

Non-technical summary
Voltage-dependent L-type calcium (CaV1.2) channels are critical gateways for Ca2+ entry into excitable cells such as heart myocytes and neurons. This Ca2+ signal controls many essential physiological responses including triggering the heartbeat and regulating gene expression in nerve cells. CaV1.2 channels are multi-subunit proteins, comprising α1C, β, and α2δ subunits, and must target to the cell surface to function. Association of a pore-forming α1C and cytosolic β is necessary for targeting CaV1.2 channels to the cell surface through poorly understood mechanisms. Here, using a chimeric channel strategy, we provide data that suggest β binding to the α1C intracellular I–II loop causes a global rearrangement of intracellular domains, shifting a balance of power between export signals on the I–II loop and retention signals elsewhere on the channel. The results provide novel insights into the mechanism of a protein–protein interaction that is vital for forming functional CaV1.2 channels.
Abstract
Abstract
Ca2+ influx via CaV1/CaV2 channels drives processes ranging from neurotransmission to muscle contraction. Association of a pore-forming α1 and cytosolic β is necessary for trafficking CaV1/CaV2 channels to the cell surface through poorly understood mechanisms. A prevalent idea suggests β binds the α1 intracellular I–II loop, masking an endoplasmic reticulum (ER) retention signal as the dominant mechanism for CaV1/CaV2 channel membrane trafficking. There are hints that other α1 subunit cytoplasmic domains may play a significant role, but the nature of their potential contribution is unclear. We assessed the roles of all intracellular domains of CaV1.2-α1C by generating chimeras featuring substitutions of all possible permutations of intracellular loops/termini of α1C into the β-independent CaV3.1-α1G channel. Surprisingly, functional analyses demonstrated α1C I–II loop strongly increases channel surface density while other cytoplasmic domains had a competing opposing effect. Alanine-scanning mutagenesis identified an acidic-residue putative ER export motif responsible for the I–II loop-mediated increase in channel surface density. β-dependent increase in current arose as an emergent property requiring four α1C intracellular domains, with the I–II loop and C-terminus being essential. The results suggest β binding to the α1C I–II loop causes a C-terminus-dependent rearrangement of intracellular domains, shifting a balance of power between export signals on the I–II loop and retention signals elsewhere.
Introduction
The entry of calcium ions into excitable cells through voltage-dependent CaV1 (CaV1.1 − 1.4) and –2 (CaV2.1 − 2.3) channels constitutes a prevalent and versatile signal transduction paradigm in biology. This basic mechanism is used to evoke neurotransmitter release that underlies synaptic transmission (Catterall & Few, 2008), control neuronal excitability by coupling to Ca2+-sensitive K+ channels (Fakler & Adelman, 2008), trigger excitation–contraction coupling in heart muscle (Bers, 2002), and regulate gene expression (Deisseroth et al. 2003). Hence, myriad biological processes critically depend on the proper cell surface targeting and function of CaV1/CaV2 channels. Functional CaV1/CaV2 channels are multi-subunit protein complexes containing a membrane-spanning α1 subunit assembled with auxiliary proteins that include β (β1−β4) and α2δ (α2δ1−3) subunits, and calmodulin (Catterall & Few, 2008). While the α1 subunit contains the voltage sensor and channel pore, its subcellular localization and biophysical properties are profoundly influenced by the accessory proteins. In particular, β subunits are essential determinants of channel behaviour, affecting both trafficking and gating (Dolphin, 2003; Buraei & Yang, 2010). Auxiliary βs induce a hyperpolarizing shift in the voltage dependence of channel activation, increase channel open probability (Po), and determine channel inactivation properties (Perez-Reyes et al. 1992; Neely et al. 1993; De Waard & Campbell, 1995; Colecraft et al. 2002; Dolphin, 2003; Takahashi et al. 2004; Buraei & Yang, 2010). In addition to the biophysical modifications, a cardinal feature of CaV1/CaV2 channels is their reliance on association with a β for effectual targeting to the cell surface (Gao et al. 1999; Dolphin, 2003; Kanevsky & Dascal, 2006; Buraei & Yang, 2010; Obermair et al. 2010; Yang et al. 2010). Because this trafficking step is fundamental to the formation of functional CaV1/CaV2 channels, much effort has focused on elucidating how β subunits promote membrane-targeting of CaV channels.
CaVβs contain a src homology 3 (SH3)/guanylate kinase (GK) structural module (Chen et al. 2004; Opatowsky et al. 2004; Van Petegem et al. 2004), identifying them as members of the membrane-associated guanylate kinase (MAGUK) family of scaffold proteins (Funke et al. 2005). The β GK domain binds with high affinity to a conserved 18-residue region (termed the α1 interaction domain, or AID) in the α1 subunit intracellular I–II loop (Pragnell et al. 1994; Chen et al. 2004; Opatowsky et al. 2004; Van Petegem et al. 2004). Mutations that selectively disrupt the β-AID interaction prevent β-induced channel targeting to the membrane, suggesting a dominant role for this association in regulating CaV channel trafficking (Van Petegem et al. 2008; Bourdin et al. 2010; Buraei & Yang, 2010; Obermair et al. 2010). One prevalent idea, based on experiments carried out on CaV2.1 channels, is that CaV1/CaV2 α1 subunits possess an ER retention signal on the I–II loop that is masked upon β-binding, thus allowing forward trafficking of the channel to proceed (Bichet et al. 2000). However, several observations challenge the sufficiency and generality of this model to account for β-induced membrane targeting of CaV1/CaV2 channels (Buraei & Yang, 2010). First, the putative ER retention sequence on the α1 I–II loop has not been identified in any CaV1/CaV2 channel (Bichet et al. 2000). Second, in contrast to CaV2.1, neither the CaV1.2 nor CaV2.2 α1 subunit I–II loop displayed ER retention properties when fused to CD4 (Altier et al. 2011). Third, deletions in the CaV1.2 α1C C-terminus that do not impact β binding to the channel can, nevertheless, severely diminish membrane trafficking (Gao et al. 2000). Fourth, point mutations in the α1C C-terminus that disrupt apo-calmodulin, but not β subunit binding, also significantly impair channel trafficking (Wang et al. 2007; Bourdin et al. 2010). These discrepancies underscore a clear need for a unifying model that accounts for both the essential role of β binding to the I–II loop and the apparent importance of the C-terminus for CaV1.2 channel membrane trafficking. Furthermore, the potential contribution of other α1 subunit intracellular domains (N-terminus, II–III loop, and III–IV loop) to β-induced trafficking has not been rigorously explored in any CaV1/CaV2 channel. This omission critically compromises the necessary dataset required to formulate a more complete model of β-induced trafficking of CaV1/CaV2 channels.
Identifying whether and how the various intracellular domains of α1 contribute to β-dependent channel trafficking is a daunting task given their multiplicity and likely complex three-dimensional spatial arrangement. One typical approach to the problem has been to mutate or delete portions of α1 intracellular domains and assess whether they compromise channel trafficking (Bichet et al. 2000; Wang et al. 2007). While such loss-of-function approaches provide important information about channel regions that may be involved in trafficking, the dataset produced typically yield only modest insights into the mechanistic basis for β-dependent regulation of CaV channel trafficking. For example, observations that deletions in the CaV1.2 α1C C-terminus may impair channel trafficking simply informs that this intracellular domain is important for the process, but not how it is involved. An alternative reductionist approach involves splicing individual α1 subunit intracellular domains to the C-terminus of the trans-membrane protein CD8 (or CD4) and observing how they impact targeting of this protein to the cell surface (Bichet et al. 2000; Cornet et al. 2002; Wang et al. 2007; Altier et al. 2011). This is a useful method that, nevertheless, suffers from two main disadvantages. First, in the context of the channel, three of the α1 intracellular loops (the I–II, II–III, and III–IV loops) are geometrically constrained by having transmembrane regions bracketing either side. This configuration is lost when the loops are fused to CD8, and the resulting change in conformation could impact their functional properties. Second, this approach neglects possible interactions among the α1 intracellular domains that could give rise to new emergent properties.
Here, we took a gain-of-function chimeric channel approach seeking to reconstitute β-dependent trafficking in a normally β-independent channel. While the chimeric channel strategy has its own inherent limitations, the approach offered the potentially unique advantage that CaV channel α1 subunit intracellular domains could be used in a manner that preserved their conformations and spatial inter-relationships. This was achieved by systematically generating a series of 31 separate chimeric channels featuring a swap of all possible permutations of the intracellular domains of the β-dependent CaV1.2 α1C into the analogous positions in the β-independent CaV3.1 channel α1G subunit. Functional analyses of these chimeras yielded results that provided a different perspective of the determinants underlying β-mediated membrane trafficking of CaV1.2 channels, and suggest a new mechanistic model for this physiologically crucial phenomenon.
Methods
Generation of plasmid constructs
All the experiments comply with the policies and regulations of The Journal of Physiology given by Drummond (2009). Generation of plasmids encoding α1C[BBS]–yellow fluorescent protein (YFP) and β2a–cyan fluorescent protein (CFP) have been previously described (Yang et al. 2010). Chimeric channels featuring a systematic swap of intracellular domains of rabbit CaV1.2 α1C (accession no.: X15539) into the rat CaV3.1 α1G (accession no.: AF027984) backbone were generated using the in-fusion cloning technique (Clontech, Mountain View, CA, USA) according to the manufacturer's instructions. To generate chimeras in which only one intracellular domain of α1G-YFP was replaced by the corresponding intracellular domain from α1C, four specially designed primers were used – two primers for PCR amplification of the vector containing the α1G segment, and two primers for PCR amplification of the relevant α1C segment as the insert (online Supplemental Material, Table S1). The in-fusion reaction was carried out with purified vector and insert in a 1:2 molar ratio, and the resulting chimeric constructs subsequently cloned. Replacement of multiple intracellular domains was achieved by combining the in-fusion technique with traditional restriction enzyme digestion and ligation methods. The precise boundaries of the N- and C-termini, and the intracellular loops of CaV1.2 and CaV3.1 used to generate the chimeras are shown in the online Supplemental Material, Fig. S1.
