

Abstract
A series of hairpin oligomers containing benzimidazole (Bi) and imidazopyridine (Ip) rings were synthesized and screened to target 5′-WGGGGW-3′, a core sequence in the DNA binding site of NF-κB, a prolific transcription factor important in biology and disease. Five Bi and Ip containing oligomers bound to the 5′-WGGGGW-3′ site with high affinity. One of the oligomers (Im-Im-Im-Im-γ-PyBi-PyBi-β-Dp) was able to inhibit DNA binding by the transcription factor NF-κB.
1. Introduction
DNA-binding polyamides based on the architecture of the natural products netropsin and distamycin A have emerged as a promising class of gene modulators.1,2 These molecules are capable of distinguishing the four Watson-Crick base pairs in the DNA minor groove and have been the subject of intense study along with other classes of minor groove binders for interfering with specific protein-DNA interfaces.3c-f,4–5 Sequence-specific recognition of the minor groove of DNA by polyamides arises from the pairing of three different aromatic amino acids, pyrrole (Py), imidazole (Im), and hydroxypyrrole (Hp).4,5 The direct read out, or information face, on the inside of the crescent-shaped polyamide may be programmed by the incremental change of atoms on the corners of the ring pairs presented to the DNA minor groove floor. Stabilizing and, importantly, destabilizing interactions with the different edges of the four Watson-Crick bases are modulated by shape complementarity and specific hydrogen bonds.4–6,7k For example, the Im/Py pair distinguishes G·C from C·G, T·A and A·T. Im presents a lone pair of electrons to the DNA minor groove and can accept a hydrogen bond from the exocyclic amine of guanine.5 Additionally, the Hp/Py pair distinguishes T·A from A·T, G·C and C·G.4–6 Hp projects an exocyclic OH group toward the minor groove floor that is sterically accommodated in the cleft of the T·A base pair, preferring to lie over T not A.5 Since the development of these pairing rules based on the five-membered heterocyclic carboxamides Py, Im, and Hp, hundreds of DNA sequences have been successfully coded for using polyamides.3,5 Yet, due to sequence dependent alterations in the shape of the DNA minor groove, there remain DNA sequences that prove difficult to target with high affinity and specificity.
There has been an ongoing effort to broaden the repetoire of heterocycles with improved DNA recognition, stability, and cellular trafficking profiles. We recently reported that the benzimidazole ring can be an effective platform for modular paired recognition elements in the minor groove of DNA.8,9 The benzimidazole 6–5 bicyclic ring structure, while having slightly different curvature from the classic five-membered N-methyl pyrrole-carboxamides, presents an “inside edge” with a similar atomic readout to the DNA minor groove floor, effectively mimicking Py, Im, and Hp.8,9 The imidazopyridine/pyrrole pair Ip/Py distinguishes G·C from C·G and the hydroxybenzimidazole/pyrrole pair Hz/Py distinguishes T·A from A·T, providing a solution to the unanticipated hydroxypyrrole instability limitation.8,9 This second generation solution to DNA recognition can be elaborated further, deleting incrementally almost all carboxamide linkages in the backbone of the hairpin motif creating an oligomer comprised of four dimer units capable of binding the site 5′-GTAC-3′, a sequence formally containing all four Watson-Crick base pairs.10 A key strategic issue for the program is interfering with protein-DNA interfaces in the promoter of important genes. The new oligomer architecture has been successful in targeting the hypoxia inducible factor (HIF-1) binding site on the promoter of the VEGF gene.11 This recent success spurred our interest in using these new oligomers to target another prolific transcription factor, NF-κB, important in biology and disease.12 Guanine rich sequences are highly prevalent and partially conserved in the promotor region of NF-κB driven genes (Fig. 1).12,13 Previous studies from our lab have established the ability of polyamides, targeting the sequence 5′-GGGACT-3′, to inhibit DNA binding by NF-κB, however targeting the four consecutive guanines was less successful due to the modest affinity of pyrrole/imidazole polyamides for this sequence.14,15 The question arises whether a second generation oligomer architecture can target 5′-GGGG-3′ recognition sequences with improved affinity.
Figure 1.

Crystal structure of the p50/p65 NF-κB heterodimer bound to the DNA duplex 5′-TGGGGACTTTCC-3′ (ref. 13). The p50 and p65 monomers are represented as gold and dark blue ribbons, respectively. a) Top view looking down the DNA double helix. b) Side view showing the 5′-GGGG-3′ oligomer binding site. GC rich regions are shown in red and AT rich regions are shown in light blue. c) Sequence of DNA bound to NF-κB.
We report the synthesis, DNA binding properties, and NF-κB:DNA binding inhibition properties of a new class of DNA binding oligomers targeted at the DNA sequence 5′-WGGGGW-3′ containing 4-consecutive guanines (Fig. 3). Oligomers vary by incorporation of benzimidazole-pyrrole into various positions in the parent polyamide 1, resulting in oligomers 2–6. From previous studies, we expect the BiPy dimer to be a good mimic for the traditional Py-Py recognition elements.16 The IpIm moiety is introduced as a new mimic for the traditional Im-Im recognition motif (Fig. 2). Quantitative DNase I footprinting titration experiments17 were used to determine the binding affinities and specificities against single base-pair mismatch sites of oligomers 2–6 (Fig. 3) as compared to their parent polyamide 1. We found that in oligomers 2–6, the 6–5 fused rings are effective mimics of their respective five-membered ring systems and that these oligomers target the binding-site 5′-WGGGGW-3′ without loss of affinity as compared to parent hairpin polyamide 1. In addition we report the inhibition of DNA binding by the transcription factor NF-κB using this new class of minor groove binding oligomers (Fig. 3 & 4).
Figure 3.

Postulated hydrogen-bonding models for the 1:1 polyamide-DNA complexes with their matched sequence and the ball-and-stick representation for compounds 1 and 6 over the 6-base-pair matched binding site (variable region W = A or T). a) Im-Im-Im-Im-γ-Py-Py-Py-Py-β-Dp (1); b) Im-Im-Im-Im-γ-Py-Bi-Py-Bi-β-Dp (2), Im-Im-Im-Ip-γ-Py-Bi-Py-Bi-β-Dp (3), Im-Im-Ip-Im-γ-Py-Bi-Py-Bi-β-Dp (4), Im-Ip-Im-Im-γ-Py-Bi-Py-Bi-β-Dp (5), Im-Ip-Im-Ip-γ-Py-Bi-Py-Bi-β-Dp (6).
Figure 2.

Structures of the (a) pyrrole-benzimidazole internal dimer (-Py-Bi-), (b) imidazole-imidazopyridine internal dimer (-Im-Ip-), and (c) imidazole-imidazopyridine cap (Im-Ip-) in comparison with their respective five membered ring systems. Hydrogen-bonding surfaces to the DNA minor-groove floor are bolded.
Figure 4.

