

Abstract
Hypertension is a well established risk factor for cardiovascular diseases such as stroke and is the leading cause of chronic kidney failure. Although a number of pharmacologic agents are available for the treatment of hypertension including agents that affect the renin-angiotensin-aldosterone system (RAAS), unmet needs in the treatment of hypertension suggest that identification of novel pharmacological targets would be an important healthcare goal. One potential target is prostaglandin E2 (PGE2), a potent lipid mediator with a diverse and sometimes opposing range of biological effects. PGE2 signals through four subtypes of G-protein coupled receptors designated EP1 through EP4. PGE2 functions primarily as a vasodepressor; under certain conditions PGE2 administration mediates vasopressor activity. This review focuses on the current understanding of the roles of PGE2 receptors in vascular reactivity, hypertension and end-organ damage.
Keywords: Prostaglandin E2, hypertension, GPCR, mouse, rat
Introduction
Hypertension increases the risk of stroke, heart attack and end-organ damage including kidney failure. Current therapies to reduce blood pressure and diminish the incidence of complications include blockade of the renin-angiotensin-aldosterone axis, calcium channels, and beta adrenergic receptors. Nonetheless there is significant unmet need for novel therapeutic agents [1].
Prostaglandins (PGs) are cyclooxygenase metabolites of arachidonic acid, and mediate an array of physiologic functions including the regulation of systemic blood pressure. Prostaglandin E2 (PGE2) is a major prostanoid contributing to this regulation of blood pressure, where it can exert either vasopressor or vasodepressor effects depending upon the setting [2,3,4]. These physiologically opposing effects can be explained in part by the existence of four PGE2 receptors, designated the E-Prostanoid (EP) receptors EP1 through EP4. Previous studies have determined that the EP1 and EP3 receptors primarily mediate the pressor response, while the EP2 and EP4 receptors mediate the depressor response [2,5,6,7,8,9,10].
The role of PGs in the regulation of blood pressure is highlighted by the pro-hypertensive action of non-steroidal anti-inflammatory drugs (NSAIDs) which inhibit cyclooxygenase mediated prostanoid production, suggesting that overall prostaglandins have an anti-hypertensive role [11]. However, under certain conditions NSAIDs can also have hypotensive effects [12]. The balance of functionally antagonistic prostaglandin action fine-tunes blood pressure homeostasis. The remainder of this review will focus on the actions of PGE2 and its receptors in blood pressure regulation and its impact on downstream consequences of blood pressure dysregulation.
Isomerization of PGH2 to PGE2 by microsomal PGE synthase (mPGES) plays a key role in the regulation of blood pressure by regulating vascular tone, sodium balance and/or renin release [13]. Inhibitors of mPGES are currently being pursued for treatment of cancer, pain and inflammation; however, unwanted pro-hypertensive effects may result from this strategy. In rodents, deletion of mPGES-1 has been shown to further increase blood pressure in several models of hypertension, including deoxycorticosterone-salt water-induced hypertension and acute or chronic treatment with angiotensin II [14,15,16]. In humans, where there is a great deal of phenotypic heterogeneity, the contribution of PGE2 towards hypertension is more controversial. In some cases, patients with essential hypertension have been shown to have low urinary excretion of PGE2; however, in other cases patients have been shown to have high urinary PGE2 excretion [17]. This inter-patient variability may result in a wide range of untoward side effects for mPGES inhibitors. In order to gain a better understanding of the effects of PGE2 on blood pressure homeostasis, attention has been focused on the actions of its receptors using knockout mouse models.
