

Abstract
Injectable thermoresponsive hydrogels are of interest for a variety of biomedical applications, including regional tissue mechanical support as well as drug and cell delivery. Within this class of materials there is a need to provide options for gels with stronger mechanical properties as well as variable degradation profiles. To address this need, the hydrolytically labile monomer, methacrylate-polylactide (MAPLA), with an average 2.8 lactic acid units, was synthesized and copolymerized with N-isopropylacrylamide (NIPAAm) and 2-hydroxyethyl methacrylate (HEMA) to obtain bioabsorbable thermally responsive hydrogels. Poly(NIPAAm-co-HEMA-co-MAPLA) with three monomer feed ratios (84/10/6, 82/10/8 and 80/10/10) was synthesized and characterized with NMR, FTIR and GPC. The copolymers were soluble in saline at reduced temperature (<10°C), forming clear solutions that increased in viscosity with the MAPLA feed ratio. The copolymers underwent sol-gel transition at lower critical solution temperatures of 12.4, 14.0 and 16.2°C respectively and solidified immediately upon being placed in a 37°C water bath. The warmed hydrogels gradually excluded water to reach final water contents of ~45%. The hydrogels as formed were mechanically strong, with tensile strengths as high as 100 kPa and shear moduli of 60 kPa. All three hydrogels were completely degraded (solubilized) in PBS over a 6–8 month period at 37°C, with a higher MAPLA feed ratio resulting in a faster degradation period. Culture of primary vascular smooth muscle cells with degradation solutions demonstrated a lack of cytotoxicity. The synthesized hydrogels provide new options for biomaterial injection therapy where increased mechanical strength and relatively slow resorption rates would be attractive.
Keywords: thermally responsive material, hydrogel, biodegradation, cytotoxicity, cardiac tissue engineering
Introduction
Injectable thermally responsive hydrogels with a lower critical solution temperature (LCST) below body temperature represent promising biomaterials for a variety of biomedical applications, including regional tissue mechanical support as well as drug and cell delivery applications. 1–4 Generally, the LCST-based phase transition occurs upon warming in situ as a result of entropically-driven dehydration of polymer components, leading to polymer collapse. 5 Various naturally derived and synthetic polymers exhibiting this behavior have been utilized. Natural polymers include elastin-like peptides 6 and polysaccharide derivatives 3, while notable synthetic polymers include those based on poly(N-isopropylacrylamide) (PNIPAAm), 7 and amphiphilic block copolymers, often containing poly(ethylene glycol) 3,8. The structure of PNIPAAm, containing both hydrophilic amide bonds and hydrophobic isopropyl groups, leads to a sharp phase transition at the LCST. 9 Studies suggest that the average number of hydrating water molecules per NIPAAm group falls from 11 to ~2 upon the hydrophobic collapse above the LCST (32°C). 5,9,10.
PNIPAAm-based polymers have been extensively studied as injectable biomaterials for tissue regeneration and drug delivery 11–20, yet PNIPAAm homopolymer is nondegradable due to a constant LCST, which prevents its clearance from the body at physiologic temperature. This limitation of PNIPAAm has provided the motivation for developing biodegradable NIPAAm-based polymers by conjugating the PNIPAAm with natural biodegradable segments such as MMP-susceptible peptide, 14 gelatin, 21 collagen, 22 hyaluronic acid 23,24 and dextran 25. However, these may be only partially bioabsorbable since sufficiently long PNIPAAm segments would remain non-soluble following removal of the natural segments. Another disadvantage of these materials is that the natural polymers often function as crosslinking agents and only a small range of low crosslinking density can be used before the systems are no longer injectable.
Copolymers formed from NIPAAm and monomers with degradable side chains comprise another category of NIPAAm-based bioabsorbable, thermally responsive hydrogels. Here, hydrolytic removal of hydrophobic side chains increases the hydrophilicity of the copolymer, raising the LCST above body temperature and making the polymer backbone soluble.26 Also, NIPAAm-based copolymers with enzyme-susceptible peptide side chains could be soluble following removal of the peptide side chains.27 Due to the relative simplicity of the synthetic process, the most investigated biodegradable monomers have been HEMA-based monomers, such as 2-hydroxyethyl methacrylate-polylactide (HEMA-PLA), 18–20 2-hydroxyethyl methacrylate-polycaprolactone (HEMA-PCL), 25,28 and 2-hydroxyethyl methacrylate-polytrimethylene carbonate (HEMA-PTMC), 11,12. However, the backbone remnant following hydrolysis, HEMA, presents hydroxyethyl side groups (-CH2CH2-OH), which have a relatively limited effect on remnant polymer hydrophilicity. 17 In previous studies, such hydrogels have been found to be either partially bioabsorbable, 28 or completely bioabsorbable, but have required the inclusion of considerably hydrophilic co-monomers such as acrylic acid (AAc) in the hydrogel synthesis.11,12,20
To overcome the limitations associated with previous NIPAAm copolymers utilizing HEMA-based co-monomers for hydrolytic lability, our objective was to synthesize a non-HEMA-based biodegradable monomer, methacrylate-polylactide (MAPLA), which would present highly hydrophilic carboxylate groups upon hydrolysis of the PLA segments. The MAPLA monomer was then used as the basis for the development of bioabsorbable and thermally responsive NIPAAm-based copolymer hydrogels. Copolymers were synthesized using monomer NIPAAm, HEMA and MAPLA at three different monomer ratios. The resulting hydrogels were characterized in terms of their composition and thermal, mechanical, hydrolytic and cytocompatibility properties.
Materials and Methods
Materials
All chemicals were purchased from Sigma-Aldrich unless otherwise stated. NIPAAm was purified by recrystallization from hexane and vacuum dried. 2-hydroxyethyl methacrylate (HEMA) was purified by vacuum distillation. Lactide was purified by recrystallization from ethyl acetate. Benzoyl peroxide (BPO), sodium methoxide (NaOCH3) and methacryloyl chloride were used as received.