Illustration of NF-kB:DNA binding inhibition by oligomer 2.
2. Results and Discussion
2.1. Heterocycle Synthesis
Dimeric units Im-Ip-OH (9) and Boc-Im-Ip-OH (12) were synthesized by oxidative condensation of aldehydes 7 and 10, respectively, with previously reported diaminopyridine 8. Mixing 7 and 8 in DMF at 80°C for 1 hr followed by 12 hrs of heating at 100°C in the presence of FeCl3 and air affforded 9 after purification by precipitation from water and saponification using a mixture of KOH (4M) in MeOH at 40°C (Scheme 1). Nitroimidazole (10) was synthesized from 7 using a mixture of H2SO4+SO3 and neat red fuming nitric acid.18 10 was then added to a mixture of 8 in nitrobenzene and refluxed at 140 °C open to the atmosphere overnight to provide NO2-Im-Ip-OMe (11) after precipitation from water. Compound 11 was reduced using Pd/C in the presence of hydrogen followed by Boc protection using a mixture of Boc2O and DMAP in DMF to provided the final dimer Boc-Im-Ip-OH (12) after saponification and precipitation (Scheme 1).
Scheme 1.

Synthesis of imidazopyridine-imidazole dimers Im-Ip-OH (9) and Boc-Im-Ip-OH (12). i) 8, PhNO2, 140°C; ii) Oleum, red-fuming HNO3; iii) 8, PhNO2, 140°C; iv) H2, Pd/C, DMF; (Boc)2O, DIEA, DMAP, DMF; 4N KOH, MeOH.
2.2. Oligomer Synthesis
Oligomers 1–6 were synthesized using manual solid phase synthesis methodology on commercially available β-Ala-PAM resin as previously described (Scheme 2).19 Starting from base resin (BR1), monomeric and dimeric heterocyclic units were appended onto the resin in stepwise fashion using HBTU activation. Couplings were allowed to proceed for several hours between 25–40 °C. Unreacted amines were acylated between coupling rounds using acetic anhydride. Deprotection of the Boc-protected amines was accomplished using 80% TFA in DCM. After completion of solid phase synthesis, the resin was treated with dimethylaminopropylamine (Dp) and the oligomers were purified by reverse phase preparatory HPLC: Im-Im-Im-Im-γ-Py-Py-Py-Py-β-Dp (1), Im-Im-Im-Im-γ-PyBi-PyBi-β-Dp (2), Im-Im-ImIp-γ-PyBi-Py-Bi-β-Dp (3), Im-ImIp-Im-γ-PyBi-PyBi-β-Dp (4), ImIp-Im-Im-γ-PyBi-PyBi-β-Dp (5), ImIp-ImIp-γ-PyBi-PyBi-β-Dp (6). Oligomers were characterized using MALDI-TOF mass spectrometry.
Scheme 2.

Representative solid-phase synthesis of polyamide 6 along with a table of the amino acid building blocks used for the synthesis. Reaction conditions: (i) 80% TFA/DCM; (ii) Boc-Py-Bi-OH (13), HBTU, DIEA, DMF; (iii) Ac2O, DIEA, DMF; (iv) repeat i – iii; (v) 80% TFA/DCM; (vi) Boc-γ-OH (14), HBTU, DIEA, DMF; (vii) Ac2O, DIEA, DMF to provide BR1; (viii) 80% TFA/DCM; (ix) Boc-Im-Ip-OH (12), HBTU, DIEA, DMF; (x) 80% TFA/DCM; (xi) Im-Ip-OH, HBTU, DIEA, DMF; (xii) dimethylaminopropylamine (Dp), 80 °C 2 h; (xiii) preparative HPLC to give 6.
2.3. DNA Affinity and Sequence Specificity
Quantitative DNaseI footprinting titrations were carried out for oligomers 1–6 on the PCR product of plasmid pEF16 (Fig. 5 & 6). Plasmid pEF16 was constructed containing two designed match sites (5′-XGGGGT-3′ X = A, T) and two mismatch sites (5′-AGGGAT-3′ and 5′-AGAGGT-3′ (Fig. 5 & 6). The first two match sites 5′-TGGGGT-3′ and 5′-AGGGGT-3′ were included to determine if there was an energetic penalty associated with a 5′-AG-3′ step. Previously, DNA sequences containing multiple 5′-AG-3′ transitions have proven more difficult to target at high affinity and it has been shown that changes in flanking sequence can affect binding at a proximal site.20 The second two binding sites, 5′-AGGGAT-3′ and 5′-AGAGGT-3′ were designed to elucidate the energetic penalty for the loss of a favorable hydrogen bond between the exocyclic amine of guanine and the lone pair nitrogen on the oligomer in question.
Figure 5.

Quantitative DNase I footprinting experiments in the hairpin motif for polyamides 1, 2, and 3 respectively, on the 278 bp, 5′-end-labelled PCR product of plasmid pEF16: lane 1, intact DNA; lane 2, G and A sequencing reaction; lane 3, DNase I standard; lanes 5–15, 1 pM, 3 pM, 10 pM, 30 pM, 100 pM, 300 pM, 1 nM, 3 nM, 10 nM, 30 nM, 100 nM polyamide concentration, respectively. Each footprinting gel is accompanied by the following: (top) Ball-and-stick models of the parent polyamide; and (bottom) Binding isotherms for the four designed sites. θnorm values were obtained according to published methods. A binding model for the hairpin motif is shown centered at the top as a ball-and-stick model with the polyamide bound to its target DNA sequence. Imidazoles and pyrroles are shown as filled and non-filled circles, respectively; Dimer units are represented as rectangles containing either Bi enclosed in a circle representing benzimidazole or Ip in a black box representing imidazopyridine; Beta alanine is shown as a diamond; the gamma-aminobutyric acid turn residue is shown as a semicircle connecting the two subunits.
Figure 6.

Quantitative DNase I footprinting experiments in the hairpin motif for polyamides 4, 5, and 6 respectively, on the 278 bp, 5′-end-labelled PCR product of plasmid pEF16: lane 1, intact DNA; lane 2, G and A sequencing reaction; lane 3, DNase I standard; lanes 5–15, 1 pM, 3 pM, 10 pM, 30 pM, 100 pM, 300 pM, 1 nM, 3 nM, 10 nM, 30 nM, 100 nM polyamide concentration, respectively. Each footprinting gel is accompanied by the following: (top) Ball-and-stick models of the parent polyamide; and (bottom) Binding isotherms for the four designed sites. θnorm values were obtained according to published methods. A binding model for the hairpin motif is shown centered at the top as a ball-and-stick model with the polyamide bound to its target DNA sequence. Imidazoles and pyrroles are shown as filled and non-filled circles, respectively; Dimer units are represented as rectangles containing either Bi enclosed in a circle representing benzimidazole or Ip in a black box representing imidazopyridine; Beta alanine is shown as a diamond; the gamma-aminobutyric acid turn residue is shown as a semicircle connecting the two subunits.
Control compound 1 bound both 4-G match sequences (5′-XGGGGT-3′, X = A,T) with comparably low affinity (Ka ~ 108 M−1), showing no bias for either site (Table 1). Compound 1 distinguished against mismatch sequences (5′-AGGGAT-3′ and 5′-AGAGGT-3′) with roughly 10-fold specificity (Table 1). Oligomer 2 demonstrated a large increase in affinity (Ka ~ 1010 M−1) as compared to 1 for both match sites, but showed lower specificity (4-fold) over the mismatch sites. Oligomer 5 showed a moderate increase in affinity (Ka ~ 109 M−1) and demonstrated minor selectivity (3 fold) over the mismatch 5′-atGGGAt-3′, however showed an 11 fold specificity over the mismatch 5′-atGAGGt-3′. Oligomer 4 demonstrated high affinity for the 4-G match sequences (Ka ~ mid 109 M−1) with reasonable 5-fold selectivity over the mismatch sequences (5′-AGGGAT-3′ and 5′-AGGAGT-3′). Compounds 3 and 6 bound all designed sequences with similar affinity. Thermodynamic data for oligomers 1–6 is summarized in Table 1.
Table 1.
| Polyamide | 5′-atGGGGt-3′ | 5′-aaGGGGt-3′ | 5′-atGGGAt-3′ | 5′-atGAGGt-3′ |
|---|---|---|---|---|
1

