

Abstract
IL-23 is required for the IL-17 response to infection with Mycobacterium tuberculosis (Mtb), but is not required for the early control of bacterial growth. However, mice deficient for the p19 component of IL-23 (Il23a−/−) exhibit increased bacterial growth late in infection that is temporally associated with smaller B cell follicles in the lungs. Cxcl13 is required for B cell follicle formation and immunity during tuberculosis. The absence of IL-23 results in decreased expression of Cxcl13 within Mtb-induced lymphocyte follicles in the lungs and this deficiency was associated with increased cuffing of T cells around the vessels in the lungs of these mice. Il23a−/− mice also poorly expressed IL-17A and IL-22 mRNA. These cytokines were able to induce Cxcl13 in mouse primary lung fibroblasts suggesting that these cytokines are likely involved in B cell follicle formation. Indeed, IL-17RA-deficient mice (Il17ra−/−) generated smaller B cell follicles early in the response, whereas IL-22-deficient (Il22−/−) mice had smaller B cell follicles at an intermediate time after infection; however only Il23a−/− mice had a sustained deficiency in B cell follicle formation and reduced immunity. We propose that in the absence of IL-23, expression of long-term immunity to tuberculosis is compromised due to reduced expression of Cxcl13 in B cell follicles and reduced ability of T cells to migrate from the vessels and into the lesion. Further while IL-17 and IL-22 can both contribute to Cxcl13 production and B cell follicle formation it is IL-23 that is critical in this regard.
Introduction
Tuberculosis remains a worldwide threat to world health that resists ongoing attempts at control. The increasing threat from drug resistant strains requires that we understand the host immune response as this will allow us to promote protective responses by vaccination and to limit pathologic and regulatory responses.
B cell follicles are formed in Mycobacterium tuberculosis (Mtb)-infected lungs in both mouse and human (1, 2), however the induction and maintenance of these follicles and the role that they play in protection has not been extensively studied. B cells and immunoglobulin play a minor role in protection although there is mounting evidence that B cells can contribute to immunity to Mtb (3–5). In addition, manipulation of the B cell compartment could modulate the immune response to this intractable disease (6). It is important that the activity of B cells during tuberculosis be defined so that methods to control disease can be developed.
The requirement for IL-23 in the control of tuberculosis in the mouse model is secondary to the role of IL-12p70 (7) however, its role in chronic infection has not been investigated. While IL-23 can drive the protective IFN-γ response, its primary role is in the maintenance of the IL-17A response (7) however excess IL-17A and IL-23 contribute to the pathologic response to tuberculosis in the mouse lung (8). In this report we show that IL-23 is required for long-term containment of Mtb as well as expression of Cxcl13 within- and the maintenance of- B cell follicles within the lung lesions. We also show that IL-17RA and IL-22 are involved in B cell follicle development at distinct times during infection and that IL-23 is necessary for the expression of both these cytokines in the lung. IL-17A and IL-22 are also shown to induce Cxcl13 from lung stromal cells.
Materials and methods
Mice and Infection
Mice were bred at the Trudeau Institute or the University of Pittsburgh. IL-23 p19 deficient mice (Il23a−/−) were from Dr Nico Ghilardi and Dr Fred deSauvage, Genentech Inc. (South San Francisco, CA) (9). IL-17 receptor A deficient (Il17ra−/−) mice were from Amgen Inc. (Thousand Oaks, CA) (10). IL-22 deficient mice (Il22−/−) were from Dr Wenjun Ouyang Genentech Inc (11). C57BL6/J mice (B6) were used as wild type controls. Age and sex matched mice were used between the ages of 7–12 weeks. Mice received either isotype control antibody (clone 50104) or 100ug of anti-IL-17A (clone 50104, both from R&D Systems,) or 150ug of anti-IL-22 (a kind gift of Dr Wenjun Ouyang)(11) intraperitoneally. The H37Rv strain of Mtb was used to infect mice aerogenically, as described (12). Bacterial numbers were counted by viable colony-forming units in homogenized tissue (12). All experiments were approved by the Institutional Animal Care and Use Committees at either Trudeau Institute or the University of Pittsburgh.
Detection of IFN-γ producing cells
Lung and lung draining lymph node (MLN) tissue was prepared as described (12). Antigen-specific IFN-γ-producing IAb-restricted T cells from infected lungs or MLN were enumerated using peptide-driven ELISpot (7). Single cell suspensions were analyzed for CD4 (clone GK1.5), CD44 (Clone IM7), and IFN-γ (clone XMG1.2) (13). Cells were gated based on their forward and side scatter characteristics and the frequency of specific cell types determined using FlowJo (Tree Star Inc, CA).
Generation of primary lung fibroblast cultures
Lung fibroblast cultures were prepared by digesting lung sections in 0.2% trypsin, 0.1% Collagenase type IV (both from Invitrogen, Carlsbad, CA) and 400 ug/ml DNAse (Worthington, Lakewood, NJ) for 30 minutes. The digest was then passed through a 70uM filter and released cells cultured in 10% fetal bovine serum (Invitrogen) in DMEM (14).
