

Intravitreal administration of IGFBP-3NB preserves junctional integrity in the presence of VEGF or laser injury by reducing BRB permeability in part by modulating sphingomyelinase levels.
Abstract
Purpose.
To examine the effect of free insulin-like growth factor (IGF) binding protein-3 (IGFBP-3), independent of the effect of insulin-like growth factors, in modulating retinal vascular permeability.
Methods.
We assessed the ability of a form of IGFBP-3 that does not bind IGF-1 (IGFBP-3NB), to regulate the blood retinal barrier (BRB) using two distinct experimental mouse models, laser-induced retinal vessel injury and vascular endothelial growth factor (VEGF)-induced retinal vascular permeability. Additionally, in vitro studies were conducted. In the animal models, BRB permeability was quantified by intravenous injection of fluorescein labeled serum albumin followed by digital confocal image analysis of retinal flat-mounts. Claudin-5 and vascular endothelial-cadherin (VE-cadherin) localization at interendothelial junctions was studied by immunofluorescence. In vitro changes in transendothelial electrical resistance (TEER) and flux of fluorescent dextran in bovine retinal endothelial monolayers (BREC) were measured after IGFBP-3NB treatment. Acid (ASMase) and neutral (NSMase) sphingomyelinase mRNA levels and activity were measured in mouse retinas.
Results.
Four days postinjury, laser-injured mouse retinas injected with IGFBP-3NB plasmid demonstrated reduced vascular permeability compared with retinas of laser-injured mouse retinas injected with control plasmid. IGFBP-3NB administration resulted in a significant decrease in laser injury-associated increases in ASMase and NSMase mRNA and activity when compared with laser alone treated mice. In vivo, intravitreal injection of IGFBP-3NB reduced vascular leakage associated with intravitreal VEGF injection. IGFBP-3NB partially restored VEGF-induced in vivo permeability and dissociation of claudin-5 and VE-cadherin at junctional complexes. When IGFBP-3NB was applied basally to bovine retinal endothelial cells (BREC) in vitro, TEER increased and macromolecular flux decreased.
Conclusions.
Intravitreal administration of IGFBP-3NB preserves junctional integrity in the presence of VEGF or laser injury by reducing BRB permeability in part by modulating sphingomyelinase levels.
Breakdown of the blood retinal barrier (BRB) is a prominent feature of a wide range of retinal diseases including diabetic retinopathy, venous occlusive diseases, and cystoid macular edema.1,2 The inner BRB constitutes a remarkable physical and biochemical barrier between the retina and the circulation. The BRB is composed of a monolayer of nonfenestrated vascular endothelial cells, which are surrounded by pericytes and glial cells.1 Endothelial cells control the infiltration of blood proteins and circulating cells through the vessel wall into the surrounding tissues. Endothelial permeability occurs by the paracellular pathway, which is mediated by the coordinated opening and closure of endothelial cell-cell junctions.3 Paracellular increases in endothelial permeability occur by the changes in adherens junction (AJ) and tight junction (TJ)-associated proteins.3–5 Cell-cell junctions act as signaling structures which communicate cell position, limit growth, apoptosis, and regulate vascular homeostasis. Cell-cell junctions maintain endothelial integrity and prevent exposure of the subendothelial matrix.3,4 AJs are formed by the homotypic association of the extracellular segments of members of the cadherin family of adhesion proteins. Retinal microvascular endothelial cells express high levels of vascular endothelial cadherin (VE-cadherin).6 While the barrier function of the endothelium is supported by multiple intercellular adhesion systems, disruption of VE-cadherin is sufficient to disrupt all these intercellular junctions.7,8 In contrast to AJs, TJs are formed by membrane-spanning proteins (claudins, occludins, and junctional adhesion molecules), which interact with cytoplasmic proteins (AF-6 and ZO-1, -2, -3) that regulate their assembly and maintenance.9 Of the claudin family, retinal vascular endothelial cells predominantly express types 1, 3, and 5.10–13 The molecular composition of tight junctions is highly regulated and changes rapidly in response to factors that affect permeability. One of the factors implicated in disrupting the BRB integrity is vascular endothelial growth factor (VEGF), also known as the vascular permeability factor, which is typically increased in the eyes of patients with retinopathies.14–16
Ceramide, the proinflammatory and proapoptotic messenger, increases vascular permeability by a mechanism that is not yet fully understood, but involves the regulation of both endothelial Ca2+ signaling and nitric oxide (NO) formation.17–19 In response to both acute and chronic cutaneous permeability barrier disruption, sphingomyelinases (SMases) hydrolyze sphingomyelin to ceramide. Several isoforms of sphingomyelinases have been identified and are further distinguished by their catalytic pH optimum, cellular localization, primary structure, and cofactor dependence. Alkaline sphingomyelinase activity is confined to the intestinal mucosa, bile, and liver and does not participate in signal transduction.20–22 Neutral (NSMase) and acid (ASMase) sphingomyelinases, however, are crucially involved in the pathophysiology of metabolic disorders23 and play an active role in cellular signaling.24 Dysregulation of sphingolipid metabolism is believed to play a major role in many chronic diseases. Both NSMase and ASMase can be activated by tumor necrosis factor (TNF) and interleukin-1β (IL-1β) to induce the proinflammatory state in tissues.25,26 Opreanu et al.27 report that docosahexaenoic acid (DHA) downregulates basal and cytokine-induced ASMase and NSMase activity in human retinal endothelial cells and inhibition of sphingomyelinases in retinal endothelial cells prevent cytokine-induced inflammatory responses. Furthermore, ASMase is upregulated in the diabetic retina and blocking ASMase protects the retina from diabetes-induced damage.28
Previously, we and others, have examined the vascular protective effect of insulin-like growth factor (IGF) binding protein-3 (IGFBP-3) in both the early developing and adult retinal vasculature. IGFBP-3 can reduce pathologic neovascularization in the oxygen-induced retinopathy (OIR) model29–31 and more importantly serum levels of IGFBP-3 predict the outcome of retinopathy of prematurity (ROP), with increased IGFBP-3 levels associated with reduced ROP.30 IGFBP-3 increases recruitment of bone marrow-derived cells into areas of injured retina and facilitate vascular repair mechanisms31 and is neuroprotective.32 However, the effects of IGFBP-3 on permeability have not been studied. The purpose of this study was to determine whether free IGFBP-3, independent of IGF-1, can modulate the BRB. We used IGFBP-3NB in our experiments to assess the IGF-1 independent effects of IGFBP-3 in our studies, because this form of IGFBP-3 cannot bind IGF-1.33–35 We assessed permeability in vivo using two murine retinal vascular injury models. We studied the direct effects of IGFBP-3 in the retina and asked whether IGFBP-3 modulates vascular barrier properties. We found that intravitreal administration of IGFBP-3NB protected the BRB by partially restoring tight junction and adherens junction integrity of murine retinal blood vessels, which is likely due to the ability of IGFBP-3 to decrease proinflammatory sphingomyelinase levels in injured retina.