To generate C-terminus-truncated α1C[BBS]-YFP constructs, we used PCR to introduce a XhoI site immediately downstream of the sequence for the last desired residue (G1540, D1632, L1732, or K1906) in the context of α1C[BBS]. YFP was then PCR amplified and cloned in-frame and downstream of truncated α1C using XhoI and XbaI sites. Point mutations changing acidic residues (D or E) to alanines in the α1C I–II loop were accomplished using the QuikChange Lightning Site-Directed Mutagenesis Kit (Stratagene) according to manufacturer's instructions. All constructs were verified by sequencing.
Cell culture and transfection
Low-passage-number human embryonic kidney (HEK) 293 cells were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 100 μg ml−1 penicillin–streptomycin at 37°C. HEK 293 cells were transiently transfected with appropriate constructs – 6 μg wild-type or modified α1C or α1G, 6 μg β2a (as needed), and 1 μg T antigen using calcium phosphate precipitation – and cultured in supplemented DMEM at 37°C for 48–72 h before use.
Electrophysiology
Whole-cell recordings were conducted 48–72 h after transfection using an EPC-10 patch clamp amplifier (HEKA Electronics, Lambrecht, Germany) controlled by PULSE software (HEKA). Micropipettes were prepared from 1.5 mm thin-walled glass (World Precision Instruments, Sarasota, FL, USA) with a filament micropipette puller (P-97, Sutter Instrument Co., Novato, CA, USA). The internal solution contained (in mm): 135 caesium methanesulphonate (MeSO3), 5 CsCl, 5 EGTA, 1 MgCl2, 4 MgATP (added fresh) and 10 Hepes (pH 7.3). External solution contained (in mm): 140 tetraethylammonium-MeSO3, 5 BaCl2 and 10 Hepes (pH 7.4). When filled with internal solution, the resistance of the pipette was typically 1.5–2 MΩ. Whole-cell I–V curves were generated from a family of step depolarizations (−50 to +50 mV from a holding potential of −90 mV for α1C constructs, or −100 to +40 mV from a holding potential of −100 mV for wild-type α1G and chimeric constructs). Currents were sampled at 25 kHz and filtered at 5 or 10 kHz. Traces were acquired at a repetition interval of 6 s. Leak and capacitive currents were subtracted using a P/8 protocol. I−V curves from individual cells were fit using a least-squares method to the following modified Boltzmann equation:
where, G is the specific conductance, Vrev is the reversal potential, V0.5 is the potential of half-maximal activation, and k is a slope factor.
Detection and quantification of cell surface Cav1.2 channels with quantum dots
Relative surface expression of epitope-tagged α1C subunits was quantitatively determined using quantum dot labelling and flow cytometry as previously described (Yang et al. 2010). Briefly, HEK 293 cells transfected with BBS-tagged α1C-YFP constructs in six-well tissue culture dishes were washed twice with phosphate-buffered saline (PBS) containing Ca2+ and Mg2+, and sequentially incubated with 1 μm biotinylated α-bungarotoxin (BTX) in DMEM–3% BSA at room temperature for 1 h and 5 nm streptavidin-conjugated quantum dot (QD655, Invitrogen) at 4°C for 1 h in the dark. Surface labelled HEK 293 cells were harvested with trypsin, washed with PBS and assayed by flow cytometry using a BD LSRII Cell Analyzer (BD Biosciences, San Jose, CA, USA). CFP- and YFP-tagged proteins were excited at 407 and 488 nm, respectively, and red quantum dot signal was excited at 633 nm. For each group of experiments, we used isochronal untransfected and single colour controls to manually set the appropriate gain settings for each fluorophore to ensure signals remained in the linear range, and to set threshold values. The same gain settings were then used for assaying all isochronal transfection samples.
Flow cytometry data were analysed using FlowJo software. To normalize for protein expression, analysis of QD fluorescence was conducted over a window selected such that the mean YFP fluorescence intensity registered 1000 arbitrary units across all groups. In the case of α1CΔ1632-YFP and α1CΔ1540-YFP, there was an apparent decrease in protein expression. Therefore, for these constructs the comparisons to control were done over an analysis window with a mean YFP fluorescence intensity of 400 arbitrary units (Supplemental Material, Fig. S6). For each experimental condition, the ratio of the mean QD fluorescence intensity to the mean YFP fluorescence intensity (RQD/YFP) was calculated and normalized to the RQD/YFP obtained for isochronal control cells expressing α1C+β2a channels.
Fluorescence imaging
Fluorescence images of labelled (YFP and quantum dot) HEK 293 cell suspensions were obtained using an inverted Nikon Eclipse Ti microscope equipped for epifluorescence. Fluorophores were excited with the appropriate wavelength using a DeltaRAM Random Access Monochromator (Photon Technology International, Birmingham, NJ, USA) and the emitted light imaged with a QuantEM CCD camera (Roper Scientific, Trenton, NJ, USA).
Western blotting
Transiently transfected HEK 293 cells were harvested with trypsin, washed with PBS and solubilized in lysis buffer (20 mm Tris base, 1 mm EDTA, 150 mm NaCl, 1% SDS, 0.1% Triton X-100, pH 7.5), supplemented with Complete Mini protease inhibitor cocktail (Roche), by brief sonication. Protein concentrations of the cell lysates were determined with Pierce BCA Protein Assay Kit (Thermo Scientific). Following addition of sample buffer (50 mm Tris base, 10% glycerol, 2% sodium dodecyl sulfate (SDS), 50 mm dithiothreitol (DTT), 0.2 mg ml−1 bromphenol blue, pH 7.5), cell lysates were resolved by SDS–PAGE (NuPage 10% gel, Invitrogen) at a constant voltage of 200 V for 1 h. Protein bands were then electrotransferred to a 0.45 μm nitrocellulose membrane for 2.5 h at 4°C at a constant voltage of 30 V in transfer buffer (96 mm glycine, 12 mm Tris base, 0.01% SDS, 20% methanol, pH 8.3). The membranes were blocked for 1 h at room temperature with 5% milk in TBS-T buffer (20 mm Tris base, 140 mm NaCl, 0.1% Tween-20, pH 7.6) and incubated overnight at 4°C with rabbit anti-GFP antibody (1:10,000) in TBS-T. The blots were washed with TBS-T and incubated with goat anti-rabbit antibody (1:10,000) for 1 h at room temperature. After further washing, the immune complexes were visualized using SuperSignal West Pico Chemiluminescent Substrate (Thermo Scientific).
Data and statistical analyses
Electrophysiological data were analysed off-line using built in functions in PulseFit (HEKA), Microsoft Excel and Origin software. Statistically significant differences between means (P < 0.05) were determined using one-way ANOVA followed by pairwise comparisons using the Bonferroni test for multiple group comparisons, and either Student's unpaired t test or Wilcoxon's rank sum test for comparisons between two groups. Data are presented as means ± SEM.
Results
Differential impact of CaVβ on functional expression of CaV1.2 (α1C) and CaV3.1 (α1G) channels
We examined the impact of auxiliary β subunits on functional expression of CaV1.2 and CaV3.1 channels reconstituted in HEK 293 cells by transient transfection. Cells transfected with the CaV1.2 α1C-YFP subunit alone displayed barely detectable currents across the relevant range of test pulse potentials (Fig. 1A and B). Co-expression of α1C-YFP with β2a-CFP resulted in a dramatic 15-fold increase in whole-cell current amplitude, accompanied by a 10 mV hyperpolarizing shift in the voltage dependence of channel activation (Fig. 1A and B; Table 1). CaV1.2 channels activated at a threshold voltage of −30 mV and peaked at either 0 mV (with β2a) or +10 mV (no β) (Fig. 1A and B). In sharp contrast, cells transfected with CaV3.1 α1G-YFP subunit alone displayed large whole-cell currents that activated at a threshold potential of −60 mV and peaked at −30 mV (Fig. 1C and D), consistent with its classification as a low voltage-activated calcium channel (Perez-Reyes et al. 1998). Co-expression of α1G-YFP with β2a-CFP had no impact on either the whole-cell current amplitude or the voltage dependence of channel gating compared to cells expressing α1G alone (Fig. 1C and D; Table 1). These results recapitulate the well-known impact of β subunits on the functional expression of high-voltage-activated CaV1 and CaV2 channels, and the relative β independence of low voltage-activated CaV3 channels.
Figure 1. Divergent regulation of CaV1.2 and CaV3.1 channels by auxiliary β subunits.
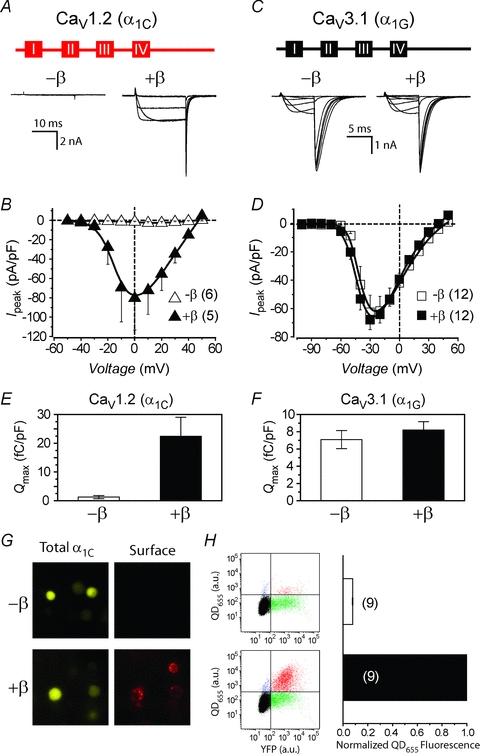
A, top, schematic diagram illustrating topology of CaV1.2 α1C subunit. Four homologous transmembrane domains (boxes labelled I–IV) are connected and flanked by five intracellular modules (red lines). Bottom, exemplar whole-cell currents from CaV1.2 channels reconstituted without (left) or with (right) auxiliary β subunits. Displayed currents were elicited by voltage steps to −30, −10, +10 and +30 mV. B, population peak current density versus voltage (I−V) relationship for CaV1.2 channels reconstituted with either α1C alone (▵, n = 6 for each point) or α1C+β2a (▴, n = 5). C, top, topological illustration of CaV3.1 α1G subunit. Bottom, exemplar currents from CaV3.1 channels reconstituted without (left) or with (right) auxiliary β subunits. D, population I−V relationship for CaV3.1 channels reconstituted with either α1G alone (□, n = 12) or α1G+β2a (▪, n = 12). E, fluorescence images of HEK 293 cells expressing either α1C[BBS]-YFP alone (top) or α1C[BBS]-YFP +β2a (bottom). Left, YFP fluorescence shows total α1C expressed in the cells. Right, quantum dot (QD655) fluorescence specifically labels α1C channels at the cell surface. F, left, representative flow cytometry results from live HEK 293 cells transiently transfected with either α1C[BBS]-YFP alone (top) or α1C[BBS]-YFP +β2a (bottom), and with surface channels labelled with QD655. Right, relative surface density of CaV1.2 channels as reported by normalized QD655 fluorescence intensity.