Immunohistochemistry
The formalin fixed caudal lobe of the lung from infected mice was processed for immunohistological analysis as described (15). Sections were probed with biotinylated rat anti-mouse B220 (BDPharmingen) and goat anti-mouse CD3 (Santa Cruz Biotechnology). Secondary antibodies or Streptavidin were labeled with Alexafluor 594-conjugated or Alexafluor 488 (both from Invitrogen). A Zeiss Axioplan 2 microscope and a Zeiss AxioCam digital camera were used to generate images. B220+ B cell follicles were counted in 4–5 different mice and results expressed as the average number of B cell follicles/lobe. All B220+ B cell follicles were outlined with the automated tool of the Zeiss Axioplan 2 microscope and average size in squared microns calculated. T cell cuffing around small blood vessels (100–150 μm diameter) was measured by outlining 3 random clusters of T cells per lobe (n= 5 lobes per sample) with the automated tool of the Zeiss Axioplan microscope and the average±SD was calculated. Samples were analyzed in a blinded fashion.
CXCL13-specific staining was performed on 6 micron cryosections of Mtb-infected mouse lung instilled with Tissue-Tek CRYO-OCT Compound (Fisher Scientific, Pittsburgh, PA) and adhered to coated slides. Sections were fixed with acetone:ethanol (75:25) for 10 minutes at room temperature and then blocked with 5% normal mouse serum in PBS (nms-PBS) for 30 minutes. The primary antibody was 1 mg/ml goat anti-mouse CXCL13 (AF470, R&D Systems) in 5% nms-PBS for 2 hours followed by the secondary donkey anti-goat Alexa-Fluor 647 (A21447, Invitrogen) at 1:250 in nms-PBS. Phycoerythrin-labelled rat anti-mouse B220 (553090, BDPharmingen) was used at 1:100 in nms-PBS to detect B cells in the same sections.
In situ hybridization
Mouse CXCL13 cDNA was RT-PCR amplified with primers BFJ.mCXCL13_F1 (5′-GAACTCCACCTCCAGGCAGA -3′) and BFJ.mCXCL13_R1 (5′-CTTTTGAGATGATAGTGGCT -3′). Rhesus macaque CXCL13 cDNA was RT-PCR amplified with primers (5′-AGACAGAATGAAGTTCATCT-3 ′ and 5 ′-GTGGAAATATCAGCATCAGGG-3′). PCR products were ligated to the pGEM-T vector (Promega) and DNA sequenced. The pGEMT-CXCL13 plasmid was linearized by restriction digest. Gene-specific riboprobes were synthesized by in vitro transcription using a Maxiscript SP6/T7 kit (Ambion) and unincorporated nucleotides were removed using RNA Mini Quick Spin Columns (Roche). Paraffin embedded tissue specimens were pretreated as described (16), following deparaffinization in xylene and rinsing in ethanol. In situ hybridization (ISH) with 35S-labeled riboprobes was performed at 50°C overnight as described (17), with 0.1M dithiothreitol included in the hybridization mix. Tissue sections were coated with NTB-2 emulsion (Kodak) and exposed at 10°C for 10 days. The sections were counterstained with hematoxylin (Vector) and mounted with Permount (Fisher). Images were visualized using an Olympus BX41 microscope and captured using a SPOT RT3 digital camera (Diagnostics Instruments, Inc).
Real Time PCR
RNA was extracted from lung tissue and analyzed by real-time PCR as previously described (7).
Statistical analysis
Differences between the means of experimental groups were analyzed using the two-tailed Student’s t-test or ANOVA as appropriate. Differences were considered significant where P ≤ 0.05. Inherently logarithmic data from bacterial growth and RT-PCR was transformed for statistical analysis.
Results
IL-23 is required for the long-term containment of bacterial growth in Mtb-infected mice
We showed that IL-23 does not impact the early control of bacterial growth in Mtb-infected mice but that it augments the IFN-γ response in the absence of IL-12 (7). To determine whether IL-23 plays a role during long-term infection, we compared bacterial burden in B6 and Il23a−/− mice over time. We found that while early bacterial burden did not differ, by day 150 bacteria had significantly increased in the lungs of Il23a−/− mice, a difference that was maintained through day 250 (Fig 1). Upon histological evaluation of the lungs, we noted that Il23a−/− mice had reduced numbers of B cell follicles surrounding the lesions (Fig 1B). In addition, the B cell follicles appeared disorganized in the lungs of Il23a−/− mice at day 200 following infection (Fig 1C). Thus, the development of inducible bronchus associated lymphoid tissue (iBALT) (18) was compromised in the absence of IL-23.
Figure 1. Mice lacking IL-23 are less able to control bacterial growth when chronically infected with Mtb.

(A) B6 (closed circles) and Il23a−/− (open circles) were infected via the aerosol route with 100 Mtb H37Rv and the number of bacteria in the lungs was determined. The graph shows the combined data of three experiments all of which showed a significant difference between B6 and Il23a−/− in bacterial burden after day 120. Data points are the mean ± standard deviation for an n of 12–15 mice per group. (B) The number of B cell follicles in the lungs of B6 (closed circles) and Il23a−/−(open circles) mice infected for 200 days was determined. Data points are from an n of 4 mice per group and represent one experiment of two total. The significance was determined by the Student’s t test with * P ≤ 0.05, ** P ≤ 0.001 or *** P ≤ 0.0001. (C) B6 mice and Il23a−/− mice were infected as in panel A for 200 days and representative sections stained with hematoxylin and eosin are shown (top panels). Sections were probed with antibodies to the B cell marker, B220 (green), the T cell marker, CD3 (red) and macrophage marker iNOS (also red) (bottom panels).