Methods
Animals
All animals were treated in accordance with the Guiding Principles in the Care and Use of Animals (NIH) and the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research. The studies were approved by the Institutional Animal Care and Use Committee at the University of Florida. C57BL/6 mice were obtained from The Jackson Laboratory (Bar Harbor, ME) and housed in the institutional animal care facilities at the University of Florida. A total of 156 C57BL/6 mice were used for in vivo permeability studies.
Retinal Vessel Occlusion by Photocoagulation
Plasmid encoding IGFBP-3 with undetectable low affinity for IGF-1 (IGFBP-3NB)34 was placed under the control of a proliferating endothelial-cell specific promoter composed of 7 × 46-mer multimerized endothelin enhancer upstream of a human Cdc6 promoter.36 IGFBP-3NB plasmid was placed into liposomes and delivered intravitreally immediately after laser injury.31 Three groups of C57BL/6 mice were used in these experiments. The groups consisted of laser only (n = 12), laser with IGFBP-3NB plasmid injection (n = 12), and IGFBP-3NB plasmid injection only (n = 12). The mice were anesthetized with ketamine/xylazine before treatment. The animals were photocoagulated by delivering approximately 35 spots at 150 watts for 0.2 seconds with a diode laser to the retinal vessels of the right eye. All three groups of mice were euthanized 4 days post laser and/or IGFBP-3 treatment.
In Vivo Permeability Assay
Recombinant human IGFBP-3NB was expressed and purified as previously described.34 In the first set of experiments, a total of five experimental groups were included and permeability assessed as previously described.37 The groups included IGFBP-3NB alone (n = 12), VEGF alone (n = 12), VEGF followed by IGFBP-3NB injected at 6 hours (n = 12), VEGF followed by IGFBP-3NB injected at 24 hours (n = 12), and the vehicle control for IGFBP-3NB consisting of 10 mM acetic acid (n = 12). One microliter of 40 ng/μL recombinant VEGF (Sigma, St. Louis, MO) was injected into the vitreous. This was followed by 1 μL of 100 ng/μL recombinant IGFBP-3NB. Recombinant VEGF was administered first to induce permeability followed by the blocking agent, in this case, IGFBP-3NB, 6 hours later as per our established protocol.37,38 After 48 hours following the initial VEGF injection, mice received a tail injection of FITC albumin. As an additional control only IGFBP-3NB was injected in each series of studies. Two hours later the mice were euthanized by cardiac perfusion with 4% paraformaldehyde (PFA) in PBS. The mice eyes were fixed in 4% PFA for 24 hours. Retinas were isolated, prepared as flat-mounts and digital images generated of random areas using a confocal microscope and FITC filter. All images were generated using identical settings within each experiment. Fluorescence intensity was determined by ImageJ software (developed by Wayne Rasband, National Institutes of Health, Bethesda, MD; available at http://rsb.info.nih.gov).
In the second set of studies, C57BL/6 mice underwent laser injury and injection within the vitreous with plasmid expressing IGFBP-3NB under control of proliferating endothelial cell-specific promoter placed in liposomes as previously described.31 There were a total of three experimental groups: laser only (n = 12), IGFBP-3 injection only (n = 12), and laser and IGFBP-3 injection (n = 12). Four days post laser treatment, the mice received a tail vein injection of FITC albumin. Two hours later the mice were euthanized by cardiac perfusion with 4% PFA. The eyes were fixed in 4% PFA for 24 hours. Retinas were harvested and processed for fluorescence analysis and confocal microscopy as described above.
In Vitro Permeability Assay
Bovine retinal endothelial cells (BREC) were isolated from bovine eyes as previously described37 and grown in M131 media (Mediatech, Mannasses, VA). For barrier assays P1 or P2 BRECs were seeded into transwell inserts (6.5 mm diameter, 0.4 μm pore size; Transwell, Corning, Cambridge, MA) and grown until transendothelial electrical resistance (TEER) stabilized at values >20 Ω · cm2. Macromolecular flux or TEER changes were measured in the absence or presence of IGFBP-3NB (100 ng/mL) during 12 hours. IGFBP-3NB was added to the basal side of the BREC monolayer to mimic the in vivo conditions as closely as possible (i.e., abluminal application through intravitreal injection). Changes in TEER were measured using a volt-ohmmeter (EVOM; World Precision Instruments, Sarasota, FL). At the indicated times, resistance readings (Ω) were obtained from each insert and multiplied by the membrane area (Ω · cm2). The resistance value of an empty culture insert (no cells) was subtracted to result in TEER of cells only. Macromolecular flux across BREC monolayers was measured by adding 1 mg/mL 40 kDa Rhodamine-dextran (Sigma) to the upper chamber of transwell inserts. At various time points 50 μL of media was removed from the lower chamber and then replaced with fresh media. The relative fluorescence was measured using a fluorescent plate reader (Biotek, Winooski, VT) with excitation at 530 nm and emission at 590 nm. Data shown represents changes of relative fluorescence between consecutive time points. For both TEER and flux measurements data were collected from triplicate inserts from separate experiments and expressed as mean ± SEM.
Immunofluoresence Histochemistry
Eyes from C57BL/6 mice were fixed with 4% paraformaldehyde/PBS overnight. The neural retina was dissected from the posterior cup and washed with PBS and then permeabilized in 10% normal goat serum with 0.2% Triton X-100 in PBS overnight at 4°C. The neural retinas were stained for detection of Claudin-5 (Abcam Inc., Cambridge, MA) at 1:3000 and VE-cadherin at 1:3000 (Cell Signaling Technology, Inc., Danvers, MA) overnight at 4°C for detection of tight or adherens junctions. The retinas were transferred to secondary antibody for 24 hours at 4°C after washing in PBS with 0.2% Triton X-100. The secondary antibody was Cy3 conjugated goat anti-rabbit IgG (1:250). The retinas were then incubated 30 minutes at room temperature in 1:500 FITC-conjugated agglutinin in 10 mM HEPES, 150 mM NaCl and 0.1% Tween-20 to visualize the retinal blood vessels. Retinas were flat-mounted onto microscope slides by making three or four radial cuts and covered in aqueous mounting medium (VectaShield; Vector Laboratories, Inc., Burlingame, CA) for observation by confocal microscopy. Digital confocal images were captured with a confocal microscope (Olympus DSU-Olympus IX81; Olympus America, Inc., Center Valley, PA). Digital images from experimental and control retinas with identical photomultiplier tube gain settings were captured by using imaging software (SlideBook4.2; Olympus America, Inc.). Maximum projections generated from z-section stacks of confocal images were processed identically in experimental and control retinas.
