

Abstract
Increased levels of inducible nitric oxide synthase (iNOS) during cardiac stress such as ischemia-reperfusion, sepsis and hypertension may display both beneficial and detrimental roles in cardiac contractile performance. However, the precise role of iNOS in the maintenance of cardiac contractile function remains elusive. This study was designed to determine the impact of chronic iNOS inhibition on cardiac contractile function and the underlying mechanism involved with a special focus on the NO downstream signaling molecule Akt. Male C57 or Akt2 knockout [Akt2(−/−)] mice were injected with the specific iNOS inhibitor 1400W (2 mg/kg/d) or saline for 7 days. Both 1400W and Akt2 knockout dampened glucose and insulin tolerance without additive effects. Treatment of 1400W decreased heart and liver weights as well as cardiomyocyte cross-sectional area in C57 but not Akt2 knockout mice. 1400W but not Akt2 knockout compromised cardiomyocyte mechanical properties including decreased peak shortening and maximal velocity of shortening/relengthening, prolonged relengthening duration, reduced intracellular Ca2+ release and decay rate, the effects of which were ablated or attenuated by Akt2 knockout. Akt2 knockout but not 1400W increased the levels of intracellular Ca2+ regulatory proteins including SERCA2a and phospholamban phosphorylation. 1400W reduced the level of anti-apoptotic protein Bcl-2, the effect of which was unaffected by Akt2 knockout. Neither 1400W nor Akt2 knockout significantly affected ER stress, autophagy, the post-insulin receptor signaling Akt, GSK3β and AMPK, as well as the stress signaling IΚB, JNK, ERK and p38 with the exception of elevated IΚB phosphorylation with jointed effect of 1400W and Akt2 knockout. Taken together, these data indicated that an essential role of iNOS in the maintenance of cardiac morphology and function possibly through an Akt2-dependent mechanism.
Keywords: Akt, nitric oxide, myocardial contractile function, intracellular Ca2+
INTRODUCTION
Production of nitric oxide (NO) is mainly carried out by three distinct isoforms of nitric oxide synthase (NOS) namely endothelial NOS (eNOS), neuronal NOS (nNOS), and inducible NOS (iNOS). eNOS and nNOS are constitutively expressed while iNOS is usually induced upon inflammatory stimuli (Palmer et al., 1987). Recent evidence has suggested a pivotal role of NO in insulin signaling (Carvalho-Filho et al., 2005) and maintenance of cardiac contractile function (Ziolo et al., 2008). Under pathological states such as insulin resistance, iNOS expression may be increased leading to S-nitrosylation of key insulin signaling proteins and dampened insulin signal transduction (Carvalho-Filho et al., 2005), a process considered to be responsible for nitrosative stress triggered by the NO reactive product peroxynitrite (ONOO−) (Ceriello et al., 2002). However, little is known with regards to the precise role for iNOS on insulin signaling and cardiac function under otherwise healthy status. To this end, this study was designed to test the hypothesis that basal iNOS activity is permissive to proper insulin signaling and maintenance of cardiac function. To delineate the role of iNOS in cardiac contractile function and insulin signaling, as well as the potential role of the NO downstream signaling Akt in iNOS-mediated responses, the specific iNOS inhibitor N-[[3-(aminomethyl)phenyl]methyl]-ethanimidamide, dihydrochloride (1400W) was administered to for 7 days prior to assessment of insulin and glucose tolerance, cardiac histology and cardiac mechanical function. A unique murine model of Akt2 knockout was employed to assess the role of Akt (with Akt2 being the predominant Akt isoform in the heart) (Hers et al., 2011) in the iNOS inhibition-induced cardiac responses, if any. Akt2 knockout was found to display normal cardiac phenotype albeit with a global insulin resistance (Cho et al., 2001). To elucidate the underlying mechanism(s) involved in 1400W and/or Akt2 knockout-induced cardiac responses, levels of apoptotic, endoplasmic reticulum (ER) stress and autophagic protein markers were examined. In addition, levels of key intracellular Ca2+ regulatory proteins including sarco(endo)plasmic reticulum-Ca2+-ATPase (SERCA) and phospholamban, post-insulin receptor signaling molecules Akt, glycogen synthase kinase 3β (GSK3β) and AMP-dependent protein kinase (AMPK) as well as the essential cell stress signaling molecules including IΚB, the specific inhibitor of nuclear factor NFΚB, c-Jun N-terminal kinase (JNK), extracellular signal-regulated kinase (ERK) and p38 mitogen-activated protein kinase (MAPK), were closely monitored in both wild-type control and Akt2 knockout mice with or without chronic 1400W treatment.
MATERIALS AND METHODS
Experimental animals, glucose and insulin tolerance tests
All animal procedures described were approved by the University of Wyoming Animal Use and Care Committee. The Akt2 knockout mice were obtained from Professor Morris Birnbaum at the University of Pennsylvania (Philadelphia, PA) and were characterized in detailed previously (Cho et al., 2001). All mice were maintained on a 12:12-hr light-dark illumination cycle and allowed food and water ad libitum. Adult age- and gender-matched Akt2 knockout mice and their littermates without Akt2 knockout (used as controls) were injected with either the iNOS inhibitor 1400W (2 mg/kg/d, s.c.) or saline for 7 consecutive days (Nagareddy et al., 2005; Nagareddy et al., 2009). Intraperitoneal glucose tolerance test (IPGTT) was performed (Dong et al., 2007). In brief, following the drug or saline treatment, mice fasted overnight were given an injection of glucose (2 mg/kg, i.p.). For intraperitoneal insulin tolerance test (IPITT), fed mice were given an injection of insulin (1.5 U/kg, i.p.) (Kandadi et al., 2011). For both IPGTT and IPITT tests, blood samples were taken from tail veins immediately before the injection and at 15, 30, 60, and 120 min thereafter. Blood glucose levels were measured using an AccuCheck glucose monitor (Roche Diagnostics, Indianapolis, IN).
