

Abstract
Hepatitis C virus (HCV) NS5B polymerase is a key target for anti-HCV therapeutics development. Here we report the synthesis and biological evaluation of a new series of α,γ-diketo acids (DKAs) as NS5B polymerase inhibitors. We initiated structure-activity relationship (SAR) optimization around the furan moiety of compound 1a [IC50 = 21.8 μM] to achieve more active NS5B inhibitors. This yielded compound 3a [IC50 = 8.2 μM] bearing the 5-bromobenzofuran-2-yl moiety, the first promising lead compound of the series. Varying the furan moiety with thiophene, thiazole and indazole moieties resulted in compound 11a [IC50 = 7.5 μM] bearing 3-methylthiophen-2-yl moiety. Finally replacement of the thiophene ring with a bioisosteric phenyl ring further improved the inhibitory activity as seen in compounds 21a [IC50 = 5.2 μM] and 24a [IC50 = 2.4 μM]. Binding mode of compound 24a using glide docking within the active site of NS5B polymerase will form the basis for future SAR optimization.
Keywords: α,γ-Diketo acid; Pyrophosphate; NS5B polymerase; Docking
Introduction
Hepatitis C virus (HCV) infection has emerged as a significant global public health problem. HCV is responsible for the development of malignant chronic liver disease, including liver cirrhosis and hepatocellular carcinoma, which frequently ends in liver failure [1–4]. An estimated 200 million cases of HCV infections exist worldwide, of which over 4.1 million are in the United States [5]. Despite its large medical and economic impact, neither a vaccine nor a therapy with effective broad spectrum mode of action against all genotypes of HCV is available [6–8]. Current HCV therapy comprising of pegylated interferon α (PEG-IFN-α) in combination with ribavirin has found limited patient compliance due to severe adverse effects [9,10]. Therefore, there is an urgent need to develop novel and efficacious antiviral therapeutics targeting HCV.
The HCV non-structural protein 5B (NS5B), a 66 kDa RNA-dependent RNA polymerase (RdRp), is an attractive therapeutic target, since it plays an important role in replicating the HCV RNA genome and the host lacks its functional equivalent [11–13]. The crystal structure of HCV NS5B polymerase reveals a classical “right hand” shape with characteristic fingers, palm and thumb subdomains. The combination of crystallographic, biochemical and mutagenesis studies has facilitated the identification of five distinct non-nucleoside inhibitor (NNI) binding sites on NS5B [14–17]. Allosteric pockets, AP-1 and AP-2 are located in the thumb subdomain, whereas AP-3 and AP-4 are partially overlapped and located in the palm subdomain, in close proximity to the active site, and AP-5 is located in the fingers subdomain [17–20]. The NS5B polymerase active site is located in the palm subdomain, which contains two Mg2+ ions that are coordinated by conserved aspartic acid residues Asp220 and Asp318 [18]. The Mg2+ ions serve the dual role of positioning/stabilizing the pyrophosphate leaving group on the incoming nucleoside triphosphate (NTP) and activating the 3′-OH of the growing RNA for nucleophilic attack on the α-phosphate group of NTP [14,15].
Derivatives of 4,5-dihydroxypyrimidine carboxylic acid [19] and α,γ-diketo acids (DKAs) have been reported previously as pyrophosphate (PPi) mimetic inhibitors of HIV-1 IN [20] and HCV NS5B polymerase [19,21]. These compounds function as product-like analogues and chelate the divalent metal ions at the active site of NS5B [22,23]. In continuation of our efforts towards identification and development of NS5B PPi mimetic inhibitors [24], we have explored the effect of various substituted aryl/heteroaryl specificity domain of DKAs on NS5B inhibition. Herein we describe the synthesis, SAR, and molecular modeling of optimized DKA analogs.
Results and discussion
Chemistry
The α,γ-diketo esters (1–13, 15–20 and 22–24) were synthesized by Claisen condensation of appropriate acetyl derivative with diethyl oxalate in the presence of freshly prepared sodium ethoxide. The resulting α,γ-diketo esters were readily hydrolyzed by treating with 1N sodium hydroxide to corresponding DKAs (1a-13a, 15a–20a and 22a–24a) (Scheme 1) [25]. Target compound 14a was prepared in good yields from the 1H-indazole-3-carboxylic acid as shown in Scheme 2. The 3-acetyl-1H-indazole intermediate was synthesized from 1H-indazole-3-carboxylic acid by initial conversion into Winreb amide using the one-pot reaction: mixed anhydride method followed by nucleophilic attack of N,O-dimethylhydroxylamine hydrochloride. The Winreb amide thus formed was further treated with methyl magnesium bromide to yield 3-acetyl-1H-indazole [26]. The 3-acetyl-1H-indazole was converted to α,γ-diketo ester 14 and further converted to compound 14a as described above. Target compound 21a was prepared from 20a by reacting it with sodium azide in the presence of the catalytic amount of ammonium chloride (Scheme 3) [27].
Scheme 1.
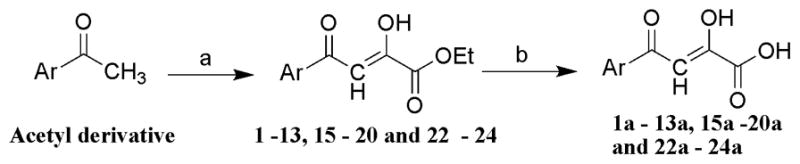
(a) diethyl oxalate, sodium ethoxide, anhydrous THF, rt, 65–86%; (b) 1 N NaOH, THF/methanol 1:1, rt, 70–86%.