|
1.4(±1.0) × 108 | 2.6(±1.1) × 108 | 2.3(±0.8) × 107 | ≤ 1.0 × 107 |
2

|
1.9(±1.4) × 1010 | 2.0(±1.1) × 1010 | 4.8(±1.1) × 109 | 3.6(±0.9) × 109 |
3

|
2.6(±0.6) × 109 | 2.7(±0.4) × 109 | 2.6(±0.2) × 109 | 2.9(±0.1) × 109 |
4

|
4.4(±0.9) × 109 | 4.1(±1.2) × 109 | 8.1(±2.3) × 108 | 8.2(±1.3) × 108 |
5

|
2.6(±0.5) × 109 | 2.9(±0.2) × 109 | 8.6(±2.1) × 108 | 2.5(±0.8) × 108 |
6

|
1.6(±0.2) × 109 | 1.1(±0.4) × 109 | 1.3(±0.1) × 109 | 1.1(±0.1) × 109 |
values reported are the mean values from at least three DNase I footprinting titration experiments with the standard deviation given in parentheses
Assays were performed at 22 °C in a buffer of 10 mM Tris.HCl, KCl, 10 mM MgCl2, and 5 mM CaCl2 at pH 7.0
In general, the global increase in affinity for these novel oligomers is not altogether unexpected. In contrast to the 5-membered heterocyclic carboxamides, the 6–5 fused benzimidazole analogues have a larger hydrophobic surface, likely promoting their placement in the DNA minor groove.21 Aromatic stacking and van der Waals interactions are also contributing factors.22,23 Furthermore, the benzimidazole derivatives are a more rigid structure with a lower degree of rotational freedom. Such pre-organization may decrease the entropic cost of DNA complexation.
2.4. NF-κB Electrophoretic Mobility Gel Shift Assay
The guanine rich region of the NF-κB binding element has been shown to be important in protein-DNA recognition (Fig. 1).13,15,24,25 Polyamides targeted to this region have previously been shown to inhibit NF-κB binding.15 The possibility of steric or allosteric inhibition exists due to numerous contacts between the p50 protein and the phosphate backbone or direct protein-DNA contacts in the major groove, opposite the minor groove polyamide binding site.13,15 To test whether the second generation oligomer architecture would be successful at targeting 5′-GGGG-3′ in a biologically relevant context we employed an NF-κB electrophoretic mobility gel shift assay.
The NF-κB binding inhibition properties of compounds 1–6 were screened against a 40 base pair DNA probe containing the 5′–WGGGGW-3′ sequence, which is part of an element from the intronic enhancer of the immunoglobulin κ light-chain gene recognized and bound by NF-κB (Fig. 7).13 In the initial screen each oligomer was tested at two concentrations, 10 nM and 100 nM, for the ability to interfere with protein binding (Fig. 7). At the higher concentration, 2 and 4 demonstrate a clear decrease in band intensity whereas compounds 1, 3, 5 and 6 have little effect. This result agrees with the footprinting data, as protein inhibition appears to scale with compound affinity for the 5′-WGGGGW-3′ site. At the 10 nM concentration compound 2 is the only compound to show significant protein inhibition, reducing band intensity by more than 80% (Fig. 7). Full titration of compound 2 over a concentration range of 500 pM to 500 nM established an EC50 of 15.7 nM for NF-κB inhibition as shown in Figure 8. In addition the identity of the NF-κB band was confirmed by antibody supershift and the data is shown in figure 9.
Figure 7.

a) Matched DNA sequence with the p50 protein of NF-κB overlapping the oligomer binding site. Asterisks indicate the location of radio-labeled nucleotides in the probe sequence. b) Mismatch DNA sequence. c) Gel shift screen for compounds 1–6 at concentrations of 10 and 100 nM. d) Plot of relative NF-κB inhibition for compounds 1–6.
Figure 8.