Absence of IL-23 does not compromise the expression of type 1 immune responses in the lung
We previously showed that IL-23 compensates for IL-12 in the response of mice to Mtb infection (7). To examine the expression of protective immunity therefore, we measured the induction of mRNA for molecules associated with protection in B6 and Il23a−/− Mtb-infected mice. While mRNA expression of IFN-γ, and the markers of macrophage anti-mycobacterial activity Lrg-47 and iNOS (not shown) was equivalent, the expression of IL-22 and IL-17A was reduced in Il23a−/− lungs (Fig 2A). In contrast, the expression of IL-21, another Th17 associated cytokine, was not significantly affected (data not shown). While induction of IL-17A and IL-22 was dramatically reduced there was some early expression of IL-17A and some late expression of IL-22 mRNA. Using flow cytometry and ELISpot assays we saw a reduction in both total numbers of IFN-γ-producing cells (Fig 2B left) and antigen-specific IFN-γ-producing (Fig 2B right) cells in the lungs of the Il23a−/− mice at the latest time point. These data suggest that while type-17 responses are reduced in the absence of IL-23, the accumulation of Th1 cells in the lung tissue is only modestly impacted.
Figure 2. Mice lacking IL-23 maintain expression of IFN-γ and LRG-47 but not IL-17 or IL-22.
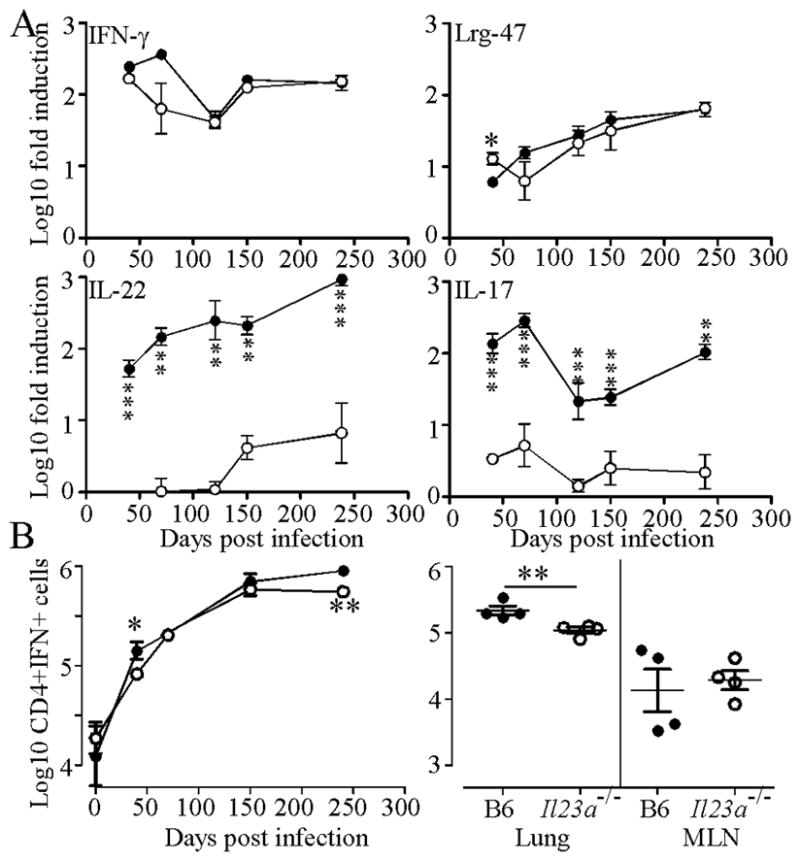
(A) B6 (filled circles) and Il23a−/− (open circles) were infected as for Fig 1 and induction of mRNA for IFN-γ, Lrg-47, IL-17 and IL-22 relative to uninfected control mice determined by RT-PCR. One of two representative experiments is shown. (B) A single cell suspension from infected mice was analyzed by flow cytometry (left panel) or ELISpot (right panel, Day 238) for IFN-γ production. Data points are the mean ±standard deviation for an n of 3–4 mice per group. Significance was determined by the Student’s t test with * P ≤ 0.05, ** P ≤ 0.01, *** P ≤ 0.001.
Neither the absence of IL-17A nor IL-22 is responsible for the increased bacterial burden in IL-23 deficient mice
To determine whether the increased bacterial growth in the IL-23 deficient mice was a result of the loss of either IL-17A or IL-22, we infected IL-22 deficient or IL-17RA deficient mice with Mtb. We found that both Il22−/− and Il17ra−/− mice controlled bacteria as well as B6 mice for up to 200 days (Fig 3A–B). We also blocked IL-22 signaling by administering anti-IL-22 antibody to B6 mice every other day for 30 days prior to harvest at either day 50, day 90, day 150 or day 200 with similar results (Fig 3C). To investigate whether combined deficiency resulted in increased bacterial growth we treated IL-17RA deficient mice with anti-IL-22 and Il22−/− mice with anti-IL-17A and observed no increase in bacterial growth (Fig 3D–E). These data suggest that neither IL-17A nor IL-22 (either alone or in combination) can directly impact the ability of Mtb-infected mice to control bacterial growth.