EP Receptors in blood pressure homeostasis
PGE2 signals through four subtypes of G-protein coupled receptors designated EP1–EP4, each having distinct tissue localization and signal transduction properties [7,18]. EP1 couples to Gq-proteins, mobilizes intracellular calcium, and stimulates phosphoinositide turnover activating protein kinase C. EP1 receptors were originally defined as constrictor receptors in smooth muscle. EP1 receptor mRNA is ubiquitously expressed; in a recent report, mRNA was detected in all 41 tissues assayed including kidney, lung and adrenal gland. This is consistent with EP1 expression throughout the vasculature [19]. EP1 receptors function as constrictors in the smooth muscle of the trachea, gastrointestinal tract, bladder and uterus. EP2 and EP4 couple to Gs-proteins, activate adenylate cyclase and increase intracellular cAMP ([cAMP]i). The EP2 receptor was initially described as a smooth muscle relaxant receptor. EP2 mRNA is most abundantly expressed in the lung, spleen and ovary while EP4 mRNA is predominantly localized the uterus, thymus, ileum, lung, spleen, adrenal gland, and kidney. EP3 couples to Gi-proteins, inhibits adenylate cyclase and decreases intracellular [cAMP]i. EP3 receptor expression is widespread, and it is found in the kidney, uterus, pancreas, stomach, thymus, spleen, smooth muscle of the gastrointestinal tract, vasculature, and CNS. In the brain, EP3 receptor mRNA has been observed in the hippocampus, preoptic area, hypothalamus, locus coeruleus and raphe nuclei (For reviews, see [7,18,20]). Although the EP receptors were initially described by their ligand binding selectivity and coupling to these well-characterized heterotrimeric G-protein mediated signaling pathways, they are now appreciated to couple to other signal transduction pathways as well, including arrestin-mediated signaling pathways [21,22]. Overall, the principal vasodepressor actions of PGE2 are mediated via the EP2 and EP4 receptors, whereas the vasopressor actions are mediated by activation of the EP1 and EP3 receptors [2,5,6,7,8,9,10] (Fig. 1).
Figure 1.
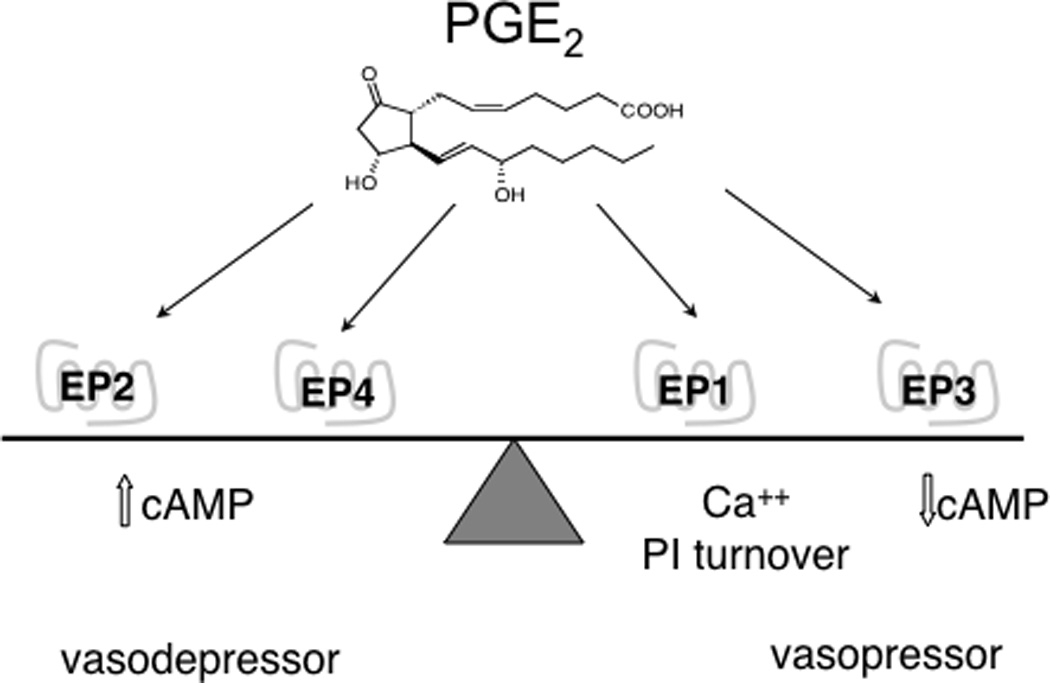
The role of EP receptors as regulators of blood pressure. The vasodepressor EP2 and EP4 receptors are functionally antagonistic to the pressor EP1 and EP3 receptors.