Reverse Transcription–Polymerase Chain Reaction
Total mRNA of mouse retinas was isolated (Total RNA Mini Kit; Bio-Rad, Hercules, CA) per the manufacturer's instructions. The mRNA was transcribed (iScript cDNA Synthesis Kit; Bio-Rad), and real-time PCR was performed (ABI Master Mix; ABI Biosystems, Foster City, CA). Primers for mouse TATBP, VEGF, and sphingomyelinase (both acid and neutral forms) were purchased directly from ABI Biosystems. All samples were normalized to TATBP (ABI Biosystems). Real-time PCR was performed (ABI 7500 Fast PCR machine; ABI Biosystems) for 60 cycles. All reactions were performed in triplicate.
ASMase Activity Assay
Mouse retinas were homogenized with acid lysis buffer consisting of 50 mM sodium acetate, pH 5; 1% Triton X-100; 1 mM EDTA with freshly added protease inhibitor cocktail (Sigma-Aldrich, St. Loius, MO). Sphingomyelinase activity was measured (Amplex Red Sphingomyelinase Assay Kit; Molecular Probes, Eugene, OR) as described in the manufacturer's protocol.
Statistical Analysis
All experiments were repeated at least three times. The TEER and paracellular permeability data at different time points were assessed using a Student's t-test plus ANOVA for multiple comparisons. Results are expressed as mean ± SEM. Statistical analysis was performed using commercial software (Prism 5; GraphPad Software, La Jolla, CA) with P < 0.05 considered statistically significant.
Results
IGFBP-3NB Expressing Plasmid Reduces Vascular Permeability in a Retinal Ischemia–Induced Laser Injury Model
We first tested the effects of IGFBP-3NB in a laser injury model associated with retinal vascular leakage. In this model, laser injury initiates retinal vascular damage and subsequent vascular leakage. Retinas injected with IGFBP-3NB expressing plasmid alone or injected with the IGFBP-3NB expressing plasmid followed by laser injury demonstrated minimal leakage compared with the control plasmid-injected laser-treated retinas, which showed a dramatic increase in retinal vascular leakage (Fig. 1).
Figure 1.
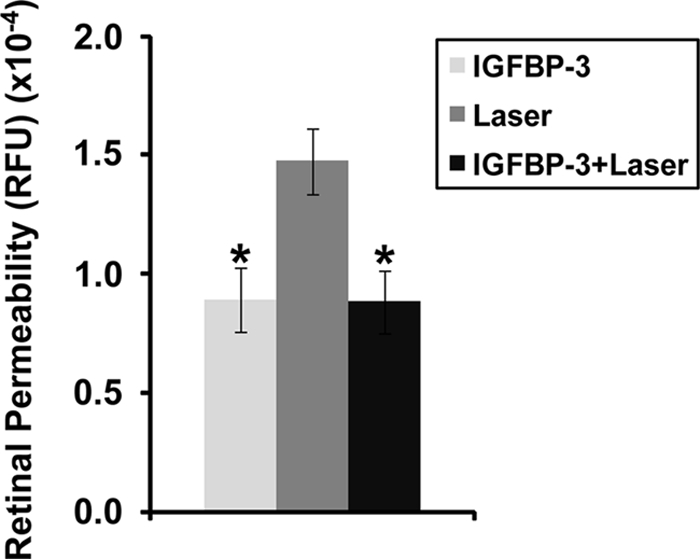
IGFBP-3NB overexpression improved retinal vascular permeability in laser-injured retinas. Graph represents FITC-conjugated albumin leakage from retinal vessels 4 days post laser treatment. The IGFBP-3NB plasmid (2 micrograms/microliter) was overexpressed in the retina for 4 days after which time the mice were subjected to laser retinal vessel occlusion. The mice were euthanized 4 days after laser treatment and retinas were isolated for analysis. *P < 0.02 when compared with laser treatment alone. RFU, relative fluorescence units.
IGFBP-3NB Reduces ASMase and NSMase mRNA and Activity Levels in a Retinal Ischemia–Induced Laser Injury Model
Previously, we showed in this laser injury model that injection with IGFBP-3-expressing plasmid reduced the levels of ceramide generated31 and reduced inflammation. Therefore, we questioned whether IGFBP-3NB reduced SMase levels in this model. As expected, laser-treated retinas showed a dramatic increase in ASMase and NSMase expression. Injection of IGFBP-3NB plasmid normalized ASMase and NSMase mRNA levels (P < 0.05) (Figs. 2A, 2B). The ASMase mRNA levels were induced more than the NSMase mRNA levels in the laser-treated retinas. This corresponded to increased ASMase activity levels in the laser-treated retinas (P < 0.05) that was brought back to normal in the IGFBP-3NB and laser-treated retinas (Fig. 2C). This suggests that IGFBP-3NB can reduce leakage in injured retinas by reducing SMase levels, particularly ASMase levels, which may promote BRB integrity.
Figure 2.

IGFBP-3NB decreases ASMase mRNA and activity levels in laser-injured mouse retina. (A, B) RT-PCR assessing ASMase (acid form) and NSMase (neutral form) levels in mouse retina. (A) Four days after treatment, when the IGFBP-3NB expressing plasmid is highly expressed in the retina, ASMase levels are significantly decreased in laser-treated retinas injected with IGFBP-3NB compared with laser-treated only retinas. (B) Four days after treatment, when the IGFBP-3NB expressing plasmid is highly expressed in the retina, NSMase levels are significantly decreased in laser-treated retinas injected with IGFBP-3NB compared with laser-treated only retinas. (C) ASMase activity levels were significantly decreased in lasered retinas injected with IGFBP-3NB after 4 days post laser treatment compared with the laser-treated control. *P < 0.05.
IGFBP-3NB Regulates Vascular Permeability Induced by Intravitreal VEGF Injection
In addition to studying vascular permeability in the laser injury model, we also tested the effects of IGFBP-3NB in a VEGF-induced permeability model. We specifically asked whether IGFBP-3NB would directly impact VEGF-induced permeability. Figure 3A shows quantification of fluorescent leakage after either VEGF (40 nanograms/microliter) followed by IGFBP-3NB (40 nanograms/microliter), VEGF alone, IGFBP-3NB alone, or the vehicle for VEGF, which was saline. VEGF injection increased permeability compared with vehicle-injected eyes. When IGFBP-3NB was injected post VEGF treatment, retinal vascular permeability was decreased. In Figures 3B–E, representative retinal whole mounts showing FITC albumin leakage from the retinal vessels of animals described in Figure 3A are shown. The vascular leakage induced by VEGF was attenuated by IGFBP-3NB administration.
Figure 3.

