

Abstract
Wegener’s granulomatosis (WG) is a systemic disease with a complex genetic background. It is characterised by inflammation of the small blood vessels leading to damage in any number of organs. The common features include granulomatous inflammation of the respiratory tract and kidneys. Most patients have measurable autoantibodies against neutrophil proteinase-3 (Antineutrophil Cytoplasmic Antibody, ANCA). Pituitary involvement is a rare complication of this disease and, when it occurs, diabetes insipidus is the most common manifestation. We describe a 38-year-old female with known long-term WG who presented with partial hypopituitarism, severe malnutrition and ANCA negative status, with a favourable response to steroid pulse therapy.
Background
Wegener’s granulomatosis (WG is an antineutrophil cytoplasmic antibody- (ANCA) associated systemic vasculitis of small and medium-sized vessels. WG typically affects a combination of ear, nose and throat, lungs and kidneys, but may also affect joints, skin, eyes and virtually any tissue or organ. Pituitary involvement is rare with only 23 previous case reports in the English literature between 1966 and 2007.1 Diabetes insipidus (DI) is the most common manifestation of WG involving the pituitary gland. There are a few case reports of WG involving the anterior pituitary gland and resulting in hyperprolactinemia or panhypopituitarism.2
We describe a case with an unusual presentation due to the severe undernutrition, exclusive anterior pituitary involvement despite the loss of posterior signal in MRI and the persistently negative ANCA status despite its former positivity.
Case presentation
A 38-year-old white female with a known history of severe WG was admitted to the hospital for evaluation of severe headache. She complained of depression, generalised weakness and a 1-year history of intense weight loss (15 Kg) with abdominal pain, nausea and vomiting. She did not complain of thirst, nor exhibit polyuria or polydipsia.
The patient had been diagnosed with WG15 years previously when she presented with pulmonary-renal syndrome and renal biopsy revealed a segmental necrotizing glomerulonephritis. Over the years, she experienced progressive renal failure despite receiving cyclophosphamide intermittent pulses (cumulative doses of 20 g) and steroid treatment, not requiring dialysis at the time of the current admission. She exhibited sinonasal involvement including chronic rhinosinusitis, septal perforation (figure 1) and saddle-nose deformity. She had a history of cyclophosphamide-induced ovarian failure and secondary amenorrhea during the last 8 years. Tests for ANCA had been positive with a cytoplasmic pattern since diagnosis (c-ANCA).
Figure 1.
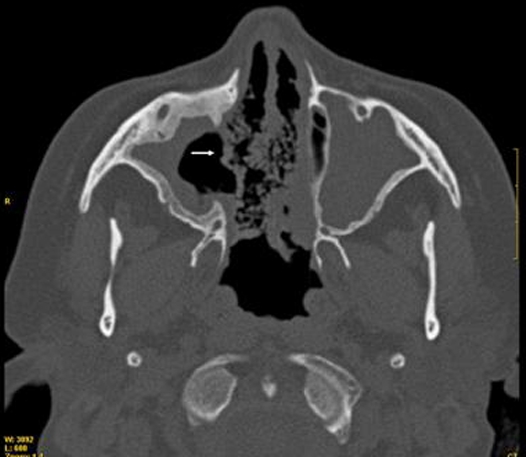
Sinus CT exam 2 years before admission. Shows septal perforation (arrow).
During the past 12 months, she was admitted to the hospital twice for severe headache, diplopia and complaints of unsteadiness. She was being diagnosed with ischemic cranial nerve VI palsy. (CT) of the brain failed to show any abnormalities in the pituitary gland, the sella turcica or the hypothalamus. An MRI showed a pituitary volume slightly higher than expected for the patient’s age and condition (figure 2). The rest of the workup, including colonoscopy, gastroscopy, gastroduodenal transit study and coeliac disease antibodies, failed to show any other organ’s involvement.
Figure 2.