Synthesis of methacrylate polylactide (MAPLA)
As shown in Scheme 1, polylactide (HO-PLA-OCH3) was synthesized by NaOCH3 initiated ring opening polymerization of lactide. Lactide (100g, 0.694mol) was dissolved in 150 mL dichloromethane, to which a NaOCH3 solution containing 2g NaOCH3 (0.037mol) in 20mL methanol (0.494 mol) was added under vigorous stirring. The reaction proceeded for 2 h at 0°C before the solution was sequentially rinsed with 0.1M HCl and deionized (DI) water. The organic phase was isolated by centrifugation and dried over anhydrous MgSO4. The solvent (dichloromethane) was removed by rotary evaporation at 60°C to obtain ~90g HO-PLA-OCH3. Biodegradable monomer MAPLA was synthesized by dropping 45ml (0.47mol) methacryloyl chloride into the HO-PLA-OCH3 (90g, 0.39mol) solution in 150 mL dichloromethane containing 65mL (0.47 mol) triethylamine. After reacting at 0°C overnight, the solution was filtered to remove precipitate, and was then rinsed sequentially with 0.2M Na2CO3, 0.1M HCl and DI water. The organic phase was isolated by centrifugation and dried over anhydrous MgSO4. The solvent (dichloromethane) was removed by rotary evaporation at 40°C to get the raw product of MAPLA, which was finally purified by flash chromatography, with yields of 60%.
Synthesis of poly(NIPAAm-co-HEMA-co-MAPLA)
Poly(NIPAAm-co-HEMA-co-MAPLA) copolymers were synthesized by free radical polymerization (Scheme 1). Monomers, NIPAAm (6g, 0.053mol), HEMA and MAPLA, with certain molar ratios (80/10/10, 82/10/8 and 84/10/6) were dissolved in 150mL 1,4-dioxane containing 150 mg BPO. The polymerization was carried out at 70°C for 24 h under argon atmosphere. The copolymer was precipitated in hexane and further purified by precipitation from THF into diethyl ether and vacuum dried, with yields of ~90%.
Characterization
1H-NMR and 13C-NMR spectra of MAPLA and the poly(NIPAAm-co-HEMA-co-MAPLA) copolymers were recorded with a 300 MHz BRUKER spectrometer using CD3Cl or DMSO-d6 as a solvent. The mass spectrum of MAPLA was recorded on an HP 1100 Series LC/MSD instrument with an API-ES positive ionization method. Fourier transform infrared (FTIR) spectra of the copolymers were obtained with a Nicolet FTIR spectrometer, with samples prepared by coating a 1 % copolymer solution onto a NaCl window. Molecular weight of the copolymers was determined by gel permeation chromatography (GPC, Waters Breeze System, Waters 1515 HPLC Pump, Waters 2414 differential refractometer). The copolymers were dissolved in THF at a concentration of 1 mg/mL and the GPC analyses were performed at 35°C. A poly(methyl methacrylate) standard kit (Fluka, ReadyCal Set Mp 500–2,700,000) was used for molecular weight-elution volume calibration.
LCSTs of the copolymer solutions in PBS (16.7 wt% and 0.3 wt%, pH 7) were studied by measuring optical absorption at 500 nm over a temperature range of 0 to 25°C. The LCST of each copolymer was determined (n=4) by determining the temperature at which the absorbance of the copolymer solution reached half of its maximal value, during the phase transition. Differential scanning calorimetry (DSC-60; Shimadzu) was also used to characterize the LCST behavior of the copolymer solutions (16.7 wt% in PBS) with a scanning rate of 5°C/min over a range of 0 to 40°C. The temperature at the maximum of the endothermal peak was recorded as the LCST 29.
Rheology studies were conducted on a TA Instrument rheometer (AR2000). Copolymer solution viscosities at 10 °C were measured with a shear rate sweep (0–10 sec−1). To observe the mechanical property change of the hydrogels during the temperature induced sol-gel transition, the polymer solutions (16.7 wt% in PBS) were placed between two parallel plates. With temperature sweep from 10 to 45°C and a heating rate of 4°C/min, the shear storage modulus G′ and the loss modulus G″ were collected as a function of temperature at a fixed strain of 2% and a frequency of 1 Hz.
To measure the mechanical properties of the hydrogels, samples were incubated in a 37°C water bath for 24 h to reach stable water contents, and then the solid hydrogels were placed between two parallel plates and the G′ and G″ were measured at 37°C with a fixed strain of 5% and a frequency sweep from 0.1 to 2 rad/s. For tensile testing of the hydrogels, samples (n=4 each) were cut into rectangular strips 1 mm thick, 4 mm wide and 25 mm long, and then loaded in a water bath test cell equilibrated to 37±2°C with preheated grips attached to each sample end. An ATS 1101 Universal Testing Machine equipped with a 10 lb load cell was utilized with a cross-head speed of 6 cm/min.
The gelation speed of the copolymer solutions (PBS, 16.7 wt%) was measured by incubating 3 mL glass vials containing ~1 mL hydrogel solution in a 37°C water bath. The water content of the hydrogel at different incubation time points was measured over 24 h. Water content was defined as (w2−w1)/w2 × 100%, where w2 and w1 are wet mass and dry mass of the hydrogel, respectively. The microstructures of the (80/10/10) hydrogel at the beginning of the gelation process (30 sec) and after 24 hr incubation at 37°C were imaged by quenching the hydrogels in liquid nitrogen followed by freeze drying, gold sputtering and scanning electron microscopic observation (SEM JEM-1011, JEOL).
Hydrogel degradation was quantified by mass loss measurements. Hydrogels with known initial dry masses (~60 mg) were immersed into 20 mL PBS (replaced biweekly to maintain a constant pH value of 7) at 37°C. At pre-defined time points over an 8 month period the hydrogels (n=3 each) were lyophilized and the relative mass loss recorded.
Cytotoxicity assay
The cytotoxicity of the hydrogel degradation products was assessed by measuring the relative metabolic viability of cells cultured with medium supplemented with degradation products, as previously described 11,20,30. The hydrogel degradation solution was prepared by hydrolysis of the hydrogel in 1.0 M NaOH, followed by removal of NaOH using a cationic ion-exchange resin (Amberlite IR-120H, Aldrich) and supplementing with a 10× EMEM culture medium (BioWhittaker, Lonza) at a volume ratio of 1:9 with respect to the hydrogel degradation solution. Rat vascular smooth muscle cells (RSMCs) were isolated according to ref. 31. These cells were chosen in consideration of potential cardiovascular applications of the hydrogels and in lieu of adult cardiomyocytes, which are less amenable to isolation and culture for first level cytotoxicity screening. Cells were cultured in Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS) to the fifth passage and seeded into a 24-well tissue culture plate at a seeding density of 15,000/well. The hydrogel degradation supplemented with EMEM was added into each well to obtain a final concentration of 5 mg/mL. Culture medium without added degradation solution was used as a control. Cell metabolic activity was measured (n=4 each) using an MTS assay kit (Promega CellTiter 96® Cell Proliferation Assay) to quantify mitochondrial activity. To qualitatively verify the results of the above test, cells were also observed under fluorescence microscopy after live/dead staining with a Promokine® Live/Dead Cell Staining Kit.