Figure 3. Mice lacking IL-17R or IL-22 are unaffected in their ability to control Mtb.
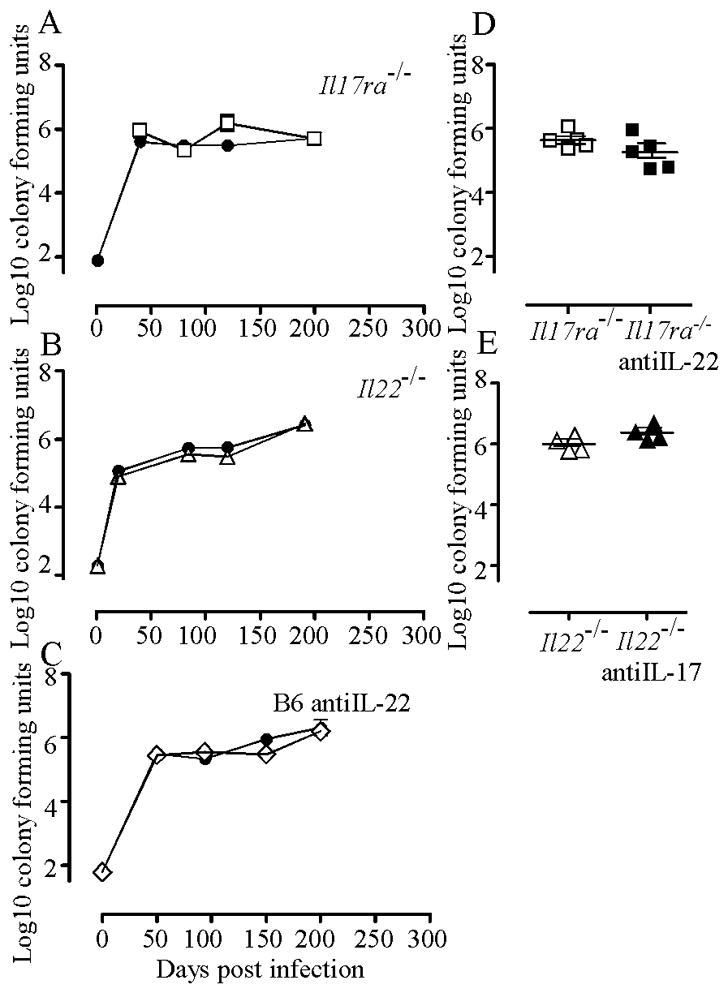
(A) B6 (closed circles) and Il17ra−/− (open squares) mice were infected as in Fig 1 and the number of bacteria in the lungs was determined. One of two representative experiments is shown. (B) B6 (closed circles) and Il22−/− (open triangles) mice were infected. One of two representative experiments is shown. (C) B6 mice were infected and treated with isotype control antibody (closed circles) or with anti-IL-22 antibody (open diamonds) for 30 days prior to each day of harvest. One of two representative experiments is shown. For A–C data points are the mean ±standard deviation for an n of 4–5 mice per group. (D) Il17ra−/− mice were infected and treated with anti-IL-22 antibody for 30 days prior to harvest on day 150. (E) Il22−/− mice were infected and treated with anti-IL-17 antibody for 30 days prior to harvest on day 150. For both D and E data represent one experiment total with a n=4. There were no significant differences.
IL-23, IL-17R and IL-22 impact the size of B cell follicles in the lungs of Mtb-infected mice at different times during infection
We investigated whether absence of IL-17R or IL-22 impacted the generation of B cell follicles. Using morphometric analysis of lesions in the lung, we found that the absence of IL-17RA resulted in reduced size of B cell follicles early after infection but not after day 50 (Fig 4A, E.i). In contrast, the absence of IL-22 had an impact on B cell follicle size at day 80 as shown both by the gene deficient mice and the anti-IL-22 treated animals (Fig 4B, E.ii). By day 150 (Fig 4C) and including day 200 (Fig 4D) only the absence of IL-23a had an impact on B cell follicle size.
Figure 4. Mice lacking IL-23, IL-22, IL-17R have altered development of B cell follicles over time.

B6 (closed circles), Il23a−/− (open circles), Il17ra−/− (open squares), B6 treated with anti-IL-22 (open diamonds) and Il22−/− (open triangles) were infected as in Fig 1 and the area of B cell follicles measured on days 50 (A), 80 (B), 150 (C) and 200 (D). The data points are the individual values for each follicle in the caudal lobe section for each mouse with an n of 3–5 mice per group. (E) Lymphocyte accumulations were characterized in (i) the lungs of B6 (top panel) and Il17ra−/− (bottom panel) mice infected for 50 days and (ii) the lungs of B6 (left panel) and Il22−/− (upper right panel) and anti-IL-22-treated (lower right) mice infected for 80 days. The lymphocyte accumulations were characterized as in Fig 1C.