Scheme 2.

(a) (i) isobutyl chloroformate, N-methylmorpholine, −20°C, 4h, (ii) N,O-dimethylhydroxylamine hydrochloride in triethylamine, rt, 6h; (b) anhydrous THF, methyl magnesium bromide (12% in THF), −78°C, 2h, rt, 5h, 55% over two steps; (c) diethyl oxalate, sodium ethoxide, anhydrous THF, rt, 78%; (d) 1 N NaOH, THF/methanol 1:1, rt, 72%.
Scheme 3.
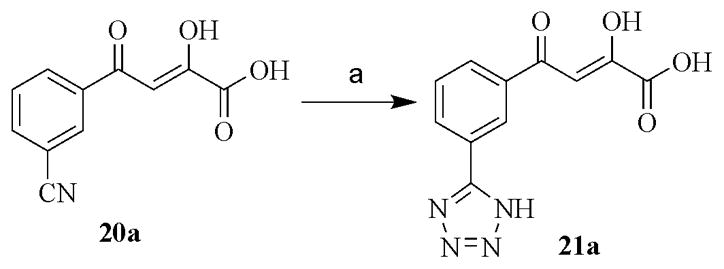
(a) ammonium chloride, sodium azide, DMF, 110°C, 6 h, 75%.
Structure-activity relationship
The HCV NS5B RdRp inhibitory activity of DKAs is reported in Table 1. All the DKAs exhibited appreciable inhibition of HCV NS5B polymerase, with IC50 values ranging from 2.4 μM to 63.9 μM. Having identified compound 1a, a modest NS5B inhibitor as the initial hit, we sought to explore SAR around the aryl/heteroaryl specificity domain while keeping the DKA metal chelating pharmacophore constant. Replacing furan moiety with benzofuran led to compound 2a which was marginally less active. Since the benzofuran ring is in conjugation with the γ-keto group, we expected that the electron withdrawing bromo group would increase the acidity of the diketo moiety and its ionized enolic form would be more suitable for strong metal chelation, thereby resulting in a relatively more active inhibitor. Consistent with this hypothesis, compound 3a bearing 5-bromo substitution on the benzofuran ring exhibited ~3.5-fold improvement in inhibitory activity over compound 2a. Bioisosteric replacement of the benzofuran moiety by benzothiophene resulted in compound 4a, with marginal loss of activity with respect to 2a. By contrast, replacement of benzothiophene (compound 4a) with thiophene (compound 5a), increased inhibitory activity by ~2.2-fold. We further explored the influence of varying substituents on the thiophene ring of compound 5a. Substitution of the methyl group at 5-position of the thiophene ring resulted in compound 6a with approximately 3-fold decrease in activity than the parent compound. Bioisosteric replacement of the methyl group with chloro (compound 7a) or bromo (compound 8a) groups resulted in ~2-fold enhancement of inhibitory activity, whereas 5-iodo derivative, compound 9a, was found to be relatively less active than the methyl analog. We also explored the effect of variation in the position of the substitution around the thiophene ring. Moving the methyl group from the 5th to the 4th-position (compound 10a), improved the activity of the compound by ~2-fold; whereas moving it to the 3rd-position yielded compound 11a with ~7-fold higher inhibitory activity. Replacement of the thiophene ring with thiazole (compound 12a) led to ~ 1.8-fold decrease in activity. Introduction of the pyrrole ring in place of the furan ring yielded compound 13a with similar activity. Introduction of the indazole moiety in place of the benzofuran (compound 14a) conferred slightly improved inhibitory activity. Among several corresponding α,γ-diketo esters tested (data not shown), only compounds 14 and 17 conferred inhibitory activity comparable to their acid counterparts, thus underscoring the observation that carboxylate anion moiety may be critical for chelating the active site divalent metal ions.
Table 1.