Statistics
Data are expressed as means with the standard deviation. Analyses utilized SPSS software (SPSS Inc, Chicago IL). Statistical analyses were performed by one-way ANOVA followed by Tukey’s post-hoc testing. Statistical significance was considered to exist at p<0.05.
Results
Synthesis
The synthesis of MAPLA was confirmed by 1H-NMR (Figure 1a) and 13C-NMR spectra (Figure 1b) which contained proton peaks and carbon peaks in agreement with the molecular structure of MAPLA. The chemical structure of MAPLA was also confirmed by the mass spectrum (API-ES positive ionization). Peaks at 267.0 (MAPLA2+Na+), 339.0 ((MAPLA3+Na+), 411.0 (MAPLA4+Na+), 483.0 (MAPLA5+Na+) and 555.2 (MAPLA6+Na+) were observed, indicating that the product was a mixture of molecules containing different PLA lengths. The number average length of PLA units per monomer was determined from 1H-NMR spectrum (Figure 1a) as 2.8 by calculation from the ratio of the integrals of hydrogen peaks from PLA (peaks c) and the peaks from the hydrogens bordering the double bond (CH2=) (peaks a and b).
Figure 1.
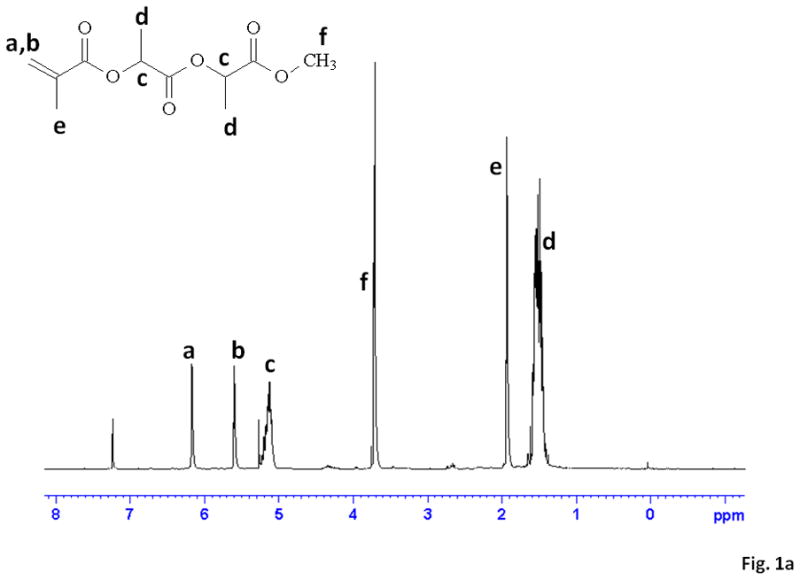

(a) 1H-NMR and (b) 13C-NMR spectra for MAPLA.
Copolymers with different monomer ratios were prepared by free radical polymerization. Table 1 summarizes poly(NIPAAm-co-HEMA-co-MAPLA) copolymers synthesized with three different MAPLA feed ratios. All of the copolymers had number average molecular weights between 20 and 30k, and a polydispersity index of 1.5–2.0. FTIR spectra of the copolymer (Figure 2) exhibited an amide I peak at 1550 and an amide II peak at 1650 cm−1 characteristic of NIPAAm and also a strong peak at 1730 cm−1 for the -C=O of the ester groups from HEMA and MAPLA. Figure 3 shows typical 1H-NMR and 13C-NMR spectra for a synthesized copolymer. Proton and carbon peaks characteristic of NIPAAm and MAPLA are seen and the ratio of MAPLA to NIPAAm in the copolymer was obtained by peak area integration of these characteristic peaks at 5.1 ppm and 3.8 ppm, respectively (Table 1). The MAPLA monomer ratios in the copolymers were found to be lower than their feed ratios, indicating that the polymerization rate of MAPLA was lower than that for NIPAAm under the polymerization conditions evaluated.
Table 1.
Poly(NIPAAm-co-HEMA-co-MAPLA) copolymers with different monomer feed ratios.
| Monomer Feed ratio NIPAAm/HEMA/MAPLA | Mn | Mw/Mn | MAPLA/NIPAAm ratio in Polymer (%) | LCST (°C) 16.7 wt% in PBS | LCST (°C) 0.3 wt% in PBS | Hydrogel Water content (%)at 37°C in PBS |
|---|---|---|---|---|---|---|
| 84/10/6 | 22K | 1.5 | 4.3 | 16.2±0.2* | 22.1±0.2* | 42±0.3 |
| 82/10/8 | 25K | 1.5 | 5.0 | 14.0±0.2* | 21.4±0.2* | 47±0.3 |
| 80/10/10 | 22K | 1.6 | 6.1 | 12.4±0.4* | 20.0±0.3* | 44±0.3 |
By optical absorption analysis, p<0.001 versus each of other copolymers.
Figure 3.


(a) 1H-NMR and (b) 13C-NMR spectra for poly(NIPAAm-co-HEMA-co-MAPLA) (80/10/10).