IL-23 is required for CXCL13 expression in the B cell follicles and to promote migration of T cells away from the blood vessels
B cell follicles are dependent upon the chemokine CXCL13 (19) and we have previously shown that there is reduced expression of immunity in Mtb-infected cxcl13−/− mice and that this is associated with inappropriate T cell accumulation around vessels in the lung (15). We therefore investigated the availability of CXCL13 and the preponderance of T cell cuffing around vessels in the lungs of the IL-23 deficient mice using microscopy. We found that expression of CXCL13 protein was abundant in the B cell follicles in lungs of Mtb-infected B6 mice (Fig 5A.i) but that this was reduced in the infected lungs of Il23a−/− mice (Fig 5A.ii). We also performed in situ hybridization for the cxcl13 mRNA on the infected lungs and found that the lymphoid aggregates contained cxcl13 mRNA (Fig 5B.i) and that this expression was lost in infected lungs of Il23a−/− mice (Fig 5B.ii). These data demonstrate that IL-23 is required for the expression of CXCL13 within the lymphoid aggregates that develop in the lungs of Mtb-infected mice.
Figure 5. Mice lacking IL-23a are unable to express CXCL13 in B cell follicles induced by Mtb infection.

(A) B6 (i) and Il23a−/− (ii) mice were infected with Mtb as described for Fig 1 and lung sections stained for the CXCL13 protein shown in green with the B220 positive cells shown in red (n=2, images representative of all tissues examined are shown). (B) B6 (i, and ii) and Il23a−/− (iii) mice were infected with Mtb and lung sections subjected to in situ hybridization with (ii, iii) the antisense probe for Cxcl13 mRNA (ii, iii) or the sense control probe (i) n=4, images representative of all tissues examined are shown, positive staining is highlighted by the arrows. (C) Formalin fixed sections from B6 (closed circles), isotype control (crosses), Il23a−/− (open circles), Il17ra−/−(open squares) and anti-IL-22 treated mice (open diamonds) were also stained for CD3 and the tendency for T cell cuffing to occur was measured as area of CD3 positive cells around blood vessels at day 200 post infection. (D) Fibroblasts from mouse lungs were exposed to IL-17 and/or IL-22 and the production of CXCL13 measured by bead array. The data points are the mean of triplicate wells pooled from two mice, one experiment of two showing an effect of IL-17 and IL-22. For F,G, significance was determined by the ANOVA with * P ≤ 0.05, ** P ≤ 0.01, *** P ≤ 0.001
We also analyzed the distribution of T cells within the lesions and found that in the absence of IL-23a there was a significant increase in the amount of T cell cuffing around the blood vessels close to the lesional site (Fig 5C). An increased level of T cell cuffing was also seen in the absence of IL-17A or IL-22 but this was significantly less for Il17ra−/− mice, and trended towards less for the anti-IL-22 treated mice, when compared to the Il23a−/− mice (Fig 5C). The similarity of the T cell distribution in this model to that seen in the Mtb-infected cxcl13−/− mice caused us to investigate the ability of IL-17A and IL-22 to induce CXCL13. To do this we exposed mouse fibroblasts to IL-17A or IL-22 and found that lung stromal cells produced the chemokine CXCL13 (Fig 5D) as well as IL-6, GM-CSF and KC (data not shown). In contrast, lymphotoxin α, CXCL12, and CCL21 which can play a role in lymphoid tissue formation were not induced in response to IL-17 or IL-22 (data not shown). These data suggest that IL-23 is required for CXCL13-dependent generation of B cell follicles as well as the development of a T cell containing granuloma during Mtb-infection. Further, IL-23-dependent IL-17 and IL-22 may contribute to the development of the granuloma via induction of CXCL13 in the stromal cells of the lung.
Discussion
We show that IL-23 is required for the long term containment of Mtb growth in mouse lungs and that it is critical for the CXCL13-dependent development of B cell follicles in infected lung tissue. We have previously reported that the lack of CXCL13 compromises immunity to tuberculosis and that this is linked to poor lymphoid follicle formation and increased accumulation of T cells around the vessels (15). In the model reported here we show that IL-23 is critical for CXCL13 expression in lymphoid follicles and that there is increased T cell cuffing in the absence of IL-23 as the disease becomes chronic. We hypothesize therefore that the increased susceptibility to bacterial growth that occurs later in disease in the absence of IL-23 is a result of poor CXCL13 expression and the subsequent reduced ability of T cells to migrate from the vessels to the infected phagocytes and activate them efficiently.
We screened a large number of genes for differences in expression between the Mtb-infected B6 and IL-23a deficient lung and found that the strongest differences noted was the loss of the type 17 inflammatory cytokines IL-17A and IL-22. The absence of these genes either individually or in concert did not however recapitulate the deficiency in bacterial control seen in the absence of IL-23. Deficiency of IL-17 signaling or IL-22 did however modestly impact the development of the B cell follicles but this was not a sustained effect. Our observation that these cytokines were able to induce production of CXCL13 from lung stromal cells suggests that while IL-23 is critical for the CXCL13 expression in the lymphoid follicles, IL-23 may act through induction of IL-17 and IL-22 to mediate its effect. The fact that absence of either cytokine only modestly affects the development of follicles suggests that they compensate for each other in vivo.