Structures and NS5B inhibitory activities of target compounds
 | |||
|---|---|---|---|
| Compd | Ar | R | IC50, μM |
| 1a | furan-2-yl | H | 21.8 ± 1.30 |
| 2a | benzofuran-2-yl | H | 29.6 ± 1.30 |
| 3a | 5-bromobenzofuran-2-yl | H | 8.2 ± 1.20 |
| 4a | benzothiophen-2-yl | H | 37.7 ± 1.30 |
| 5a | thiophen-2-yl | H | 17.3 ± 1.10 |
| 6a | 5-methylthiophen-2-yl | H | 55.5 ± 1.20 |
| 7a | 5-chlorothiophen-2-yl | H | 25.5 ± 0.40 |
| 8a | 5-bromothiophen-2-yl | H | 31.4 ± 1.20 |
| 9a | 5-iodothiophen-2-yl | H | 63.9 ± 1.30 |
| 10a | 4-methylthiophen-2-yl | H | 32.7 ± 1.30 |
| 11a | 3-methylthiophen-2-yl | H | 7.5 ± 1.30 |
| 12a | thiazol-2-yl | H | 30.4 ± 2.10 |
| 13a | pyrrol-2-yl | H | 21.7 ± 1.30 |
| 14 | indazol-3-yl | C2H5 | 38.4 ± 1.40 |
| 14a | indazol-3-yl | H | 24.7 ± 1.30 |
| 15a | para-biphenyl | H | 20.9 ± 1.90 |
| 16a | 2-nitrophenyl | H | 11.3 ± 1.10 |
| 17 | 2-fluorophenyl | C2H5 | 12.4 ± 1.50 |
| 17a | 2-fluorophenyl | H | 14.9 ± 1.20 |
| 18a | 2-methylphenyl | H | 6.9 ± 1.10 |
| 19a | 3-fluorophenyl | H | 49.9 ± 1.10 |
| 20a | 3-cyanophenyl | H | 39.7 ± 1.30 |
| 21a | 3-(tetrazol-2-yl)phenyl | H | 5.2 ± 1.20 |
| 22a | 2,6-difluorophenyl | H | 17.0 ± 1.20 |
| 23a | 2,4-dichlorophenyl | H | 49.7 ± 1.10 |
| 24a | 2,4-difluorophenyl | H | 2.4 ± 0.05 |
We next explored the influence of substituted phenyl ring versus the heterocyclic ring on NS5B inhibition. Para-biphenyl derivative 15a, the first analog explored, was ~4-fold less active than the previously reported unsubstituted phenyl analog (IC50 = 5.7 μM) by Summa and colleagues [28], thus suggesting possible steric hindrance by the second phenyl ring within the active site of NS5B polymerase. The 2-nitrophenyl and 2-fluorophenyl analogs (compounds 16a and 17a, respectively) were ~2-fold more active than 15a, while the 2-methylphenyl derivative 18a was comparable in activity to the unsubstituted phenyl analog [28]. Further, the 3-fluorophenyl (compound 19a) and 3-cyanophenyl (compound 20a) analogs were 7 to 8 fold less active, while the 3-(1H-tetrazol-2-yl)phenyl analog (compound 21a) was marginally more active than the unsubstituted phenyl analog [28]. The 2,6-difluorophenyl derivative 22a exhibited comparable activity to 2-fluorophenyl analog, thus suggesting that the second ortho-fluoro substituent may not be important for NS5B inhibition. The 2,4-dichlorophenyl analog (compound 23a) was 8-fold less active than the unsubstituted phenyl analog [28], while 2,4-difluorophenyl analog (compound 24a) proved to be the most active compound in the present investigation.
These studies thus shed light on the effect of various substituted aryl/heteroaryl groups in DKAs on NS5B inhibition, and have identified 2,4-difluorophenyl as a promising specificity domain for further development. To address the issue of poor cell membrane permeability of DKAs, current efforts are underway to synthesize tetrazole bioisosteres of the metal chelating carboxyl group in DKA pharmacophore. This endeavor may potentially develop more potent compounds with promising cell permeability and physicochemical properties. The results of this investigation will be the subject of a future communication.
Glide predicted binding mode of compound 24a
To understand the binding mechanism of these synthesized compounds, we generated a Glide docking model on the basis of the co-crystal structure of rUTP-NS5B polymerase. As expected, DKA pharmacophore chelates two Mn2+ ions through coordinate bonding network (Figure 1). Metal A is coordinated by the carboxylate anion of the DKA pharmacophore, while metal B is coordinated by the carboxylate anion in addition to the ionizable tautomeric α-hydroxy group. The other coordinate bonds to both metals are contributed by the conserved active site aspartic acid residues – Asp220, Asp318 and Asp319 and the backbone carbonyl oxygen atom of Thr221. The 2,4-difluorophenyl moiety is stabilized through hydrophobic interactions with the methylene groups of Arg158 and the side chain of Ile160. The 4-fluoro substituent is located within hydrogen bonding distance from the hydroxyl group of Ser282 (-F---HO Ser282, 2.5 Å). An intramolecular hydrogen bond between the γ-keto and α-hydroxyl groups forms a pseudo-six membered ring, a feature favorable to metal chelation due to increase in electron density at the oxygen atom of α-hydroxy group. The γ-keto group is located within hydrogen bonding interaction distance from the active site water molecule 2478 (C=O---HOH2478, 2.3 Å), which in turn forms a hydrogen bond with the carboxylate group of Asp225. Since 24a forms water mediated interaction with the strictly conserved Asp225, it is plausible that the virus would not easily acquire mutations to escape inhibition by such inhibitors. Indeed, mutations of Asp225 have been shown to completely inactivate the enzyme [29].
Figure 1.

Glide docking model of 24a within the active site of NS5B polymerase. Amino acid residues are shown in as stick model whereas inhibitor is shown as ball and stick model. The metal ions (Mn+2) A and B are shown in cyan color. Dotted black line indicates hydrogen bonding interaction whereas dotted red line indicates electrostatic interactions.
Conclusions
A new series of α,γ-diketo acids linked to varied aryl/heteroaryl rings was successfully developed as HCV NS5B polymerase inhibitors. The present SAR revealed the tolerance of various aryl/heteroaryl systems such as benzofuran, thiophene and benzene as the specificity domain. The 2,4-difluorophenyl ring as specificity domain (compound 24a) was found to be the most potent inhibitor of NS5B polymerase in the present study. Docking model of 24a within the active site of NS5B polymerase will serve as a rational basis for future SAR optimization.