(Top) Ball-and-stick model for 2 and EC50 value from gel shift experiment. (Middle) Representative NF-κB titration gel (n=3) for 2. (Bottom) Binding isotherm for 2.
Figure 9.
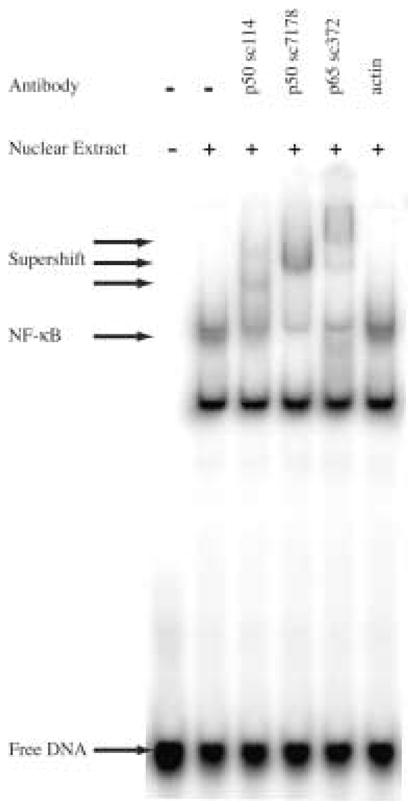
Antibody supershift on Match DNA. The NF-κB band is shifted in the presence of anti-p50 or anti-p65 antibody.
3. Conclusion
While traditional Py, Im and Hp containing polyamides have been successful at recognizing hundreds of pre-determined DNA sequences with high affinity and specificity, a host of target sequences such as 5′-GGGG-3′ have proven difficult to code for using polyamides. A series of novel oligomers containing the 6–5 fused benzimidazole (Bi) and imidazopyridine (Ip) heterocycles, were developed. These oligomers, composed of 5-membered heterocyclic carboxamides, demonstrated a substantial increase in affinity (10 to 100-fold) for the 5′-GGGG-3′ sequence. The marked increase in affinity could be attributed to a combination of oligomer properties including, a larger hydrophobic surface, a high degree of ligand pre-organization, or differential solvation/desolvation effects. The ability of this class of new oligomers to inhibit protein-DNA binding was demonstrated by the inhibition of NF-κB. We are encouraged by the fact that these oligomers demonstrate improved affinity for guanine rich DNA sequences and future work directed towards improving sequence specificity and examination of the nuclear trafficking ability is a priority.
4. Experimental
4.1. General
N,N-dimethylformamide (DMF), N,N-diisopropylethylamine (DIEA), N,N-dimethylaminopropylamine (Dp), Triethylamine (TEA), nitrobenzene (NO2Ph), 2-formyl-N-methylimidazole, red fuming nitric acid, 1,3-dichlo-4-nitroropyridine, 30% bromine in acetic acid, palladium acetate Pd(OAc)2, and 10% palladium on carbon, were purchased from Aldrich. Boc-β-alanine-(4-carbonylaminomethyl)-benzyl-ester-copoly(styrene-divinylbenzene)resin (Boc-β-Pam-resin), dicyclohexylcarbodiimide (DCC), hydroxybenzotriazole (HOBt), 2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HBTU), N,N-dimethylaminopyridine (DMAP), and Boc-β-alanine were purchased from NOVA Biochem. Trifluoroacetic acid (TFA) was purchased from Halocarbon. All other solvents were reagent grade from EM. Oligonucleotide inserts were synthesized by the Biopolymer Synthesis Center at the California Institute of Technology. Precoated silica gel plates 60F254 for TLC and silica gel 60 (40 μm) for flash chromatography were from Merck. Glycogen (20 mg/mL), dNTPs (PCR nucleotide mix), and all enzymes, unless otherwise stated, were purchased from Boehringer-Mannheim. pUC19 was purchased from New England Biolabs, and deoxyadenosine [γ-32P]triphosphate was provided by ICN. Calf thymus DNA (sonicated, deproteinized) was from Amersham Pharmacia. DNaseI (7500 units/mL, FPLC pure) was purchased from Roche. AmpliTaq DNA polymerase was from Perkin-Elmer and used with the provided buffers. Tris·HCl, DTT, RNase-free water, and 0.5 M EDTA were from United States Biochemical. Calcium chloride, potassium chloride, and magnesium chloride were purchased from Fluka. Tris-borate-EDTA was from GIBCO and bromophenol blue was from Acros. All reagents were used without further purification. NMR spectra were recorded on a Varian spectrometer at 300 MHz in DMSO-d6 or CDCl3 with chemical shifts reported in parts per million relative to residual solvent. UV spectra were measured on a Hewlett-Packard Model 8452A diode array spectrophotometer. High resolution FAB and EI mass spectra were recorded at the Mass Spectroscopy Laboratory at the California Institute of Technology. Matrix-assisted, laser desorption/ionization time of flight mass spectrometry (MALDI-TOF-MS) was conducted at the Mass Spectroscopy Laboratory at the California Institute of Technology.
4.2. Heterocycle Synthesis
Heterocyclic building blocks Boc-Im-OH and Boc-Py-Bi-OH were synthesized as reported.19,8 Im-Im-OH (1-methyl-4-(1-methyl-1H-imidazole-2-carboxamido)-1H-imidazole-2-carboxylic acid, CAS [464892-44-2]) and Boc-Im-Im-OH (4-(4-(tert-butoxycarbonylamino)-1-methyl-1H-imidazole-2-carboxamido)-1-methyl-1H-imidazole-2-carboxylic acid, CAS [502170-49-2]), are commercially available.
4.2.1. 1-methyl-4-nitro-1H-imidazole-2-carbaldehyde (NO2-Im-CHO) (10)
A cooled flask (0°C) of 1-methyl-2-imidazole-carboxaldehyde (7) (8g, 72.6 mmol, Aldrich) was treated dropwise with a precooled (0°C) solution of red fuming nitric acid (75 ml) in conc. H2SO4•SO3 (30%) (75 ml). The mixture was warmed to room temperature and stirred for 12 hours open to the atmosphere. Next, the mixture was poured over ice, neutralized with solid sodium carbonate, extracted 4 times with dichloromethane, dried over anhydrous sodium sulfate, and concentrated in vacuo to give a brownish-yellow oil. The oil was recrystallized from iPrOH/Et2O or EtOH/Et2O to give 1-methyl-4-nitro-1H-imidazole-2-carbaldehyde (10) as a tan crystalline solid (4.5 g, 40% Yield). TLC (1:1 EtOAc/Hex) Rf = 0.4; 1H NMR (300 MHz, DMSO-d6) δ 9.74 (s, 1H), 8.71 (s, 1H), 3.99 (s, 3H); 13C (75 MHz, DMSO-d6) δ 182.31, 146.14, 140.56, 127.33, 35.70; HR-MS (EI+): calcd for C5H5O3N3: 155.0330; found: 155.0350.
4.2.2. 2-(1-Methyl-4-nitroimidazol-2-yl)-3H-imidazo[4,5-b]pyridine-5-carboxylic acid methyl ester (NO2-Im-Ip-OMe) (11)
1-Methyl-4-nitro-1H-imidazole-2-carbaldehyde (2.01 g, 13.0 mmol) (10) and methyl 5,6- diaminopyridine-2-carboxylate (8) (2.17 g, 13.0 mmol) suspended in 120 ml of nitrobenzene was heated to 140°C for 48 hours open to the atmosphere. The reaction mixture was cooled to 23°C and the precipitate collected by vacuum filtration. The solid was washed with diethyl ether and dried under high vacuum to provide 2-(1-methyl-4-nitroimidazol-2-yl)-3H-imidazo[4,5-b]pyridine-5-carboxylic acid methyl ester (11) (3.6 g, 92% Yield) as a powdery tan solid. 1H NMR (300 MHz, DMSO-d6) δ 14.02 (broad s, 1H), 8.03–8.06 (m, 2H), 4.27 (s, 3H), 3.91 (s, 3H); 13C (75 MHz, DMSO-d6) δ 166.01, 146.44, 142.95, 136.59, 135.80, 130.36, 127.35, 123.81, 121.17, 121.04, 52.85, 37.43; HR-MS (EI+): calcd for C12H10N6O4: 302.0760; found: 302.0760.