IGFBP-3NB regulates vascular permeability in vivo. (A) Quantification of fluorescent leakage after either VEGF (40 ng/μL) alone, VEGF followed by IGFBP-3NB (100 nanograms/microliter), IGFBP-3NB alone, or vehicle for VEGF (saline) injection. VEGF injection increased permeability compared with vehicle (saline)-injected eyes. When IGFBP-3NB was intravitreally injected post VEGF treatment, retinal vascular permeability was decreased compared with the VEGF-injected only control. (B-E) Representative retinal flat-mounts from animals described in Fig. 2A. (B) Saline, (C) VEGF, (D) VEGF followed by IGFBP-3NB, and (E) IGFBP-3NB. Arrows indicate representative areas of vascular leakages.
IGFBP-3NB Enhances In Vitro Retinal Endothelial Cell Barrier Characteristics
To better understand vascular barrier protection by IGFBP-3 in the retina, we assessed its effect in cultures of isolated bovine retinal endothelial cells (BREC). The addition of IGFBP-3NB to BREC monolayers led to a significant increase in TEER, which peaked within 5 minutes of protein addition. Significantly higher TEER were observed for at least 12 hours in cultures treated with IGFBP-3NB, suggesting that endothelial barrier was improved (Fig. 4A). Consistent with this idea we also found that the macromolecular flux across BREC cultures was significantly reduced in the presence of IGFBP-3NB (Fig. 4B). Flux of 40 kDa dextran was reduced by 50% within half an hour of treatment and remained low during the next 12 hours. Taken together these data showed that IGFBP-3 can enhance the barrier function of the retinal vasculature by acting directly on retinal endothelial cells.
Figure 4.

IGFBP-3NB enhances barrier properties of BREC in vitro. (A) BREC were isolated and grown on permeable transwell inserts. At time 0 recombinant IGFBP-3NB (100 ng/mL) protein or vehicle (control) was added to lower chambers (i.e., the basal sides of the monolayer; to mimic the conditions of intravitreal injections as closely as possible) and changes in TEER (A) or 40 kDa macromolecular flux (B) recorded at the indicated times. Enhanced TEER occurred within 5 minutes of IGFBP-3NB application. Significantly higher TEERs were observed in IGFBP-3NB-treated cells compared with control cells for the entire length of the experiment (12 hours). This indicated improved interendothelial junction integrity in response to IGFBP-3 and in agreement a nearly twofold reduction in macromolecular flux was observed within 30 minutes of IGFBP-3 treatment, which was maintained for at least 12 hours. *P < 0.05 when compared with control.
IGFBP-3NB Prevents the Dissociation of Tight Junction and Adherens Junction Integrity after VEGF Injection
To evaluate the integrity of the BRB, immunohistochemical analysis of retinal flat-mounts were performed for claudin-5 and VE-cadherin expression. Retinal flat-mounts were prepared from mice that underwent intravitreal injection of either VEGF, IGFBP-3NB, both IGFBP-3NB and VEGF, or saline. As shown in Figure 5, saline-injected vehicle control eyes showed clear expression of claudin-5 in the retinal vessels (Figs. 5A–C), as did the IGFBP-3NB-injected retinas (Figs. 5D–F). In contrast, VEGF injected eyes displayed a significant attenuation of claudin-5 expression in the retinal vessels (Figs. 5G–L). However, in retinas in which VEGF was injected followed by injection of IGFBP-3NB, there was partial restoration of claudin-5 expression in the retinal vessels (Figs. 5M–O). VE-cadherin expression was apparent in the saline-injected vehicle control in the retinal vessels (Figs. 6A–C). Likewise, the IGFBP-3NB-injected eyes demonstrated distinct VE-cadherin staining in the retinal vessels (Figs. 6D–F). However, VEGF-injected eyes displayed a loss of VE-cadherin expression in the retinal vessels (Figs. 6G–I) and was similar in appearance to the VEGF followed by saline-injected retinas (Figs. 6J–L). VEGF followed by IGFBP-3NB injection demonstrated no dissociation of VE-cadherin (Figs. 6M–O). These results suggest that IGFBP-3NB partially prevents dissociation of adherens and tight junctions in eyes injected with VEGF.
Figure 5.

IGFBP-3NB maintains tight junction integrity by maintaining claudin-5 expression in mice injected with VEGF. (A–O) Immunohistochemical analysis of retinal flat-mounts for claudin-5 expression at magnification ×60. The red stain is claudin-5 and the green stain is FITC-conjugated albumin used to visualize the retinal blood vessels. (A–C) Saline-injected vehicle control retinas showing expression of claudin-5 levels in the retinal vessels. (D–F) IGFBP-3NB-injected retinas showing expression of claudin-5 levels in the retinal vessels. (G–I) VEGF-injected retina showing a significant loss of claudin-5 expression in the retinal vessels. (J–L) VEGF-injected retina followed by saline injection 6 hours later. There is significant loss of claudin-5 expression in the retinal vessels. (M–O) VEGF-injected retina followed by IGFBP-3NB injection 6 hours later. There is preservation of claudin-5 expression levels in the retinal vessels when IGFBP-3NB is intravitreally administered to VEGF-injured retinas.
Figure 6.

IGFBP-3NB maintains adherens junctions by preventing dissociation of VE-cadherin expression in mice injected with VEGF. Immunohistochemical analysis of retinal flat-mounts for VE-cadherin expression at magnification ×60. The red stain is VE-cadherin and the green stain is FITC-conjugated albumin used to visualize the retinal blood vessels. (A–C) Saline-injected vehicle control showing VE-cadherin expression in the retinal vessels. (D–F) IGFBP-3NB-injected showing VE-cadherin expression. (G–I) VEGF-injected depicting loss of VE-cadherin expression in the retinal vessels. (J–L) VEGF followed by saline injection showing decreased VE-cadherin expression. (M–O) VEGF followed by IGFBP-3NB injection illustrating maintenance of VE-cadherin expression by IGFBP-3NB.
IGFBP-3NB Expression Decreases SMase mRNA Levels in VEGF-Injected Mice
Our results suggest that IGFBP-3NB reduces vascular permeability induced by intravitreal injection of VEGF. To investigate a possible mechanism for this effect, mice underwent intravitreal injection with either IGFBP-3NB expressing plasmid or control plasmid and 4 days later retinas were exposed to recombinant VEGF protein. Twenty-four hours later, both ASMase and NSMase mRNA levels were measured in these retinas. Both ASMase and NSMase levels were increased by VEGF administration but were normalized in retinas of IGFBP-3NB plasmid-injected eyes compared with retinas from control plasmid-injected eyes (Fig. 7).
Figure 7.