Experimental
Chemistry-General
Melting points (mp) were determined on a Thomas-Hoover capillary melting point apparatus and are uncorrected. The reagents for organic synthesis were purchased from Aldrich Chemical Co. (Milwaukee, WI), TCI America (Portland, OR), Alfa Aesar (Ward Hill, MA), and Acros Organics (Antwerp, Belgium). All compounds were checked for homogeneity by TLC using silica gel as a stationary phase. NMR spectra were recorded on a Bruker 400 Avance DPX spectrometer (1H at 400 MHz and 13C at 100 MHz) outfitted with a z-axis gradient probe. The chemical shifts for 1H and 13C are reported in parts per million (δ ppm) downfield from the tetramethylsilane (TMS) as an internal standard. The 1H NMR data are reported as follows: chemical shift, multiplicity (s) singlet, (bs) broad singlet, (d) doublet, (t) triplet, (q) quartet, (m) multiplet. The C, H, and N analyses were performed by Atlantic Microlabs, Inc., (Norcross, GA) and the observed values were within ±0.4% of calculated values.
General procedure for the preparation of α, γ-diketo esters [25]
Following the reported procedure [25], sodium ethoxide (0.43 g, 6.30 mmol) was added into a well stirred mixture of appropriate acetyl derivatives (3.12 mmol) and diethyl oxalate (0.92 g, 6.24 mmol) in anhydrous THF (15 mL) under nitrogen atmosphere. The reaction mixture was stirred at room temperature and upon completion of the reaction, was poured into n-hexane (50 mL). The precipitates were collected and then vigorously stirred in 1N HCl (30 mL) for 30 min. The resultant solid formed was filtered, washed with water, and dried under vacuum. The crude mass obtained was further dissolved in ethyl acetate and reprecipitated with hexane to obtain desired esters 1–20 and 22–24.
General procedure for the preparation of α, γ-diketo acids [25]
The mixture of 1N NaOH (7 mL) and corresponding ester derivatives 1–20 and 22–24 (1.3 mmol) in THF/methanol 1:1 (15 mL) was stirred at room temperature for 45 min. Upon completion, the reaction mixture was concentrated under vacuum. The viscous concentrate was poured onto crushed ice and acidified with 1 N HCl to pH 1. The precipitates obtained were filtered, washed with water and dried under vacuum. The crude mass obtained was purified by crystallizing it from ethanol which yielded the desired α,γ-diketo acids 1a–20a and 22a–24a.
Ethyl 4-(furan-2-yl)-2-hydroxy-4-oxobut-2-enoate (1)
Obtained from 2-acetylfuran (0.34 g, 3.12 mmol) starting material as white solid (0.55 g, 81%); m.p. 120–122°C; Rf = 0.52 (n-hexane:EtOAc 60:40); 1H NMR (400 MHz, CDCl3, TMS) δ 14.45 (s, 1H), 7.68 (dd, J = 1.7 Hz, 0.7 Hz, 1H), 7.35 (dd, J = 3.5 Hz, 0.6 Hz, 1H), 6.94 (s, 1H), 6.62 (dd, J = 3.5 Hz, 1.6 Hz, 1H), 4.39 (q, J = 7.1 Hz, 2H), 1.41 (t, J = 7.2 Hz, 3H).
4-(Furan-2-yl)-2-hydroxy-4-oxobut-2-enoic acid (1a) [30]
Using 1 (0.27 g, 1.3 mmol) as the starting material, 1a was obtained as a pale yellow solid (0.17 g, 72%); m.p. 145–148°C; Rf = 0.42 (EtOAc: n-hexane 60:40); 1H NMR (400 MHz, DMSO-d6, TMS) δ 14.55 (bs, 2H), 8.09 (s, 1H), 7.67 (s, 1H), 6.79 (dd, J = 3.7 Hz, 1.5 Hz, 2H); 13C NMR (100 MHz; DMSO-d6; TMS) δ 99.16 (2), 113.83, 119.86, 149.48, 150.90, 163.61 (2). Anal. Calcd. for C8H6O5. 1/3H2O: C, 51.07; H, 3.57. Found: C, 51.03; H, 3.41.
Ethyl 4-(benzofuran-2-yl)-2-hydroxy-4-oxobut-2-enoate (2)
Obtained from 2-acetylbenzofuran (0.49 g, 3.12 mmol) as yellow solid (0.65 g, 80%); m.p. 80–83°C; Rf = 0.43 (n-hexane:EtOAc 70:30); 1H NMR (400 MHz, DMSO-d6, TMS) δ 12.35 (bs, 1H), 7.89 (s, 1H), 7.79 (d, J = 7.8 Hz, 1H), 7.72 (d, J = 8.2 Hz, 1H), 7.52 (t, J = 7.7 Hz, 1H), 7.35 (t, J = 7.5 Hz, 1H), 6.80 (s, 1H), 4.27 (q, J = 7.0 Hz, 2H), 1.30 (t, J = 7.1 Hz, 3H).
4-(Benzofuran-2-yl)-2-hydroxy-2-oxobuten-2-enoic acid (2a) [30]
Obtained from 2 (0.34 g, 1.3 mmol) as a bright yellow solid (0.25 g, 83%); m.p. 145–147°C; Rf = 0.31 (EtOAc: n-hexane: 60:40); 1H NMR (400 MHz, DMSO-d6, TMS) δ 14.12 (bs, 2H), 8.12 (s, 1H), 7.84 (d, J = 7.9 Hz, 1H), 7.77 (d, J = 7.4 Hz, 1H), 7.58 (t, J = 7.8 Hz, 1H), 7.39 (t, J = 7.5 Hz, 1H), 6.03 (s, 1H); 13C NMR (100 MHz, DMSO-d6, TMS) δ 99.62, 123.57, 124.61, 127.31, 127.69, 128.03, 135.61, 146.63, 155.82, 160.56, 161.59, 163.65. Anal. Calcd. for C12H8O5. 2/5 H2O: C, 60.21; H, 3.71. Found: C, 60.05; H, 3.36.