Vasodepressor Receptors
Upon acute infusion, PGE2 is a vasodepressor in both humans and mice [2,5,23]. This observation underscores the pro-hypertensive effects of blockade of all prostaglandin production and subsequent receptor activation by NSAIDs [11,24,25,26,27] consistent with the loss of a tonic vasodepressor PG effect. Although no selective EP2 antagonists are available, the pharmacology of EP vasodepressor response has been addressed with EP2 knockout mice. It has been shown that the depressor effect is primarily due to the activation of the EP2 receptor. When the EP2 receptor is deleted, the depressor response to PGE2 is lost. Moreover, the loss of the depressor response unmasks a PGE2 pressor response [2]. In addition, EP2 −/− mice fed a high-salt diet experienced an increase in blood pressure consistent with a protective role for EP2 activation in salt-sensitive hypertension [2]. Intravenous infusion of the EP2 agonist ONO-AE1-259 into Wistar rats increases retinal arteriolar and venous diameter and substantially reduces mean arterial pressure [28]. EP2 and EP4 receptors evoke an increase in [cAMP]i through a Gs coupled pathway, a classical mechanism for smooth muscle relaxation. Hristovska et al. demonstrated a dose-dependent relaxation in response to PGE2 in aortic rings which was lost in tissue from EP4 −/− mice but remained intact in EP2 −/− tissue. The EP4 dilator effect was dependent upon endothelium-derived nitric oxide production via eNOS [29]. Acute blood pressure studies are challenging in EP4 −/− mice because the mice exhibit near complete perinatal lethality in inbred strains as a result of persistent patent ductus arteriosus [30]. Analysis of studies performed with surviving EP4 −/− animals on a mixed-strain background may not be straightforward as their survival may be dependent on modifier genes. Nonetheless, deletion of EP4 resulted in a diminished vasodepressor response to PGE2 [5]. In rats, infusion of the EP4 selective agonist ONO-AE1-329 significantly reduces blood pressure; it does not alter retinal vessel diameter [28]. Taken together is consistent with EP4 vasodilator action in a subset of vascular beds.
Vasopressor Receptors
As described above, the pressor effects of systemic PGE2 infusion are only observed in the absence of the predominant depressor receptor EP2 [2]. In contrast, infusion of EP3 receptor selective agonists such as sulprostone, MB28767 or SC46275 in wildtype mice results in an acute and substantial rise in mean arterial pressure [8]. It would be of interest to determine the result of systemic EP3 agonists on heart rate, as presynaptic inhibitory EP3 receptors are believed to mediate the reduced release of norepinephrine by PGE2 [31]. The EP3 mediated pressor effect undergoes desensitization with repeated administration of EP3 agonists. In EP2 −/− mice after desensitization of EP3 responses, the depressor action of EP4 in response to PGE2 infusion is then apparent [8]. Thus, upon systemic infusion of PGE2 in mice the depressor action of EP2 predominates, followed by the pressor action of EP3, and then the depressor action of EP4. Importantly, the order of expression of EP receptor mRNA does not mirror the phenotypic effects of the EP receptors. RNA levels determined by RNAse protection identified expression levels of EP3>>EP4>EP1≥EP2 in both renal resistance vessels and the aorta [8]. It is unclear whether changes in EP receptor density underlie changes in vascular tone in the hypertensive state. Because messenger RNA levels do not correlate with receptor function, and anti-receptor antibodies are of questionable value, this remains an important unanswered question.
In contrast to the depressor effects of systemic administration, when PGE2 is administered intracerebroventricularly (ICV) a rise in mean arterial pressure occurs, accompanied with tachycardia and enhanced renal sympathetic nerve activity [32]. These effects were ascribed to the EP3 receptor using ICV infusion of subtype selective EP receptor agonists [32]. Taken together, these data demonstrate that the centrally mediated pressor actions of PGE2 are EP3-mediated.
Although the EP1 receptor does not appear to play a significant role in the blood pressure effects of systemically administered PGE2, it has been shown to be a significant contributor to hypertension, particularly in cases with enhanced renin-angiotensin system activity. Genetic deletion of the EP1 receptor in mice has been shown to significantly decrease systolic blood pressure, an effect amplified when mice are fed a low sodium diet [33]. Importantly, EP1 −/− mice have blunted pressor responses to both acute and chronic angiotensin II administration [10]. In isolated vascular preparations of preglomerular arterioles and mesenteric arteries, pre-treatment with SC51322, an EP1/3 antagonist, was able to abolish any angiotensin II-mediated vasoconstriction [10]. Furthermore, treatment of spontaneously hypertensive rats, a multifactorial model of essential hypertension, with SC51322 significantly reduces blood pressure [10], indicating the EP1 receptor and/or EP3 receptor may be novel targets for the treatment of hypertension.
