765–776. doi: 10.1016/0190-9622(95)91457-9. [DOI] [PubMed] [Google Scholar]
- Smith WL, DeWitt DL, Garavito RM. Cyclooxygenases: structural, cellular, and molecular biology. Annu Rev Biochem. 2000;69:145–182. doi: 10.1146/annurev.biochem.69.1.145. [DOI] [PubMed] [Google Scholar]
- Stabler SP, Morton RL, Winski SL, Allen RH, White CW. Effects of parenteral cysteine and glutathione feeding in a baboon model of severe prematurity. Am J Clin Nutr. 2000;72:1548–1557. doi: 10.1093/ajcn/72.6.1548. [DOI] [PubMed] [Google Scholar]
- Tewari-Singh N, Agarwal C, Huang J, Day BJ, White CW, Agarwal R. Efficacy of glutathione in ameliorating sulfur mustard analog-induced toxicity in cultured skin epidermal cells and in SKH-1 mouse skin in vivo. J Pharmacol Exp Ther. 2011 doi: 10.1124/jpet.110.173708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tewari-Singh N, Gu M, Agarwal C, White CW, Agarwal R. Biological and molecular mechanisms of sulfur mustard analogue-induced toxicity in JB6 and HaCaT cells: possible role of ataxia telangiectasia-mutated/ataxia telangiectasia-Rad3-related cell cycle checkpoint pathway. Chem Res Toxicol. 2010;23:1034–1044. doi: 10.1021/tx100038b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tewari-Singh N, Rana S, Gu M, Pal A, Orlicky DJ, White CW, Agarwal R. Inflammatory biomarkers of sulfur mustard analog 2-chloroethyl ethyl sulfide-induced skin injury in SKH-1 hairless mice. Toxicol Sci. 2009;108:194–206. doi: 10.1093/toxsci/kfn261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai JC, Sheu HM, Hung PL, Cheng CL. Effect of barrier disruption by acetone treatment on the permeability of compounds with various lipophilicities: implications for the permeability of compromised skin. J Pharm Sci. 2001;90:1242–1254. doi: 10.1002/jps.1077. [DOI] [PubMed] [Google Scholar]
- Valent P, Sillaber C, Baghestanian M, Bankl HC, Kiener HP, Lechner K, Binder BR. What have mast cells to do with edema formation, the consecutive repair and fibrinolysis? Int Arch Allergy Immunol. 1998;115:2–8. doi: 10.1159/000023823. [DOI] [PubMed] [Google Scholar]
- Van den Steen PE, Dubois B, Nelissen I, Rudd PM, Dwek RA, Opdenakker G. Biochemistry and molecular biology of gelatinase B or matrix metalloproteinase-9 (MMP-9) Crit Rev Biochem Mol Biol. 2002;37:375–536. doi: 10.1080/10409230290771546. [DOI] [PubMed] [Google Scholar]
- Wormser U. Toxicology of mustard gas. Trends Pharmacol Sci. 1991;12:164–167. doi: 10.1016/0165-6147(91)90534-y. [DOI] [PubMed] [Google Scholar]
- Wormser U, Langenbach R, Peddada S, Sintov A, Brodsky B, Nyska A. Reduced sulfur mustard-induced skin toxicity in cyclooxygenase-2 knockout and celecoxib-treated mice. Toxicol Appl Pharmacol. 2004;200:40–47. doi: 10.1016/j.taap.2004.03.013. [DOI] [PubMed] [Google Scholar]
- Yourick JJ, Dawson JS, Mitcheltree LW. Reduction of erythema in hairless guinea pigs after cutaneous sulfur mustard vapor exposure by pretreatment with niacinamide, promethazine and indomethacin. J Appl Toxicol. 1995;15:133–138. doi: 10.1002/jat.2550150213. [DOI] [PubMed] [Google Scholar]