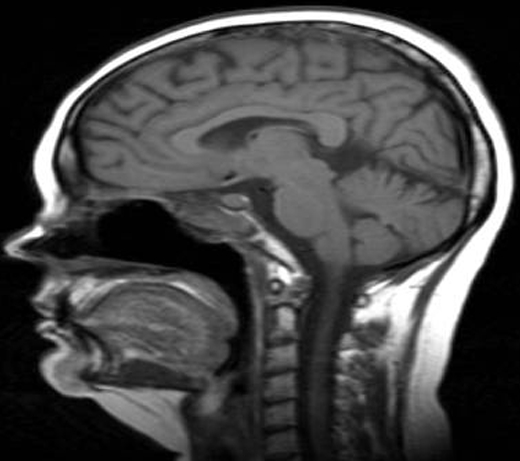
MRI scan of the pituitary gland the previous year. Slight increase in pituitary volume with respect to the patient’s age and condition when the headaches started, 16 months before the diagnosis of pituitary involvement.
During the current admission, physical examination revealed severe undernutrition with body mass index of 14 kg/m2. Her blood pressure was 110/70 mmHg and her pulse was 80 bpm. Her face was expressionless at rest and puffy. A neurological examination revealed no signs of intracranial hypertension. The visual acuity and fields were preserved. Her previous prescriptions included prednisolone 10 mg, mycophenolate, atorvastatin, vitamin D, folic acid, vitamin B12, enalapril, bisoprolol, losartan, torasemide, triflusal, pantoprazol, darbepoetin α, risedronate and sodium bicarbonate. The patient could not tolerate estrogen replacement due to menorrhagia.
Investigations
Biochemical results (table 1) were consistent with secondary hypothyroidism (low FT4 with normal thyrotrophin-stimulating hormone ) and secondary hypogonadism (low estrogen with low gonadotrophins). Cortisol levels and adrenocorticotropic hormone levels were taken while the patient was still taking prednisolone. c-ANCA was repeatedly negative. Her plasma sodium levels and plasma osmolarity were within the range. A brain MRI (figure 3A), performed without gadolinium contrast due to her kidney condition and the risk of nephrogenic systemic fibrosis3 showed a marked infundibular thickening and a 22 × 14 × 14 mm sellar mass with hypointensity and the loss of the characteristic hyperintensity signal from the posterior pituitary on the T1-weighted image. Non-specific white matter lesions were seen with high-signal intensity on intermediate-weighted images in the frontoparietal regions. There was heterogeneous soft tissue in the sphenoid, maxillary ethmoid sinuses and the sphenoid and ethmoid bones.
Table 1.
Patient’s investigation results.
| Test | On admission | 2 weeks later | 4 weeks later | 10 weeks later | Reference range |
|---|---|---|---|---|---|
| Haemoglobin | 11.1 | 12.6 | 13.9 | 12.8 | 12–16 g/dl |
| Platelel count | 279 × 103 | 184 × 10 | 204 × 10 | 65 × 10 | 150–400 × 103/mcl |
| White cell count | 9.5 × 103 | 9.1 × 103 | 11.1 | 14.1 | 4–10 × 103/ mcl |
| Creatinine | 2.9 | 3.2 | 3.6 | 3.1 | 0.6–1.1 mg/dl |
| Cortisol | <27.6 | 157 | NR | NR | 138–690 nmol/l |
| Adrenocorticotropic hormone | NR | < 5 | < 5 | 9.98 | < 46 pg/ml |
| Estradiol | <10 | <10 | 12 | <10 | 11–165 pg/mlL (FP) |
| 146–526 pg/ml (O) | |||||
| 33–196 pg/ml (LP) | |||||
| < 37 mUI/ml (PM) | |||||
| Luteinizing hormone | <0.07 | 0.,54 | < 0.07 | <0.07 | 1.9–12.5 mUI/ml(FP) |
| 8.7–76.3 mUI/ml (O) | |||||
| 0.5–16.9 mUI/ml (LP) | |||||
| 15.9–54 mUI/ml (PM) | |||||
| Follicle-stimulating hormone | 2.53 | 2.24 | 0.93 | 0.58 | 2.5–10.2 mUI/ml (FP) |
| 3.4–33.4 mUI/ml (O) | |||||
| 1.5–9.1 mUI/ml (LP) | |||||
| 23–116 mUI/ml (PM) | |||||
| Thyroid stimulating hormone | 1.22 | 0.39* | 0.29* | 3.09* | 0.35–5.35 mcU/ml |
| Free thyroxine | 0.53 | 0.74* | 0.88* | 1.09* | 0.81–1.76 ng/dl |
| Prolactin | 102.7 | 108.9 | 62.9 | 88.2 | 2.8–29.2 ng/dl |
| Growth hormone | 6.61 | 0.35 | 0.94 | NR | 0.06–5 ng/ml |
| Insulin-like growth factor-1 | 138 | 72 | 73 | 81 | 101–133 ng/ml |
| ANCA | Negative | Negative | Negative | Negative |
ANCA: antineutrophil cytoplasmic antibody, FP: follicular phase, LP: luteal phase, NR: not reported, O: ovulation, PM: postmenopausal.