Ethyl 4-(5-bromobenzofuran-2-yl)-2-hydroxy-4-oxobut-2-enoate (3)
Obtained from 2-acetyl-5-bromobenzofuran (0.74 g, 3.12 mmol) as a yellow solid (0.79 g, 76%); m.p. 260–262°C; Rf = 0.72 (n-hexane:EtOAc 70:30); 1H NMR (400 MHz, DMSO-d6, TMS) δ 14.43 (s, 1H), 7.85 (s, 1H), 7.59 (d, J = 7.1 Hz, 1H), 7.55 (d, J = 7.0 Hz, 1H), 7.48 (d, J = 7.9 Hz, 1H), 7.09 (s, 1H), 4.42 (q, J = 7.1 Hz, 2H), 1.43 (t, J = 7.1 Hz, 3H).
4-(5-Bromobenzofuran-2-yl)-2-hydroxy-4-oxobut-2-enoic acid (3a)
Obtained from 3 (0.44 g, 1.3 mmol) as a yellow solid (0.32 g, 80%); m.p. 270–273°C; Rf = 0.40 (EtOAc: n-hexane 60:40); 1H NMR (400 MHz, DMSO-d6, TMS) δ 13.74 (bs, 2H), 8.03 (d, J = 6.8 Hz, 2H), 7.72 (d, J = 7.7 Hz, 1H), 7.60 (d, J = 7.6 Hz, 1H), 7.00 (s, 1H); 13C NMR (100 MHz, DMSO-d6, TMS) δ 99.53, 114.08, 114.85, 116.93, 126.31, 129.63, 131.74, 152.27, 154.66, 163.36, 168.28, 179.94. Anal. Calcd. for C12H7BrO5: C, 46.33; H, 2.27. Found: C, 46.45; H, 2.40.
Ethyl 4-(benzo[b]thiophen-2-yl)-2-hydroxy-4-oxobut-2-enoate (4)
Obtained from 2-acetylbenzo[b]thiophene (0.55 g, 3.12 mmol) as a yellow solid (0.68 g, 79%); m.p. 190–194°C; Rf = 0.65 (n-hexane:EtOAc 60:40); 1H NMR (400 MHz, DMSO-d6, TMS) δ 13.96 (bs, 1H), 8.71 (s, 1H), 8.08 (d, J = 7.2 Hz, 1H), 8.01 (d, J = 7.2 Hz, 1H), 7.54 (t, J = 7.1 Hz, 1H), 7.50 (t, J = 7.1 Hz, 1H), 7.21 (s, 1H), 4.34 (q, J = 7.1 Hz, 2H), 1.33 (t, J = 7.1 Hz, 3H).
4-(Benzo[b]thiophen-2-yl)-2-hydroxy-4-oxobut-2-enoic acid (4a) [30]
Obtained as a yellow solid (0.24 g, 74%) from 4 (0.36 g, 1.3 mmol) as the starting material; m.p. 212–215°C; Rf = 0.42 (EtOAc: n-hexane 60:40); 1H NMR (400 MHz, DMSO-d6, TMS) δ 14.14 (bs, 2H), 8.65 (s, 1H), 8.09 (d, J = 7.7 Hz, 1H), 8.02 (d, J = 7.6 Hz, 1H), 7.56 (t, J = 7.4 Hz, 1H), 7.48 (t, J = 7.4 Hz, 1H), 7.16 (s, 1H); 13C NMR (100 MHz, DMSO-d6, TMS) δ 99.77, 123.65, 125.91, 127.02, 128.61, 132.03, 139.64, 142.15, 142.54, 163.53, 165.01, 187.37. Anal. Calcd. for C12H8O4S: C, 58.06; H, 3.25. Found: C, 58.33; H, 3.28.
2-Hydroxy-4-(thiophen-2-yl)-4-oxobut-2-enoic acid (5a) [31]
Obtained as pink flakes (0.54 g, 76%) using 2-acetylthiophene (0.39 g, 3.12 mmol) as starting material, followed by subsequent hydrolysis of the diketoester; m.p. 170 – 173°C; Rf = 0.40 (n-hexane:EtOAc 60:40); 1H NMR (400 MHz, DMSO-d6, TMS) δ 14.40 (bs, 2H), 8.22 (d, J = 3.8 Hz, 1H), 8.15 (d, J = 4.9 Hz, 1H), 7.30 (t, J = 4.4 Hz, 1H), 7.02 (s, 1H); 13C NMR (100 MHz, DMSO-d6, TMS) δ 115.3, 126.3, 129.5, 136.7, 141.8, 169.2, 178.2, 191.3 Anal. Calcd. For C8H6O4S: C, 48.48; H, 3.05. Found: C, 48.57; H, 3.09.
Ethyl-2-hydroxy-4-(5-methylthiophen-2-yl)-4-oxobut-2-enoate (6)
Obtained from 2-acetyl-5-methylthiophene (0.44 g, 3.12 mmol) as yellow solid (0.61 g, 82%); m.p. 160–162°C; Rf = 0.70 (n-hexane:EtOAc 80:20); 1H NMR (400 MHz; CDCl3; TMS ) δ 14.66 (s, 1H), 7.68 (d, J = 3.8 Hz, 1H), 6.88 – 6.84 (m, 2H; due to overlap of allylic (s) and thiophene ring (d) protons), 4.38 (q, J = 7.1 H, 2H), 2.58 (s, 3H), 1.40 (t, J = 7.1 Hz, 3H).