Echocardiographic assessment
Cardiac geometry and function were evaluated in anesthetized (Avertin 2.5%, 10 μl/g bw, i.p.) mice using a 2-D guided M-mode Sonos 5500 echocardiography (Phillips Medical Systems, Andover, MD) equipped with a 15–6 MHz linear transducer. Hearts were imaged in 2-D mode in the parasternal long-axis view with a depth of 2 cm. A M-mode cursor was then positioned perpendicular to interventricular septum and posterior wall of the left ventricular (LV) at the level of the papillary muscles in the 2-D mode. The sweep speed was 100 mm/sec at the M-mode. Diastolic wall thickness, LV end diastolic dimension (EDD) and LV end systolic dimension (ESD) were measured from leading edge to leading edge in accordance with the Guidelines of the American Society of Echocardiography (Manning et al., 1994). LV fractional shortening was calculated as [(EDD-ESD)/EDD] × 100. Heart rates were averaged over 10 cardiac cycles (Turdi et al., 2011).
Myocardial histology
Paraffin slides were prepared as described (Doser et al., 2009). In brief, following anesthesia, hearts were excised, washed briefly in PBS, and placed in 10% buffered formalin at room temperature for 24 hrs. Hearts were then embedded in paraffin and cut into 5-μm sections. Slides were depariffinized, rinsed once with PBS, and incubated with 0.1 mg/ml Lectin-FITC conjugate (Sigma-Aldrich) for 2 hrs at room temperature in the dark (Wang et al., 2009). Slides were washed with PBS and mounted with the Prolong® Gold Antifade Reagent (Invitrogen, Carlsbad, CA). Cross sectional area was measured using Image J software.
Isolation of murine cardiomyocytes
Cardiomyocytes were enzymatically isolated as described (Turdi et al., 2011). Briefly, hearts were removed and perfused (37°C) with oxygenated (5% CO2–95% O2) Krebs–Henseleit bicarbonate (KHB) buffer containing (in mM) 118 NaCl, 4.7 KCl, 1.2 MgSO4, 1.2 KH2PO4, 25 NaHCO3, 10 HEPES and 11.1 glucose. Hearts were subsequently perfused with a Ca2+-free KHB-buffer that containing Liberase Blendzyme (10 mg/ml; Roche, Indianapolis, IN) for 15 min. After perfusion, left ventricles were removed and minced to disperse the individual cardiomyocytes in Ca2+-free KHB-buffer. Extracellular Ca2+ was added incrementally to 1.25 mM. Myocytes with obvious sarcolemmal blebs or spontaneous contractions were not used. Only rod-shaped myocytes with clear edges were selected for mechanical study.
Cell shortening/relengthening
Mechanical properties of cardiomyocytes were assessed using a SoftEdge MyoCam system (IonOptix Corporation, Milton, MA) (Turdi et al., 2011). Cardiomyocytes were placed in a chamber mounted on the stage of an inverted microscope (Olympus, IX-70) and superfused at 25°C with a buffer containing (in mmol/l): 131 NaCl, 4 KCl, 1 CaCl2, 1 MgCl2, 10 Glucose and 10 HEPES, at pH 7.4. Cells were field-stimulated with suprathreshold voltage at the frequency of 0.5 Hz unless otherwise stated, with a 3 msec duration, using a pair of platinum wires placed on opposite sides of the chamber and connected to an electrical stimulator (FHC Inc, Brunswick, NE). The myocyte being studied was displayed on a computer monitor using an IonOptix MyoCam camera. IonOptix SoftEdge software was used to capture changes in cell length during shortening and relengthening. Cell shortening and relengthening were assessed using the following indices: peak shortening (PS), maximal velocities of cell shortening and relengthening (± dL/dt), time-to-PS (TPS), time-to-90% relengthening (TR90).
Intracellular Ca2+ transients
Separate cohorts of myocytes were loaded with fura-2/AM (0.5 μM) for 15 min, and fluorescence intensity was measured with a dual-excitation fluorescence photomultiplier system (IonOptix). Myocytes were placed on an inverted Olympus microscope and imaged through a Fluor 40x-oil objective. Cells were exposed to light emitted by a 75 W mercury lamp and passed through either a 360 nm or a 380 nm filter. The myocytes were stimulated to contract at 0.5 Hz. Fluorescence emissions were detected between 480 nm and 520 nm by a photomultiplier tube after cells were first illuminated at 360 nm for 0.5 sec and then at 380 nm for the duration of the recording protocol (333 Hz sampling rate). The 360 nm excitation scan was repeated at the end of the protocol, and qualitative changes in intracellular Ca2+ concentration were inferred from the ratio of the fluorescence intensity at two wavelengths. Intracellular Ca2+ decay rate was calculated from single exponential curve fitting (Turdi et al., 2011).