4.2.3. 2-{4-amino-1-methylimidazol-2-yl)-3H-imidazo[4,5-b]pyridine-5-carboxylic acid methyl ester (H2N-Im-Ip-OMe)
2-(1-methyl-4-nitroimidazol-2-yl)-3H-imidazo[4,5-b]pyridine-5-carboxylic acid methyl ester (11) (3 g, 9.9 mmol) was dissolved in anhydrous DMF (150 ml) and the solution was degassed with Ar. After the addition of Pd/C (10 wt. %, 600 mg) the reaction mixture was purged 3 times with hydrogen and then left to stir at 23°C for 9 hours under a hydrogen balloon atmosphere. After filtering through a pad of Celite and washing with copious amounts EtOAc the filtrate was concentrated in vacuo to give 2-{4-amino-1-methylimidazol-2-yl)-3H-imidazo[4,5-b]pyridine-5-carboxylic acid methyl ester, without further purification (2.7 g, 100% Yield). 1H NMR (300 MHz, DMSO-d6) δ; 13C (75 MHz, DMSO-d6) δ 166.17, 149.03, 148.21, 141.40, 131.50, 120.12, 107.77; HR-MS (EI+): calcd for C12H12N6O2: 272.1020; found: 272.1030.
4.2.4. 2-{4-[(tert-Butoxy)carbonylamino]-1-methylimidazol-2-yl)-3H-imidazo[4,5-b]pyridine-5-carboxylic acid methyl ester (Boc-Im-Ip-OMe)
2-{4-amino-1-methylimidazol-2-yl)-3H-imidazo[4,5-b]pyridine-5-carboxylic acid methyl ester (11) (2.0 g, 7.3 mmol) dissolved in DMF (25 ml) was treated with Boc2O (5.3 g, 24.3 mmol), DIEA (5.2 ml), and DMAP (95 mg, 0.73 mmol). The reaction mixture was then heated to 80°C for 72 hours, cooled to 23°C, and flashed through a plug of silica gel eluting with EtOAc to give a mixture of mono- and di-bocked (2-{4-[(tert-Butoxy)carbonylamino]-1-methylimidazol-2-yl)-3-[(tert-Butoxy)carbonylamino]-imidazo[4,5-b]pyridine-5-carboxylic acid methyl ester) products which were carried on for saponification.
4.2.5. 2-{4-[(tert-Butoxy)carbonylamino]-1-methylimidazol-2-yl)-3H-imidazo[4,5-b]pyridine-5-carboxylic (Boc-Im-Ip-OH) (12)
2-{4-[(tert-Butoxy)carbonylamino]-1-methylimidazol-2-yl)-3-[(tert-Butoxy)carbonylamino]-imidazo[4,5-b]pyridine-5-carboxylic acid methyl ester dissolved in MeOH (10 ml) and NaOH (1 N, 25 ml) was heated to 50°C for 4 hours. The reaction mixture was cooled to 0°C and the pH adjusted slowly to pH = 4 with 1 N HCl. The reaction mixture was then extracted with ethyl acetate (4 times), dried over anhydrous sodium sulfate, concentrated in vacuo, and dried under high vacuum to give 2-{4-[(tert-Butoxy)carbonylamino]-1-methylimidazol-2-yl)-3H-imidazo[4,5-b]pyridine-5-carboxylic (12) (258 mg, 60% Yield) as a light yellow solid. 1H NMR (300 MHz, DMSO-d6) δ 13.19 (broad s, 2H), 9.53 (s, 1H), 8.00 (m, 2H), 7.36 (s, 1H), 4.15 (s, 3H), 1.47 (s, 9H); 13C (75 MHz, DMSO-d6) δ 166.61, 152.92, 147.81, 142.48, 138.38, 132.01, 128.91, 119.74, 113.50, 79.07, 35.35, 28.12; HR-MS (EI+): calcd for C16H18N6O4: 358.1390; found: 358.1370.
4.2.6. 2-(1-Methylimidazol-2-yl)-3H-imidazo[4,5-b]pyridine-5-carboxylic acid methyl ester (Im-Ip-OMe)
1-Methylimidazole-2-carbaldehyde (7) (214 mg, 1.85 mmol) and methyl 5,6-diaminopyridine-2-carboxylate (8) (310 mg, 1.85 mmol) were suspended in 17 ml of DMF and heated to 80°C for 1 hour open to the atmosphere. Next, FeCl3-6H2O (24mg, 0.09 mmol) was added and the reaction mixture was heated to 100°C for 12 hrs while air was bubbled through the reaction mixture. The reaction mixture was then cooled to room temperature and poured over ice. The precipitate was collected by filtration and washed with cold diethyl ether. The material was dissolved in hot iso-propanol, cooled to room temperature and re-precipitated with diethyl ether. The solid was washed with diethyl ether and dried under high vacuum to provide 2-(1-methylimidazol-2-yl)-3H-imidazo[4,5-b]pyridine-5-carboxylic acid methyl ester (Im-Ip-OMe) (240 mg, 50% Yield) as s tan solid. 1H NMR (300 MHz, DMSO-d6) δ 7.99 (s, 2 H), 7.51 (s, 1H), 7.20 (s, 1H), 4.18 (s, 3H), 3.88 (s, 3H); 13C (75 MHz, DMSO-d6) δ 147.7, 142.0, 137.1, 131.8, 129.6, 126.9, 119.9, 109.3, 52.8, 35.8; HR-MS (EI+): calcd for C12H11N5O2: 257.0910; found: 257.0920.
4.2.7. 2-(1-Methylimidazol-2-yl)-3H-imidazo[4,5-b]pyridine-5-carboxylic acid (Im-Ip-OH) (9)
2-(1-methylimidazol-2-yl)-3H-imidazo[4,5-b]pyridine-5-carboxylic acid methyl ester (Im-Ip-OMe) (250 mg, 0.97 mmol) dissolved in MeOH (2 ml) and KOH (4 N, 3 ml) was heated to 50°C for 4 hours. The methanol was removed in vacuo and the aqueous layer washed with EtOAc (2 × 10 mL) to remove any starting material and trace impurities. The pH of the aqueous layer was then adjusted slowly to pH = 4 with 1 N HCl upon which time a cloudy beige precipitate formed. The mixture was placed in a falcon tube and the precipitate concentrated by centrifugation. The supernatant was decanted and the solid dried under high vacuum to give 2-(1-Methylimidazol-2-yl)-3H-imidazo[4,5-b]pyridine-5-carboxylic acid (9) (154 mg, 65% Yield) as a brown solid. 1H NMR (300 MHz, DMSO-d6) δ 7.99 (s, 2H), 7.51 (d, 1H, J = 0.9 Hz), 7.20 (d, 1H, J = 0.9 Hz), 4.18 (s, 3H); HR-MS (EI+): calcd for C11H9N5O2: 243.0750; found: 243.0740.
4.3. Oligomer Synthesis
Oligomers were synthesized on solid support using Boc-β-PAM resin (0.59 meq/g). Stepwise elongation of the oligomers was done according to previously published protocols.19 The synthesis of compound 1 has been previously reported.14
4.3.1. Preparation of Base Resin R-β-BiPy-BiPy-γ-NHBoc (BR1)
To a manual solid phase synthesis vessel was added Boc-β-PAM resin (0.3 g). The resin was washed with DMF (15 mL) and allowed to swell for 15 minutes while shaking at room temperature. The resin was then washed with DCM (~30 mL), followed by 80% TFA in DCM (~30 mL) to remove the Boc-group. The resin was then agitated at room temperature in 80% TFA/DCM for another 25 minutes to provide the deprotected resin bound amine (R–β-NH2). Following Boc-deprotection, the resin was washed with DCM and 10% DIEA in DMF to neutralize and prepare for coupling. Simultaneously, in a separate reaction vessel, Boc-PyBi-OH (189 mg, 531 μM), HBTU (191 mg, 504 μM), DIEA (137 mg, 185 μL, 1.06 mM) and DMF (1.2 mL) was mixed and allowed to activate at room temperature for 25 minutes. This mixture was then added to the solid phase synthesis vessel containing R–β-NH2. Coupling was allowed to proceed at room temperature with agitation for 3–6 hours. Initial loading of the resin requires elongated coupling times. Following coupling, the resin was acylated by the addition of acetic anhydride to the mixture and shaking for 15 minutes. The addition of the next Boc-PyBi-OH (25) dimer was incorporated and deprotected as described above to provide the resin bound fragment (R-β-BiPy-Bi-Py-NH2). To this fragment was added a preactivated mixture of Boc-γ-OH (180 mg, 885 μM), HBTU (319 mg, 841 μM), DIEA (229 mg, 308 μL, 1.77 mM). Coupling was allowed to proceed for 3 h at room temperature with agitation. The resin was then capped with acetic anhydride as described above to provide the base resin R-β-BiPy-Bi-Py-γ-NHBoc (BR1). BR1 was then washed with DCM followed by MeOH and Et2O. The resin was then dried under high vacuum and stored for subsequent use.