Gelation process
The phase transition behavior at the LCST was examined through real-time screening of the optical, mechanical and thermal properties of the hydrogel solutions (16.7 wt% in PBS) during temperature changes. Representative optical absorption curves of the hydrogel solutions (80/10/10, 82/10/8 and 84/10/6) for both 16.7 and 0.3 wt% solutions are shown in Figure 4a and the calculated LCSTs are summarized in Table 1. Hydrogel LCSTs decreased with increasing MAPLA feed ratios. The same phenomenon and relative order of response was also observed by measuring the mechanical property (G′ and G″) change of the hydrogels with increasing temperature, with typical curves shown in Figure 4b. While G″ values exceeded G′ values, the sol-gel transition was defined based on the abrupt change of several physical properties (optical, mechanical and thermal). Typical DSC curves (Figure 4c) of hydrogel solutions (16.7 wt% in PBS) showed broad but obvious endothermal peaks at 21–22°C, corresponding to the LCSTs, but the relative order of these curves varied and responses between the hydrogels were not consistent. DSC analysis of hydrogel solutions at a lower concentration (0.3 wt% in PBS) was also conducted. However, no endothermal peaks could be identified due to the low signal strength of the heat absorption of the samples (data not shown).
Figure 4.


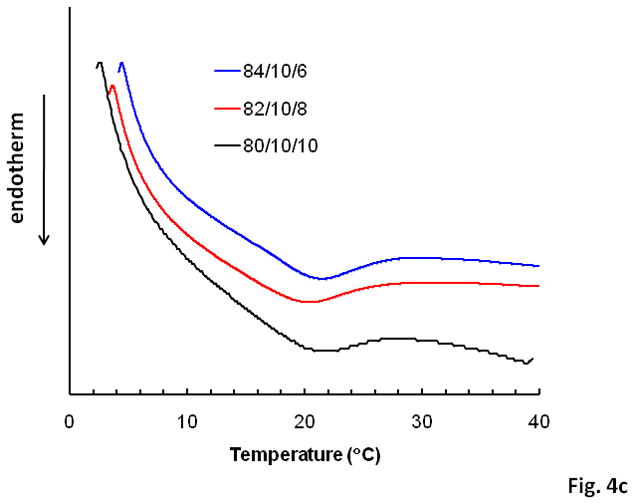
LCST determination by (a) measurement of copolymer solution optical absorption; (b) measurement of shear modulus on a rheometer, 1 Hz, 2% strain; (c) DSC analysis, 5°C/min.
A macroscopic perspective of the poly(NIPAAm-co-HEMA-co-MAPLA) (80/10/10) gelation process is presented in Figure 5. Immediate sol-gel transition occurred upon the incubation of the solution into a 37°C water bath within 30 sec (Figure 5). After that, the hydrogel continued to exclude water gradually to reach a stable size after several hr. The final equilibrated water contents of the hydrogels were measured at ~45% after 24 hr (Table 1). The relative speed of water exclusion during the gelation process is evident by plotting the water content of the hydrogels against the time of the incubation at 37°C, as shown in Figure 6. Microstructures of the (80/10/10) hydrogel at the beginning of the gelation process (30 sec) and after 24 hr incubation at 37°C are shown in Figure 5. The loss of microporosity is evident at the later time point.
Figure 5.

Gelation process of the poly(NIPAAm-co-HEMA-co-MAPLA) (80/10/10) hydrogel (PBS, 16.7 wt %) and the microstructure of the hydrogel formed at 37°C after 30 sec and 1 day. Samples were quenched with liquid nitrogen and freeze dried at −40°C.
Figure 6.

Gelation of the poly(NIPAAm-co-HEMA-co-MAPLA) hydrogels (PBS, 16.7 wt %). Water contents were plotted against the incubation time of the hydrogels in a 37°C water bath.
By decreasing the temperature below the LCSTs, the gelation process of the thermoresponsive hydrogels could be reversed and the solidified hydrogels could be re-dissolved to form solutions again. The speed of this gel-sol process depended on the degree of the dehydration (water exclusion) of the hydrogel, which depended on the time spent above the LCST. For a hydrogel already dehydrated to its stable water content (~45% at 37°C), the re-dissolving process at 5°C required more than 10 hr, while for a hydrogel which had formed for less than 1 min at 37°C, re-dissolving the hydrogel at 5°C was complete within 5 min.
Mechanical properties
At a temperature below the hydogels’ LCST (10 °C), the clear, viscous solutions were observed, with the solution viscosity increasing with the MAPLA feed ratio, as shown in Figure 7. Since the copolymers all had similar molecular weights (Table 1), the viscosity differences were attributed to the varying hydrophobicity in the different samples, with increased viscosity driven by increased intermolecular hydrophobic interactions. Figure 7 also indicates that the viscosities were more shear rate-dependent with increased MAPLA feed ratio.
Figure 7.

Viscosity of the poly(NIPAAm-co-HEMA-co-MAPLA) hydrogel solutions (PBS, 16.7 wt %) at 10°C.
The final equilibrated hydrogels incubated in PBS at 37°C after 24 hr were highly flexible gum-like materials with plastic deformation occurring beyond the maximum tensile strength at approximately 60–100% strain, as shown in the tensile curves in Figure 8. The dynamic shear modulus of the hydrogels at different angular frequencies is shown in Figure 9. Both the tensile and the shear modulus test showed that the hydrogels’ stiffness increased with the MAPLA feed ratio in the copolymer.
Figure 8.

Tensile curves (a) and maximum tensile strength (b) of the poly(NIPAAm-co-HEMA-co-MAPLA) hydrogels at 37°C. Hydrogels were formed in 37°C water bath for 24h.
Figure 9.

Dynamic shear modulus of the poly(NIPAAm-co-HEMA-co-MAPLA) hydrogels at 37°C. Strain, 5%. Hydrogels were formed in a 37°C water bath for 24 hrs.
Degradation
The in vitro degradation properties of the hydrogels with different MAPLA feed ratios were first evaluated by hydrolysis in NaOH (1M) at room temperature for 1 day to theoretically cleave the PLA residues, representing extensive degradation of the copolymer. The H1-NMR spectrum of the hydrolyzed copolymer did not show the peak characteristic of PLA at 5.1 ppm which was found in the NMR spectrum before degradation (Figure 10). The hydrolyzed copolymers gave clear solutions at 37°C, demonstrating that the LCSTs were well above 37°C. A second evaluation of in vitro degradation of the hydrogel was performed in PBS at 37°C with the resulting mass loss curve shown in Figure 11. The hydrogels were gradually solubilized at a much lower rate than in the NaOH solution. For all the three hydrogels, mass loss at 100 days was only about 20%, followed by an accelerated mass reduction during the next 100 days before complete dissolution of the hydrogels in PBS.
Figure 10.