Western blot analysis
Expression of the essential autophagic markers Beclin-1 and LC3, the intracellular Ca2+ regulatory proteins sarco(endo)plasmic reticulum Ca2+-ATPase 2a (SERCA2a) and phospholamban, the endoplasmic reticulum (ER) stress marker Bip, the apoptosis marker BCL-xL, and Bcl-2 and the post-insulin receptor signaling Akt, GSK3β and AMPK as well as the stress signaling molecules IΚB, JNK, ERK and p38 were examined by Western blot analysis. The protein was prepared as described (Turdi et al., 2011). Samples containing equal amount of proteins were separated on 10% SDS-polyacrylamide gels in a minigel apparatus (Mini-PROTEAN II, Bio-Rad, Hercules, CA) before being transferred to nitrocellulose membranes. The membranes were blocked with 5% milk in TBS-T, and were incubated overnight at 4°C with anti-Beclin-1, anti-LC3, anti-SERCA2a, anti-phospholamban (PLB), anti-phosphorylated phospholamban (pPLB), anti-Bip, anti-BCL-xL, anti-Bcl-2, anti-Akt (total), anti-phosphorylated Akt (pAkt), anti-GSK3β, anti-phosphorylated GSK3β (pGSK3β), anti-AMPK, anti-phosphorylated AMPK (pAMPK), anti-IκB, anti-phosphorylated IκB (pIΚB), anti-JNK, anti-phosphorylated JNK (pJNK), anti-ERK, anti-phosphorylated ERK (pERK), anti-p38, anti-phosphorylated p38 (pp38) and anti-GAPDH (loading control) antibodies. Anti-SERCA2a was purchased from Affinity BioReagents (Golden, CO). Anti-phospholamban antibodies were purchased from Abcam (Cambridge, MA). All other antibodies were obtained from Cell Signaling Technology (Beverly, MA). After immunoblotting, the film was scanned and the intensity of immunoblot bands was detected with a Bio-Rad Calibrated Densitometer. GAPDH was used as the loading control.
Statistical analysis
Data were Mean ± SEM. Statistical significance (p < 0.05) for each variable was determined by a one-way ANOVA followed by the Tukey’s post hoc test.
RESULTS
Effect of iNOS inhibition on insulin and glucose homeostasis and biometrics
Given that iNOS is considered to play a somewhat controversial role in the regulation of insulin sensitivity and glucose metabolism (Bai-Feng et al., 2010; Lu et al., 2010), insulin and glucose tolerance were evaluated in control and Akt2 knockout mice treated with 1400W or saline. Data shown in Fig. 1 exhibited that both Akt2 knockout and iNOS inhibition with 1400W overtly compromised the glucose and insulin tolerance (shown in time-dependent curve and area underneath curve, p < 0.05 vs. control group) with little additive effect. While body weight and fasting blood glucose levels were comparable between the control and Akt2(−/−) mice, heart and liver weights as well as heart- or liver-to-body weight ratios were significantly decreased by 1400W treatment (p < 0.05 vs. control group). Akt2 knockout ablated the 1400W-induced changes in heart and liver weight or ratio (p < 0.05 vs. 1400W group) without eliciting any effect by itself. To the contrary, Akt2 knockout induced a subtle although significant decrease in kidney weight and size (kidney-to-body weight ratio, p < 0.05 vs. control group). The iNOS inhibitor 1400W significantly attenuated Akt2 knockout-induced effect on kidney weight and size without eliciting any effect by itself (Fig. 2, p > 0.05 between Akt2(−/−)+1400W and control groups).
Fig. 1.
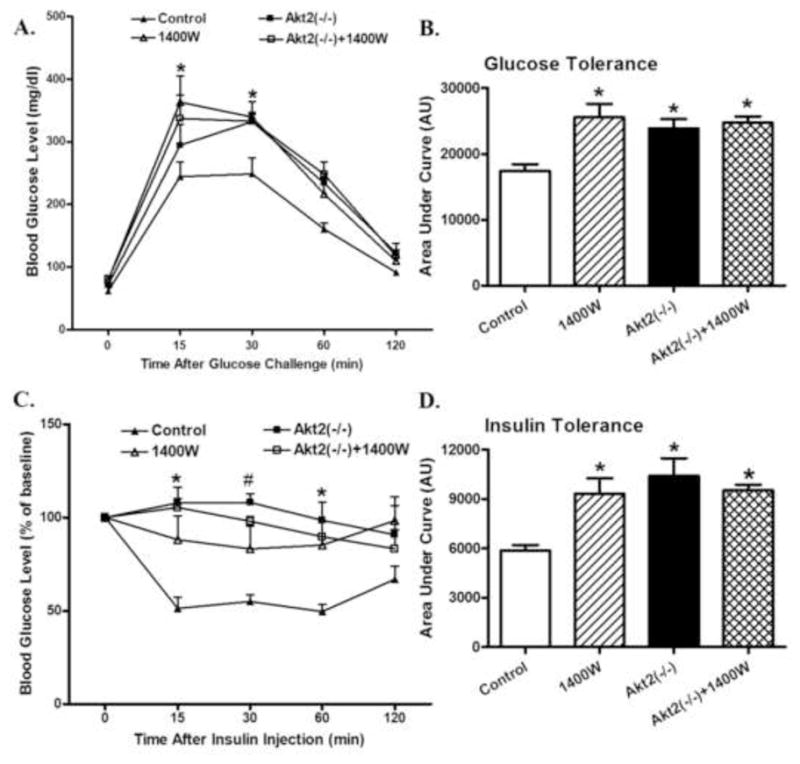
Effect of chronic iNOS inhibition using 1400W (2 mg/kg/d for 7 days, s.c.) on glucose homeostasis in control and Akt2 deficient mice. A: IPGTT following glucose challenge (2 g/kg, b.w.); B: AUC for IPGTT C: IPITT following insulin challenge (1.5 U/kg, b.w.); and D: AUC for IPITT. Mean ± SEM, n = 6–7 mice per group, * p < 0.05 vs. Control group.
Fig. 2.