2-Hydroxy-4-(5-methylthiophen-2-yl)-4-oxobut-2-enoic acid (6a)
Obtained from 6 (0.31 g, 1.3 mmol) as yellow solid (0.20 g, 74%); m.p. 172–174°C; Rf = 0.53 (EtOAc: n-hexane 60:40); 1H NMR (400 MHz, DMSO-d6, TMS) δ 14.12 (bs, 2H), 8.06 (d, J = 3.8 Hz, 1H), 7.03 (d, J = 3.8 Hz, 1H), 6.96 (s, 1H), 2.54 (s, 3H); 13C NMR (100 MHz, DMSO-d6, TMS) δ 16.26, 99.42, 128.85, 135.46, 140.12, 152.57, 163.58, 164.48, 186.31. Anal. Calcd. for C9H8O4S: C, 50.94; H, 3.80. Found: C, 50.67; H, 3.73.
Ethyl 2-hydroxy-4-(5-chlorothiophene-2-yl)-4-oxobut-2-enoate (7)
Obtained as buff powder (0.62 g, 76%) using 2-acetyl-5-chlorothiophene (0.49 g, 3.12 mmol) as the starting material; m.p. 185–188°C; Rf = 0.52 (EtOAc : n-hexane 60:40); 1H NMR (400 MHz; CDCl3; TMS ) δ 14.33 (s, 1H), 7.64 (d, J = 4.1 Hz, 1H), 7.02 (d, J = 4.1 Hz, 1H), 6.83 (s, 1H), 4.39 (q, J = 7.1 Hz, 2H), 1.40 (t, J = 7.1 Hz, 3H).
2-Hydroxy-4-(5-chlorothiophene-2-yl)-4-oxobut-2-enoic acid (7a) [32]
Obtained as yellow solid (0.25 g, 82%) using 7 (0.34 g, 1.3 mmol) as the starting material; m.p. 210–213°C; Rf = 0.35 (EtOAc: n-hexane 60:40); 1H NMR (400 MHz, DMSO-d6, TMS) δ 14.52 (bs, 2H), 8.16 (d, J = 4.1 Hz, 1H), 7.34 (d, J = 4.2 Hz, 1H), 7.00 (s, 1H); 13C NMR (100 MHz, DMSO-d6, TMS) δ 99.07, 130.04, 134.81, 139.65, 141.70, 163.46, 164.66, 185.69. Anal. Calcd. for C8H5ClO4S. 1/3 H2O: C, 40.34; H, 2.40. Found: C, 40.64; H, 2.46.
Ethyl 4-(5-bromothiophen-2-yl)-2-hydroxy-4-oxobut-2-enoate (8)
Obtained from 2-acetyl-5-bromothiophene (0.64 g, 3.12 mmol) as yellow solid (0.76 g, 81%); m.p. 180–183°C; Rf = 0.65 (n-hexane:EtOAc 70:30); 1H NMR (400 MHz, DMSO-d6, TMS) δ 11.11 (bs, 1H), 8.09 (s, 1H), 7.45 (d, J = 3.4 Hz, 1H), 6.98 (s, 1H), 4.29 (q, J = 6.8 Hz, 2H), 1.30 (t, J = 7.0 Hz, 3H).
4-(5-Bromothiophen-2-yl)-2-hydroxy-4-oxobut-2-enoic acid (8a)
Obtained as yellow solid (0.29 g, 82%) using 8 (0.39 g, 1.3 mmol) as the starting material; m.p. 210–212°C; Rf = 0.41 (EtOAc: n-hexane 80:20); 1H NMR (400 MHz, DMSO-d6, TMS) δ 14.07 (bs, 2H), 8.09 (d, J = 4.0 Hz, 1H), 7.45 (d, J = 4.0 Hz, 1H), 6.99 (s, 1H); 13C NMR (100 MHz, DMSO-d6, TMS) δ 99.14, 124.08, 133.42, 135.33, 144.31, 163.45, 164.90, 185.43. Anal. Calcd. for C8H8BrO4S. 1/4 C3H6O: C, 35.37; H, 2.27. Found: C, 35.33; H, 1.93.
Ethyl-2-hydroxy-4-(5-iodothiophen-2-yl)-4-oxobut-2-enoate (9)
Obtained from 2-acetyl-5-iodothiophene (0.78 g, 3.12 mmol) as a brown solid (0.76 g, 69%); m.p. 225–229°C; Rf = 0.70 (n-hexane : EtOAc 70:30); 1H NMR (400 MHz, CDCl3, TMS) δ 14.43 (s, 1H), 7.46 (d, J = 3.9 Hz, 1H), 7.35 (d, J = 3.9 Hz, 1H), 6.82 (s, 1H), 4.39 (q, J = 7.1 Hz, 2H), 1.40 (t, J = 7.1 Hz, 3H).
2-Hydroxy-4-(5-iodothiophen-2-yl)-4-oxobut-2-enoic acid (9a)
Obtained as brown solid (0.32 g, 76%) using 9 (0.46 g, 1.3 mmol) as the starting material; m.p. 235–238°C; Rf = 0.40 (EtOAc : n-hexane 60:40); 1H NMR (400 MHz, DMSO-d6, TMS) δ 14.36 (bs, 2H), 7.91 (d, J = 3.5 Hz, 1H), 7.55 (d, J = 3.2 Hz, 1H), 6.97 (s, 1H); 13C NMR (100 MHz, DMSO-d6, TMS) δ 91.78, 99.33, 135.92, 139.67, 148.14, 163.51 (2), 165.33. Anal. Calcd. for C8H8IO4S. 1/6 C4H8O: C, 30.97; H, 1.90. Found: C, 30.64; H, 1.75.