(a) 1H-NMR of poly(NIPAAm-co-HEMA-co-MAPLA) (80/10/10) before (a) and after (b) the removal of PLA by hydrolysis in 1M NaOH.
Figure 11.

Mass loss curves of poly(NIPAAm-co-HEMA-co-MAPLA) hydrogels in PBS at 37°C.
Cytotoxicity
With RSMC mitochondrial activity serving as an indirect index for cell viability, Figure 12 demonstrates a lack of toxic effect of medium containing degradation and solubilized products on RSMC culture. This result was further verified by fluorescent live/dead staining of RSMC cultured under control or degradation product-containing culture medium (Figure 13). For both culture media, dead cells (stained red) were seen in low numbers and no difference was found in the relative number of dead cells viewed over several culture wells.
Figure 12.

Cytotoxity assay of poly(NIPAAm-co-HEMA-co-MAPLA) (80/10/10) hydrogel degradation products by cell metabolic activity assessment (MTS assay) for RSMCs cultured on tissue culture polystyrene. Hydrolyzed hydrogel solution was supplemented into cell culture medium at a final concentration of 5.0 mg/mL.
Figure 13.
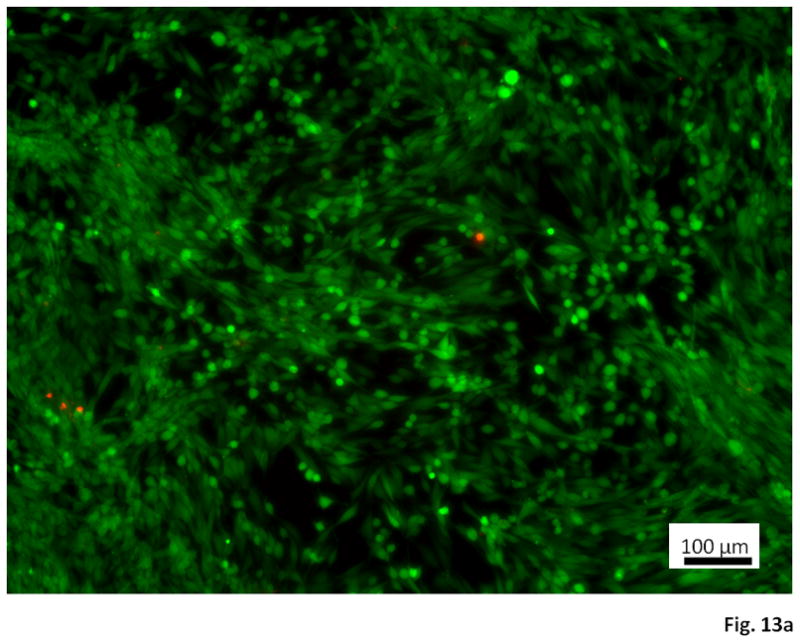

Live/dead staining of RSMC cultured on tissue culture polystyrene using (a) untreated culture medium and (b) culture medium containing 5.0 mg/mL hydrolyzed poly(NIPAAm-co-HEMA-co-MAPLA) (80/10/10). Observations were recorded at a culture time of 3 days. Scale bar: 100 μm.
Discussion
NIPAAm based thermally responsive and bioabsorbable hydrogels have been extensively investigated as injectable materials for applications in regenerative medicine including as temporary matrices to facilitate localized cell and drug delivery 1–3,32–35. Injectable biomaterials are also increasingly of interest as bulking agents to provide mechanical support in failing, traumatized and diseased tissues, such as urinary sphincters for urinary incontinence,36,37 nucleus pulposus for degenerative disc disease,38,39 and the cardiac wall for ischemic cardiomyopathy4. For mechanical support applications, fewer investigations have been performed evaluating NIPAAm-based materials. One area where there has been notable recent interest has been in cardiac wall injection therapy. 11, 25 Although naturally derived materials including alginate,40 fibrin,41,42 alginate-fibrin composites,43 collagen,42 chitosan44 and self-assembling peptides45,46 have been tried with different degrees of beneficial effects observed, synthetic injectable hydrogels have the advantage of being well defined in chemical structure and thus amenable to manipulation on the molecular level to achieve desired properties such as a increased mechanical strength and a targeted degradation profile. Appropriate control of these functional parameters may result in improved outcomes in the post-infarction cardiac wall remodeling process.
One of our previously developed thermoresponsive and biodegradable hydrogels, poly(NIPAAm-co-AAc-co-Acryloxysuccinimide-co-HEMAPLA),20 was found degrade too fast (2 days in vivo), making it non-ideal for in vivo application. A more recently developed hydrogel poly(NIPAAm-co-AAc-co-HEMAPTMC) showed much slower degradation process (80% mass loss in 4 month in PBS) and was demonstrated to produce beneficial effect in altering the post-infarction remodeling and heart failure process in a rat model.11 A drawback of this hydrogel, however, is that it has a pH-sensitive mechanical strength. Although the hydrogel has a tensile strength of 6 kPa at pH 3.6, at physiologic neutral pH value (7.4) the hydrogel is weakened. The mechanism for this weakening is the deprotonation of the AAc carboxyl groups, resulting in increased hydration at neutral pH, and weakening of the hydrophobic interactions between polymer chains that contributed to the mechanical strength. Utilization of the monomer AAc, however, is necessary for the hydrogel to be bio-absorbable. Without AAc to increase polymer hydrophilicity, cleavage of the PTMC residues to reveal HEMA does not increase the copolymer’s hydrophilicity adequately to make the copolymer soluble at 37°C,11 because there are no charged residues produced on the polymer chains upon the hydrolysis of the degradable side chains of the HEMA-based biodegradable monomers. 17
In this work, a novel biodegradable monomer, MAPLA, was designed and synthesized (Scheme 1). This monomer generates highly hydrophilic carboxylate groups upon hydrolytic cleavage of the PLA residues, making the copolymer adequately hydrophilic to allow dissolution with appropriate co-monomers other than AAc. Random copolymers of poly(NIPAAm-co-HEMA-co-MAPLA) formed thermoresponsive biodegradable hydrogels of relatively high mechanical strength at neutral pH (Figure 8). This polymer design strategy is somewhat similar to that reported by Cui et al., who used dimethyl-gamma-butyrolactone acrylate (DBA) as a biodegradable monomer and formed a copolymer, poly(NIPAAm-co-DBA) with an LCST well above the body temperature upon hydrolytic opening of the ring structure of DBA to generate carboxylate groups. 17
The ratio of the biodegradable monomer MAPLA in the hydrogel needs to be controlled for the hydrogel to be both bioabsorbable and injectable. First, the MAPLA content must be high enough in the copolymer so that there can be enough carboxylate groups produced after cleavage of the PLA residues to achieve copolymer solubilization at 37°C. On the other hand, since MAPLA is a hydrophobic monomer, too much MAPLA content before cleavage will make the copolymer insoluble in aqueous solution, even at low temperature. This effect is also seen in the increase in the viscosity of the copolymer solution with increasing MAPLA content (Figure 7). A high viscosity will make hydrogel injection difficult, particularly in the case of cardiac injection therapy where very small diameter needles (23G or smaller) are desirable for injection to minimize tissue trauma and bleeding risk. Furthermore, if catheter-based delivery is to be considered, viscosity limits become of increasing concern, as does heating of the delivery pathway. In results not presented, it was found that if the feed ratio of MAPLA was 12%, the copolymer (78/10/12) could not be completely solubilized at 5°C and the resulting viscous cloudy mixture was not able to be injected through a 23G needle. Therefore, in this work poly(NIPAAm-co-HEMA-co-MAPLA) with monomer ratios of 84/10/6, 82/10/8 and 80/10/10 were synthesized and characterized.