We know that IL-23 can compensate for the absence of IL-12 in the IFN-γ-response during mouse tuberculosis (7) and we see here a minor decrease in the IFN-γ producing cells in the chronically infected Il23a−/− mice. Important recent data has shown that during chronic inflammatory conditions such as examined here, the development of antigen-specific cells from an IL-17-producing phenotype to an IFN-γ producing phenotype is dependent upon the IL-23a subunit (20). Together with our data, this suggests that in the absence of IL-23 there may be a chronic, if small, inability to develop an IFN-γ-producing phenotype over time and this may also contribute to the increase in bacterial growth over the long term.
IL-17 has been implicated in germinal center formation (21) and promotion of IgG2a and IgG3 isotypes (22) as well as being associated with lymphoid neogenesis in graft rejection (23). In a model of neonatal pulmonary inflammation wherein LPS treatment results in tertiary lymphoid tissues in the lung a clear dependence on IL-17-producing CXCR5-expressing TFH cells was demonstrated (24). We propose that as we see an early, albeit low, level of IL-23a independent IL-17A and delayed initiation of B cell follicle formation in the IL-17RA deficient mice that a similar process of IL-17 induction of CXCL13 production by lung stromal cells occurs during the early response to Mtb. In contrast to the data reported here however, the size of tertiary lymphoid tissues in the neonatal inflammation model was not impacted by the absence of IL-23a, although there were fewer lymphoid accumulations seen. Together these two different models suggest that while IL-17 and IL-23 play significant roles in the development of tertiary lymphoid structures, their relative roles may vary depending upon the nature of the stimulus. A critical issue may be the length of time and nature of the stimulus and the actual nature of the induced lymphoid follicle. We further show that IL-22 can augment the induction of CXCL13 in lung stromal cells and as infection progresses, it may be that there is a requirement for IL-22 in driving and maintaining CXCL13 production and this is why we see a requirement for IL-22 in B cell follicle formation in Mtb-infected lungs by day 80 of our study. As disease progresses however, B cell follicle growth in the Il17ra−/− and Il22−/− mice becomes equivalent with the B6 mice and it is the absence of IL-23 that has the greatest effect on the maintenance of the B cell follicle. Recent data show that IL-23 is critical for the expansion of infection-associated lymphoid tissue inducer (LTi) cells (25) and that IL-22 is a critical product of these cells (26). While these cells are associated with the development of lymphoid tissues they are also able to mediate protection against bacterial infection in the gut (25, 26) however preliminary studies do not show any appreciable differences in the LTi populations between infected control and Il23a−/− mice (not shown).
While we propose that IL-23 is required to drive CXCL13 production and focus T cells to the granuloma, the possibility that IL-17 and IL-22 may be mediating direct effects should be considered. In more acute models of bacterial infection (27) and specifically in high dose intratracheal BCG infection, IL-17 activity has been shown to be required for the early inflammatory response as well as protection; these activities are dependent upon IL-17 derived from γδ T cells (28, 29). We do not see a requirement for IL-17RA in protection in the low dose aerosol model suggesting that rapid expression of IL-17 activity within the lung is a requirement that depends upon dose and acuteness of challenge. Others have also reported little role for IL-22 in protection against Mtb infection (30). We have not directly investigated the impact of the loss of IL-23 on the B cell function here but others have shown that blockade of CXCL13 does not impact B cell activation in tertiary lymphoid follicles (31).
Our data support the need to investigate the role of IL-23 in the containment of tuberculosis. Humans lacking the IL-12p40 subunit that contributes to both IL-12p70 and IL-23 cytokine (32) are particularly susceptible to mycobacterial diseases and this deficiency may reflect a role of both IL-12 and IL-23 in control of infection. In contrast to a protective role for IL-23, we recently published that excess IL-23 is associated with increased pathologic consequences (8) and this highlights that, for a chronic disease such as tuberculosis, a balance in the level of this cytokine is essential. Too much and there is pathology, too little and there is a loss of protection.
Footnotes
This work was supported by the Trudeau Institute, Inc.; a Career Development Award from AI057158 (North East Biodefense Center-Lipkin), AI083541 and HL-105427 to S.A.K. and Children’s Hospital of Pittsburgh of UPMC to S.A.K., Y.L and S.S; NIH grants HL69409 to T.D.R and AI46530 and AI069121 to A.M.C.