Table 1.
Gating parameters for reconstituted channels
| Construct | Gmax (pS) | V0.5 (mV) | k | n |
|---|---|---|---|---|
| α1C | 0.19 ± 0.12 | 13.91 ± 11.36 | 13.59 ± 3.78 | 6 |
| α1C+β2a | 1.98 ± 0.70† | −7.81 ± 2.95 | 6.19 ± 0.83 | 5 |
| α1G | 0.96 ± 0.15 | −38.86 ± 0.90 | 5.10 ± 0.49 | 12 |
| α1G+β2a | 1.04 ± 0.09 | −43.45 ± 1.59 | 4.56 ± 0.23 | 12 |
| α1G[cgggg] | 0.23 ± 0.10* | −35.09 ± 4.47 | 13.57 ± 7.20 | 6 |
| α1G[cgggg]+β2a | 0.19 ± 0.07 | −41.92 ± 3.30 | 6.68 ± 1.37 | 4 |
| α1G[gcggg] | 4.08 ± 0.70* | −68.61 ± 1.75* | 1.25 ± 0.35* | 8 |
| α1G[gcggg]+β2a | 1.35 ± 0.23† | −69.76 ± 2.37 | 1.24 ± 0.53 | 5 |
| α1G[ggcgg] | 0.55 ± 0.12 | −40.39 ± 0.52 | 5.59 ± 0.20 | 10 |
| α1G[ggcgg]+β2a | 0.35 ± 0.09 | −39.07 ± 1.16 | 5.67 ± 0.24 | 10 |
| α1G[gggcg] | 0.17 ± 0.06* | −33.26 ± 1.55* | 7.71 ± 0.94* | 7 |
| α1G[gggcg]+β2a | 0.23 ± 0.11 | −33.82 ± 1.55 | 6.57 ± 1.05 | 6 |
| α1G[ggggc] | 0.30 ± 0.12* | −38.65 ± 2.08 | 7.89 ± 1.33* | 8 |
| α1G[ggggc]+β2a | 0.21 ± 0.11 | −44.43 ± 2.64 | 7.40 ± 0.33 | 4 |
P < 0.05 compared to the corresponding –β data, Wilcoxon's rank sum test.
P < 0.05 compared to α1G data using Wilcoxon's rank sum test.
In mammalian cells, β subunits markedly enhance the membrane trafficking of CaV1/CaV2 channels to the plasma membrane, and this represents a major mechanistic contributor to the β-induced increase in whole-cell current amplitude. This effect of β on CaV1.2 channels can be readily demonstrated in our reconstituted system in two independent ways. First, the maximal gating current recorded in cells expressing α1C either in the absence or the presence of β provides a measure of the number of channels with moveable voltage sensors in the membrane (Neely et al. 1993; Takahashi et al. 2004). Hence, based on the assumption that β subunits do not alter the unitary gating charge required to open the channel, the time integral of the maximum gating charge (Qmax) provides an index of the number of channels in the membrane. Using this metric, cells expressing α1C-YFP +β2a-CFP display a significantly larger Qmax, and, therefore, more surface channels than cells transfected with α1C-YFP alone (Fig. 1E). By contrast, β2a-CFP had no effect on Qmax obtained from cells expressing α1G subunits (Fig. 1F), fitting with the lack of impact of the auxiliary subunit on whole-cell current amplitude in CaV3.1 channels. A second, more direct way of visualizing CaV1.2 channels at the membrane involves labelling cell surface channels with quantum dots followed by quantification of signals using flow cytometry (Yang et al. 2010). Here, a 13-residue bungarotoxin-binding site (BBS) (Sekine-Aizawa & Huganir, 2004) is introduced into the extracellular domain II S5–S6 loop of α1C-YFP, and surface channels are selectively labelled in non-permeabilized cells by sequential exposure to biotinylated bungarotoxin and streptavidin-conjugated quantum dot (Fig. 1G). The relative cell surface density of CaV1.2 channels is then quantified in a high throughput manner by flow cytometry. Using this method, the presence of β2a-CFP resulted in a 12-fold increase in QD655 fluorescence intensity compared to cells expressing BBS epitope-tagged α1C-YFP alone (Fig. 1H).
The stark contrast between CaV1.2 α1C and CaV3.1 α1G subunits could potentially be exploited to identify the important determinants and mechanisms underlying β-dependent trafficking. Given that the β subunit is entirely intracellular, we hypothesized that cytoplasmic domains of the α1C subunit play a prominent role in CaV1.2 channel ER retention and β-dependent ER export. Accordingly, we systematically generated 31 separate chimeric channels featuring all possible permutations of the five α1C intracellular domains (N-terminus, I–II loop, II–III loop, III–IV loop and C-terminus) swapped into the analogous regions of α1G and assessed the resulting functional outcomes.
Functional outcomes of single intracellular domain-substituted chimeras
We first investigated whether a single α1C subunit intracellular domain could potentially confer ER retention and β-dependent ER export when transplanted into α1G. We generated five individual chimeras (termed α1G[cgggg], α1G[gcggg], α1G[ggcgg], α1G[gggcg] and α1G[ggggc]) in which just one intracellular region of α1C was swapped into the α1G backbone (Fig. 2). We adopted a naming convention in which the backbone channel subunit is followed by a square bracket containing letters that denote the configuration of the intracellular domains. Hence, for example, α1G[cgggg] refers to the chimeric channel in which the N-terminus of α1G is replaced with the analogous segment from α1C. To assess the functional impact of the single-domain substitutions, we transiently expressed the chimeric channels in HEK 293 cells in either the absence or the presence of β2a-CFP and recorded whole-cell currents (Fig. 2). All chimeras, here and throughout, were tagged with YFP enabling direct visual confirmation of expression to be used as a criterion for cell selection. All the chimeras expressed full-length channels as assessed by Western blot using an anti-GFP antibody (Fig. 2K, inset). Cells expressing α1G[cgggg] alone displayed whole-cell currents that were significantly smaller at all test voltages compared to α1G alone channels (Fig. 2A and B, open symbols; Table 1). When peak amplitudes are compared at −30 mV to ensure a similar driving force, α1G[cgggg] channels exhibited a substantive fourfold decrease in current amplitude (Fig. 2K). Co-expressing β2a had no impact on either current amplitude or voltage dependence in this chimeric channel (Fig. 2A, B and K).
Figure 2. Functional outcomes of chimeras featuring substitutions of individual intracellular domains from CaV1.2 α1C into CaV3.1 α1G subunit.

A, top, topological illustration of the chimeric channel α1G[cgggg], which features the sole substitution of the α1C N-terminus into α1G. Bottom, exemplar whole-cell currents from cells expressing either α1G[cgggg] alone (left) or α1G[cgggg]+β2a (right). Displayed currents were elicited by voltage steps to −50, −30, −10 and +10 mV. B, I−V curves for channels reconstituted with α1G[cgggg] alone (□) or α1G[cgggg]+β2a (▪). Data for wild-type α1G (cyan trace) have been reproduced to facilitate direct visual comparison. C−J, topological illustrations, exemplar currents and population I−V relationships for the remaining single intracellular domain substituted chimeras α1G[gcggg], α1G[ggcgg], α1G[gggcg] and α1G[ggggc], respectively. Same format as A and B. K, comparison of peak current densities acquired at −30 mV for the various chimeras in the absence and presence of β2a. The average current density at −30 mV for wild-type α1G alone channels is represented (cyan dashed line) to facilitate visual comparison. **P < 0.005 for grouped data (±β2a) compared to α1G (±β2a) using one-way ANOVA followed by means comparisons using the Bonferroni test. *P < 0.001 compared to α1G (±β2a), one-way ANOVA followed by Bonferroni test. #P < 0.05, Wilcoxon's rank sum test. Inset, Western blot: 1-α1G; 2-α1G[cgggg]; 3-α1G[gcggg]; 4-α1G[ggcgg]; 5-α1G[gggcg]; 6-α1G[ggggc].
In striking contrast to the results obtained with α1G[cgggg], cells expressing α1G[gcggg], a chimera featuring a swap of the entire α1C I–II loop into α1G, demonstrated a dramatic increase in current amplitude and a 30 mV leftward shift in the I−V relationship (Fig. 2C and D, open symbols; Table 1). When compared at a −30 mV test pulse, α1G[gcggg] channels exhibited an over threefold increase in current density compared to wild-type α1G-alone channels (Fig. 2F; Ipeak = 62.6 ± 12.1 pA pF−1, n = 12, for α1G, and Ipeak = 197.4 ± 23.0 pA pF−1, n = 8 for α1G[gcggg] channels, P < 0.05, one-way ANOVA). The surprising result with the α1G[gcggg] chimera confirms recent findings (Arias et al. 2005; Fan et al. 2010), and are seemingly at odds with the notion that the I–II loop of CaV1 and CaV2 channels harbours a dominant ER retention signal. Co-expressing β2a unexpectedly diminished, rather than increased α1G[gcggg] currents, without affecting the voltage dependence of channel gating (Fig. 2C, D and K; Table 1).
Similar to the results obtained with α1G[cgggg], cells transfected with α1G[ggcgg], α1G[gggcg], or α1G[ggggc] displayed significantly smaller currents compared to wild-type α1G, and were β independent (Fig. 2E–K; Table 1). Additionally, these other chimeras had only modest, if any, effects on the voltage dependence of channel activation (Table 1). These functional results were not correlated with differences in protein expression (Fig. 2K, inset). Overall, these results suggested that no single α1C intracellular domain could confer β-dependent plasma membrane targeting to the α1G backbone. Moreover, the data also raised the radical possibility that the α1C I–II loop could actually facilitate ER export rather than retention in the context of CaV channel α1 subunits. We next sought to test this possibility.