Effect of chronic iNOS inhibition using 1400W (2 mg/kg/d for 7 days, s.c.) on biometric parameters in control and Akt2 deficient mice. A: Body weight; B: Fasting blood glucose; C: Heart weight; D: Heart-to- body weight ratio; E: Liver weight; F: Liver-to-body weight ratio; G: Kidney weight; and H: Kidney-to-body weight ratio. Mean ± SEM, n = 5–6 mice per group, * p < 0.05 vs. Control group, # p < 0.05 vs. 1400W group.
Effect of Akt2 knockout on iNOS inhibition-induced change in myocardial histology
To assess the impact of Akt2 knockout on myocardial histology in response to iNOS inhibition, cardiomyocyte cross-sectional area was examined using lectin staining. Data shown in Fig. 3 revealed a subtle but significant decrease in cardiomyocyte cross-sectional area following 1400W treatment (p < 0.05 vs. control group), consistent with the reduced heart weight and heart size in 1400W-treated mice. Akt2 knockout significantly attenuated 1400W-induced cardiomyocyte atrophy (p < 0.05 vs. 1400W group) without eliciting any effect by itself.
Fig. 3.

Effect chronic iNOS inhibition using 1400W (2 mg/kg/d for 7 days, s.c.) on cardiac morphology in control and Akt2 deficient mice. A: Representative images of transverse cardiac sections stained with a Lectin-FitC conjugate; and B: Quantitative analysis of cardiomyocyte cross-sectional area, Mean ± SEM, n = 150 images from 3 – 4 mice per group, * p < 0.05 vs. Control group, # p < 0.05 vs. 1400W group.
Effect of Akt2 knockout and iNOS inhibition on mechanical and intracellular Ca2+ properties of cardiomyocytes
The resting cell length was comparable among all 4 groups regardless of 1400W or Akt2 conditions. 1400W reduced peak shortening and maximal velocity of shortening/relengthening (± dL/dt), prolonged TR90 associated with similar TPS (p < 0.05 vs. control group), the effects of which were ablated or significantly attenuated by Akt2 knockout (p < 0.05 vs. 1400W group for the ablation effect). Akt2 knockout itself did not exert any notable effect on cardiomyocyte mechanics (Fig. 4). To better understand the mechanism(s) underneath Akt2 knockout-offered beneficial effects against iNOS inhibition-induced changes in cardiomyocyte contractile function, fura-2 fluorescence was monitored to evaluate intracellular Ca2+ handling properties. Cardiomyocytes from 1400W-treated mice exhibited subtle although significant drop in intracellular Ca2+ release in response to electrical stimuli (ΔFFI) and prolonged intracellular Ca2+ decay (p < 0.05 vs. control group) along with unchanged resting and peak intracellular Ca2+ levels (resting and peak FFI), the effect of which was reconciled or significantly attenuated by Akt2 knockout with the exception of reduced resting and peak FFI in 1400W-treated Akt2(−/−) mice (p < 0.05 vs. 1400W group for the reconciliation effect). Akt2 knockout itself did not alter intracellular Ca2+ homeostasis (Fig. 5).
Fig. 4.

Contractile properties of cardiomyocytes in control and Akt2 deficient mice treated with or without the iNOS inhibitor 1400W (2 mg/kg/d for 7 days, s.c.). A: Resting cell length; B: Peak shortening (PS, normalized to resting cell length); C: Maximal velocity of shortening (+ dL/dt); D: Maximal velocity of relengthening (−dL/dt); E: Time-to-PS (TPS); and F: Time-to-90% relengthening (TR90). Mean ± SEM, n = 88–110 cells per group, * p < 0.05 vs. Control group, # p < 0.05 vs. 1400W group.
Fig. 5.

Intracellular Ca2+ properties in cardiomyocytes from control and Akt2 deficient mice treated with or without the iNOS inhibitor 1400W (2 mg/kg/d for 7 days, s.c.). A: Resting fura-2 fluorescence intensity (FFI); B: Peak FFI (360/380 Ratio); C: Electrically-stimulated rise in FFI (ΔFFI); and D: Intracellular Ca2+ decay. Mean ± SEM, n = 53–55 cells per group, * p < 0.05 vs. Control group, # p < 0.05 vs. 1400W group.
Effects of Akt2 knockout and iNOS inhibition on intracellular Ca2+ regulatory, apoptotic autophagic and ER stress proteins
Western blot analysis revealed overtly upregulated levels of intracellular Ca2+ regulatory protein SERCA2a, phospholamban phosphorylation and SERCA2a-to-phospholamban ratio in myocardium from Akt2 knockout mice (p < 0.05 vs. control group). 1400W did not alter the levels of these intracellular Ca2+ regulatory proteins nor did it affect Akt2 knockout-induced responses in these proteins. Assessment of cardiac apoptotic proteins revealed that neither 1400W nor Akt2 knockout affected the expression of BCL-xL although combination of the two significantly downregulated BCL-xL expression (p < 0.05 vs. control group). 1400W overtly downregulated the level of the anti-apoptotic protein Bcl-2 (p < 0.05 vs. control group), the effect of which was unaffected by Akt2 knockout (p < 0.05 between Akt2(−/−)+1400W and control groups). Akt2 knockout itself did not affect the level of Bcl-2. Neither 1400W nor Akt2 knockout, nor combination of the both, affected the levels of the ER stress marker Bip or the autophagy markers Beclin-1 and LC3-II-to-LC3-I ratio (Fig. 6).
Fig. 6.