4.3.2. Im-Im-Im-Im-γ-PyBi-Py-Bi-β-Dp (2)
BR1 (50 mg) was added to a manual solid phase synthesis vessel. The resin was washed with DCM (~15 mL), followed by deprotection with 80% TFA in DCM. The resin was shaken at room temperature in the 80% TFA solution for 25 minutes. The resin was then drained, washed with DCM, and neutralized with 10% DIEA in DMF. A pre-activated mixture of Boc-Im-Im-OH (54 mg, 148 μM), HBTU (53 mg, 140 μM), DIEA (38 mg, 52 μL, 295 μM) and DMF (400 μL) was then added to the reaction vessel and coupling was allowed to proceed for three hours at room temperature, followed by capping with acetic anhydride as described for BR1 to give R-β-BiPy-BiPy-γ-Im-Im-NHBoc. Following resin deprotection as described above, Im-Im-OH was activated as described for Boc-Im-Im-OH. Coupling of Im-Im-OH to the resin was allowed to proceed overnight at room temperature to provide R-β-BiPy-BiPy-γ-Im-Im-Im-Im. The resin was treated with the cleavage protocol outlined below to provide Im-Im-Im-Im-γ-PyBi-Py-Bi-β-Dp 2 in 5% Yield. MALDI-TOF-MS: calcd for C58H66N23O8: 1212.55; found 1212.50 [M+H]+.
4.3.3. ImIp-Im-Im-γ-PyBi-Py-Bi-β-Dp (5)
BR1 (50 mg) was added to a manual solid phase synthesis vessel and R-β-BiPy-BiPy-γ-Im-Im-NHBoc was prepared as described above for 2. Following deprotection, washing and neutralization as described above, a pre activated mixture of ImIp-OH (14) (21.5 mg, 88.5 μM), HBTU (32 mg, 84 μM), DIEA (23 mg, 31μL, 177 μM), DMF (400 μL) was added to the vessel containing R-β-BiPy-BiPy-γ-Im-Im-NH2. Coupling was allowed to proceed overnight at room temperature to provide R-β-BiPy-BiPy-γ-Im-Im-IpIm. The resin was treated with the cleavage protocol outlined below to provide ImIp-Im-Im-γ-PyBi-Py-Bi-β-Dp 5 in 2.2% Yield. MALDI-TOF-MS: calcd for C59H64N23O7: 1206.54; found 1206.60 [M+H]+.
4.3.4. Im-Im-ImIp-γ-PyBi-PyBi-β-Dp (3)
BR1 (50 mg, 0.81 meq/g) was added to a manual solid phase synthesis vessel. The resin was treated with the cleavage protocol outlined below to provide Im-Im-ImIp-γ-PyBi-Py-Bi-β-Dp 3 in 3% Yield. MALDI-TOF-MS: calcd for C59H64N23O7: 1206.50; found 1206.50 [M+H]+.
4.3.5. Im-ImIp-Im-γ-PyBi-PyBi-β-Dp (4)
BR1 (70 mg, 0.59 meq/g) was added to a manual solid phase synthesis vessel. The resin was washed with DCM (~15 mL), followed by deprotection with 80% TFA in DCM. The resin was shaken at room temperature in the 80% TFA solution for 25 minutes. The resin was then drained, washed with DCM, neutralized with 50% DIEA in DCM, and washed with DMF. A pre-activated mixture of Boc-Im-OH (50 mg, 207 μmol), HBTU (79 mg, 208 μmol), DIEA (53 mg, 72 μL, 413 μmol) and DMF (900 μL) was then added to the reaction vessel and coupling was allowed to proceed for 12 hours at room temperature, followed by capping with acetic anhydride as described for BR1 to give R-β-BiPy-BiPy-γ-Im-NHBoc. Following resin deprotection as described above, Boc-ImIp-OH (12) was activated as described for Boc-Im-OH. Coupling of 12 to the resin was allowed to proceed overnight at room temperature to provide R-β-BiPy-BiPy-γ-Im-IpIm-NHBoc. Following resin deprotection as described above, Im-CCl3 (2-Trichloroacetyl-1-methylpyrrole) (47 mg, 207 μmol) and DIEA (53 mg, 72 μL, 413 μmol) were dissolved in NMP (900 μL) and added to the reaction vessel. Coupling of Im-CCl3 to the resin was allowed to proceed overnight at 32°C to provide R-β-BiPy-BiPy-γ-Im-IpIm-Im. The resin was treated with the cleavage protocol outlined below to provide Im-ImIp-Im-γ-PyBi-Py-Bi-β-Dp 4 in 3% Yield. MALDI-TOF-MS calcd for C59H64N23O7: 1206.54; found 1206.50 [M+H]+.
4.3.6. ImIp-ImIp-γ-PyBi-PyBi-β-Dp (6)
BR1 (50 mg) was added to a manual solid phase synthesis vessel. The resin was treated with the cleavage protocol outlined below to provide ImIp-ImIp-γ-PyBi-Py-Bi-β-Dp 6 in 2% Yield. MALDI-TOF-MS calcd for C60H62N23O6: 1200.52; found 1200.50 [M+H]+.
4.4. Resin Cleavage Procedure
A sample of resin (20–100 mg) was washed with DCM followed by the addition of dimethylaminopropylamine (Dp) (1 mL). The mixture was heated to 80°C for 2 h with occasional agitation. The resin was then filtered and washed with 0.1% TFA in water (7 mL). The combined filtrate was collected and subjected to purification by reverse phase preparatory HPLC using a Waters C18 column and 0.1% TFA/ACN solvent system. Appropriate fractions from the HPLC purification were checked for purity by analytical HPLC and characterized by MALDI-TOF spectroscopy. Pure fractions were then pooled, flash frozen using liquid nitrogen and lyophilized to a dry solid for later use.
4.5. Footprinting Experiments
Plasmids pEF16 was constructed using standard methods. DNaseI I footprint titrations were performed according to standard protocols.17
4.6. NF-κB Electrophoretic Mobility Shift Assay
4.6.1. Materials
Jurkat Nuclear Extract containing activated NF-κB was purchased from Active Motif (36013) and diluted as necessary just prior to use with Buffer C (20 mM Hepes pH 7.9, 420 mM KCl, 1.5 mM MgCl2, 25% glycerol, 0.2 mM EDTA, 0.5 mM DTT, 0.2 mM PMSF, 1 μg/ml aprotinin). The final working concentration was 0.5–2.5 μg/μl. The identity of the NF-κB band was confirmed by antibody supershift (Fig. 9) using antibodies against p50 (sc-7178, sc-114) and p65 (sc-372) from SantaCruz Biotechnology.
4.6.2. Antibody Supershift