All three of the synthesized copolymers readily formed solutions in PBS at 5°C and formed opaque solid hydrogels when the temperature was increased above 37°C. The LCST of the hydrogels decreased with MAPLA content due to the enhanced hydrophobicity, as shown by optical analysis (Figure 4a). The LCST measured by optical analysis was also found to be polymer concentration-dependant (Figure 4a). As summarized in Table 1, the LCSTs of the copolymer solutions with a concentration of 0.3 wt % were about 6–8° higher than those with a concentration of 16.7 wt%. Enhanced intermolecular hydrophobic interactions in a higher concentration hydrogel solution may facilitate aggregation of the molecular chains and formation of light scattering micelles at a lower temperature. Mechanical property analysis also showed that the LCST of the hydrogels increased with MAPLA content (Figure 4b). This trend, however, was not observed in the DSC analysis (Figure 4c). It is noteworthy that LCSTs measured by different methods can vary due to differing definitions of the LCST. The LCSTs measured by mechanical property change (16, 20, 22°C as shown in Figure 4b) were higher than those measured optically (as shown Table 1), while the DSC curves of the three hydrogels showed broad endothermal peaks centered around 22, 21 and 22°C, respectively. Optical absorption of the hydrogel solution jumps when micelles are formed in the solution at a certain temperature to give a considerable amount of light scattering, while for mechanical analysis a higher transition temperature is necessary for a load-bearing 3-D gel network to form. In DSC analysis, the endothermal peaks appear when the temperature is high enough to induce the endothermal process of hydrogen bond breaking in the ice-like water molecule clusters (clathrate stuctures) around the hydrophobic domains and between the water molecules and amide bonds in the copolymers, together with the collapsing of the molecules from coils into globules 7,47,48.
Although the LCST-based transition of the NIPAAm-based polymer is a first-order thermodynamic transition, experimentally it is often observed as a slowly occurring continuous transition controlled by the kinetics of water diffusion out of the gel. 47,49,50 In this work, the gelation of the three hydrogels at 37°C also appeared as a slow process in which the polymer chains collapsed and the water diffused out of the hydrogel gradually until the hydrogels stabilized at smaller volumes (Figure 5) with final water contents of ~45% (Table 1). The hydrogels therefore exhibited different microstructures during the gelation process, in which a relatively uniform and loose structure was found at the beginning (30 sec), while the final hydrogel formed had a much more condensed polymer phase, as shown by SEM micrographs (Figure 5). The hydrogels containing higher MAPLA content had stronger hydrophobic interactions between molecular chains and faster polymer collapsing and water exclusion rates, as characterized by plotting the water content of the hydrogels as a function of incubation time (Figure 6). Also, due to the stronger hydrophobic interactions, the hydrogel with a higher MAPLA content showed stronger tensile and shear mechanical strength (Figure 8 and 9).
Upon removal of the PLA side chains by hydrolysis, all three copolymers became soluble at 37°C (LCST >37°C), verified both by the fast degradation study in 1M NaOH, and by degradation under more physiological conditions in PBS at 37°C over a period of 8 months (Figure 11). The degradation curves shown in Figure 11 all followed the same pattern, in which an initial “burst” mass loss of ~10–20% over the first 3 weeks occurred, followed by ~3 months with almost no mass change, and then followed by an accelerated mass loss process over ~ 3 months until the hydrogels were completely dissolved. The accelerated degradation in the latter stage could be explained by positive feedback between PLA ester bond cleavage leading to increased copolymer hydrophilicity, leading to increased water content to facilitate further ester bond cleavage.17 The initial mass loss was possibly caused by the dissolution of the lower molecular weight fractions in the copolymers, while the main mass loss occurred only after enough carboxylate groups were produced by PLA side chain cleavage. The degradation rate of the hydrogel was found to be influenced by the MAPLA feed ratio. A higher MAPLA feed ratio would result in more carboxylate groups being produced in the degradation process, leading to faster copolymer dissolution. While hydrogels containing less MAPLA would require longer periods for enough carboxylate to be produced to facilitate solubilization. Interestingly, the initial mass loss speed of the hydrogels decreased with the increasing MAPLA content in the hydrogel, due to the fact that hydrogel with more MAPLA is more hydrophobic before degradation, slowing down the water diffusion into the hydrogel and the diffusion and dissolution of the low molecular weight fractions of the copolymer. This trend in rates reverses for the latter stage, which can be explained by the relative availability of carboxylates.