Identification of a putative ER export signal in the α1C I–II loop
It was possible that the variance in current density between wild-type α1G and the single-domain-swapped chimeras was due to changes in channel gating (such as distinctions in channel open probability) rather than differences in channel trafficking. As such, it was important to determine whether the different chimeras displayed divergent propensities to target to the cell surface. Unfortunately, the quantum dot labelling method for surface channels was unsuccessful in the context of CaV3.1, probably due to geometric constraints that limited accessibility to the surface epitope tag. Fortunately, however, the α1G subunit exhibits robust gating currents, thus rendering it possible to measure Qmax as an index of channel trafficking to the membrane. Compared to wild-type α1G, α1G[gcggg] displayed a significantly larger gating current and Qmax, while all the other single-domain-swap chimeras (except α1G[ggcgg]) exhibited a diminished Qmax (Fig. 3A). This result indicates that α1G[gcggg] shows markedly improved membrane targeting, whereas the other chimeras show a relative retention compared to wild-type α1G.
Figure 3. Identification of an acidic sequence that functions as an ER export signal in α1C I–II loop.
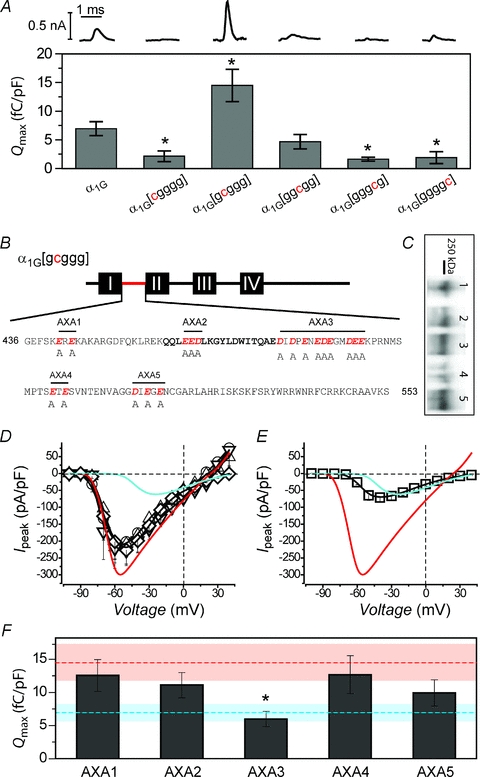
A, top, exemplar gating currents for wild-type α1G and single domain-substituted chimeric channels. Bottom, comparison of maximal gating charge (Qmax) for the different channel types. *P < 0.05 compared to α1G. B, sequence of the α1C I–II loop showing candidate acidic-residue ER export signals that were mutated to alanines in the context of the α1G[gcggg] chimera to generate five distinct mutants, AXA1−AXA5. C, Western blot: 1, AXA1; 2, AXA2; 3, AXA3; 4, AXA4; 5, AXA5. D, population I−V curves for chimeric mutant channels AXA1 (○, n = 9), AXA2 (▵, n = 6), AXA4 (▿, n = 6) and AXA5 (⋄, n = 6). Data for wild-type α1G (cyan trace) and α1G[gcggg] (red trace) are reproduced to facilitate visual comparison. E, I−V curve for AXA3 mutant chimeric channels (□, n = 8). F, comparison of Qmax for the different AXA mutant chimeric channels. Mean data for wild-type α1G (dashed cyan line) and α1G[gcggg] (dashed red line) are represented. *P < 0.05 compared to α1G[gcggg], Wilcoxon's rank sum test.
We hypothesized that the α1C I–II loop contains an ER export signal that could potentially explain the enhanced membrane targeting of α1G[gcggg]. Di-acidic motifs (DXD, DXE, EXD and EXE, where X is any amino acid) have been found to act as ER export signals in many membrane proteins (Ma et al. 2001; Ma et al. 2002; Mikosch & Homann, 2009). We searched for possible acidic residue ER export signals by scanning the α1C I–II loop sequence (Fig. 3B; Supplemental Material, Fig. S2). We identified five candidate regions with acidic residue motifs and generated five distinct mutants (termed AXA1–AXA5) in which acidic amino acids (D or E) in the α1C I–II loop were changed to alanine, all within the context of the α1G[gcggg] chimera (Fig. 3B). All five chimeras expressed full-length channels (Fig. 3C). Whole-cell currents were recorded from HEK 293 cells transfected with each mutant in the absence of β, and I−V curves were generated (Fig. 3D). The mutant chimeras AXA1, AXA2, AXA4 and AXA5 displayed current densities that were only modestly reduced compared to α1G[gcggg] (Fig. 3D), indicating that the acidic residues present in the associated regions do not account for the ER export capabilities of the α1C I–II loop. By contrast, the mutant chimera AXA3 exhibited a current density that was essentially identical to wild-type α1G (Fig. 3E), indicating that the nine acidic residues mutated in this cluster accounted wholly for the increased current density observed with the α1G[gcggg] chimera. Significantly, AXA3 still displayed a V1/2 that was substantially left-shifted compared to wild-type α1G (Fig. 3E). Therefore, the increase in current density and leftward-shift in activation are separable functions conferred by the α1C I–II loop in the context of α1G[gcggg]. Gating charge analyses (Fig. 3F) demonstrated that AXA3 displayed a Qmax that was significantly smaller than obtained with α1G[gcggg] (Qmax = 6.01 ± 1.16 fC pF−1, n = 8 for AXA3, and Qmax = 14.48 ± 2.83 fC pF−1, n = 7, for α1G[gcggg]-alone channels, P < 0.05), and essentially identical to wild-type α1G (Qmax = 6.95 ± 1.22 fC pF−1, n = 7). The remaining mutated chimeras (AXA1, AXA2, AXA4 and AXA5), by contrast, exhibited Qmax values that were not significantly different from α1G[gcggg] (Fig. 3F). Taken together, the results suggest that the acidic residue cluster mutated in AXA3 is a putative ER export region (PEER; Supplemental Material, Fig. S2) in the α1C I–II loop, and that the other α1C intracellular domains may either confer ER retention propensities or increase the rate of channel removal from the cell surface.
Functional interactions among putative α1C ER export and retention modules probed with double and triple domain-substituted chimeras
How do the putative ER retention and export signals functionally interact with each other, and what is the minimum combination of domains required to confer β dependency to channel regulation? To address these questions we generated chimeric channels featuring multiple intracellular domains of α1C swapped into α1G, starting with the 10 possible chimeras containing double-domain substitutions (Fig. 4). For these double-domain chimeras, there were two types of questions of primary interest. First, how did the putative ER retention characteristics of the other intracellular domains functionally interact with the export capabilities of the I–II loop? Second, were the putative ER retention properties of individual intracellular domains synergistic when present on the same molecule? Examination of the electrophysiological properties of the four chimeras in which α1C I–II loop was paired with each of the other intracellular domains suggested varying strengths among the distinct ER retention regions (Fig. 4). Cells expressing α1G[ccggg] alone displayed an intermediate current density that lay between wild-type α1G and α1G[gcggg] (Fig. 4A and E), suggesting α1C N-terminus moderately opposed the ability of the α1C I–II loop to increase channel surface density. Cells expressing α1G[gccgg] alone displayed large currents that were indistinguishable from α1G[gcggg] channels (Fig. 4B and E), indicating that the α1C II–III loop is unable by itself to diminish the trafficking function of α1C I–II loop. At the other extreme, α1G[gcgcg] and α1G[gcggc] channels exhibited relatively small currents similar in amplitude to wild-type α1G channels (Fig. 4C–E). Hence, α1C III–IV loop and C-terminus completely neutralized the α1C I–II loop enhanced trafficking effect. All other chimeras that contained two α1C intracellular domains, exclusive of the I–II loop, exhibited exceptionally small currents (Fig. 4E; Supplemental Material, Fig. S3). None of the double domain-substituted chimeras showed a β-dependent increase in current density –α1G[gccgg] channels displayed a β-dependent decrease in current density (Fig. 4B and E) similar to that observed with α1G[gcggg]. These results were not due to differences in protein expression among the distinct chimeric channels (Fig. 4E, inset). Overall, these results indicated that the putative ER retention signals on the α1C intracellular loops acted synergistically, and could counteract the propensity of α1C I–II loop to increase channel surface density, but with differing efficacies. Moreover, no two α1C intracellular domains were sufficient to reconstitute β-dependent increased channel trafficking.
Figure 4. Functional outcomes of chimeras featuring two intracellular domains from α1C substituted into α1G.

A−D, topological illustrations and population I−V curves (±β) for those double intracellular domain swapped chimeras that include the α1C I–II loop. Data for wild-type α1G (cyan trace) and α1G[gcggg] (red trace) are reproduced for comparison. E, peak current densities acquired at −30 mV for all double intracellular domain swapped chimeras compared to wild-type α1G (dashed cyan line). **P < 0.005 for grouped data (±β2a) compared to α1G (±β2a) using one-way ANOVA followed by means comparisons using the Bonferroni test. *P < 0.001 compared to α1G (±β2a), one-way ANOVA followed by the Bonferroni test. Inset, Western blot: 1, α1G[ccggg]; 2, α1G[cgcgg]; 3, α1G[cggcg]; 4, α1G[cgggc]; 5, α1G[gccgg]; 6, α1G[gcgcg]; 7, α1G[gcggc]; 8, α1G[ggccg]; 9, α1G[ggcgc]; 10, α1G[gggcc].
These observations were consolidated and extended by considering the functional outcomes of chimeras in which three α1C intracellular domains were swapped into α1G (Fig. 5). All the triple-substituted chimeras which included the α1C I–II loop exhibited currents with amplitudes that were either on par with or less than wild-type α1G (Fig. 5). When the α1C I–II loop was absent, the triple mutant chimeras did not register discernible currents (Fig. 5G; Supplemental Material, Fig. S4), consistent with the idea that the discrete putative ER retention signals acted synergistically to retain channels inside the cell. All the triple-substituted chimeras expressed full-length proteins (Fig. 5G, inset), and the functional results were not correlated with differences in protein expression. Finally, none of the triple-substituted chimeras displayed β-dependent increase in current density, further emphasizing the surprisingly complex nature of this phenomenon.
Figure 5. Functional outcomes of chimeras featuring three intracellular domains from α1C substituted into α1G.