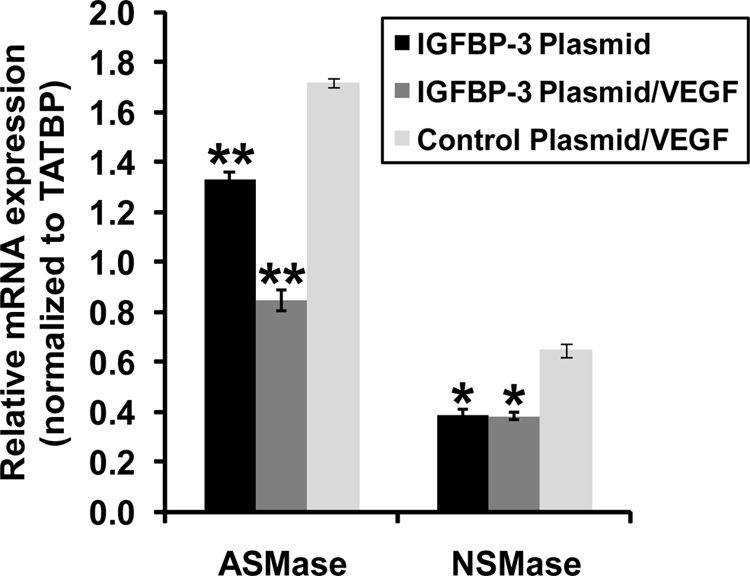
IGFBP-3NB expressing plasmid decreases VEGF-induced increases in ASMase and NSMase mRNA levels in mouse retina. The IGFBP-3NB plasmid (2 micrograms/microliter) was overexpressed in the retina for 4 days after which time the mice were intravitreally injected with recombinant VEGF (40 nanograms/microliter). The mice were euthanized 48 hours post VEGF injection and retinas were isolated for analysis. *P < 0.05, **P < 0.005 when compared with control plasmid/VEGF-injected control.
Discussion
In this study, we used either recombinant IGFBP-3NB or plasmid expressing IGFBP-3NB, under the direction of an endothelial cell specific promoter, to examine the effects of free IGFBP-3. Our results show that IGFBP-3 influences BRB integrity and does so independent of IGF binding. In vivo, IGFBP-3 reduces permeability induced by either direct VEGF administration or VEGF expression by laser occlusion-induced ischemia.
The deleterious effect of intravitreal VEGF on vascular permeability in normal rats has been successfully blocked with an inhibitor of protein kinase C (LY 333531) and by pigment epithelium-derived factor (PEDF) and placenta growth factor-1 (PLGF) in mice.37,39 LY 333531 also reduced BRB permeability in short-term diabetic rats. While we focused on the effects of IGFBP-3 on changes in the anterior blood retinal barrier, studies already exist demonstrating that VEGF exposure of ARPE-19 cells results in increased expression of IGFBP-3,40,41 which we believe represents a direct protective response to VEGF-induced injury and suggests a protective role for IGFBP-3 in the retinal pigment epithelial cells (RPE), which compose the posterior blood retinal barrier. Slomiany and Rosenzweig40 also report an autocrine loop of IGF-1-induced VEGF secretion with subsequent IGFBP-3 secretion in RPE cells. This suggests that under physiological conditions, there exists a delicate balance between the VEGF-IGF systems. Mukherjee and Guidry42 and Ainscough et al.43 further corroborated the beneficial effect of IGFBP-3 secretion in response to IGF-induced VEGF secretion in human RPE cells. In our in vivo laser studies, we infer that the prolonged IGFBP-3 expression generated by the plasmid acts to dampen the endogenous VEGF response to laser injury and likely explains the decrease in vascular permeability we observed.
In agreement with our in vivo findings, IGFBP-3 reduced monolayer permeability of highly purified BREC cultures in vitro, indicating that its effect on vascular integrity could be attributed to direct action on retinal endothelial cells, rather than any perivascular cells. Barrier enhancement in response to IGFBP-3 occurred rapidly and was observed 5 minutes post treatment and was sustained for at least 12 hours. Barrier improvement increased TEER and a substantially reduced macromolecular flux, indicating that IGFBP-3 altered interendothelial junctions rather than vesicular transport in BREC. Moreover, we demonstrated IGFBP-3 prevented dissociation of TJ and AJ in retinal blood vessels by partially restoring expression levels of claudin-5 and VE-cadherin, respectively.
It is unclear whether in vivo IGFBP-3 that does bind to IGF-1 would act the same way as the nonbinder mutant; however, there are several possibilities. The response to the mutant IGFBP-3 could be considered as the response to “free” IGFBP-3 (not complexed to IGF-1), which has physiological effects. However, the nonbinder mutant IGFBP-3 is not simply IGFBP-3 without IGF-1 on it. This particular IGFBP-3 is mutated at residues 80, 81, 217, and 223 and residues 217 and 223 are involved in nuclear receptor binding (Robert Baxter, unpublished). Because the mutant is active in reducing vascular permeability, this action does not require nuclear receptor binding or IGF binding, nonetheless wild type IGFBP-3 could still be modulated by nuclear receptor binding or IGF-1 binding. Also, because IGF-1 is known to affect vascular permeability by stimulating VEGF, IGFBP-3 may oppose this by binding IGF-1. This study shows that IGFBP-3 has a separate, IGF-independent action on VEGF-stimulated permeability. Therefore, IGFBP-3 could act in two ways: inhibiting VEGF expression by an IGF-binding-mediated mechanism or inhibiting VEGF action by an IGF-binding-independent mechanism.
Previously, we have examined the effect of PEDF and PLGF on VEGF-induced vascular permeability using the identical experimental design and time course as in the present study.37,38 In this study, we show that IGFBP-3NB can also block VEGF-induced vascular permeability. The in vivo effect of IGFBP-3NB was supported by our in vitro studies. IGFBP-3NB applied to the basal side of the endothelium in vitro demonstrated increased TEER and reduced macromolecular flux across the BREC monolayers. This basal administration represents ablumenal administration as would occur during an intravitreal injection. We also showed that IGFBP-3NB reduced vascular permeability induced by VEGF administration or by laser-induced injury. Furthermore, we demonstrate that IGFBP-3NB facilitates the reassociation of tight junctions and adherens junctions in retinal blood vessels after their disassociation by VEGF.
Using GFP+ chimeric mice that underwent the laser injury model, we showed that exposure of retinal vessels to IGFBP-3 increases recruitment of hematopoietic stem cells to sites of injury and their differentiation into endothelial cells, pericytes, and astrocytes.31,32 Pericyte and astrocyte ensheathment serve to strengthen the BRB after ischemia-induced injury,31,32 which is consistent with our current findings. Astrocytes are known to play a role in maintaining the BRB,44,45 while glial dysfunction has been linked to BRB breakdown.46 Astrocytes surround capillary endothelial cells and regulate retinal capillaries by paracrine interactions.46,47 Equally important, IGFBP-3 can reduce inflammation as we previously reported, after vessel occlusion by laser injury, and was associated with a reduced retinal ceramide/sphingomyelin ratio, which is an indicator of the inflammatory or proapoptotic state of cells.31 Our present study supports this as we show that IGFBP-3 reduces sphingomyelinase mRNA expression and activity levels in the laser-occluded retinas. This in turn would result in a decrease in the ceramide/sphingomyelin ratio, since sphingomyelinases are the enzymes responsible for the generation of ceramide from sphingomyelin (Fig. 8). We have shown previously IGFBP-3 can reduce the number of activated microglial cells after retinal injury and can increase apoptosis of these cells lending support that IGFBP-3 is anti-inflammatory.32 In this same setting, IGFBP-3 reduces levels of activated macrophages, while these studies are supportive of the effect of IGFBP-3 they were performed using the form of IGFBP-3 that binds IGF-1 making it difficult to exclude the contribution of IGF-1 to these findings.31 However, our current studies are in agreement with our earlier work showing IGFBP-3 can reduce inflammation in the retina, which can provide vascular stability to injured retinal vessels.31,32
Figure 8.