Consequences of hypertension
Hypertension is an established risk factor for cardiovascular diseases including stroke, myocardial infarction, heart failure, arterial aneurysm and is the leading cause of chronic kidney failure. Current anti-hypertensive therapies reduce the risk of the related cardiovascular sequelae, though not to baseline risk observed in normotensive subjects. There is an unmet need for treatment which will reduce blood pressure and maximize target organ protection [1]. In considering whether PGE2 and its receptors make viable drug targets for hypertension, determination of their ability to reduce end-organ damage will be important. While it has been shown that genetic deletion of mPGES-1 in mice results in increased blood pressure [14,15,16], deletion of mPGES-1 has also been shown to protect against aortic aneurysm formation and vascular injury [34,35]. Angiotensin II infusion into hyperlipidemic mice produced fewer and less severe aneurysms, and reduced oxidative stress on a mPGES-1 −/− background compared to wildtype mice [36]. However, these results were complicated by the observed increase in PGI2 and PGD2 production accompanying the reduction in PGE2 [36]. It is yet to be determined whether potentially beneficial substrate diversion is a consequence specific to genetic mPGES-1 deletion, or would be recapitulated with chronic use of an mPGES inhibitor.
Blockade of individual PGE2 receptors might result in a reduction in end-organ damage while being less likely to produce unwanted side effects. In addition, GPCRs are demonstrably “druggable” and are one of the most common targets of currently developed therapeutic agents. Antagonism of EP1 receptors has been shown to preserve renal function, reducing tubulointerstitial damage, proliferative lesions, fibrotic area and proteinuria in stroke-prone spontaneously hypertensive rats [37], as well as cerebrovascular dysfunction induced by angiotensin II [38], making the EP1 receptor a potential target for treatment of hypertension and protection from end-organ damage. In the study performed in stroke-prone hypertensive rats tail cuff blood pressure was modestly reduced two weeks post-treatment with an EP1 antagonist, but this reduction was not maintained past five weeks of treatment. Nonetheless treatment with the EP1 antagonist provided end-organ protection. In contrast to the deleterious actions of the EP1 receptor, EP2 and EP4 receptors have been shown to be cardioprotective; it would seem important to maintain function of these receptors. For example, deletion of the EP4 receptor in a mouse model of ischemia reperfusion of the heart significantly increased infarct size, while treatment of wildtype mice with an EP4 agonist, ONO-4819, reduced infarct size [39]. Therefore, EP4 agonists could be useful for reducing blood pressure and afford cardioprotective benefits. Selective blockade of EP1 and/or EP3 receptors, while EP2 and EP4 signaling remains intact may be preferable to the loss of signaling in all four receptors resulting from inhibition of PGE2 ligand production.
Conclusions
In summary, PGE2 plays a dynamic role in regulation of blood pressure homeostasis. The existence of multiple receptors with diverse signaling abilities allows for modulation both positively and negatively. The development and availability of additional highly selective agonists and antagonists for EP receptors is fundamental to the advancement of the field. Unwanted side effects resulting from inhibition of the cyclooxygenase enzymes upstream of prostanoid production demonstrated the value of selective targeting as proximal to the pathophysiological action as possible. Development of new therapeutics targeting specific PGE2 receptors could reduce blood pressure and provide end-organ protection, while minimizing side effects.
Highlights.