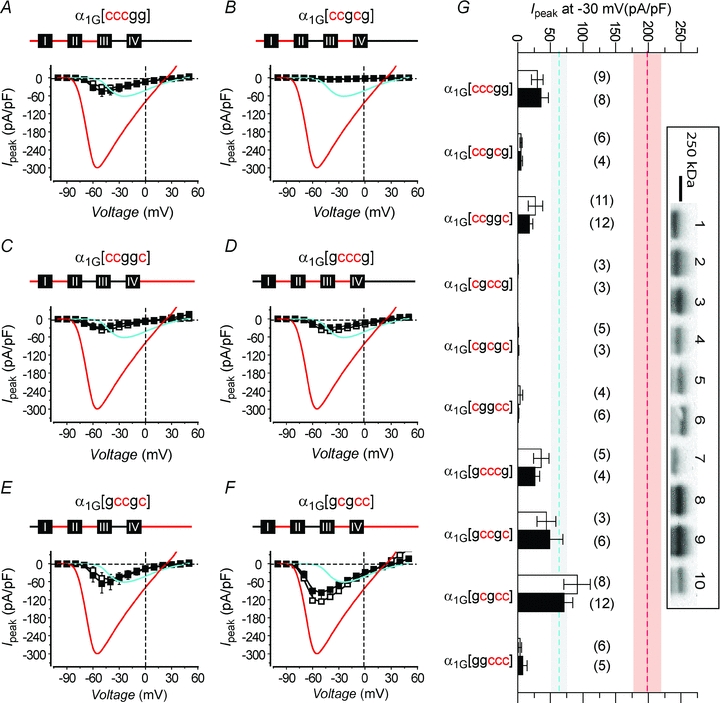
A−F, topological illustrations and population I−V curves (±β) for those triple intracellular domain-swapped chimeras that include the α1C I–II loop. Data for wild-type α1G (cyan trace) and α1G[gcggg] (red trace) are reproduced for comparison. G, peak current densities acquired at −30 mV for all triple intracellular domain-swapped chimeras compared to wild-type α1G (dashed cyan line) and α1G[gcggg] channels (dashed red line). **P < 0.005 for grouped data (±β2a) compared to α1G (±β2a) using one-way ANOVA followed by means comparisons using the Bonferroni test. Inset, Western blot: 1, α1G[cccgg]; 2, α1G[ccgcg]; 3, α1G[ccggc]; 4, α1G[cgccg]; 5, α1G[cgcgc]; 6, α1G[cggcc]; 7, α1G[gcccg]; 8, α1G[gccgc]; 9, α1G[gcgcc]; 10, α1G[ggccc].
Chimeras displaying successful reconstitution of β-dependent channel regulation
Arguably, the most profound insights into the mechanistic bases of β-dependent channel regulation may be provided by successfully reconstituting this phenomenon in a normally β-independent channel background. As such, it was instructive that three distinct chimeras (α1G[cccgc], α1G[ccgcc] and α1G[gcccc]) characterized by four intracellular domains of α1C substituted into the α1G background recapitulated a β-dependent increase in current density that is normally only seen in CaV1/CaV2 channels (Fig. 6). Two other quadruple domain-substituted chimeras, α1G[cgccc] and α1G[ccccg], displayed no current in either the absence or presence of β (Fig. 6A, D and F). The fact that α1G[cgccc] produced no currents was not surprising based on the emerging concept that this chimera essentially contains four α1C ER retention modules while lacking the robust ER export services of the α1C I–II loop. Less predictable was the absence of current observed with the α1G[ccccg] chimera. Together with results from other chimeras, this finding suggests that the α1C C-terminus is absolutely essential for channel trafficking to the membrane and β-dependent channel regulation, but only when it is assembled with at least three other α1C intracellular domains on the same channel molecule. A chimera in which all five intracellular domains of α1C were placed into α1G also displayed currents which trended higher in the presence of β, although this effect did not reach statistical significance (Fig. 6F and G). All the chimeric channels expressed well (Fig. 6G, inset), ruling out differences in protein expression as a potential trivial explanation for the results.
Figure 6. Reconstitution of β regulation of current density in specific chimeric channels containing four α1C intracellular domains swapped into α1G.

A−F, topological illustrations and population I−V curves (±β) for quadruple and quintuple intracellular domain-swapped chimeras. Data for wild-type α1G (cyan trace) and α1G[gcggg] (red trace) are reproduced for comparison. *P < 0.05 compared to corresponding −β data, Wilcoxon's rank sum test. G, peak current densities acquired at −30 mV for quadruple and quintuple intracellular domain-swapped chimeras compared to wild-type α1G (dashed cyan line) channels. Inset, Western blot: 1, α1G[ccccg]; 2, α1G[cccgc]; 3, α1G[ccgcc]; 4, α1G[cgccc]; 5, α1G[ccccc].
Overall, these results demonstrate that a minimum of four α1C intracellular domains that must include the I–II loop and C-terminus are necessary and sufficient to reconstitute β-dependent regulation of current density in α1G/α1C chimeric channels.
Trafficking role of the I–II loop and C-terminus in the α1C subunit context
Two remaining questions were: (1) does the newly identified PEER in the α1C I–II loop regulate channel surface density in the context of wild-type CaV1.2 α1C subunit? and (2) how and why is the C-terminus important for CaV1.2 channel targeting? To address the hypothesized ER export function of the α1C I–II loop in the native channel, we introduced the distinct AXA1–AXA5 mutations (Fig. 3B) in the context of α1C[BBS]-YFP (Fig. 7A), and monitored channel targeting to the cell surface using the quantum dot labelling method (Fig. 7B). Compared to wild-type α1C[BBS]-YFP +β2a, the α1C[AXA3] mutant (+β2a) exhibited a 60% decrease in QD655 fluorescence intensity (Fig. 7C), supporting the idea that the PEER is important for channel trafficking within the context of the native CaV1.2 channel. The fact that β-dependent channel trafficking was not completely abolished in the AXA3 mutant channel may indicate that other ER export signals reside elsewhere on the α1C subunit. Channels bearing mutations of other acidic residues in the I–II loop (AXA2, AXA4 and AXA5) also displayed moderate decreases in trafficking (Fig. 7C). However, combining mutations (AXA345) did not decrease the trafficking beyond what was observed with AXA3 alone (data not shown).
Figure 7. Role of the I–II loop and C-terminus in β-dependent trafficking of CaV1.2 α1C subunit.

A, topological illustration of α1C with AXA1–AXA5 mutations in the I–II loop. B, exemplar flow cytometry results examining surface expression of α1C[AXA1] (top) and α1C[AXA3] (bottom) ±β2a using the quantum dot labelling method. C, relative surface expression of (normalized QD655 fluorescence intensities) for the distinct α1C AXA mutants. *P < 0.05 compared to α1C[BBS]-YFP +β2a. #P < 0.05 compared to the corresponding –β2a data. D, left, topological illustration of serial C-terminus truncation mutants of α1C showing the relative position of CaM-binding pre-IQ and IQ sites. Right, impact of C-terminus truncations on relative surface expression of α1C subunits ±β2a. E and F, impact of combining C-terminus truncations and AXA1–AXA5 mutations on relative surface expression of α1C subunits, n = 3−4. Dashed lines represent means of data for α1CΔ1906 +β2a and α1CΔ1732 +β2a, respectively, and the corresponding 95% confidence intervals (shaded regions). *P < 0.05 compared to α1CΔ1906 +β2a and α1CΔ1732 +β2a, respectively. #P < 0.05 compared to the corresponding –β2a data.
The α1C C-terminus is a hub of protein–protein interactions that modulate the CaV1.2 channel (Supplemental Material, Fig. S5). In particular, the proximal C-termini of CaV1 and 2 channels contain pre-IQ and IQ regions (Fig. 7D; Supplemental Material, Fig. S5) which serve as binding sites for apo-calmodulin (CaM) and Ca2+–CaM (Romanin et al. 2000; Pitt et al. 2001; Erickson et al. 2003; Kim et al. 2004; Van Petegem et al. 2005), and are critical for feedback regulation of CaV channels (inactivation or facilitation) by Ca2+ ions (Lee et al. 1999; Peterson et al. 1999; Zuhlke et al. 1999). Point mutations in the pre-IQ region that ablate apo-CaM binding to the C-terminus also markedly depress β-dependent targeting of the CaV1.2 channel to the cell surface (Wang et al. 2007; Bourdin et al. 2010). We sought to gain a deepened appreciation of the role of the α1C C-terminus in CaV1.2 channel targeting by determining whether the important determinants of β-dependent trafficking were pervasively distributed throughout the C-terminus, or were localized to the pre-IQ/IQ region. Accordingly, we tested the impact of a series of C-terminus truncations on CaV1.2 channel trafficking to the plasma membrane. Serial truncations at the α1C C-terminus led to graded decreases in CaV1.2 channel targeting to the cell surface – channels truncated at residues 1906 and 1732 displayed a 60% and 80% reduction in surface density, respectively (Fig. 7D). Longer truncations at residues 1632 and 1540, respectively, virtually eliminated β-dependent membrane targeting (Supplemental Material, Fig. S6). Importantly, QD655 fluorescence was normalized for YFP expression under all conditions, explicitly ruling out the potential trivial explanation that differences in trafficking propensity among the distinct α1C species could be due to variations in protein expression. Both α1CΔ1906 and α1CΔ1732 functionally retain Ca2+-dependent inactivation (CDI) gating (Erickson et al. 2001; Kim et al. 2004), as might be expected given that the pre-IQ and IQ regions remain intact (Supplemental Material, Fig. S5). The results indicate that critical determinants of β-dependent trafficking of CaV1.2 channels are widely distributed throughout the C-terminus, rather than being locally limited to the proximal CaM-binding preIQ/IQ region.
Finally, we examined the impact of combining the α1C I–II loop AXA1–AXA5 mutations and the C-terminus truncations on CaV1.2 channel trafficking to the cell surface (Fig. 7E and F). In both α1CΔ1906 and α1CΔ1732, only AXA3 caused a further significant decrease in channel surface density, while the other AXA mutations either caused no change, or even slightly enhanced channel trafficking (Fig. 7E and F). These results offer further evidence that the PEER in the α1C I–II loop is an important determinant of CaV1.2 channel surface density.