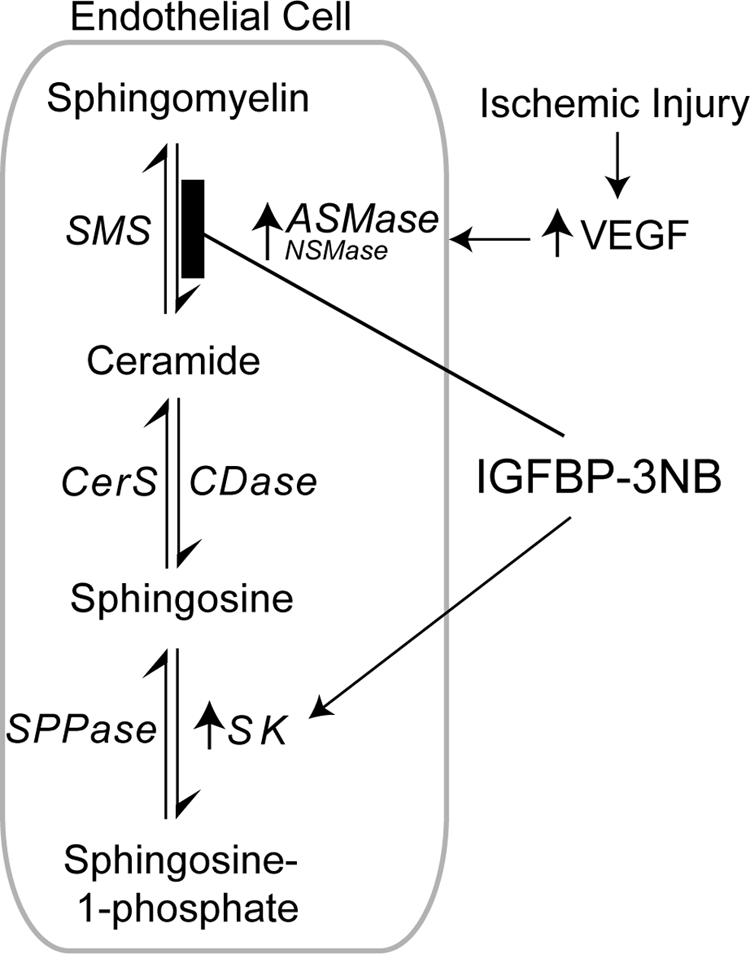
Possible mechanism of IGFBP-3's effects on VEGF and laser injury-induced retinal vascular permeability. Acid sphingomyelinase (ASMase) and neutral sphingomyelinase (NSMase) activation is an integral part of VEGF or laser-induced injury, which leads to disruption of the BRB and increased permeability. ASMase and NSMase are the principle enzymes responsible for the generation of ceramide from sphingomyelin. IGFBP-3 reduces ASMase and NSMase mRNA expression and activity levels after injury, which leads to a decrease in ceramide/sphingomyelin ratio. IGFBP-3 also increases the expression of sphingosine kinase (SK), which may serve to reduce ceramide further by using ceramide as a substrate for sphingosine and generation of sphingosine-1-phosphate (S1P), which has also been shown to improve blood retinal barrier integrity.
Another novel finding of this study is that ASMase and NSMase activation is an integral part of VEGF or laser-induced increase in permeability. Inhibition of ASMase and/or NSMase may represent an important part of the protective effects of IGFBP-3. Activation of SMases leads to an increase in ceramide production and/or another possible mediator, sphingosine-1-phosphate (S1P).
Previously, we and others, have shown that IGFBP-3 increases the expression of sphingosine kinase 1 (SphK1) in both endothelial cells and epithelial cells.31,48,49 This increase in SphK1 serves to generate S1P. There are several studies showing S1P regulates endothelial cell barrier function. Depending on the cellular milieu, the concentration of S1P and the type of injury or vascular bed, S1P can regulate vascular permeability.50 Zhang et al.51 report S1P prevents permeability increases via activation of endothelial S1P receptor-1 in rat venules and Lee et al.52 show a balance of S1P1 and S1P2 signaling regulates peripheral microvascular permeability in rat muscle vasculature. Tauseef et al.53 show that activation of Sphk1 reverses the increase in lung vascular permeability through S1P receptor signaling in endothelial cells. Also, Camerer and colleagues54 show S1P in the plasma compartment regulates basal and inflammation-induced vascular leak in mice. S1P can also influence tight junction protein integrity as Sun et al.55 reported an enhanced interaction between focal adhesion and adherens junction proteins after S1P exposure. Also, noteworthy are the observations by Jensen et al.56 that show NSMase is critically involved in modulation of skin edema.
In summary, we showed that IGFBP-3 can regulate BRB integrity as schematically illustrated in Figure 8. Thus, by enhancing the BRB, IGFBP-3 can provide vascular stabilization to injured retinal vessels and preserve vascular integrity. IGFBP-3, due to its beneficial effects on the restoration of tight junctions and adherens junctions, may represent a potential therapeutic agent in the treatment of eye diseases, such as retinopathies, where the BRB is commonly compromised.
Footnotes
Supported by NIH Grants EY007739 (MBG), EY012601 (MBG), R01DK090730 (MBG), U01 HL087366 (MBG), RO1 EY018358 (MEB), and EY016077 (JVB).