The thermally responsive hydrogels showed no negative effects on the metabolic activity and live/dead ratio of the cells (RSMC) that were co-cultured with hydrogel degradation products (Figure 12 and 13), confirming the potential applicability of the material for cardiac injection therapy and possibly for cell or drug delivery. While the in vitro toxicity data are encouraging and the degradation product concentration tested appeared to be a high estimate for the local concentrations that would be experienced locally over a long degradation period, ultimately in vivo testing will be required to better examine local and systemic effects. Furthermore, a limitation of the material as a cell delivery carrier is that the hydrogel contains only ~45% water at 37°C (Table 1). With lower diffusion rates of water and nutrients likely for a denser material, this hydrogel may not provide a hospitable environment for long term cell culture. The shrinkage observed over relatively short periods of time in vitro also may limit cell delivery potential. Although simple inclusion of hydrophilic monomers into the copolymer can increase the water content and improve the cell encapsulation suitability, it can also decrease the hydrophobic interactions between the molecular chains and compromise the mechanical strength of the material. The trade-off between these properties can be dictated by the planned application. In the cardiac biomaterial injection literature it is clear that the injection of acellular materials is associated with improved functional outcomes 51,52, so cell survival in the early stages of injection may not be critical and cell migration into a looser injected material following a period of in situ degradation may be appropriate.
Conclusion
A novel biodegradable monomer, MAPLA, was synthesized and copolymerized with NIPAAm and HEMA to develop bioabsorbable and thermally responsive hydrogels. Poly(NIPAAm-co-HEMA-co-MAPLA) formed from three monomer feed ratios, 84/10/6, 82/10/8 and 80/10/10 were synthesized, with a higher MAPLA feed ratio giving rise to a lower LCST, higher mechanical strength and faster degradation speed. All three of the hydrogels had LCSTs below body temperature and formed mechanically strong hydrogels at 37°C. These hydrogels, upon cleavage of PLA residues by hydrolysis, became completely soluble at 37°C and exhibited no cytotoxicity associated with degradation products. This novel hydrogel design represents an injectable biomaterial that is suitable for mechanical support applications in regenerative medicine, such as for ventricular bulking following myocardial infarction.
Supplementary Material
FTIR spectra of poly(NIPAAm) and poly(NIPAAm-co-HEMA-co-MAPLA).
Synthetic scheme for MAPLA and poly(NIPAAm-co-HEMA-co-MAPLA).
References
- 1.Ruel-Gariepy E, Leroux JC. Eur J Pharm Biopharm. 2004;58:409–426. doi: 10.1016/j.ejpb.2004.03.019. [DOI] [PubMed] [Google Scholar]
- 2.Jeong B, Kim SW, Ba YH. Adv Drug Deliv Rev. 2002;54:37–51. doi: 10.1016/s0169-409x(01)00242-3. [DOI] [PubMed] [Google Scholar]
- 3.Klouda L, Mikos AG. Eur J Pharm Biopharm. 2008;68:34–45. doi: 10.1016/j.ejpb.2007.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wall ST, Walker JC, Healy KE, Ratcliffe MB, Guccione JM. Circulation. 2006;114:2627–2635. doi: 10.1161/CIRCULATIONAHA.106.657270. [DOI] [PubMed] [Google Scholar]
- 5.Ahmed Z, Gooding EA, Pimenov KV, Wang L, Asher SA. J Phys Chem B. 2009;113:4248–56. doi: 10.1021/jp810685g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wright ER, Conticello VP. Adv Drug Deliv Rev. 2002;54:1057–1073. doi: 10.1016/s0169-409x(02)00059-5. [DOI] [PubMed] [Google Scholar]
- 7.Rzaev ZMO, Dinc-er S, Pis-kin E. Prog Polym Sci. 2007;32:534–595. [Google Scholar]
- 8.He C, Kim SW, Lee DS. J Control Release. 2008;127:189–207. doi: 10.1016/j.jconrel.2008.01.005. [DOI] [PubMed] [Google Scholar]
- 9.Ono O, Shikata T. J Am Chem Soc. 2006;128:10030. doi: 10.1021/ja063990i. [DOI] [PubMed] [Google Scholar]
- 10.Ono Y, Shikata T. J Phys Chem B. 2007;111:1511–1513. doi: 10.1021/jp068954k. [DOI] [PubMed] [Google Scholar]
- 11.Fujimoto KL, Ma Z, Nelson DM, Hashizume R, Guan J, Tobita K, Wagner WR. Biomaterials. 2009;30:4357–4368. doi: 10.1016/j.biomaterials.2009.04.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang F, Li Z, Khan M, Tamama K, Kuppusamy P, Wagner WR, Sen CK, Guan J. Acta Biomater. 2009;5:2901. doi: 10.1016/j.actbio.2009.12.011. [DOI] [PubMed] [Google Scholar]
- 13.Li Z, Wang F, Roy S, Sen CK, Guan J. Biomacromolecules. 2009;10:3306–3316. doi: 10.1021/bm900900e. [DOI] [PubMed] [Google Scholar]
- 14.Kim S, Chung EH, Gilbert M, Healy KE. J Biomed Mater Res A. 2005;75:73–88. doi: 10.1002/jbm.a.30375. [DOI] [PubMed] [Google Scholar]
- 15.Su J, Wall ST, Healy KE, Wildsoet CF. Tissue Eng Part A. 2010;16:905–916. doi: 10.1089/ten.tea.2009.0488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Henderson E, Lee BH, Cui Z, McLemore R, Brandon TA, Vernon BL. J Biomed Mater Res A. 2009;90:1186–1197. doi: 10.1002/jbm.a.32179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cui Z, Lee BH, Vernon BL. Biomacromolecules. 2007;8:1280–1286. doi: 10.1021/bm061045g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee BH, Vernon B. Polym Int. 2005;54:418–22. [Google Scholar]