Discussion
The fundamental importance of auxiliary β subunits to the functional evolution of CaV1/CaV2 channels is underscored by the severe phenotypes of β-null mice: β1– lethal at birth due to asphyxiation (Gregg et al. 1996); β2– embryonic lethal due to a compromised heartbeat (Ball et al. 2002); and β4– development of a lethargic epileptic phenotype (Burgess et al. 1997). A prominent function of β subunits is to promote the cell surface trafficking of CaV1/CaV2 channels. In this work we have re-examined the important molecular determinants and mechanism underlying β-subunit-mediated plasma membrane targeting of CaV channels. There are three major new findings: (1) the CaV1.2 α1C subunit I–II loop is a putative ER export rather than retention module; (2) the α1 C-terminus plays a dual role in channel trafficking; and (3) β-dependent increase in channel surface density is an emergent property that requires multiple α1C intracellular domains inclusive of the I–II loop and C-terminus. We discuss these aspects of the work in the context of previous results.
Putative ER export versus retention role of the α1C I–II loop in CaV channel trafficking
Auxiliary β subunits bind with nanomolar affinity to the AID in the I–II loop of CaV1/CaV2 α1 subunits (Pragnell et al. 1994; Canti et al. 2001; Opatowsky et al. 2003; Butcher et al. 2006; Van Petegem et al. 2008). High-resolution crystal structures show that β subunits interact with the AID using an α1-binding pocket formed from non-contiguous residues localized in the GK domain (Chen et al. 2004; Opatowsky et al. 2004; Van Petegem et al. 2004). Point mutations within the AID that disrupt the interaction with β subunits prevent β-induced trafficking of α1 subunits to the plasma membrane in mammalian cells (Bourdin et al. 2010; Obermair et al. 2010) and Xenopus oocytes (Van Petegem et al. 2008). How does β binding to the I–II loop promote trafficking of α1 subunits to the cell surface? The prevailing notion, based on experiments carried out in CaV2.1 channels, is that the α1 subunit I–II loop contains an ER retention signal that becomes masked once β binds, thereby permitting forward trafficking (Bichet et al. 2000). The evidence presented in this work suggests that in CaV1.2 channels the I–II loop does not act as an ER retention module in the context of the CaV channel α1 subunit. Rather, the opposite situation was discovered – i.e. within the context of the channel, the α1C I–II loop may have a net ER export function. A cluster of acidic residues situated just downstream of the AID fully accounted for the capability of the I–II loop to enhance chimeric channel surface density, and reprised a similar function within the context of the native CaV1.2 channel. It is important to note here that the loss of CaV channel trafficking resulting from mutations in the PEER differ fundamentally from previously identified mutations in the I–II loop that also prevent β-induced channel targeting to the cell surface. The previous loss-of-function mutations are within the AID, and prevent channel trafficking by abolishing the α1−β interaction. By contrast, the AXA3 mutations prevent channel trafficking in a β-independent manner, as uniquely demonstrated in the α1G[gcggg] chimeric channel context. Di-acidic ER export motifs have been demonstrated in membrane proteins from plant and animal cells (Ma et al. 2001, 2002; Mikosch & Homann, 2009). The basis of their ER export function is believed to be their recognition by components of the coat protein II (COP II) complexes that mediate anterograde ER-to-Golgi vesicular transport (Mikosch & Homann, 2009). We hypothesize that the acidic sequence identified in the α1 subunit I–II loop may act in such a fashion. This hypothesis will need to be tested in future experiments.
The CaV2.1 α1 subunit I–II loop was proposed as an ER retention module based on its ability to retard membrane targeting of either Shaker K+ channels or CD8, when fused to their intracellular C-termini (Bichet et al. 2000). However, in similar experiments, neither the CaV1.2 nor CaV2.2 α1 subunit I–II loop displayed ER retention properties, suggesting there may be a genuine difference among distinct CaV1/CaV2 channel isoforms (Altier et al. 2011). Given that the experiments described in this work have focused exclusively on trafficking determinants in CaV1.2 channels, it is worth considering whether the I–II loop may function similarly in other CaV1/CaV2 channel types. In this regard, it is reassuring that the I–II loops of both CaV2.2 (α1B) and CaV2.1 (α1A) result in a substantial increase in current density when transplanted into to α1G (Arias et al. 2005; Fan et al. 2010). Moreover, similar to α1C, the α1A and α1B I–II loops contain an acidic residue rich region immediately downstream of the AID (Supplemental Material, Fig. S2). Based on these similarities we speculate that the I–II loop may serve to enhance channel surface density similarly in all CaV1/CaV2 channels. This prediction will need to be verified experimentally in future experiments.
Role of the α1 C-terminus in CaV channel trafficking
In addition to the I–II loop, there has been accruing evidence that the α1C C-terminus plays a crucial role in CaV channel trafficking. Deletion of residues 1623−1666 in the α1C C-terminus abolishes β-dependent trafficking of CaV1.2 channels to the surface membrane, despite an enduring interaction between the two channel subunits (Gao et al. 2000; Bourdin et al. 2010). The stretch of deleted amino acids encompasses a demonstrated Ca2+–CaM binding IQ domain (Erickson et al. 2003; Kim et al. 2004; Van Petegem et al. 2005), leading to suggestions that Ca2+–CaM binding to the C-terminus may be necessary for CaV1.2 channel trafficking to the membrane (Wang et al. 2007). However, this idea is challenged by the finding that β-dependent CaV1.2 channel membrane trafficking remained intact when the α1C subunit featured a more restricted, but still complete, deletion of the IQ domain (residues 1643–1666) (Bourdin et al. 2010). On the other hand, point mutations in an upstream pre-IQ site that disrupt apo-CaM binding to the α1C C-terminus also interfere with β-induced membrane targeting of CaV1.2 channels (Wang et al. 2007; Bourdin et al. 2010). Hence, apo-CaM, rather than Ca2+−CaM, binding to the α1 C-terminus may be critical for CaV channel membrane trafficking. Irrespective of which CaM species is paramount, the question arises as to how CaM molecules can promote β-dependent forward trafficking of CaV1.2 channels. It has been previously suggested that the α1C C-terminus may contain an ER retention signal that is masked upon CaM binding, thereby permitting channel movement to the plasma membrane (Wang et al. 2007). Further, experiments in which α1C C-terminus fragments were fused to CD4 identified three separate areas in the proximal C-terminus overlapping the EF-hand/pre-IQ/IQ motifs (Supplemental Material, Fig. S5) that exhibited ER retention properties (Altier et al. 2011). Aspects of our results support the idea that the α1C C-terminus can function as a net ER retention module. Specifically, when swapped for the analogous domain in α1G, the α1C C-terminus caused a decrease in the surface density of CaV3.1 channels. Moreover, in the doubly substituted chimera α1G[gcggc], the α1C C-terminus completely neutralized the strong forward trafficking propensity conferred by the α1C I–II loop. Nevertheless, the data also indicate a more complex role of the C-terminus in CaV1.2 channel trafficking beyond a simple masking of an ER retention sequence by CaM. Truncating α1C at residue 1732 in the C-terminus results in an 80% decrease in channel trafficking even though both CaVβ and CaM still associate with the channel (Gao et al. 2000; Erickson et al. 2001). This result argues against the simple explanation that CaM merely masks a local ER retention signal, and that this in concert with β binding is sufficient to promote channel trafficking to the membrane. Rather, the data support a more pervasive distribution of determinants on the C-terminus that are important for CaV1.2 channel trafficking. It is noteworthy that although α1CΔ1906 and α1CΔ1732 target less well than wild-type α1C to the plasma membrane, they typically give rise to larger whole-cell currents in functional assays (Wei et al. 1994; Hulme et al. 2006). This discrepancy is due to an auto-inhibitory role of the distal C-terminus on channel gating, such that deletion of this region results in a large increase in CaV1.2 channel Po (Hulme et al. 2006).
Interestingly, the C-termini of the distinct CaV1 and 2 channels display little sequence homology beyond the residues corresponding to L1732 in CaV1.2 (Supplemental Material, Fig. S5). Hence, it is possible that the C-terminus may play divergent roles in β-dependent trafficking among the different channel types. This will be an important and interesting question to address in future studies.
β-dependent trafficking as an emergent property supported by multiple α1 intracellular domains
A striking result was that the double chimera, α1G[gcggc], which featured both the α1C I–II loop and the C-terminus swapped into α1G, failed to display β-dependent channel targeting to the cell surface. Instead, β-dependent regulation of current density required the presence of at least four intracellular domains of α1C that included the I–II loop and C-terminus. Therefore, the α1C I–II loop and C-terminus are necessary but not sufficient to reconstitute β-dependent channel trafficking to the membrane. A limitation of our study is that the magnitude of β-dependent up-regulation of current density reconstituted in the chimeric channels (∼three-fold increase) is less than the 10- to 15-fold increase typically observed with wild-type CaV1.2. A likely contributing factor to this discrepancy is that β subunits increase the single-channel Po of CaV1.2 channels (ranging from 2- to 8-fold in different studies) in addition to promoting channel trafficking to the plasma membrane (Kanevsky & Dascal, 2006). The increase in Po relies on a β-dependent formation of a rigid helix spanning the AID and domain IS6 (Vitko et al. 2008; Findeisen & Minor, 2009). Our results do not support a similar β-dependent increase in single channel Po in the chimeric channels since we do not observe a β-dependent enhancement of current in the majority of chimeras that contain the α1C I–II loop. Hence, the magnitude of β-dependent increase in current density may be quite comparable between the relevant chimeras and CaV1.2 if the enhanced single-channel Po effect on the latter is taken into account.