Disclosure: J.L. Kielczewski, None; S. Li Calzi, None; L.C. Shaw, None; J. Cai, None; X. Qi, None; Q. Ruan, None; L. Wu, None; L. Liu, None; P. Hu, None; T. Chan-Ling, None; R.N. Mames, None; S. Firth, None; R.C. Baxter None; P. Turowski, None; J.V. Busik, None; M.E. Boulton, None; M.B. Grant, None
References
- 1. Kaur C, Foulds WS, Ling EA. Blood-retinal barrier in hypoxic ischaemic conditions: basic concepts, clinical features and management. Prog Retin Eye Res. 2008;27:622–647 [DOI] [PubMed] [Google Scholar]
- 2. Zhang X, Bao S, Lai D, Rapkins RW, Gillies MC. Intravitreal triamcinolone acetonide inhibits breakdown of the blood-retinal barrier through differential regulation of VEGF-A and its receptors in early diabetic rat retinas. Diabetes. 2008;57:1026–1033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Dejana E, Orsenigo F, Lampugnani MG. The role of adherens junctions and VE-cadherin in the control of vascular permeability. J Cell Sci. 2008;121:2115–2122 [DOI] [PubMed] [Google Scholar]
- 4. Taddei A, Giampietro C, Conti A, et al. Endothelial adherens junctions control tight junctions by VE-cadherin-mediated upregulation of claudin-5. Nat Cell Biol. 2008;10:923–934 [DOI] [PubMed] [Google Scholar]
- 5. Ronaldson PT, Demarco KM, Sanchez-Covarrubias L, Solinsky CM, Davis TP. Transforming growth factor-beta signaling alters substrate permeability and tight junction protein expression at the blood-brain barrier during inflammatory pain. J Cereb Blood Flow Metab. 2009;29:1084–1098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Blecharz KG, Drenckhahn D, Forster CY. Glucocorticoids increase VE-cadherin expression and cause cytoskeletal rearrangements in murine brain endothelial cEND cells. J Cereb Blood Flow Metab. 2008;28:1139–1149 [DOI] [PubMed] [Google Scholar]
- 7. Corada M, Mariotti M, Thurston G, et al. Vascular endothelial-cadherin is an important determinant of microvascular integrity in vivo. Proc Natl Acad Sci U S A. 1999;96:9815–9820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. May C, Doody JF, Abdullah R, et al. Identification of a transiently exposed VE-cadherin epitope that allows for specific targeting of an antibody to the tumor neovasculature. Blood. 2005;105:4337–4344 [DOI] [PubMed] [Google Scholar]
- 9. Morita K, Sasaki H, Fujimoto K, Furuse M, Tsukita S. Claudin-11/OSP-based tight junctions of myelin sheaths in brain and Sertoli cells in testis. J Cell Biol. 1999;145:579–588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Liebner S, Fischmann A, Rascher G, et al. Claudin-1 and claudin-5 expression and tight junction morphology are altered in blood vessels of human glioblastoma multiforme. Acta Neuropathol. 2000;100:323–331 [DOI] [PubMed] [Google Scholar]
- 11. Liebner S, Kniesel U, Kalbacher H, Wolburg H. Correlation of tight junction morphology with the expression of tight junction proteins in blood-brain barrier endothelial cells. Eur J Cell Biol. 2000;79:707–717 [DOI] [PubMed] [Google Scholar]
- 12. Morcos Y, Hosie MJ, Bauer HC, Chan-Ling T. Immunolocalization of occludin and claudin-1 to tight junctions in intact CNS vessels of mammalian retina. J Neurocytol. 2001;30:107–123 [DOI] [PubMed] [Google Scholar]
- 13. Erickson KK, Sundstrom JM, Antonetti DA. Vascular permeability in ocular disease and the role of tight junctions. Angiogenesis. 2007;10:103–117 [DOI] [PubMed] [Google Scholar]
- 14. Simo R, Hernandez C. Advances in the medical treatment of diabetic retinopathy. Diabetes Care. 2009;32:1556–1562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Schlingemann RO, van Hinsbergh VW. Role of vascular permeability factor/vascular endothelial growth factor in eye disease. Br J Ophthalmol. 1997;81:501–512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Senger DR, Galli SJ, Dvorak AM, Perruzzi CA, Harvey VS, Dvorak HF. Tumor cells secrete a vascular permeability factor that promotes accumulation of ascites fluid. Science. 1983;219:983–985 [DOI] [PubMed] [Google Scholar]
- 17. Goggel R, Winoto-Morbach S, Vielhaber G, et al. PAF-mediated pulmonary edema: a new role for acid sphingomyelinase and ceramide. Nat Med. 2004;10:155–160 [DOI] [PubMed] [Google Scholar]
- 18. Goggel R, Uhlig S. The inositol trisphosphate pathway mediates platelet-activating-factor-induced pulmonary oedema. Eur Respir J. 2005;25:849–857 [DOI] [PubMed] [Google Scholar]
- 19. Uhlig S, Jestoi M, Kristin Knutsen A, Heier BT. Multiple regression analysis as a tool for the identification of relations between semi-quantitative LC-MS data and cytotoxicity of extracts of the fungus Fusarium avenaceum (syn. F. arthrosporioides). Toxicon. 2006;48:567–579 [DOI] [PubMed] [Google Scholar]
- 20. Duan RD. Alkaline sphingomyelinase: an old enzyme with novel implications. Biochim Biophys Acta. 2006;1761:281–291 [DOI] [PubMed] [Google Scholar]
- 21. Kolesnick R. The therapeutic potential of modulating the ceramide/sphingomyelin pathway. J Clin Invest. 2002;110:3–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Pavoine C, Pecker F. Sphingomyelinases: their regulation and roles in cardiovascular pathophysiology. Cardiovasc Res. 2009;82:175–183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Holland WL, Summers SA. Sphingolipids, insulin resistance, and metabolic disease: new insights from in vivo manipulation of sphingolipid metabolism. Endocr Rev. 2008;29:381–402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Marchesini N, Hannun YA. Acid and neutral sphingomyelinases: roles and mechanisms of regulation. Biochem Cell Biol. 2004;82:27–44 [DOI] [PubMed] [Google Scholar]
- 25. Jensen JM, Schutze S, Neumann C, Proksch E. Impaired cutaneous permeability barrier function, skin hydration, and sphingomyelinase activity in keratin 10 deficient mice. J Invest Dermatol. 2000;115:708–713 [DOI] [PubMed] [Google Scholar]
- 26. Wiegmann K, Schutze S, Machleidt T, Witte D, Kronke M. Functional dichotomy of neutral and acidic sphingomyelinases in tumor necrosis factor signaling. Cell. 1994;78:1005–1015 [DOI] [PubMed] [Google Scholar]