Western blot analysis of SERCA2a, phospholamban (pan and phosphorylated), BCL-xL, Bcl-2, Bip, Beclin-1, and LC3 in myocardium from control and Akt2 deficient mice treated with or without the iNOS inhibitor 1400W (2 mg/kg/d for 7 days, s.c.). A: Representative gel blots of SERCA2a, pan and phosphorylated phospholamban, BCL-xL, Bcl-2, Bip, Beclin-1, LC3 and GAPDH (loading control) using specific antibodies; B: SERCA2a; C: SERCA2a-to-PLB ratio; D: phosphorylated PLB (pPLB)-to-PLB ratio; E: BCL-xL; F: Bcl-2; G: Bip; H: Beclin-1; and I: LC3-II-to-LC3-I ratio. Mean ± SEM, n = 3–4 mice per group, * p < 0.05 vs. Control group, # p < 0.05 vs. 1400W group.
Effects of Akt2 knockout and iNOS inhibition on post-insulin receptor and stress signaling
Given that iNOS is implicated to affect metabolic signaling (Carvalho-Filho et al., 2005), levels of the post-insulin receptor signaling cascades including Akt, GSK3β and AMPK were examined. Our results revealed that neither chronic iNOS inhibition nor Akt2 knockout, nor combination of both, significantly affected the pan and phosphorylated levels of Akt, GSK3β and AMPK (Fig. 7). We further evaluated the effect of chronic iNOS inhibition and/or Akt2 knockout on stress signaling pathways. Data exhibited in Fig. 8 indicated that pan and phosphorylation levels of IΚB, JNK, ERK and p38 were all unaffected by 1400W, akt2 knockout, or both, with the exception of an increased IΚB phosphorylation with the combined effect of 1400W and Akt2 knockout.
Fig. 7.

Western blot analysis of pan and phosphorylated Akt, GSK3β and AMPK in myocardium from control and Akt2 deficient mice treated with or without the iNOS inhibitor 1400W (2 mg/kg/d for 7 days). A: Representative gel blots of Akt, pAkt, GSK3β, pGSK3β, AMPK, pAMPK, and GAPDH (loading control) using specific antibodies; B: pAkt-to-Akt ratio; C: pGSK3β-to-GSK3β ratio; and D: pAMPK-to-AMPK ratio. Mean ± SEM, n = 3 – 4 mice per group.
Fig. 8.

Western blot analysis of pan and phosphorylated IΚB, JNK, ERK, and p38 in myocardium from control and Akt2 deficient mice treated with or without the iNOS inhibitor 1400W (2 mg/kg/d for 7 days). A: pIΚB-to-IΚB ratio; B: pJNK-to-JNK ratio; C: pERK-to-ERK ratio; and D: pp38-to-p38 ratio. Mean ± SEM, n = 3–4 mice per group, * p < 0.05 vs. Control group, # p < 0.05 vs. 1400W group.
DISCUSSION
The salient findings from this study indicated that chronic iNOS inhibition using 1400W interrupts glucose and insulin homeostasis, disturbs cardiac histology (atrophy), cardiomyocyte contractile and intracellular Ca2+ properties. Disturbed insulin and glucose homeostasis as well as cardiac histology were accompanied by loss of heart and liver weights. Interestingly, despite the fact that Akt2 knockout itself displayed an insulin resistant phenotype as reported previously (Cho et al., 2001), it overtly attenuated or mitigated iNOS inhibition-elicited cardiac atrophy, cardiomyocyte contractile and intracellular Ca2+ anomalies without affecting overall insulin or glucose intolerance. Assessment of the essential intracellular Ca2+ cycling proteins SERCA2a and phospholamban revealed minor role of these proteins in 1400W-induced cardiomyocyte mechanical defects although Akt2 knockout itself enhanced SERCA2a levels and SERCA2a-to-phospholamban ratio, as well as phosphorylation of the SERCA lock phospholamban (favoring a better SERCA activity). Meanwhile, evaluation of protein markers for apoptosis, ER stress and autophagy did not favor a major role of these cell death and self-repair machineries in iNOS inhibition- and Akt2 knockout-induced changes in cardiac histology, contractile and intracellular Ca2+ properties. Along the same line, neither post-insulin receptor signaling (Akt, GSK3β and AMPK) nor cell stress signaling molecules (IΚB, JNK, ERK and p38) appears to play a critical role in the iNOS inhibition- and/or Akt2 knockout-elicited cardiac histological, mechanical and intracellular Ca2+ responses.