The NF-κB antibody supershift data is shown in figure 9. NF-κB antibodies (sc-7178 and sc-114 against p50 and sc-372 against p65) came from Santa Cruz Biotechnology. The actin antibody was purchased from Sigma-Aldrich (A5441). Antibodies were diluted to 0.1 μg/μl just prior to use with ice cold PBS. Complete binding reactions contained 0.1 ng labeled probe, 10 mM Tris·HCl pH 7.5, 50 mM NaCl, 10% glycerol, 1% NP-40, 1 mM EDTA, 0.1 mg/ml polydIdC, 1 μl antibody (0.1 μg), and 4 μg (2 μl) nuclear extract in a total volume of 10 μl. Nuclear extract was incubated with antibody at 4° C for 1 hour followed by addition of binding buffer and probe. The complete reactions were allowed to incubate a further 30 minutes at room temperature, loaded onto pre-run 5% acrylamide, 5% glycerol gels, and resolved for 2 hours at 150 volts. Gels were pre-run for 15–45 minutes prior to loading. The gel and running buffer was 24.8 mM Tris Base, 190 mM glycine, 1 mM 0.5 M EDTA.
4.6.3. Sequence of Gel Shift Probes
Oligonucleotides were purchased from Integrated DNA Technologies. The size of both probes following labeling was 40 basepairs. The match probe contained an imbedded κB site from the intronic enhancer of the immunoglobulin κ light-chain gene (underlined below). As a control, the sequence of the mismatch probe contained a mutated κB that prevented NF- κB binding (changes bolded below).
Match Probe:
5′ AATTCATGCAGTTGAGGGGACTTTCCCAGGCATGCA 3′
Match Complementary Sequence:
5′ AGCTTGCATGCCTGGGAAAGTCCCCTCAACTGCATG 3′
Mismatch Probe:
5′ AATTCATGCAGTTGAGGTTACTTTCCCAGGCATGCA 3′
Mismatch Complementary Sequence:
5′ AGCTTGCATGCCTGGGAAAGTAACCTCAACTGCATG 3′
4.6.4. Preparation of 3′ Labeled Probes
In separate reactions, 1.1 pmol of match or mismatch oligonucleotide was annealed to an equal amount of its complementary sequence by heating at 95°C for 1 minute and cooling slowly to room temperature. The 3′ ends were radiolabeled with 32P using Sequenase Version 2.0 (Amersham) and α32P dATP (PerkinElmer). The resulting labeled probes were purified with G-50 Microspin Columns (Amersham). Probe concentration was estimated at 0.8 ng/μl assuming 100% recovery based on a calculated molecular weight of 24,662 g/mol. Just prior to use, labeled probes were diluted in water to 0.1 ng/μl.
4.6.5. Gelshift Screen
Dry HPLC purified aliquots of polyamide were dissolved in water and their concentration determined by measuring the Absorbance at 310 nm (ε = 69,520 M−1 C−1). 10× solutions (1 μM and 100 nM) of each polyamide were prepared from serial dilutions of the concentrated stock. Binding reactions contained 0.1 ng labeled probe, 10 mM Tris·HCl pH 7.5, 50 mM NaCl, 10% glycerol, 1% NP-40, 1 mM EDTA, 0.1 mg/ml polydIdC, 1 μl 10× polyamide (final concentration 10 nM or 100 nM), and 3 μg (1–2 μl) nuclear extract in a total volume of 10 μl. Reactions were allowed to equilibriate at room temperature without nuclear extract for 3 hours, followed by addition of protein. After a further 30 min at room temperature, the reactions were loaded and resolved on non-denaturing 5% acrylamide, 5% glycerol gels for 2–2.5 hours at 150 volts. The gels were immediately dried and exposed to a phosphoimage storage plate for at least 8 hours. The gel and running buffer was 24.8 mM Tris Base, 190 mM glycine, 1 mM 0.5 M EDTA. Gels were pre-run for 15 to 45 minutes prior to loading.
4.6.6. Gelshift Titration
Reactions were prepared as described above with a constant 0.1 ng probe and 3 μg nuclear extract, except that 10× solutions of polyamide were prepared to give a final concentration range of 500 pM to 500 nM.
4.6.7. Data Analysis
A Typhoon 8600 Variable Mode Imager was used to visualize the gels and band intensity was quantified using ImageQuant software version 5.1 from Molecular Dynamics. The fraction of DNA bound to NF-κB was calculated as the intensity ratio of the NF-κB shifted band to the sum total of all bands, shifted and unshifted. EC50 values were determined graphically as the polyamide concentration required to reduce NF-κB band intensity to half its value. The data was plotted vs. polyamide concentration and fit to the Hill equation (n=1) using Kaleidagraph software.
Acknowledgments
We thank Eric J. Fechter for the preparation of plasmid pEF16 and Thomas Leung for valuable discussions. We thank The National Institutes of Health for grant support and a NRSA predoctoral training grant to J.A.P. We also thank the Parsons Foundation for a fellowship to M.A.M.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.(a) Olenyuk BZ, Zhang G, Klco JM, Nickols NG, Kaelin WG, Jr, Dervan PB. Proc Natl Acad Sci USA. 2004;101:16768. doi: 10.1073/pnas.0407617101. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Darnell JE. Nat Rev Cancer. 2002;2:740–748. doi: 10.1038/nrc906. [DOI] [PubMed] [Google Scholar]; (c) Pandolfi PP. Oncogene. 2001;20:3116–3127. doi: 10.1038/sj.onc.1204299. [DOI] [PubMed] [Google Scholar]; (d) Dervan PB, Edelson BS. Curr Opin Struct Biol. 2003;13:284–299. doi: 10.1016/s0959-440x(03)00081-2. [DOI] [PubMed] [Google Scholar]
- 2.(a) Finlay AC, Hochstein FA, Sobin BA, Murphy FX. J Am Chem Soc. 1951;73:341–343. [Google Scholar]; (b) Arcamone FNV, Penco S, Orezzi P, Nicolella V, Pirelli A. Nature. 1964;203:1064–1065. doi: 10.1038/2031064a0. [DOI] [PubMed] [Google Scholar]
- 3.(a) Kopka ML, Yoon C, Goodsell D, Pjura P, Dickerson RE. Proc Natl Acad Sci USA. 1985;82:1376–1380. doi: 10.1073/pnas.82.5.1376. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Pelton JG, Wemmer DE. Proc Natl Acad Sci USA. 1989;86:5723–5727. doi: 10.1073/pnas.86.15.5723. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Wemmer DE. Annu Rev Biomol Struct. 2000;29:439–461. doi: 10.1146/annurev.biophys.29.1.439. [DOI] [PubMed] [Google Scholar]; (d) Buchmueller KL, Staples AM, Howard CM, Horick SM, Uthe PB, Minh Le N, Cox KK, Nguyen B, Pacheco KAO, Wilson DW, Lee M. J Am Chem Soc. 2004;127:742–750. doi: 10.1021/ja044359p. [DOI] [PubMed] [Google Scholar]; (e) Baraldi PG, Bovero A, Fruttarolo F, Preti D, Tabrizi MA, Pavani MG, Romagnoli R. Med Res Rev. 2004;24:475–528. doi: 10.1002/med.20000. [DOI] [PubMed] [Google Scholar]; (f) Reddy PM, Toporowski JW, Kahane AL, Bruice TC. Bioorg Med Chem Lett. 2005;15:5531–5536. doi: 10.1016/j.bmcl.2005.08.076. [DOI] [PubMed] [Google Scholar]