- 19.Lee BH, Vernon B. Macromol Biosci. 2005;5:629–635. doi: 10.1002/mabi.200500029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guan J, Hong Y, Ma Z, Wagner WR. Biomacromolecules. 2008;9:1283–92. doi: 10.1021/bm701265j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ohya S, Matsuda T. J Biomater Sci Polym Ed. 2005;16:809–827. doi: 10.1163/1568562054255736. [DOI] [PubMed] [Google Scholar]
- 22.Li F, Carlsson D, Lohmann C, Suuronen E, Vascotto S, Kobuch K, Sheardown H, Munger R, Nakamura M, Griffith M. Proc Nat Acad Sci USA. 2003;100:15346–15351. doi: 10.1073/pnas.2536767100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ohya S, Nakayama Y, Matsuda T. Biomacromolecules. 2001;2:856–863. doi: 10.1021/bm010040a. [DOI] [PubMed] [Google Scholar]
- 24.Ohya S, Sonoda H, Nakayama Y, Matsuda T. Biomaterials. 2005;26:655–659. doi: 10.1016/j.biomaterials.2004.03.002. [DOI] [PubMed] [Google Scholar]
- 25.Wang T, Wu D, Jiang X, Zhang X, Li X, Zhang J, Zheng Z, Zhuo R, Jiang H, Huang C. Eur J Heart Fail. 2009;11:14–19. doi: 10.1093/eurjhf/hfn009. [DOI] [PubMed] [Google Scholar]
- 26.Neradovic D, Hinriches WLJ, Kettenes-van den Bosch JJ, Hennink WE. Macromol Rapid Commun. 1999;20:577–581. [Google Scholar]
- 27.Overstreet DJ, Dhruv HD, Vernon BL. Biomacromolecules. 2010;11:1154–1159. doi: 10.1021/bm100035f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wu D, Qiu F, Wang T, Jiang X, Zhang X, Zhuo R. ACS Appl Mater Interf. 2009;2:312–327. doi: 10.1021/am8000456. [DOI] [PubMed] [Google Scholar]
- 29.Feil H, Bae YH, Feijen J, Kim SW. Macromolecules. 1993;26:2496–2500. [Google Scholar]
- 30.Vihola H, Laukkanen A, Valtola L, Tenhu H, Hirvonen J. Biomaterials. 2005;26:3055–3064. doi: 10.1016/j.biomaterials.2004.09.008. [DOI] [PubMed] [Google Scholar]
- 31.Ray JL, Leach R, Herbert JM, Benson M. Methods Cell Sci. 2001;23:185–188. doi: 10.1023/a:1016357510143. [DOI] [PubMed] [Google Scholar]
- 32.Stile RA, Burghardt WR, Healy KE. Macromolecules. 1999;32:7370–7379. [Google Scholar]
- 33.Qiu Y, Park K. Adv Drug Deliv Rev. 2001;53:321–339. doi: 10.1016/s0169-409x(01)00203-4. [DOI] [PubMed] [Google Scholar]
- 34.Gil ES, Hudson SM. Prog Polym Sci. 2004;29:1173–1222. [Google Scholar]
- 35.Peppas NA, Kim B. J Drug Del Sci Tech. 2006;16:11–18. [Google Scholar]
- 36.Appell RA. Urol Clin North Am. 1994;21:177–82. [PubMed] [Google Scholar]
- 37.Ghoniem GM, Elsergany R, Lewis V. Urol Nurs. 1998;18:125–128. [PubMed] [Google Scholar]
- 38.Vernengo J, Fussell GW, Smith NG, Lowman AM. J Biomed Mater Res B. 2008;84:64–69. doi: 10.1002/jbm.b.30844. [DOI] [PubMed] [Google Scholar]
- 39.Cloyd JM, Malhotra NR, Weng L, Chen W, Mauck RL, Elliott DM. Eur Spine J. 2007;16:1892–1898. doi: 10.1007/s00586-007-0443-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Landa N, Miller L, Feinberg MS, Holbova R, Shachar M, Freeman I, Cohen S, Leor J. Circulation. 2008;117:1388–1396. doi: 10.1161/CIRCULATIONAHA.107.727420. [DOI] [PubMed] [Google Scholar]
- 41.Christman KL, Fok HH, Sievers RE, Fang QH, Lee RJ. Tissue Eng. 2004;10:403–409. doi: 10.1089/107632704323061762. [DOI] [PubMed] [Google Scholar]
- 42.Huang NF, Yu J, Sievers R, Li S, Lee RJ. Tissue Eng. 2005;11:1860–1866. doi: 10.1089/ten.2005.11.1860. [DOI] [PubMed] [Google Scholar]
- 43.Mukherjee R, Zavadzkas JA, Saunders SM, McLean JE, Jeffords LB, Beck C, Stroud RE, Leone AM, Koval CN, Rivers WT, Basu S, Sheehy A, Michal G, Spinale FG. Ann Thorac Surg. 2008;86:1268–1277. doi: 10.1016/j.athoracsur.2008.04.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lu W, Lu S, Wang H, Li D, Duan C, Liu Z, Hao T, He W, Xu B, Fu Q, Song Y, Xie X, Wang C. Tissue Eng. 2009;15:1437–1447. doi: 10.1089/ten.tea.2008.0143. [DOI] [PubMed] [Google Scholar]
- 45.Davis ME, Motion JP, Narmoneva DA, Takahashi T, Hakuno D, Kamm RD, Zhang S, Lee RT. Circulation. 2005;111:442–450. doi: 10.1161/01.CIR.0000153847.47301.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Davis ME, Hsieh PC, Takahashi T, Song Q, Zhang S, Kamm RD, Grodzinsky AJ, Anversa P, Lee RT. Proc Natl Acad Sci USA. 2006;103:8155–8160. doi: 10.1073/pnas.0602877103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tian J, Seery TAP, Weiss RA. Macromolecules. 2004;37:9994–10000. [Google Scholar]
- 48.Bae YH, Kim SW. In: Polymeric materials encyclopedia: F-G. Salamone JC, editor. Vol. 4. CRC Press; p. 3492. [Google Scholar]
- 49.Wu C, Zhou S. Macromolecules. 1997;30:574–576. [Google Scholar]
- 50.Wu C. Polymer. 1998;39:4609–4619. [Google Scholar]
- 51.Jiang XJ, Wang T, Li XY, Wu DQ, Zheng ZB, Zhang JF, Chen JL, Peng B, Jiang H, Huang C, Zhang XZ. J Biomed Mater Res, A. 2009;90:472–477. doi: 10.1002/jbm.a.32118. [DOI] [PubMed] [Google Scholar]
- 52.Yu J, Christman KL, Chin E, Sievers RE, Saeed M, Lee RJ. J Thorac Cardiovasc Surg. 2009;137:180–187. doi: 10.1016/j.jtcvs.2008.08.036. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
FTIR spectra of poly(NIPAAm) and poly(NIPAAm-co-HEMA-co-MAPLA).
Synthetic scheme for MAPLA and poly(NIPAAm-co-HEMA-co-MAPLA).