On levothyroxine 50 mcg a day.
Figure 3.

MRI scan of the pituitary gland (A) before and (B) after corticosteroid treatment. Enlarged pituitary gland with infundibular thickening (arrow) and loss of the hyperintense signal of the posterior pituitary on T1-weighted images (A) that showed a marked reduction of the pituitary size 10 weeks after 3 days of intravenous methylprednisolone bolus (B).
In view of these findings, a diagnosis of granulomatous pituitary lesion due to WG was made.
Treatment
The patient started treatment with 3 days of intravenous methylprednisolone bolus adjusted to body mass index (500 mg a day) with partial resolution of the headache within 1 week. She also required levothyroxine treatment (50 μg a day) and remained on a low prednisolone dose, 5 mg a day, after the bolus. Dialytic support with intermittent haemodialysis was started in combination with intradialysis parenteral nutrition in order to improve the nutritional status.
Outcome and follow-up
Four weeks after treatment, the patient’s clinical condition rapidly improved, with no headache and increased appetite, gaining 3 kg during that period. Pituitary function after 12 weeks was consistent with partial recovery (table 1). A pituitary MRI showed a significant reduction of the pituitary size, with dimensions of 5 × 9 × 14 mm, including the infundibular thickening (figure 3B). At the time of this report, the patient continues to require levothyroxine and steroids at maintenance dosage.
Discussion
Pituitary involvement in WG is rare and scarcely represented in the literature. It affects predominantly the posterior pituitary causing Central DI. Infrequently, partial or total anterior pituitary abnormality can be present. The suggested causes of this complication include vasculitis of pituitary vessels, granulomatous lesions in situ or extension to the nervous system of adjacent nasal or paranasal granuloma.
In the review by Yong et al,1 pituitary involvement occurred between 2 months and 15 years after the diagnosis of WG in 56% of the patients. In 34% of patients, clinical features related to pituitary involvement were present at onset. The same study illustrated that the extent of the pituitary involvement is variable. Three cases (13%) had only anterior pituitary involvement, 12 (52%) had only central DI (with or without hyperprolactinemia) and 8 (35%) had both anterior and posterior pituitary involvement including three with panhypopituitarism. The patient we reported had been diagnosed with WG 15 years earlier. During the follow-up, she had experienced paranasal granulomas with progressive nasal destruction and sinusitis, thus spread from the adjacent anatomical areas to the pituitary was considered to be the most plausible mechanism of the pituitary involvement. Her case had several uncommon features. First, her main clinical manifestations were partial hypopituitarism and severe malnutrition, the less common ones in the series mentioned previously. In addition, her ANCA levels were persistently negative, which has been unexpected.
Antibodies directed at ANCA play an important role in the diagnosis of relapse. Indeed, rising ANCA titres or ANCA positivity after a period of ANCA negativity is a risk factor for relapse. In a review by Yong et al, out of 17 cases in which ANCA results were reported, 16 were positive. According to the information reported in a prospective study,4 if a patient is ANCA positive during a period of active disease, a persistently ANCA-negative status is consistent with remission, as only 8% of the patients in that study were ANCA negative at the time of the relapse. A possible explanation of the ANCA negative status in our case could be the severe malnutrition and the chronic treatment with low doses of corticosteroids over the past 12 months. This fact was partly responsible for the delay in the diagnosis of this patient.
Moreover, the clinical picture was not the one that has been previously described in the literature in association with these types of MRI findings. Murphy et al5 reported the MRI findings in brain and meninges in 19 patients with WG and only 2 patients had an enlarged pituitary gland (craniocaudal dimension > 11 mm). In both patients, enhancement of the pituitary gland was homogeneous, and there was thickening of the infundibulum. There also was a large, enhanced inflammatory mass in the sphenoid sinus in both patients. In the other study,1 the most common finding was an enlarged pituitary gland (80%), enhanced heterogeneously or homogenously on MRI. Other findings included cystic changes in the pituitary and infundibular thickening with contrast enhancement. An important finding on MRI is the loss of the characteristic hyperintensity signal from the posterior pituitary on the T1-weighted image, in 88% of the cases with central DI as clinical manifestation.1 In our patient, MRI without contrast showed enlargement of the pituitary gland, thickening of the infundibulum and absence of posterior pituitary lobe hyperintensity, without developing DI as clinical manifestation.