- 4.White S, Szewczyk JW, Turner JM, Baird EE, Dervan PB. Nature. 1998;391:468–471. doi: 10.1038/35106. [DOI] [PubMed] [Google Scholar]
- 5.(a) Kielkopf CL, White S, Szewcyzk JW, Turner JM, Baird EE, Dervan PB, Rees DC. Science. 1998;282:111–115. doi: 10.1126/science.282.5386.111. [DOI] [PubMed] [Google Scholar]; (b) Kielkopf CL, Bremer RE, White S, Szewczyk JW, Turner JM, Baird EE, Dervan PB, Rees DC. J Mol Biol. 2000;295:557–567. doi: 10.1006/jmbi.1999.3364. [DOI] [PubMed] [Google Scholar]
- 6.(a) Urbach AR, Szewczyk JW, White S, Turner JM, Baird EE, Dervan PB. J Am Chem Soc. 1999;121:11621–11629. [Google Scholar]; (b) White S, Turner JM, Szewezyk JW, Baird EE, Dervan PB. J Am Chem Soc. 1999;121:260–261. [Google Scholar]
- 7.(a) Hays FA, Teegarden A, Jones ZJR, Harms M, Raup D, Watson J, Cavaliere E, Shing Ho P. Proc Natl Acad Sci USA. 2005;102:7157–7162. doi: 10.1073/pnas.0409455102. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Beveridge DL, Barreiro G, Byun KS, Case DA, Cheatham TE, 3rd, Dixit SB, Giudice E, Lankas F, Lavery R, Maddocks JH, Osman R, Seibert E, Sklenar H, Stoll G, Thayer KM, Varnai P, Young MA. Biophys J. 2004;87:3799–3813. doi: 10.1529/biophysj.104.045252. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Dixit SB, Beveridge DL, Case DA, Cheatham TE, 3rd , Giudice E, Lankas F, Lavery R, Maddocks JH, Osman R, Sklenar H, Thayer KM, Varnai P. Biophys J. 2005;89:3721–3740. doi: 10.1529/biophysj.105.067397. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Wu H, Crothers DM. Nature. 1984;308:509. doi: 10.1038/308509a0. [DOI] [PubMed] [Google Scholar]; (e) Steitz TA. Annu Rev Biophys. 1990;23:205. doi: 10.1017/s0033583500005552. [DOI] [PubMed] [Google Scholar]; (f) Goodsell DS, Kopka ML, Cascio D, Dickerson RE. Proc Natl Sci USA. 1993;90:2930. doi: 10.1073/pnas.90.7.2930. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Paolella DN, Palmer R, Schepartz A. Science. 1994;264:1130. doi: 10.1126/science.8178171. [DOI] [PubMed] [Google Scholar]; (h) Kahn JD, Yun E, Crothers DM. Nature. 1994;368:163. doi: 10.1038/368163a0. [DOI] [PubMed] [Google Scholar]; (h) Geierstanger BH, Wemmer DE. Annu Rev Biochem. 1995;24:463. doi: 10.1146/annurev.bb.24.060195.002335. [DOI] [PubMed] [Google Scholar]; (i) Hansen MR, Hurley LH. Acc Chem Res. 1996;29:249. [Google Scholar]; (j) Turner JM, Swalley SE, Baird EE, Dervan PB. J Am Chem Soc. 1998;120:6219–6226. [Google Scholar]; (k) Marques MA, Doss RM, Urbach AR, Dervan PB. Helv Chim Acta. 2002;85:4485–4517. [Google Scholar]
- 8.Briehn CA, Weyermann P, Dervan PB. Chem Eur J. 2003;9:2110–2122. doi: 10.1002/chem.200204689. [DOI] [PubMed] [Google Scholar]
- 9.(a) Renneberg D, Dervan PB. J Am Chem Soc. 2003;125:5707–5716. doi: 10.1021/ja0300158. [DOI] [PubMed] [Google Scholar]; (b) Marques MA, Doss RM, Foister S, Dervan PB. J Am Chem Soc. 2004;126:10339–10349. doi: 10.1021/ja0486465. [DOI] [PubMed] [Google Scholar]
- 10.Doss RM, Marques MA, Foister S, Chenoweth DM, Dervan PB. J Am Chem Soc. 2006;128:9074–9079. doi: 10.1021/ja0621795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Viger A, Dervan PB. Bioorg Med Chem. 2006 doi: 10.1016/j.bmc.2006.08.028. In Press. [DOI] [PubMed] [Google Scholar]
- 12.(a) Natoli G, Saccani S, Bosisio D, Marazzi I. Nat Immunol. 2005;6:439. doi: 10.1038/ni1196. [DOI] [PubMed] [Google Scholar]; (b) Karin M, Greten FR. Nature Rev Immunol. 2005;5:749. doi: 10.1038/nri1703. [DOI] [PubMed] [Google Scholar]; (c) Aggarwal BB. Cancer Cell. 2004;6:203. doi: 10.1016/j.ccr.2004.09.003. [DOI] [PubMed] [Google Scholar]; (d) Pande V, Ramos MJ. Curr Med Chem. 2005;12:357. doi: 10.2174/0929867053363180. [DOI] [PubMed] [Google Scholar]
- 13.Chen FE, Huang DB, Chen YQ, Ghosh G. Nature. 1998;391(6665):410–413. doi: 10.1038/34956. [DOI] [PubMed] [Google Scholar]
- 14.(a) Swalley SE, Baird EE, Dervan PB. J Am Chem Soc. 1996;118(35):8198–8206. [Google Scholar]; (b) Swalley SE, Baird EE, Dervan PB. J Am Chem Soc. 1997;119(30):6953–6961. [Google Scholar]
- 15.Wurtz NR, Pomerantz JL, Baltimore D, Dervan PB. Biochemistry. 2002;41(24):7604–7609. doi: 10.1021/bi020114i. [DOI] [PubMed] [Google Scholar]
- 16.(a) Briehn CA, Weyermann P, Dervan PB. Chem Eur J. 2003;9:2110. doi: 10.1002/chem.200204689. [DOI] [PubMed] [Google Scholar]; (b) Renneberg D, Dervan PB. J Am Chem Soc. 2003;125:5707. doi: 10.1021/ja0300158. [DOI] [PubMed] [Google Scholar]; (c) Marques MA, Doss RM, Foister S, Dervan PB. J Am Chem Soc. 2004;126:10339. doi: 10.1021/ja0486465. [DOI] [PubMed] [Google Scholar]; (d) Foister S, Marques MA, Doss RM, Dervan PB. Bioorg Med Chem. 2003;11:4333. doi: 10.1016/s0968-0896(03)00502-9. [DOI] [PubMed] [Google Scholar]
- 17.Trauger JW, Dervan PB. Methods Enzymol. 2001;340:450. doi: 10.1016/s0076-6879(01)40436-8. [DOI] [PubMed] [Google Scholar]
- 18.Austin MW, Blackbor, Ridd JH, Smith BV. J Chem Soc. 1965:1051. [Google Scholar]
- 19.Baird EE, Dervan PB. J Am Chem Soc. 1996;118:6141. [Google Scholar]
- 20.(a) Qu XG, Ren JS, Riccelli PV, Benight AS, Chaires JB. Biochemistry. 2003;42:11960. doi: 10.1021/bi0347813. [DOI] [PubMed] [Google Scholar]; (b) Urbach AR, Love JJ, Ross SA, Dervan PB. J Mol Biol. 2002;320(1):55. doi: 10.1016/S0022-2836(02)00430-8. [DOI] [PubMed] [Google Scholar]; (c) Melander C, Herman DM, Dervan PB. Chem Eur J. 2000;6:4487. doi: 10.1002/1521-3765(20001215)6:24<4487::aid-chem4487>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- 21.Haq I, Ladbury JE, Chowdhry BZ, Jenkins TC, Chaires JB. J Mol Biol. 1997;271:244. doi: 10.1006/jmbi.1997.1170. [DOI] [PubMed] [Google Scholar]
- 22.McKay SL, Haptonstall B, Gellman SH. J Am Chem Soc. 2001;123:1244. doi: 10.1021/ja003256d. [DOI] [PubMed] [Google Scholar]
- 23.Ji YH, Bur D, Hasler W, Schmitt VR, Dorn A, Bailly C, Waring MJ, Hochstrasser R, Leupin W. Bioorg Med Chem. 2001;9:2905. doi: 10.1016/s0968-0896(01)00170-5. [DOI] [PubMed] [Google Scholar]
- 24.Tisne C, Hartmann B, Delepierre M. Biochemistry. 1999;38:3883. doi: 10.1021/bi982402d. [DOI] [PubMed] [Google Scholar]
- 25.Huang D, Phelps CB, Fusco AJ, Ghosh G. J Mol Biol. 2005;346:147. doi: 10.1016/j.jmb.2004.11.042. [DOI] [PubMed] [Google Scholar]