Overall, our results suggest the following hypothesized model for β-dependent regulation of CaV channel trafficking (Fig. 8). The α1C I–II loop contains a putative ER export signal while the other intracellular loops and termini have net ER retention characteristics. In the absence of β, the intracellular domains are configured such that the multiple ER retention signals are exposed and functionally dominant, leading to channels being retained in the ER. Upon β-binding to the α1C I–II loop, we propose that a conformational rearrangement of the intracellular domains occurs that diminishes the strength of ER retention signals relative to I–II loop export signals, leading to channel transport to the cell surface (Fig. 8). It is interesting to contemplate the possible nature of the proposed β-induced conformational change in intracellular domains. Our results do not support a model where the β subunit itself physically masks spatially distinct ER retention signals on the channel. If this were the case then it would be expected that β subunits would enhance trafficking in double and triple chimeras that included the I–II loop. However, this was not observed. Rather, the data invite speculation that β-binding to the I–II loop initiates a concerted motion of the intracellular segments that is coordinated by the C-terminus. CaM may participate in this process by promoting a permissive ternary conformation of the C-terminus. This model intimates that the α1C intracellular domains are not independent entities, but rather engage in intra-molecular interactions among themselves. In this regard, it is noteworthy that in CaV2.1 channels the I–II loop has been demonstrated to interact with the N- and C-termini, and the III–IV loop (Restituito et al. 2000; Geib et al. 2002). The exact details of the β-induced rearrangement of α1C subunit intracellular segments that we propose will need to be explored in future experiments, including structural studies.
Figure 8. Conceptual model of mechanism underlying β-mediated trafficking of CaV1.2 channels.

In the absence of β, an ER export signal present on the α1 subunit I–II loop is functionally overcome by discrete ER retention signals present in the other intracellular domains, leading to channels being retained in the ER. Upon β-binding to the α1C I–II loop, a C-terminus-dependent conformational rearrangement of the intracellular domains occurs that diminishes the strength of ER retention signals relative to I–II loop export signals, leading to channel transport to the cell surface.
Recently, two groups independently demonstrated that CaVβ binding to either CaV1.2 (Altier et al. 2011) or CaV2.2 (Waithe et al. 2011) protects the respective α1 subunit from ubiquitination and subsequent proteasomal degradation. In one study, blocking the proteasomal degradation pathway with MG132 was sufficient to rescue surface expression of CaV1.2 channels even in the absence of β (Altier et al. 2011). By contrast, MG132 did not lead to increased surface expression of CaV2.2 channels in the absence of CaVβ (Waithe et al. 2011). Possibly, the β-dependent rearrangement of α1C subunit intracellular segments we envision is necessary to prevent channel ubiquitination and targeting to the proteasome. Further work is clearly needed to reconcile these new results and to develop a more detailed mechanistic model for β regulation of CaV1/CaV2 channel trafficking.
Acknowledgments
The authors thank Ming Chen and Timothy Kernan for technical assistance; Dr Ed Perez-Reyes (University of Virginia) for the rat α1G clone; and Drs Prakash Subramanyam and Xianghua Xu for comments on the manuscript. This work was supported by grants from the National Institutes of Health (RO1 HL069911 and RO1 HL084332) to H.M.C. H.M.C. is an Established Investigator of the American Heart Association.
Glossary
Abbreviations
- AID
α interaction domain
- BBS
bungarotoxin binding site
- CaV channel
voltage-dependent calcium channel
- CaVβ
voltage-dependent calcium channel auxiliary β subunit
- CaM
calmodulin
- CCD
charge-coupled device
- CFP
cyan fluorescent protein
- C-terminus
carboxyl terminus
- ER
endoplasmic reticulum
- GK
guanylate kinase
- ICa
calcium channel current
- PEER
putative endoplasmic reticulum export region
- QD
quantum dot
- Qmax
maximal gating charge
- YFP
yellow fluorescent protein
Author contributions
K.F. designed and conducted experiments in all areas of the work, analysed results, interpreted data, and helped write the paper. H.M.C. conceived of and designed experiments, analysed results, interpreted data, and wrote the paper. Both authors approved the final version of the manuscript.
Supplementary material
Figure S1
Figure S2
Figure S3
Figure S4
Figure S5
Figure S6
Table S1
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer-reviewed and may be re-organized for online delivery, but are not copy-edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors
References
- Altier C, Garcia-Caballero A, Simms B, You H, Chen L, Walcher J, Tedford HW, Hermosilla T, Zamponi GW. The Cavβ subunit prevents RFP2-mediated ubiquitination and proteasomal degradation of L-type channels. Nat Neurosci. 2011;14:173–180. doi: 10.1038/nn.2712. [DOI] [PubMed] [Google Scholar]
- Arias JM, Murbartian J, Vitko I, Lee JH, Perez-Reyes E. Transfer of β subunit regulation from high to low voltage-gated Ca2+ channels. FEBS Lett. 2005;579:3907–3912. doi: 10.1016/j.febslet.2005.06.008. [DOI] [PubMed] [Google Scholar]
- Ball SL, Powers PA, Shin HS, Morgans CW, Peachey NS, Gregg RG. Role of the β2 subunit of voltage-dependent calcium channels in the retinal outer plexiform layer. Invest Ophthalmol Vis Sci. 2002;43:1595–1603. [PubMed] [Google Scholar]
- Bers DM. Cardiac excitation-contraction coupling. Nature. 2002;415:198–205. doi: 10.1038/415198a. [DOI] [PubMed] [Google Scholar]
- Bichet D, Cornet V, Geib S, Carlier E, Volsen S, Hoshi T, Mori Y, De Waard M. The I-II loop of the Ca2+ channel α1 subunit contains an endoplasmic reticulum retention signal antagonized by the β subunit. Neuron. 2000;25:177–190. doi: 10.1016/s0896-6273(00)80881-8. [DOI] [PubMed] [Google Scholar]
- Bourdin B, Marger F, Wall-Lacelle S, Schneider T, Klein H, Sauve R, Parent L. Molecular determinants of the Cavβ-induced plasma membrane targeting of the Cav1.2 channel. J Biol Chem. 2010;285:22853–22863. doi: 10.1074/jbc.M110.111062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buraei Z, Yang J. The β subunit of voltage-gated Ca2+ channels. Physiol Rev. 2010;90:1461–1506. doi: 10.1152/physrev.00057.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess DL, Jones JM, Meisler MH, Noebels JL. Mutation of the Ca2+ channel β subunit gene Cchb4 is associated with ataxia and seizures in the lethargic (lh) mouse. Cell. 1997;88:385–392. doi: 10.1016/s0092-8674(00)81877-2. [DOI] [PubMed] [Google Scholar]
- Butcher AJ, Leroy J, Richards MW, Pratt WS, Dolphin AC. The importance of occupancy rather than affinity of Cavβ subunits for the calcium channel I–II linker in relation to calcium channel function. J Physiol. 2006;574:387–398. doi: 10.1113/jphysiol.2006.109744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canti C, Davies A, Berrow NS, Butcher AJ, Page KM, Dolphin AC. Evidence for two concentration-dependent processes for β-subunit effects on α1B calcium channels. Biophys J. 2001;81:1439–1451. doi: 10.1016/S0006-3495(01)75799-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catterall WA, Few AP. Calcium channel regulation and presynaptic plasticity. Neuron. 2008;59:882–901. doi: 10.1016/j.neuron.2008.09.005. [DOI] [PubMed] [Google Scholar]
- Chen YH, Li MH, Zhang Y, He LL, Yamada Y, Fitzmaurice A, Shen Y, Zhang H, Tong L, Yang J. Structural basis of the α1-β subunit interaction of voltage-gated Ca2+ channels. Nature. 2004;429:675–680. doi: 10.1038/nature02641. [DOI] [PubMed] [Google Scholar]
- Colecraft HM, Alseikhan B, Takahashi SX, Chaudhuri D, Mittman S, Yegnasubramanian V, Alvania RS, Johns DC, Marban E, Yue DT. Novel functional properties of Ca2+ channel β subunits revealed by their expression in adult rat heart cells. J Physiol. 2002;541:435–452. doi: 10.1113/jphysiol.2002.018515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornet V, Bichet D, Sandoz G, Marty I, Brocard J, Bourinet E, Mori Y, Villaz M, De Waard M. Multiple determinants in voltage-dependent P/Q calcium channels control their retention in the endoplasmic reticulum. Eur J Neurosci. 2002;16:883–895. doi: 10.1046/j.1460-9568.2002.02168.x. [DOI] [PubMed] [Google Scholar]
- De Waard M, Campbell KP. Subunit regulation of the neuronal α1A Ca2+ channel expressed in Xenopus oocytes. J Physiol. 1995;485:619–634. doi: 10.1113/jphysiol.1995.sp020757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deisseroth K, Mermelstein PG, Xia H, Tsien RW. Signaling from synapse to nucleus: the logic behind the mechanisms. Curr Opin Neurobiol. 2003;13:354–365. doi: 10.1016/s0959-4388(03)00076-x. [DOI] [PubMed] [Google Scholar]
- Dolphin AC. β subunits of voltage-gated calcium channels. J Bioenerg Biomembr. 2003;35:599–620. doi: 10.1023/b:jobb.0000008026.37790.5a. [DOI] [PubMed] [Google Scholar]
- Drummond GB. Reporting ethical matters in The Journal of Physiology: standards and advice. J Physiol. 2009;587:713–719. doi: 10.1113/jphysiol.2008.167387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erickson MG, Alseikhan BA, Peterson BZ, Yue DT. Preassociation of calmodulin with voltage-gated Ca2+ channels revealed by FRET in single living cells. Neuron. 2001;31:973–985. doi: 10.1016/s0896-6273(01)00438-x. [DOI] [PubMed] [Google Scholar]
- Erickson MG, Liang H, Mori MX, Yue DT. FRET two-hybrid mapping reveals function and location of L-type Ca2+ channel CaM preassociation. Neuron. 2003;39:97–107. doi: 10.1016/s0896-6273(03)00395-7. [DOI] [PubMed] [Google Scholar]