PGE2 receptors are key modulators of blood pressure control, where EP receptors have functionally antagonistic actions
Activation of EP2 and EP4 receptors generally lowers blood pressure
Activation of EP1 and EP3 receptors generally raises blood pressure
Systemic infusion of PGE2 leads to a fall in mean arterial pressure by activation of the EP2 receptor
Intracerebroventricular infusion of PGE2 leads to a rise in mean arterial pressure by activation of the EP3 receptor
Acknowledgments
This work was supported by a Merit Award from the Department of Veterans Affairs and NIH grants DK46205, DK37097, GM015431.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Flack JM. Epidemiology and unmet needs in hypertension. J Manag Care Pharm. 2007;13:2–8. doi: 10.18553/jmcp.2007.13.s8-a.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kennedy CR, Zhang Y, Brandon S, Guan Y, Coffee K, Funk CD, Magnuson MA, Oates JA, Breyer MD, Breyer RM. Salt-sensitive hypertension and reduced fertility in mice lacking the prostaglandin EP2 receptor. Nat Med. 1999;5:217–220. doi: 10.1038/5583. [DOI] [PubMed] [Google Scholar]
- 3.Schaaf TK, Hess HJ. Synthesis and biological activity of carboxyl-terminus modified prostaglandin analogues. J Med Chem. 1979;22:1340–1346. doi: 10.1021/jm00197a012. [DOI] [PubMed] [Google Scholar]
- 4.Hockel GM, Cowley AW. Prostaglandin E2-induced hypertension in conscious dogs. Am J Physiol. 1979;237:H449–H454. doi: 10.1152/ajpheart.1979.237.4.H449. [DOI] [PubMed] [Google Scholar]
- 5.Audoly LP, Tilley SL, Goulet J, Key M, Nguyen M, Stock JL, McNeish JD, Koller BH, Coffman TM. Identification of specific EP receptors responsible for the hemodynamic effects of PGE2. Am J Physiol. 1999;277:H924–H930. doi: 10.1152/ajpheart.1999.277.3.H924. [DOI] [PubMed] [Google Scholar]
- 6.Tilley SL, Audoly LP, Hicks EH, Kim HS, Flannery PJ, Coffman TM, Koller BH. Reproductive failure and reduced blood pressure in mice lacking the EP2 prostaglandin E2 receptor. J Clin Invest. 1999;103:1539–1545. doi: 10.1172/JCI6579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Breyer MD, Breyer RM. Prostaglandin E receptors and the kidney. Am J Physiol Renal Physiol. 2000;279:F12–F23. doi: 10.1152/ajprenal.2000.279.1.F12. [DOI] [PubMed] [Google Scholar]
- 8.Zhang Y, Guan Y, Schneider A, Brandon S, Breyer RM, Breyer MD. Characterization of murine vasopressor and vasodepressor prostaglandin E(2) receptors. Hypertension. 2000;35:1129–1134. doi: 10.1161/01.hyp.35.5.1129. [DOI] [PubMed] [Google Scholar]
- 9.Audoly LP, Ruan X, Wagner VA, Goulet JL, Tilley SL, Koller BH, Coffman TM, Arendshorst WJ. Role of EP(2) and EP(3) PGE(2) receptors in control of murine renal hemodynamics. Am J Physiol Heart Circ Physiol. 2001;280:H327–H333. doi: 10.1152/ajpheart.2001.280.1.H327. [DOI] [PubMed] [Google Scholar]
- 10.Guan Y, Zhang Y, Wu J, Qi Z, Yang G, Dou D, Gao Y, Chen L, Zhang X, Davis L, Wei M, Fan X, Carmosino M, Hao C, Imig J, Breyer R, Breyer M. Antihypertensive effects of selective prostaglandin E2 receptor subtype 1 targeting. J Clin Invest. 2007;117:2496–2505. doi: 10.1172/JCI29838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gurwitz JH, Avorn J, Bohn RL, Glynn RJ, Monane M, Mogun H. Initiation of antihypertensive treatment during nonsteroidal anti-inflammatory drug therapy. JAMA. 1994;272:781–786. [PubMed] [Google Scholar]
- 12.Imanishi M, Kawamura M, Akabane S, Matsushima Y, Kuramochi M, Ito K, Ohta M, Kimura K, Takamiya M, Omae T. Aspirin lowers blood pressure in patients with renovascular hypertension. Hypertension. 1989;14:461–468. doi: 10.1161/01.hyp.14.5.461. [DOI] [PubMed] [Google Scholar]