The dosage of 1400W used in our study was chosen essentially based on the previous report that the ED50 of 1400W ranges between 0.2 – 0.6 mg/kg in major organs. 1400W exhibits high isoform specificity against iNOS over other NOS isoforms (by a couple ranks of orders) and a dosage of 2 mg/kg was found sufficient to suppress iNOS while preserving isoform specificity (Garvey et al., 1997). In our hand, iNOS inhibition using 1400W elicits overt insulin resistance as evidence by IPGTT and IPITT findings. This is, however, somewhat contradictory to the finding from iNOS knockout mice where iNOS deficiency exerted little effect on insulin sensitivity (Perreault and Marette, 2001). Although it is still unclear with regards to the mechanisms underneath discrepant insulin responses between pharmacological (using 1400W) and genetic disruption of iNOS, it is possible to speculate that the sole effect of iNOS knockout in mice may be complicated by compensatory mechanisms from other NOS isoforms. Therefore, our pharmacological inhibition of iNOS using 1400W should greatly minimize the confounding effects of other NOS isoforms. Our results did not note any additive effect in glucose or insulin intolerance between Akt2 knockout and iNOS inhibition, indicating the existence of possible redundant mechanism(s) for compromised insulin signaling, such as nitrosative stress, for these two conditions (Cho et al., 2001). The decreased heart and liver weights as well as cardiomyocyte cross-sectional area may be resulted from 1400W-induced insulin resistance. Although the mechanism of action behind iNOS inhibition-induced cardiac atrophy is unclear, acute onset of insulin resistance as a result of elevated cytokines and catabolic hormones may cause cachexia and muscle wasting (Pasini et al., 2003). To the contrary, the Akt2 knockout-induced insulin resistance occurs possibly due to a mechanism distinct from that induced by iNOS inhibition. Therefore, interruption of the post-insulin receptor signaling Akt2 may not necessarily lead to a cardiac cachexia or cardiac atrophic state. Our current finding revealed contractile and intracellular Ca2+ defects in cardiomyocytes following 1400W treatment, consistent with the previous findings of cardiomyocyte contractile and intracellular Ca2+ anomalies under insulin resistance (Fang et al., 2005; Dong et al., 2006). Interestingly, levels of SERCA2a and phospholamban were not altered following 1400W treatment, not favoring a major role of these intracellular Ca2+ regulatory proteins in the compromised cardiomyocyte mechanical properties following iNOS inhibition. Despite the apparent insulin resistant phenotype, our results indicated that Akt2 knockout exerted a rather paradoxical effect on cardiac histology, cardiomyocyte contractile and intracellular Ca2+ properties following iNOS inhibition. Certain speculation may be considered for Akt2 knockout-offered protection against iNOS inhibition-induced cardiac anomalies. Our data revealed an increased phospholamban phosphorylation (favoring SERCA activation) and elevated SERCA2a expression in Akt2 knockout mice treated with or without 1400W. The augmented SERCA function should allow the Akt2 knockout mice to overcome insulin resistance-induced intracellular Ca2+ mishandling and ultimately dampened cardiomyocyte contractile function. This data are corroborated by a recent report where Akt2 knockout mouse hearts are hypersensitive to β-adrenergic stimulation (Etzion et al. 2010), which is directly responsible for the enhanced phospholamban phosphorylation (Sulakhe and Vo, 1995).
Autophagy is a mechanism to remove aberrant and dysfunctional intracellular organelles (De Meyer et al., 2010). Our data did not favor a major role of autophagy in iNOS inhibition- and/or Akt2 knockout-elicited cardiac responses. Apoptosis, on the other hand, is a hallmark of insulin resistance-induced cardiomyopathy (Ren et al., 2010). We observed reduced levels of the anti-apoptotic protein Bcl-2 (although not that of BCL-xL), favoring a pro-apoptotic effect of iNOS inhibition and thus development of cardiac atrophy, as reported earlier (Rajesh et al., 2010; Xie et al., 2011). However, given that Bcl-2 levels remained downregulated (along with reduced BCL-xL levels) in 1400W-treated Akt2 knockout mice, no conclusive role of apoptosis can be determined for Akt2 knockout-offered beneficial responses against iNOS inhibition-induced on cardiac histological, contractile and intracellular Ca2+ anomalies. Data from our study did not revealed any change in the ER stress marker Bip among all mouse groups, indicating a minimal role of ER stress in iNOS inhibition- and/or Akt2 knockout-induced cardiac responses. Moreover, data from this study failed to identify any alterations in Akt, GSK3β and AMPK signaling among mice with iNOS inhibition, Akt2 knockout, or both. GSK3β is a downstream signal of Akt while AMPK usually displays a reciprocal relationship with Akt in the regulation of glucose and insulin homeostasis under both physiological and pathological conditions (Penumathsa et al., 2008; Leng et al., 2010; Turdi et al., 2011). Although activation of the stress pathways p38, JNK, ERK, and IΚB (negatively regulates the nuclear factor NFΚB) has been demonstrated to play pivotal role in the onset and development of insulin resistance (Pitocco et al., 2010; Ren et al., 2010). Nonetheless, our results did not favor a main role of these stress-signaling molecules in iNOS inhibition- and/or Akt2 knockout-induced cardiac geometric, histological, functional and intracellular Ca2+ responses. It is possible that the negative findings from 1400W may be associated with the relatively short duration of 1400W treatment while it may require longer duration of iNOS inhibition to initiate notable effect on these cell signaling pathways.
In summary, findings from our study demonstrated for the first time that iNOS plays an essential role in the maintenance of normal insulin signaling, cardiac geometry and mechanical function. Inhibition of iNOS creates an insulin resistant status, resulting in abnormalities in heart weight, histology, cardiomyocyte contractile and intracellular Ca2+ properties. Interestingly, ablation of these cardiac abnormalities by Akt2 knockout was accompanied by increased SERCA expression and phospholamban phosphorylation, suggesting a role of SERCA2a-phospholamban mediated intracellular Ca2+ cycling in Akt2 knockout-induced beneficial effect under iNOS inhibition. It is commonly accepted that enhanced iNOS expression, which occurs in metabolic diseases such as diabetes, may lead to increased production of NO and thus reactive intermediate peroxynitrite (ONOO−), leading to cardiac geometric and contractile defects (Soliman et al., 2008; Song et al., 2008). Observations from our present study favor a role of iNOS in the regulation of insulin signaling and cardiac function, thus prompting to a role of “double-edged sword“ for iNOS in insulin resistance. Further study is warranted to better elucidate the mechanism of action for iNOS-elicited regulation of insulin signaling and heart function.
Highlights.
iNOS inhibition promotes insulin resistance, anomalies in cardiac contractile function and intracellular Ca2+ handling.
Akt2 knockout attenuates iNOS inhibition-induced cardiac contractile and intracellular Ca2+ derangement.
Akt2 knockout promotes intracellular Ca2+ regulatory proteins SERCA2a and phospholamban phosphorylation.
Neither iNOS inhibition nor Akt2 knockout significantly affects post-insulin receptor and stress signaling.