- 27. Opreanu M, Lydic TA, Reid GE, McSorley KM, Esselman WJ, Busik JV. Inhibition of cytokine signaling in human retinal endothelial cells through downregulation of sphingomyelinases by docosahexaenoic acid. Invest Ophthalmol Vis Sci. 2010;51:3253–3263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Opreanu M, Tikhonenko M, Bozack S, et al. The unconventional role of Acid sphingomyelinase in regulation of retinal microangiopathy in diabetic human and animal models. Diabetes. 2011;60:2370–2378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chang KH, Chan-Ling T, McFarland EL, et al. IGF binding protein-3 regulates hematopoietic stem cell and endothelial precursor cell function during vascular development. Proc Natl Acad Sci USA. 2007;104:10595–10600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lofqvist C, Chen J, Connor KM, et al. IGFBP3 suppresses retinopathy through suppression of oxygen-induced vessel loss and promotion of vascular regrowth. Proc Natl Acad Sci USA. 2007;104:10589–10594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kielczewski JL, Jarajapu YP, McFarland EL, et al. Insulin-like growth factor binding protein-3 mediates vascular repair by enhancing nitric oxide generation. Circ Res. 2009;105:897–905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kielczewski JL, Hu P, Shaw LC, et al. Novel protective properties of IGFBP-3 result in enhanced pericyte ensheathment, reduced microglial activation, increased microglial apoptosis, and neuronal protection after ischemic retinal injury. Am J Pathol. 2011;178:1517–1528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Schedlich LJ, Graham LD, O'Han MK, et al. Molecular basis of the interaction between IGFBP-3 and retinoid X receptor: role in modulation of RAR-signaling. Arch Biochem Biophys. 2007;465:359–369 [DOI] [PubMed] [Google Scholar]
- 34. Yan X, Forbes BE, McNeil KA, Baxter RC, Firth SM. Role of N- and C-terminal residues of insulin-like growth factor (IGF)-binding protein-3 in regulating IGF complex formation and receptor activation. J Biol Chem. 2004;279:53232–53240 [DOI] [PubMed] [Google Scholar]
- 35. Firth SM, Ganeshprasad U, Baxter RC. Structural determinants of ligand and cell surface binding of insulin-like growth factor-binding protein-3. J Biol Chem. 1998;273:2631–2638 [DOI] [PubMed] [Google Scholar]
- 36. Shaw LC, Pan H, Afzal A, et al. Proliferating endothelial cell-specific expression of IGF-I receptor ribozyme inhibits retinal neovascularization. Gene Ther. 2006;13:752–760 [DOI] [PubMed] [Google Scholar]
- 37. Cai J, Wu L, Qi X, et al. Placenta growth factor-1 exerts time-dependent stabilization of adherens junctions following VEGF-induced vascular permeability. PLoS One. 2011;6:e18076. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 38. Cai J, Wu L, Qi X, et al. PEDF regulates vascular permeability by a gamma-secretase-mediated pathway. PLoS One. 2011;6:e21164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Aiello LP, Bursell SE, Clermont A, et al. Vascular endothelial growth factor-induced retinal permeability is mediated by protein kinase C in vivo and suppressed by an orally effective beta-isoform-selective inhibitor. Diabetes. 1997;46:1473–1480 [DOI] [PubMed] [Google Scholar]
- 40. Slomiany MG, Rosenzweig SA. Autocrine effects of IGF-I-induced VEGF and IGFBP-3 secretion in retinal pigment epithelial cell line ARPE-19. Am J Physiol Cell Physiol. 2004;287:C746–C753 [DOI] [PubMed] [Google Scholar]
- 41. Slomiany MG, Rosenzweig SA. IGF-1-induced VEGF and IGFBP-3 secretion correlates with increased HIF-1 alpha expression and activity in retinal pigment epithelial cell line D407. Invest Ophthalmol Vis Sci. 2004;45:2838–2847 [DOI] [PubMed] [Google Scholar]
- 42. Mukherjee S, Guidry C. The insulin-like growth factor system modulates retinal pigment epithelial cell tractional force generation. Invest Ophthalmol Vis Sci. 2007;48:1892–1899 [DOI] [PubMed] [Google Scholar]
- 43. Ainscough SL, Feigl B, Malda J, Harkin DG. Discovery and characterization of IGFBP-mediated endocytosis in the human retinal pigment epithelial cell line ARPE-19. Exp Eye Res. 2009;89:629–637 [DOI] [PubMed] [Google Scholar]
- 44. Kim JH, Park JA, Lee SW, Kim WJ, Yu YS, Kim KW. Blood-neural barrier: intercellular communication at glio-vascular interface. J Biochem Mol Biol. 2006;39:339–345 [DOI] [PubMed] [Google Scholar]
- 45. Gardner TW, Lieth E, Khin SA, et al. Astrocytes increase barrier properties and ZO-1 expression in retinal vascular endothelial cells. Invest Ophthalmol Vis Sci. 1997;38:2423–2427 [PubMed] [Google Scholar]
- 46. Shen W, Li S, Chung SH, Gillies MC. Retinal vascular changes after glial disruption in rats. J Neurosci Res. 2010;88:1485–1499 [DOI] [PubMed] [Google Scholar]
- 47. Abukawa H, Tomi M, Kiyokawa J, et al. Modulation of retinal capillary endothelial cells by Muller glial cell-derived factors. Mol Vis. 2009;15:451–457 [PMC free article] [PubMed] [Google Scholar]
- 48. Granata R, Trovato L, Lupia E, et al. Insulin-like growth factor binding protein-3 induces angiogenesis through IGF-I- and SphK1-dependent mechanisms. J Thromb Haemost. 2007;5:835–845 [DOI] [PubMed] [Google Scholar]
- 49. Martin JL, Lin MZ, McGowan EM, Baxter RC. Potentiation of growth factor signaling by insulin-like growth factor-binding protein-3 in breast epithelial cells requires sphingosine kinase activity. J Biol Chem. 2009;284:25542–25552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wang L, Dudek SM. Regulation of vascular permeability by sphingosine 1-phosphate. Microvasc Res. 2009;77:39–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Zhang G, Xu S, Qian Y, He P. Sphingosine-1-phosphate prevents permeability increases via activation of endothelial sphingosine-1-phosphate receptor 1 in rat venules. Am J Physiol Heart Circ Physiol. 2010;299:H1494–H1504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Lee JF, Gordon S, Estrada R, et al. Balance of S1P1 and S1P2 signaling regulates peripheral microvascular permeability in rat cremaster muscle vasculature. Am J Physiol Heart Circ Physiol. 2009;296:H33–H42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Tauseef M, Kini V, Knezevic N, et al. Activation of sphingosine kinase-1 reverses the increase in lung vascular permeability through sphingosine-1-phosphate receptor signaling in endothelial cells. Circ Res. 2008;103:1164–1172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Camerer E, Regard JB, Cornelissen I, et al. Sphingosine-1-phosphate in the plasma compartment regulates basal and inflammation-induced vascular leak in mice. J Clin Invest. 2009;119:1871–1879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Sun X, Shikata Y, Wang L, et al. Enhanced interaction between focal adhesion and adherens junction proteins: involvement in sphingosine 1-phosphate-induced endothelial barrier enhancement. Microvasc Res. 2009;77:304–313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Jensen JM, Schutze S, Forl M, Kronke M, Proksch E. Roles for tumor necrosis factor receptor p55 and sphingomyelinase in repairing the cutaneous permeability barrier. J Clin Invest. 1999;104:1761–1770 [DOI] [PMC free article] [PubMed] [Google Scholar]