Finally, the response to treatment was remarkable. The majority of the patients respond to medical management, such as corticosteroids and immunosuppressants (cyclophosphamide) associated with hormonal replacement.6 The conventional combination of high-dose corticosteroid and cyclophosphamide induced remission successfully in all patients, however, 27% relapsed after a median of 10.5 months. High-dose corticosteroids has been used with clinical improvement in the 100% of the cases, with the 33% relapsed when the corticosteroid dose is reduced. With ‘non-conventional’ treatments, 50% relapsed after a median of 4.5 months.1 These findings are consistent with our patient’s response. The management with low doses of steroids and mycophenolate during the 12 months before admission does not prevent the progression of the symptoms nor the pituitary involvement. However, the 3 days of intravenous methylprednisolone bolus was a highly successful therapy in achieving partial remission.
In the review by Yong et al, 17% of patients regained their pituitary function completely after treatment. About 48% had persistent residual deficits. Pituitary function was not reported or unknown in the remaining percentage of patients (35%). The patient we reported partially regained her pituitary function, still requiring levothyroxine treatment.
Accurate diagnosis of the pituitary involvement in WGrequires a high degree of suspicion and correlation with clinical signs and symptoms, due to the large variety of the clinical manifestations that can appear during the course of the disease. One case has described sudden death due to hypothalamopituitary inflammation resulting from WG.7
We propose to evaluate pituitary axis in patients with WG and severe sinus involvement in order to avoid pitfalls in the diagnosis of adenohypophysis or neurohypophysis damage, even in patients with ANCA-negative status.
Learning points.
-
▶
Pituitary involvement in WG, predominantly affecting posterior pituitary, has been reported in only a small proportion of cases, invariably associated with other systemic manifestations of the disease.
-
▶
Hypopituitarism and severe malnutrition are not frequently documented in literature as the main clinical manifestations of pituitary involvement in WG.
-
▶
The patient showed ANCA-negative status despite its former positivity since WG diagnosis, differing from other similar cases.
-
▶
Medical treatment with steroid pulse therapy and hormonal replacement achieved a favourable clinical and radiological response in our patient, as has been reported in previous studies.
Acknowledgments
The authors thank Dr Miguel Aguirre Sanchez-Covisa for his professional support.
Footnotes
Competing interests None.
Patient consent Obtained.
References
- 1.Yong TY, Li JY, Amato L, et al. Pituitary involvement in Wegener’s granulomatosis. Pituitary 2008;11:77–84 [DOI] [PubMed] [Google Scholar]
- 2.Lury KM. Inflammatory and infectious processes involving the pituitary gland. Top Magn Reson Imaging 2005;16:301–6 [DOI] [PubMed] [Google Scholar]
- 3.Perazella MA. Current status of gadolinium toxicity in patients with kidney disease. Clin J Am Soc Nephrol 2009;4:461–9 [DOI] [PubMed] [Google Scholar]
- 4.Boomsma MM, Stegeman CA, van der Leij MJ, et al. Prediction of relapses in Wegener’s granulomatosis by measurement of antineutrophil cytoplasmic antibody levels: a prospective study. Arthritis Rheum 2000;43:2025–33 [DOI] [PubMed] [Google Scholar]
- 5.Murphy JM, Gomez-Anson B, Gillard JH, et al. Wegener granulomatosis: MR imaging findings in brain and meninges. Radiology 1999;213:794–9 [DOI] [PubMed] [Google Scholar]
- 6.Carpinteri R, Patelli I, Casanueva FF, et al. Pituitary tumours: inflammatory and granulomatous expansive lesions of the pituitary. Best Pract Res Clin Endocrinol Metab 2009;23:639–50 [DOI] [PubMed] [Google Scholar]
- 7.Woywodt A, Knoblauch H, Kettritz R, et al. Sudden death and Wegener’s granulomatosis of the pituitary. Scand J Rheumatol 2000;29:264–6 [DOI] [PubMed] [Google Scholar]