References
- 1.Tsai M, Chakravarty S, Zhu G, Xu J, Tanaka K, Koch C, Tufariello J, Flynn J, Chan J. Characterization of the tuberculous granuloma in murine and human lungs: cellular composition and relative tissue oxygen tension. Cell Microbiol. 2006;8:218–232. doi: 10.1111/j.1462-5822.2005.00612.x. [DOI] [PubMed] [Google Scholar]
- 2.Ulrichs T, Kosmiadi G, Trusov V, Jörg S, Pradl L, Titukhina M, Mishenko V, Gushina N, Kaufmann S. Human tuberculous granulomas induce peripheral lymphoid follicle-like structures to orchestrate local host defence in the lung. J Pathol. 2004;204:217–228. doi: 10.1002/path.1628. [DOI] [PubMed] [Google Scholar]
- 3.Maglione P, Xu J, Chan J. B cells moderate inflammatory progression and enhance bacterial containment upon pulmonary challenge with Mycobacterium tuberculosis. J Immunol. 2007;178:7222–7234. doi: 10.4049/jimmunol.178.11.7222. [DOI] [PubMed] [Google Scholar]
- 4.Maglione P, Xu J, Casadevall A, Chan J. Fc{gamma} receptors regulate immune activation and susceptibility during Mycobacterium tuberculosis infection. J Immunol. 2008;180:3329–3338. doi: 10.4049/jimmunol.180.5.3329. [DOI] [PubMed] [Google Scholar]
- 5.Maglione P, Chan J. How B cells shape the immune response against Mycobacterium tuberculosis. Eur J Immunol. 2009;39:676–686. doi: 10.1002/eji.200839148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cooper AM. Cell mediated immune responses in tuberculosis. Annu Rev Immunol. 2009;27:393–422. doi: 10.1146/annurev.immunol.021908.132703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Khader S, Pearl J, Sakamoto K, Gilmartin L, Bell G, Jelley-Gibbs D, Ghilardi N, deSauvage F, Cooper A. IL-23 compensates for the absence of IL-12p70 and is essential for the IL-17 response during tuberculosis but is dispensable for protection and antigen-specific IFN-gamma responses if IL-12p70 is available. J Immunol. 2005;175:788–795. doi: 10.4049/jimmunol.175.2.788. [DOI] [PubMed] [Google Scholar]
- 8.Cruz A, Fraga A, Fountain J, Rangel-Moreno J, Torrado E, Saraiva M, Pereira D, Randall T, Pedrosa J, Cooper A, Castro A. Pathological role of Interleukin 17 in mice subjected to repeated BCG vaccination after infection with Mycobacterium tuberculosis. J Exp Med. 2010;207:1609–1616. doi: 10.1084/jem.20100265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ghilardi N, Kljavin N, Chen Q, Lucas S, Gurney A, de Sauvage F. Compromised humoral and delayed-type hypersensitivity responses in IL-23-deficient mice. J Immunol. 2004;172:2827–2833. doi: 10.4049/jimmunol.172.5.2827. [DOI] [PubMed] [Google Scholar]
- 10.Ye P, Rodriguez FH, Kanaly S, Stocking KL, Schurr J, Schwarzenberger P, Oliver P, Huang W, Zhang P, Zhang J, Shellito JE, Bagby GJ, Nelson S, Charrier K, Peschon JJ, Kolls JK. Requirement of Interleukin-17 receptor signalling for lung CXC chemokine and granulocyte colony-stimulating factor expression, neutrophil recriutment, and host defense. J Exp Med. 2001;194:519–527. doi: 10.1084/jem.194.4.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zheng Y, Danilenko D, Valdez P, Kasman I, Eastham-Anderson J, Wu J, Ouyang W. Interleukin-22, a T(H)17 cytokine, mediates IL-23-induced dermal inflammation and acanthosis. Nature. 2007;445:648–651. doi: 10.1038/nature05505. [DOI] [PubMed] [Google Scholar]
- 12.Roberts A, Cooper A, Belisle J, Turner J, Gonzalez-Juarerro M, Orme I. Murine models of tuberculosis. In: Kaufmann S, Kabelitz D, editors. Methods in Microbiology. 2. Academic Press; London: 2002. pp. 433–462. [Google Scholar]
- 13.Pearl JE, Shabaana AK, Solache A, Gilmartin L, Ghilardi N, deSauvage F, Cooper AM. IL-27 signaling compromises control of bacterial growth in mycobacteria-infected mice. J Immunol. 2004;173:7490–7496. doi: 10.4049/jimmunol.173.12.7490. [DOI] [PubMed] [Google Scholar]
- 14.Alcorn JF, Guala AS, van der Velden J, McElhinney B, Irvin CG, Davis RJ, Janssen-Heininger YMW. Jun N-terminal kinase 1 regulates epithelial-to-mesenchymal transition induced by TGF-{beta}1. J Cell Science. 2008;121:1036–1045. doi: 10.1242/jcs.019455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Khader S, Rangel-Moreno J, Fountain J, Martino C, Reiley W, Pearl J, Winslow G, Woodland D, Randall T, Cooper A. In a murine tuberculosis model, the absence of homeostatic chemokines delays granuloma formation and protective immunity. J Immunol. 2009;183:8004–8014. doi: 10.4049/jimmunol.0901937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Aujla SJ, Chan YR, Zheng M, Fei M, Askew DJ, Pociask DA, Reinhart TA, McAllister F, Edeal J, Gaus K, Husain S, Kreindler JL, Dubin PJ, Pilewski JM, Myerburg MM, Mason CA, Iwakura Y, Kolls JK. IL-22 mediates mucosal host defense against Gram-negative bacterial pneumonia. Nat Med. 2008;14:275–281. doi: 10.1038/nm1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fallert B, Reinhart T. Improved detection of simian immunodeficiency virus RNA by in situ hybridization in fixed tissue sections: combined effects of temperatures for tissue fixation and probe hybridization. J Virol Methods. 2002;99:23–32. doi: 10.1016/s0166-0934(01)00378-0. [DOI] [PubMed] [Google Scholar]