Acknowledgments
NDR was supported by a graduate assistantship from the University of Wyoming IDeA Network for Biomedical Research Excellence (NIH/NCRR P20 RR016474). This work was presented in part at the 2011 Experimental Biology Meeting held in Washington, D.C.
Footnotes
DISCLOSURES - None
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bai-Feng L, Yong-Feng L, Ying C. Silencing inducible nitric oxide synthase protects rat pancreatic islet. Diabetes Res Clin Pract. 2010;89:268–275. doi: 10.1016/j.diabres.2010.05.013. [DOI] [PubMed] [Google Scholar]
- Carvalho-Filho MA, Ueno M, Hirabara SM, Seabra AB, Carvalheira JB, de Oliveira MG, Velloso LA, Curi R, Saad MJ. S-nitrosation of the insulin receptor, insulin receptor substrate 1, and protein kinase B/Akt: a novel mechanism of insulin resistance. Diabetes. 2005;54:959–967. doi: 10.2337/diabetes.54.4.959. [DOI] [PubMed] [Google Scholar]
- Ceriello A, Quagliaro L, D’Amico M, Di Filippo C, Marfella R, Nappo F, Berrino L, Rossi F, Giugliano D. Acute hyperglycemia induces nitrotyrosine formation and apoptosis in perfused heart from rat. Diabetes. 2002;51:1076–1082. doi: 10.2337/diabetes.51.4.1076. [DOI] [PubMed] [Google Scholar]
- Cho H, Mu J, Kim JK, Thorvaldsen JL, Chu Q, Crenshaw EB, 3rd, Kaestner KH, Bartolomei MS, Shulman GI, Birnbaum MJ. Insulin resistance and a diabetes mellitus-like syndrome in mice lacking the protein kinase Akt2 (PKB beta) Science. 2001;292:1728–1731. doi: 10.1126/science.292.5522.1728. [DOI] [PubMed] [Google Scholar]
- De Meyer GR, De Keulenaer GW, Martinet W. Role of autophagy in heart failure associated with aging. Heart Fail Rev. 2010;15:423–430. doi: 10.1007/s10741-010-9166-6. [DOI] [PubMed] [Google Scholar]
- Dong F, Yang X, Sreejayan N, Ren J. Chromium (D-phenylalanine)3 improves obesity-induced cardiac contractile defect in ob/ob mice. Obesity (Silver Spring) 2007;15:2699–2711. doi: 10.1038/oby.2007.322. [DOI] [PubMed] [Google Scholar]
- Dong F, Zhang X, Yang X, Esberg LB, Yang H, Zhang Z, Culver B, Ren J. Impaired cardiac contractile function in ventricular myocytes from leptin-deficient ob/ob obese mice. J Endocrinol. 2006;188:25–36. doi: 10.1677/joe.1.06241. [DOI] [PubMed] [Google Scholar]
- Doser TA, Turdi S, Thomas DP, Epstein PN, Li SY, Ren J. Transgenic overexpression of aldehyde dehydrogenase-2 rescues chronic alcohol intake-induced myocardial hypertrophy and contractile dysfunction. Circulation. 2009;119:1941–1949. doi: 10.1161/CIRCULATIONAHA.108.823799. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Etzion S, Etzion Y, DeBosch B, Crawford PA, Muslin AJ. Akt2 deficiency promotes cardiac induction of Rab4a and myocardial beta-adrenergic hypersensitivity. J Mol Cell Cardiol. 49:931–940. doi: 10.1016/j.yjmcc.2010.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang CX, Dong F, Ren BH, Epstein PN, Ren J. Metallothionein alleviates cardiac contractile dysfunction induced by insulin resistance: role of Akt phosphorylation, PTB1B, PPARgamma and c-Jun. Diabetologia. 2005;48:2412–2421. doi: 10.1007/s00125-005-1940-y. [DOI] [PubMed] [Google Scholar]
- Garvey EP, Oplinger JA, Furfine ES, Kiff RJ, Laszlo F, Whittle BJ, Knowles RG. 1400W is a slow, tight binding, and highly selective inhibitor of inducible nitric-oxide synthase in vitro and in vivo. J Biol Chem. 1997;272:4959–4963. doi: 10.1074/jbc.272.8.4959. [DOI] [PubMed] [Google Scholar]
- Hers I, Vincent EE, Tavare JM. Akt signalling in health and disease. Cell Signal. 2011;23:1515–1527. doi: 10.1016/j.cellsig.2011.05.004. [DOI] [PubMed] [Google Scholar]
- Kandadi MR, Unnikrishnan MK, Warrier AK, Du M, Ren J, Sreejayan N. Chromium (D-phenylalanine)3 alleviates high fat-induced insulin resistance and lipid abnormalities. J Inorg Biochem. 2011;105:58–62. doi: 10.1016/j.jinorgbio.2010.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leng S, Zhang W, Zheng Y, Liberman Z, Rhodes CJ, Eldar-Finkelman H, Sun XJ. Glycogen synthase kinase 3 beta mediates high glucose-induced ubiquitination and proteasome degradation of insulin receptor substrate 1. J Endocrinol. 2010;206:171–181. doi: 10.1677/JOE-09-0456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu M, Li P, Pferdekamper J, Fan W, Saberi M, Schenk S, Olefsky JM. Inducible nitric oxide synthase deficiency in myeloid cells does not prevent diet-induced insulin resistance. Mol Endocrinol. 2010;24:1413–1422. doi: 10.1210/me.2009-0462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning WJ, Wei JY, Katz SE, Litwin SE, Douglas PS. In vivo assessment of LV mass in mice using high-frequency cardiac ultrasound: necropsy validation. Am J Physiol. 1994;266:H1672–1675. doi: 10.1152/ajpheart.1994.266.4.H1672. [DOI] [PubMed] [Google Scholar]