- 13.Jakobsson PJ, Thorén S, Morgenstern R, Samuelsson B. Identification of human prostaglandin E synthase: a microsomal, glutathione-dependent, inducible enzyme, constituting a potential novel drug target. Proc Natl Acad Sci U S A. 1999;96:7220–7225. doi: 10.1073/pnas.96.13.7220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Facemire CS, Griffiths R, Audoly LP, Koller BH, Coffman TM. The impact of microsomal prostaglandin e synthase 1 on blood pressure is determined by genetic background. Hypertension. 2010;55:531–538. doi: 10.1161/HYPERTENSIONAHA.109.145631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang DJ, Chen LH, Zhang YH, Yang GR, Dou D, Gao YS, Zhang XY, Kong XM, Zhao P, Pu D, Wei MF, Breyer MD, Guan YF. Enhanced pressor response to acute Ang II infusion in mice lacking membrane-associated prostaglandin E2 synthase-1. Acta Pharmacol Sin. 2010;31:1284–1292. doi: 10.1038/aps.2010.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jia Z, Aoyagi T, Yang T. mPGES-1 protects against DOCA-salt hypertension via inhibition of oxidative stress or stimulation of NO/cGMP. Hypertension. 2010;55:539–546. doi: 10.1161/HYPERTENSIONAHA.109.144840. [DOI] [PubMed] [Google Scholar]
- 17.Ruilope L, Garcia Robles R, Barrientos A, Bernis C, Alcazar J, Tresguerres JA, Mancheño E, Millet VG, Sancho J, Rodicio JL. The role of urinary PGE2 and renin-angiotensin-aldosterone system in the pathogenesis of essential hypertension. Clin Exp Hypertens A. 1982;4:989–1000. doi: 10.3109/10641968209060767. [DOI] [PubMed] [Google Scholar]
- 18.Coleman RA, Smith WL, Narumiya S. International Union of Pharmacology classification of prostanoid receptors: properties, distribution, and structure of the receptors and their subtypes. Pharmacol Rev. 1994;46:205–229. [PubMed] [Google Scholar]
- 19.Regard JB, Sato IT, Coughlin SR. Anatomical profiling of G protein-coupled receptor expression. Cell. 2008;135:561–571. doi: 10.1016/j.cell.2008.08.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sugimoto Y, Narumiya S. Prostaglandin E receptors. J Biol Chem. 2007;282:11613–11617. doi: 10.1074/jbc.R600038200. [DOI] [PubMed] [Google Scholar]
- 21.Kim JI, Lakshmikanthan V, Frilot N, Daaka Y. Prostaglandin E2 promotes lung cancer cell migration via EP4-betaArrestin1-c-Src signalsome. Mol Cancer Res. 2010;8:569–577. doi: 10.1158/1541-7786.MCR-09-0511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chun KS, Lao HC, Langenbach R. The prostaglandin E2 receptor, EP2, stimulates keratinocyte proliferation in mouse skin by G protein-dependent and {beta}-arrestin1-dependent signaling pathways. J Biol Chem. 2010;285:39672–39681. doi: 10.1074/jbc.M110.117689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Eklund B, Carlson LA. Central and peripheral circulatory effects and metabolic effects of different prostaglandins given I.V. to man. Prostaglandins. 1980;20:333–347. doi: 10.1016/s0090-6980(80)80051-7. [DOI] [PubMed] [Google Scholar]
- 24.Murray MD, Greene PK, Brater DC, Manatunga AK, Hall SD. Effects of flurbiprofen on renal function in patients with moderate renal insufficiency. Br J Clin Pharmacol. 1992;33:385–393. doi: 10.1111/j.1365-2125.1992.tb04056.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Johnson AG. NSAIDs and increased blood pressure. What is the clinical significance? Drug Saf. 1997;17:277–289. doi: 10.2165/00002018-199717050-00001. [DOI] [PubMed] [Google Scholar]
- 26.Cheng HF, Harris RC. Cyclooxygenases, the kidney, and hypertension. Hypertension. 2004;43:525–530. doi: 10.1161/01.HYP.0000116221.27079.ea. [DOI] [PubMed] [Google Scholar]
- 27.Solomon DH, Schneeweiss S, Levin R, Avorn J. Relationship between COX-2 specific inhibitors and hypertension. Hypertension. 2004;44:140–145. doi: 10.1161/01.HYP.0000136134.31846.83. [DOI] [PubMed] [Google Scholar]