Ethyl-2-hydroxy-4-(4-methylthiophen-2-yl)-4-oxobut-2-enoate (10)
Obtained as yellow solid (0.54 g, 73%) using 2-acetyl-4-methylthiophene (0.44 g, 3.12 mmol) as the starting material; m.p. 155–158°C; Rf = 0.67 (n-hexane:EtOAc 60:40); 1H NMR (400 MHz, DMSO-d6, TMS) δ 14.32 (bs, 1H), 8.09 (s, 1H), 7.77 (s, 1H), 6.99 (s, 1H), 4.31 (q, J = 7.1 Hz, 2H), 2.27 (s, 3H), 1.31 (t, J = 7.1 Hz, 3H).
2-Hydroxy-4-(4-methylthiophen-2-yl)-4-oxobut-2-enoic acid (10a)
Obtained as yellow solid (0.19 g, 72%) using 10 (0.31 g, 1.3 mmol) as the starting material; m.p. 170–173°C; Rf = 0.44 (EtOAc : n-hexane 60:40); 1H NMR (400 MHz, DMSO-d6, TMS) δ 14.14 (bs, 2H), 8.05 (s, 1H), 7.75 (s, 1H), 6.96 (s, 1H), 2.25 (s, 3H); 13C NMR (100 MHz, DMSO-d6, TMS) δ 15.57, 99.57, 132.93, 136.46, 140.06, 141.73, 163.52, 164.94, 186.46. Anal. Calcd. for C9H8O4S: C, 50.94; H, 3.80. Found: C, 51.11; H, 3.80.
2-Hydroxy-4-(3-methylthiophen-2-yl)-4-oxobut-2-enoic acid (11a)
Obtained as brown solid (0.23 g, 86%) using 2-acetyl-3-methylthiophene (0.44 g, 3.12 mmol) as the starting material followed by alkaline hydrolysis of the ester using the procedure as above; m.p. 147–151°C; Rf = 0.49 (EtOAc : n-hexane 90:10); 1H NMR (400 MHz, DMSO-d6, TMS) δ 14.15 (bs, 2H), 7.98 (d, J = 4.9 Hz, 1H), 7,17 (d, J = 4.9 Hz, 1H), 6.69 (s, 1H), 2.55 (s, 3H); 13C NMR (100 MHz, DMSO-d6, TMS) δ 17.24, 100.93, 133.96, 134.07, 134.20, 146.80, 163.48, 165.98, 186.76. Anal. Calcd. for C9H8O4S: C, 50.94; H, 3.80. Found: C, 51.11; H, 3.80.
Ethyl 2-hydroxy-4-(4-thiazole-2-yl)-4-oxobut-2-enoate (12)
Obtained as brown flakes (0.50 g, 71%) using 2-acetylthiazole (0.39 g, 3.12 mmol) as starting material; m.p. 190–192°C; Rf = 0.61 (n-hexane : EtOAc 70:30); 1H NMR (400 MHz; CDCl3; TMS) δ 14.08 (s, 1H), 8.08 (d, J = 3.0 Hz, 1H), 7.78 (d, J = 3.0 Hz, 1H), 7.40 (s, 1H), 4.40 (q, J = 7.1 Hz, 2H), 1.41 (t, J = 7.1 Hz, 3H).
2-Hydroxy-4-(4-thiazole-2-yl)-4-oxobut-2-enoic acid (12a)
Obtained as brown solid (0.19 g, 72%) using 12 (0.29 g, 1.3 mmol) as the starting material; m.p. 210–213°C; Rf = 0.45 (EtOAc : n-hexane 60:40); 1H NMR (400 MHz; CDCl3; TMS) δ 13.41 (bs, 2H), 8.28 (d, J = 3.1 Hz, 1H), 8.19 (d, J = 2.9 Hz, 1H), 7,15 (s, 1H); 13C NMR (100 MHz, DMSO-d6, TMS) δ 99.63, 129.90, 134.72, 137.34, 163.54, 165.14, 186.61. Anal. Calcd. for C7H5NO4S. 1/5 CH3COOC2H5: C, 43.21; H, 3.07; N, 6.86. Found: C, 42.89; H, 2.67; N, 7.14.
Ethyl 2-hydroxy-4-oxo-4-(1H-pyrrol-2-yl)-but-2-enoate (13)
Obtained as green solid (0.46 g, 70%) using 2-acetyl-1H-pyrrole (0.34 g, 3.12 mmol) as the starting material; m.p. 130–134°C; Rf = 0.65 (n-hexane:EtOAc 80:20); 1H NMR (400 MHz; CDCl3; TMS) δ 12.75 (bs, 1H), 10.25 (s, 1H), 6.98 (s, 2H), 6.03 (s, 1H), 5.29 (s, 1H), 4.23 (q, J = 7.2 Hz, 2H), 1.21 (t, J = 7.1 Hz, 3H).