- Nagareddy PR, McNeill JH, MacLeod KM. Chronic inhibition of inducible nitric oxide synthase ameliorates cardiovascular abnormalities in streptozotocin diabetic rats. Eur J Pharmacol. 2009;611:53–59. doi: 10.1016/j.ejphar.2009.03.061. [DOI] [PubMed] [Google Scholar]
- Nagareddy PR, Xia Z, McNeill JH, MacLeod KM. Increased expression of iNOS is associated with endothelial dysfunction and impaired pressor responsiveness in streptozotocin-induced diabetes. American journal of physiology. 2005;289:H2144–2152. doi: 10.1152/ajpheart.00591.2005. [DOI] [PubMed] [Google Scholar]
- Palmer RM, Ferrige AG, Moncada S. Nitric oxide release accounts for the biological activity of endothelium-derived relaxing factor. Nature. 1987;327:524–526. doi: 10.1038/327524a0. [DOI] [PubMed] [Google Scholar]
- Pasini E, Aquilani R, Gheorghiade M, Dioguardi FS. Malnutrition, muscle wasting and cachexia in chronic heart failure: the nutritional approach. Ital Heart J. 2003;4:232–235. [PubMed] [Google Scholar]
- Penumathsa SV, Thirunavukkarasu M, Zhan L, Maulik G, Menon VP, Bagchi D, Maulik N. Resveratrol enhances GLUT-4 translocation to the caveolar lipid raft fractions through AMPK/Akt/eNOS signalling pathway in diabetic myocardium. J Cell Mol Med. 2008;12:2350–2361. doi: 10.1111/j.1582-4934.2008.00251.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perreault M, Marette A. Targeted disruption of inducible nitric oxide synthase protects against obesity-linked insulin resistance in muscle. Nat Med. 2001;7:1138–1143. doi: 10.1038/nm1001-1138. [DOI] [PubMed] [Google Scholar]
- Pitocco D, Zaccardi F, Di Stasio E, Romitelli F, Santini SA, Zuppi C, Ghirlanda G. Oxidative stress, nitric oxide, and diabetes. Rev Diabet Stud. 2010;7:15–25. doi: 10.1900/RDS.2010.7.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajesh M, Mukhopadhyay P, Batkai S, Patel V, Saito K, Matsumoto S, Kashiwaya Y, Horvath B, Mukhopadhyay B, Becker L, Hasko G, Liaudet L, Wink DA, Veves A, Mechoulam R, Pacher P. Cannabidiol attenuates cardiac dysfunction, oxidative stress, fibrosis, and inflammatory and cell death signaling pathways in diabetic cardiomyopathy. J Am Coll Cardiol. 2010;56:2115–2125. doi: 10.1016/j.jacc.2010.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren J, Pulakat L, Whaley-Connell A, Sowers JR. Mitochondrial biogenesis in the metabolic syndrome and cardiovascular disease. J Mol Med (Berl) 2010;88:993–1001. doi: 10.1007/s00109-010-0663-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soliman H, Craig GP, Nagareddy P, Yuen VG, Lin G, Kumar U, McNeill JH, Macleod KM. Role of inducible nitric oxide synthase in induction of RhoA expression in hearts from diabetic rats. Cardiovasc Res. 2008;79:322–330. doi: 10.1093/cvr/cvn095. [DOI] [PubMed] [Google Scholar]
- Song D, Kuo KH, Yao R, Hutchings SR, Pang CC. Inducible nitric oxide synthase depresses cardiac contractile function in Zucker diabetic fatty rats. Eur J Pharmacol. 2008;579:253–259. doi: 10.1016/j.ejphar.2007.09.043. [DOI] [PubMed] [Google Scholar]
- Sulakhe PV, Vo XT. Regulation of phospholamban and troponin-I phosphorylation in the intact rat cardiomyocytes by adrenergic and cholinergic stimuli: roles of cyclic nucleotides, calcium, protein kinases and phosphatases and depolarization. Mol Cell Biochem. 1995;149–150:103–126. doi: 10.1007/BF01076569. [DOI] [PubMed] [Google Scholar]
- Turdi S, Kandadi MR, Zhao J, Huff AF, Du M, Ren J. Deficiency in AMP-activated protein kinase exaggerates high fat diet-induced cardiac hypertrophy and contractile dysfunction. J Mol Cell Cardiol. 2011;50:712–722. doi: 10.1016/j.yjmcc.2010.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D, Patel VV, Ricciotti E, Zhou R, Levin MD, Gao E, Yu Z, Ferrari VA, Lu MM, Xu J, Zhang H, Hui Y, Cheng Y, Petrenko N, Yu Y, FitzGerald GA. Cardiomyocyte cyclooxygenase-2 influences cardiac rhythm and function. Proc Natl Acad Sci U S A. 2009;106:7548–7552. doi: 10.1073/pnas.0805806106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Z, Lau K, Eby B, Lozano P, He C, Pennington B, Li H, Rathi S, Dong Y, Tian R, Kem D, Zou MH. Improvement of cardiac functions by chronic metformin treatment is associated with enhanced cardiac autophagy in diabetic OVE26 mice. Diabetes. 2011;60:1770–1778. doi: 10.2337/db10-0351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziolo MT, Kohr MJ, Wang H. Nitric oxide signaling and the regulation of myocardial function. J Mol Cell Cardiol. 2008;45:625–632. doi: 10.1016/j.yjmcc.2008.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]