- 28.Mori A, Saito M, Sakamoto K, Narita M, Nakahara T, Ishii K. Stimulation of prostanoid IP and EP(2) receptors dilates retinal arterioles and increases retinal and choroidal blood flow in rats. Eur J Pharmacol. 2007;570:135–141. doi: 10.1016/j.ejphar.2007.05.052. [DOI] [PubMed] [Google Scholar]
- 29.Hristovska AM, Rasmussen LE, Hansen PB, Nielsen SS, Nüsing RM, Narumiya S, Vanhoutte P, Skøtt O, Jensen BL. Prostaglandin E2 induces vascular relaxation by E-prostanoid 4 receptor-mediated activation of endothelial nitric oxide synthase. Hypertension. 2007;50:525–530. doi: 10.1161/HYPERTENSIONAHA.107.088948. [DOI] [PubMed] [Google Scholar]
- 30.Nguyen M, Camenisch T, Snouwaert JN, Hicks E, Coffman TM, Anderson PA, Malouf NN, Koller BH. The prostaglandin receptor EP4 triggers remodelling of the cardiovascular system at birth. Nature. 1997;390:78–81. doi: 10.1038/36342. [DOI] [PubMed] [Google Scholar]
- 31.Jensen TJ, Nedergaard OA. Modulation of norepinephrine release from sympathetic neurons of the rabbit aorta by prejunctional prostanoid receptors. J Pharmacol Exp Ther. 1999;291:7–11. [PubMed] [Google Scholar]
- 32.Ariumi H, Takano Y, Masumi A, Takahashi S, Hirabara Y, Honda K, Saito R, Kamiya HO. Roles of the central prostaglandin EP3 receptors in cardiovascular regulation in rats. Neurosci Lett. 2002;324:61–64. doi: 10.1016/s0304-3940(02)00174-x. [DOI] [PubMed] [Google Scholar]
- 33.Stock JL, Shinjo K, Burkhardt J, Roach M, Taniguchi K, Ishikawa T, Kim HS, Flannery PJ, Coffman TM, McNeish JD, Audoly LP. The prostaglandin E2 EP1 receptor mediates pain perception and regulates blood pressure. J Clin Invest. 2001;107:325–331. doi: 10.1172/JCI6749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang M, Ihida-Stansbury K, Kothapalli D, Tamby MC, Yu Z, Chen L, Grant G, Cheng Y, Lawson JA, Assoian RK, Jones PL, Fitzgerald GA. Microsomal prostaglandin e2 synthase-1 modulates the response to vascular injury. Circulation. 2011;123:631–639. doi: 10.1161/CIRCULATIONAHA.110.973685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang M, Lee E, Song W, Ricciotti E, Rader DJ, Lawson JA, Puré E, FitzGerald GA. Microsomal prostaglandin E synthase-1 deletion suppresses oxidative stress and angiotensin II-induced abdominal aortic aneurysm formation. Circulation. 2008;117:1302–1309. doi: 10.1161/CIRCULATIONAHA.107.731398. [DOI] [PubMed] [Google Scholar]
- 36.Wang M, Song WL, Cheng Y, Fitzgerald GA. Microsomal prostaglandin E synthase-1 inhibition in cardiovascular inflammatory disease. J Intern Med. 2008;263:500–505. doi: 10.1111/j.1365-2796.2008.01938.x. [DOI] [PubMed] [Google Scholar]
- 37.Suganami T, Mori K, Tanaka I, Mukoyama M, Sugawara A, Makino H, Muro S, Yahata K, Ohuchida S, Maruyama T, Narumiya S, Nakao K. Role of prostaglandin E receptor EP1 subtype in the development of renal injury in genetically hypertensive rats. Hypertension. 2003;42:1183–1190. doi: 10.1161/01.HYP.0000101689.64849.97. [DOI] [PubMed] [Google Scholar]
- 38.Capone C, Faraco G, Anrather J, Zhou P, Iadecola C. Cyclooxygenase 1-derived prostaglandin E2 and EP1 receptors are required for the cerebrovascular dysfunction induced by angiotensin II. Hypertension. 2010;55:911–917. doi: 10.1161/HYPERTENSIONAHA.109.145813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xiao CY, Yuhki K, Hara A, Fujino T, Kuriyama S, Yamada T, Takayama K, Takahata O, Karibe H, Taniguchi T, Narumiya S, Ushikubi F. Prostaglandin E2 protects the heart from ischemia-reperfusion injury via its receptor subtype EP4. Circulation. 2004;109:2462–2468. doi: 10.1161/01.CIR.0000128046.54681.97. [DOI] [PubMed] [Google Scholar]