- 18.Randall T. Bronchus-associated lymphoid tissue (BALT) structure and function. Adv Immunol. 2010;107:187–241. doi: 10.1016/B978-0-12-381300-8.00007-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ansel KM, V, Ngo N, Hyman PL, Luther SA, Forster R, Sedgwick JD, Browning JL, Lipp M, Cyster JG. A chemokine-driven positive feedback loop organizes lymphoid follicles. Nature. 2000;406:309–314. doi: 10.1038/35018581. [DOI] [PubMed] [Google Scholar]
- 20.Hirota K, Duarte JH, Veldhoen M, Hornsby E, Li Y, Cua DJ, Ahlfors H, Wilhelm C, Tolaini M, Menzel U, Garefalaki A, Potocnik AJ, Stockinger B. Fate mapping of IL-17-producing T cells in inflammatory responses. Nat Immunol. 2011;12:255–263. doi: 10.1038/ni.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hsu H, Yang P, Wang J, Wu Q, Myers R, Chen J, Yi J, Guentert T, Tousson A, Stanus A, Le T, Lorenz R, Xu H, Kolls J, Carter R, Chaplin D, Williams R, Mountz J. Interleukin 17-producing T helper cells and interleukin 17 orchestrate autoreactive germinal center development in autoimmune BXD2 mice. Nat Immunol. 2008;9:166–175. doi: 10.1038/ni1552. [DOI] [PubMed] [Google Scholar]
- 22.Mitsdoerffer M, Lee Y, Jäger A, Kim H, Korn T, Kolls J, Cantor H, Bettelli E, Kuchroo V. Proinflammatory T helper type 17 cells are effective B-cell helpers. Proc Natl Acad Sci U S A. 2010;107:14292–14297. doi: 10.1073/pnas.1009234107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Deteix C, Attuil-Audenis V, Duthey A, Patey N, McGregor B, Dubois V, Caligiuri G, Graff-Dubois S, Morelon E, Thaunat O. Intragraft Th17 infiltrate promotes lymphoid neogenesis and hastens clinical chronic rejection. J Immunol. 2010;184:5344–5351. doi: 10.4049/jimmunol.0902999. [DOI] [PubMed] [Google Scholar]
- 24.Rangel-Moreno J, Carragher DM, de la Luz Garcia-Hernandez M, Hwang JY, Kusser K, Hartson L, Kolls JK, Khader SA, Randall TD. The development of inducible bronchus-associated lymphoid tissue depends on IL-17. Nat Immunol. 2011;12:639–646. doi: 10.1038/ni.2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sonnenberg GF, Monticelli LA, Elloso MM, Fouser LA, Artis D. CD4+ lymphoid tissue-inducer cells promote innate immunity in the gut. Immunity. 2011;34:122–134. doi: 10.1016/j.immuni.2010.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tumanov Alexei V, Koroleva Ekaterina P, Guo X, Wang Y, Kruglov A, Nedospasov S, Fu Y-X. Lymphotoxin Controls the IL-22 Protection Pathway in Gut Innate Lymphoid Cells during Mucosal Pathogen Challenge. Cell host & microbe. 2011;10:44–53. doi: 10.1016/j.chom.2011.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Riol-Blanco L, Lazarevic V, Awasthi A, Mitsdoerffer M, Wilson B, Croxford A, Waisman A, Kuchroo V, Glimcher L, Oukka M. IL-23 receptor regulates unconventional IL-17-producing T cells that control bacterial infections. J Immunol. 2010;184:1710–1720. doi: 10.4049/jimmunol.0902796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Umemura M, Yahagi A, Hamada S, Begum M, Watanabe H, Kawakami K, Suda T, Sudo K, Nakae S, Iwakura Y, Matsuzaki G. IL-17-mediated regulation of innate and acquired immune response against pulmonary Mycobacterium bovis bacille Calmette-Guerin Infection. J Immunol. 2007;178:3786–3796. doi: 10.4049/jimmunol.178.6.3786. [DOI] [PubMed] [Google Scholar]
- 29.Okamoto Yoshida Y, Umemura M, Yahagi A, O’Brien R, Ikuta K, Kishihara K, Hara H, Nakae S, Iwakura Y, Matsuzaki G. Essential role of IL-17A in the formation of a mycobacterial Infection-induced granuloma in the lung. J Immunol. 2010;184:4414–4422. doi: 10.4049/jimmunol.0903332. [DOI] [PubMed] [Google Scholar]
- 30.Wilson MS, Feng CG, Barber DL, Yarovinsky F, Cheever AW, Sher A, Grigg M, Collins M, Fouser L, Wynn TA. Redundant and pathogenic roles for IL-22 in mycobacterial, protozoan, and helminth infections. J Immunol. 2010;184:4378–4390. doi: 10.4049/jimmunol.0903416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Henry RA, Kendall PL. CXCL13 Blockade Disrupts B Lymphocyte Organization in Tertiary Lymphoid Structures without Altering B Cell Receptor Bias or Preventing Diabetes in Nonobese Diabetic Mice. J Immunol. 2010;185:1460–1465. doi: 10.4049/jimmunol.0903710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Filipe-Santos O, Bustamante J, Chapgier A, Vogt G, de Beaucoudrey L, Feinberg J, Jouanguy E, Boisson-Dupuis S, Fieschi C, Picard C, Casanova J. Inborn errors of IL-12/23- and IFN-gamma-mediated immunity: molecular, cellular, and clinical features. Semin Immunol. 2006;18:347–361. doi: 10.1016/j.smim.2006.07.010. [DOI] [PubMed] [Google Scholar]