- Fakler B, Adelman JP. Control of KCa channels by calcium nano/microdomains. Neuron. 2008;59:873–881. doi: 10.1016/j.neuron.2008.09.001. [DOI] [PubMed] [Google Scholar]
- Fan M, Buraei Z, Luo HR, Levenson-Palmer R, Yang J. Direct inhibition of P/Q-type voltage-gated Ca2+ channels by Gem does not require a direct Gem/Cavβ interaction. Proc Natl Acad Sci U S A. 2010;107:14887–14892. doi: 10.1073/pnas.1007543107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Findeisen F, Minor DL., Jr Disruption of the IS6-AID linker affects voltage-gated calcium channel inactivation and facilitation. J Gen Physiol. 2009;133:327–343. doi: 10.1085/jgp.200810143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funke L, Dakoji S, Bredt DS. Membrane-associated guanylate kinases regulate adhesion and plasticity at cell junctions. Annu Rev Biochem. 2005;74:219–245. doi: 10.1146/annurev.biochem.74.082803.133339. [DOI] [PubMed] [Google Scholar]
- Gao T, Bunemann M, Gerhardstein BL, Ma H, Hosey MM. Role of the C terminus of the α1C (CaV1.2) subunit in membrane targeting of cardiac L-type calcium channels. J Biol Chem. 2000;275:25436–25444. doi: 10.1074/jbc.M003465200. [DOI] [PubMed] [Google Scholar]
- Gao T, Chien AJ, Hosey MM. Complexes of the α1C and β subunits generate the necessary signal for membrane targeting of class C L-type calcium channels. J Biol Chem. 1999;274:2137–2144. doi: 10.1074/jbc.274.4.2137. [DOI] [PubMed] [Google Scholar]
- Geib S, Sandoz G, Cornet V, Mabrouk K, Fund-Saunier O, Bichet D, Villaz M, Hoshi T, Sabatier JM, De Waard M. The interaction between the I-II loop and the III-IV loop of Cav2.1 contributes to voltage-dependent inactivation in a β-dependent manner. J Biol Chem. 2002;277:10003–10013. doi: 10.1074/jbc.M106231200. [DOI] [PubMed] [Google Scholar]
- Gregg RG, Messing A, Strube C, Beurg M, Moss R, Behan M, Sukhareva M, Haynes S, Powell JA, Coronado R, Powers PA. Absence of the β subunit (cchb1) of the skeletal muscle dihydropyridine receptor alters expression of the α1 subunit and eliminates excitation-contraction coupling. Proc Natl Acad Sci U S A. 1996;93:13961–13966. doi: 10.1073/pnas.93.24.13961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulme JT, Yarov-Yarovoy V, Lin TW, Scheuer T, Catterall WA. Autoinhibitory control of the CaV1.2 channel by its proteolytically processed distal C-terminal domain. J Physiol. 2006;576:87–102. doi: 10.1113/jphysiol.2006.111799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanevsky N, Dascal N. Regulation of maximal open probability is a separable function of Cavβ subunit in L-type Ca2+ channel, dependent on NH2 terminus of α1C (Cav1.2α) J Gen Physiol. 2006;128:15–36. doi: 10.1085/jgp.200609485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Ghosh S, Nunziato DA, Pitt GS. Identification of the components controlling inactivation of voltage-gated Ca2+ channels. Neuron. 2004;41:745–754. doi: 10.1016/s0896-6273(04)00081-9. [DOI] [PubMed] [Google Scholar]
- Lee A, Wong ST, Gallagher D, Li B, Storm DR, Scheuer T, Catterall WA. Ca2+/calmodulin binds to and modulates P/Q-type calcium channels. Nature. 1999;399:155–159. doi: 10.1038/20194. [DOI] [PubMed] [Google Scholar]
- Ma D, Zerangue N, Lin YF, Collins A, Yu M, Jan YN, Jan LY. Role of ER export signals in controlling surface potassium channel numbers. Science. 2001;291:316–319. doi: 10.1126/science.291.5502.316. [DOI] [PubMed] [Google Scholar]
- Ma D, Zerangue N, Raab-Graham K, Fried SR, Jan YN, Jan LY. Diverse trafficking patterns due to multiple traffic motifs in G protein-activated inwardly rectifying potassium channels from brain and heart. Neuron. 2002;33:715–729. doi: 10.1016/s0896-6273(02)00614-1. [DOI] [PubMed] [Google Scholar]
- Mikosch M, Homann U. How do ER export motifs work on ion channel trafficking? Curr Opin Plant Biol. 2009;12:685–689. doi: 10.1016/j.pbi.2009.09.020. [DOI] [PubMed] [Google Scholar]
- Neely A, Wei X, Olcese R, Birnbaumer L, Stefani E. Potentiation by the β subunit of the ratio of the ionic current to the charge movement in the cardiac calcium channel. Science. 1993;262:575–578. doi: 10.1126/science.8211185. [DOI] [PubMed] [Google Scholar]
- Obermair GJ, Schlick B, Di Biase V, Subramanyam P, Gebhart M, Baumgartner S, Flucher BE. Reciprocal interactions regulate targeting of calcium channel β subunits and membrane expression of α1 subunits in cultured hippocampal neurons. J Biol Chem. 2010;285:5776–5791. doi: 10.1074/jbc.M109.044271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opatowsky Y, Chen CC, Campbell KP, Hirsch JA. Structural analysis of the voltage-dependent calcium channel β subunit functional core and its complex with the α1 interaction domain. Neuron. 2004;42:387–399. doi: 10.1016/s0896-6273(04)00250-8. [DOI] [PubMed] [Google Scholar]
- Opatowsky Y, Chomsky-Hecht O, Kang MG, Campbell KP, Hirsch JA. The voltage-dependent calcium channel β subunit contains two stable interacting domains. J Biol Chem. 2003;278:52323–52332. doi: 10.1074/jbc.M303564200. [DOI] [PubMed] [Google Scholar]
- Perez-Reyes E, Castellano A, Kim HS, Bertrand P, Baggstrom E, Lacerda AE, Wei XY, Birnbaumer L. Cloning and expression of a cardiac/brain β subunit of the L-type calcium channel. J Biol Chem. 1992;267:1792–1797. [PubMed] [Google Scholar]
- Perez-Reyes E, Cribbs LL, Daud A, Lacerda AE, Barclay J, Williamson MP, Fox M, Rees M, Lee JH. Molecular characterization of a neuronal low-voltage-activated T-type calcium channel. Nature. 1998;391:896–900. doi: 10.1038/36110. [DOI] [PubMed] [Google Scholar]
- Peterson BZ, DeMaria CD, Adelman JP, Yue DT. Calmodulin is the Ca2+ sensor for Ca2+-dependent inactivation of L-type calcium channels. Neuron. 1999;22:549–558. doi: 10.1016/s0896-6273(00)80709-6. [DOI] [PubMed] [Google Scholar]
- Pitt GS, Zuhlke RD, Hudmon A, Schulman H, Reuter H, Tsien RW. Molecular basis of calmodulin tethering and Ca2+-dependent inactivation of L-type Ca2+ channels. J Biol Chem. 2001;276:30794–30802. doi: 10.1074/jbc.M104959200. [DOI] [PubMed] [Google Scholar]
- Pragnell M, De Waard M, Mori Y, Tanabe T, Snutch TP, Campbell KP. Calcium channel β-subunit binds to a conserved motif in the I-II cytoplasmic linker of the α1-subunit. Nature. 1994;368:67–70. doi: 10.1038/368067a0. [DOI] [PubMed] [Google Scholar]
- Restituito S, Cens T, Barrere C, Geib S, Galas S, De Waard M, Charnet P. The β2a subunit is a molecular groom for the Ca2+ channel inactivation gate. J Neurosci. 2000;20:9046–9052. doi: 10.1523/JNEUROSCI.20-24-09046.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romanin C, Gamsjaeger R, Kahr H, Schaufler D, Carlson O, Abernethy DR, Soldatov NM. Ca2+ sensors of L-type Ca2+ channel. FEBS Lett. 2000;487:301–306. doi: 10.1016/s0014-5793(00)02361-9. [DOI] [PubMed] [Google Scholar]
- Sekine-Aizawa Y, Huganir RL. Imaging of receptor trafficking by using α-bungarotoxin-binding-site-tagged receptors. Proc Natl Acad Sci U S A. 2004;101:17114–17119. doi: 10.1073/pnas.0407563101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi SX, Miriyala J, Colecraft HM. Membrane-associated guanylate kinase-like properties of β-subunits required for modulation of voltage-dependent Ca2+ channels. Proc Natl Acad Sci U S A. 2004;101:7193–7198. doi: 10.1073/pnas.0306665101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Petegem F, Chatelain FC, Minor DL., Jr Insights into voltage-gated calcium channel regulation from the structure of the CaV1.2 IQ domain-Ca2+/calmodulin complex. Nat Struct Mol Biol. 2005;12:1108–1115. doi: 10.1038/nsmb1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Petegem F, Clark KA, Chatelain FC, Minor DL., Jr Structure of a complex between a voltage-gated calcium channel β-subunit and an α-subunit domain. Nature. 2004;429:671–675. doi: 10.1038/nature02588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Petegem F, Duderstadt KE, Clark KA, Wang M, Minor DL., Jr Alanine-scanning mutagenesis defines a conserved energetic hotspot in the CaVα1 AID-CaVβ interaction site that is critical for channel modulation. Structure. 2008;16:280–294. doi: 10.1016/j.str.2007.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitko I, Shcheglovitov A, Baumgart JP, Arias O, II, Murbartian J, Arias JM, Perez-Reyes E. Orientation of the calcium channel b relative to the α12.2 subunit is critical for its regulation of channel activity. PLoS ONE. 2008;3:e3560. doi: 10.1371/journal.pone.0003560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waithe D, Ferron L, Page KM, Chaggar K, Dolphin AC. β-subunits promote the expression of CaV2.2 channels by reducing their proteasomal degradation. J Biol Chem. 2011;286:9598–9611. doi: 10.1074/jbc.M110.195909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HG, George MS, Kim J, Wang C, Pitt GS. Ca2+/calmodulin regulates trafficking of CaV1.2 Ca2+ channels in cultured hippocampal neurons. J Neurosci. 2007;27:9086–9093. doi: 10.1523/JNEUROSCI.1720-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei X, Neely A, Lacerda AE, Olcese R, Stefani E, Perez-Reyes E, Birnbaumer L. Modification of Ca2+ channel activity by deletions at the carboxyl terminus of the cardiac α1 subunit. J Biol Chem. 1994;269:1635–1640. [PubMed] [Google Scholar]
- Yang T, Xu X, Kernan T, Wu V, Colecraft HM. Rem, a member of the RGK GTPases, inhibits recombinant CaV1.2 channels using multiple mechanisms that require distinct conformations of the GTPase. J Physiol. 2010;588:1665–1681. doi: 10.1113/jphysiol.2010.187203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuhlke RD, Pitt GS, Deisseroth K, Tsien RW, Reuter H. Calmodulin supports both inactivation and facilitation of L-type calcium channels. Nature. 1999;399:159–162. doi: 10.1038/20200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.