2-Hydroxy-4-oxo-4-(1H-pyrrol-2-yl)-but-2-enoic acid (13a) [30]
Obtained as white solid (0.19 g, 83%) using 13 (0.27 g, 1.3 mmol) as the starting material; m.p. 157–160°C; Rf = 0.52 (EtOAc:n-hexane 70:30); 1H NMR (400 MHz; CDCl3; TMS) δ 14.00 (bs, 2H), 12.26 (s, 1H), 7.28 (s, 2H), 6.87 (s, 1H), 6.31 (s, 1H). Anal. Calcd. for C8H7NO4.2/5 H2O: C, 51.31; H, 4.17; N, 7.44. Found: C, 51.61; H, 4.51; N, 7.73.
3-Acetyl-1H-indazole [26]
Following a prenominal variation in the reported procedure [26], isobutyl chloroformate (0.79 g, 5.88 mmol) and N-methylmorpholine (0.59 g, 5.88 mmol) were added to a solution of 1H-indazole-3-carboxylic acid (0.6 g, 3.70 mmol) in anhydrous THF (15 mL) under nitrogen atmosphere at −20°C, and the mixture was stirred for 4 h. To this mixture was added N,O-dimethylhydroxylamine hydrochloride (0.54 g, 5.55 mmol) suspended in 5 mL triethylamine. The reaction was then stirred at room temperature for 6 h, concentrated under vacuum and suspended in 20 mL n-hexane. The white precipitates (Winreb amide) formed were filtered off, dried and immediately transferred to a three neck flask containing 10 mL anhydrous THF cooled to −78°C. To this was added methyl magnesium bromide (12% in THF) (19 mL, 18.5 mmol). The reaction was allowed to stir at −78°C for 2 h and then at room temperature for 5 h. The completion of the reaction was monitored by TLC. The reaction was quenched by slow addition of saturated aqueous solution of ammonium chloride followed by extraction with ethyl acetate (3 × 20 mL). The combined organic layer was dried over magnesium sulfate and concentrated under vacuum. The resulting viscous mass was purified by column chromatography using hexane: ethyl acetate (85:15) solvent system to obtain desired acetyl derivative (0.32 g, 55%); m.p. 182–185°C (Lit. m.p. 184 – 186° C) [26]; Rf = 0.71 (n-hexane:EtOAc 70:30); 1H NMR (400 MHz; CDCl3; TMS) δ 13.85 (s, 1H), 8.18 (d, J = 7.2 Hz, 1H), 7.67 (d, J = 7.2 Hz, 1H), 7.46 (t, J = 7.3 Hz, 1H), 7.32 (t, J = 7.3 Hz, 1H), 2.65 (s, 3H).
Ethyl 2-hydroxy-4-(1H-indazol-3-yl)-4-oxobut-2-enoate (14)
Obtained as yellow solid (0.63 g, 78%) by using 3-acetyl-1H-indazole (0.49 g, 3.12 mmol) as the starting material; m.p. 182–184°C; Rf = 0.60 (n-hexane:EtOAc 70:30); 1H NMR (400 MHz; DMSO; TMS) δ 14.26 (s, 1H), 14.04 (s, 1H), 8.25 (d, J = 8.1 Hz, 1H), 7.74 (d, J = 8.2 Hz, 1H), 7.52 (t, J = 7.6 Hz, 1H), 7.39 (t, J = 7.5 Hz, 1H), 7.35 (s, 1H), 4.33 (q, J = 7.1 Hz, 2H), 1.32 (t, J = 7.1 Hz, 3H). Anal. Calcd. for C13H12N2O4.1/4H2O : C, 58.98; H, 4.76; N, 10.58. Found: C, 59.08; H, 4.70; N, 10.56.
2-Hydroxy-4-(1H-indazole-3-yl)-4-oxobut-2-enoic acid (14a)
Obtained as yellow solid (0.22 g, 72%) using 14 (0.34 g, 1.3 mmol) as the starting material; m.p. 224–226°C; Rf = 0.42 (EtOAc: n-hexane: 60:40); 1H NMR (400 MHz; DMSO-d6; TMS) δ 14.24 (s, 3H), 8.24 (d, J = 8.1 Hz, 1H), 7.72 (d, J = 8.4 Hz, 1H), 7.50 (t, J = 7.6 Hz, 1H), 7.38 (t, J = 7.4 Hz, 1H), 7.33 (s, 1H). 13C NMR (100 MHz, DMSO-d6, TMS) δ 100.50, 111.89, 121.89 (2), 124.38, 127.71, 141.58, 141.90, 163.46, 163.70, 189.63. Anal. Calcd. for C11H8N2O4: C, 56.90; H, 3.47; N, 12.06. Found: C, 56.78; H, 3.51; N, 11.96.
Ethyl 2-hydroxy-4-(biphenyl-4-yl)-4-oxobut-2-enoate (15)
Obtained as white solid (0.66 g, 72%) using 4-acetylbiphenyl (0.61 g, 3.12 mmol) as the starting material; m.p. 168–170°C; Rf = 0.54 (n-hexane:EtOAc 60:40); 1H NMR (400 MHz; CDCl3; TMS) δ 15.39 (s, 1H), 8.07 (d, J = 8.5 Hz, 2H), 7.73 (d, J = 8.5 Hz, 2H), 7.67-7.62 (m, 2H), 7.54 – 7.39 (m, 3H), 7.12 (s, 1H), 4.41 (q, J = 7.1 Hz, 2H), 1.42 (t, J = 7.1 Hz, 3H).
2-Hydroxy-4-(biphenyl-4-yl)-4-oxobut-2-enoic acid (15a) [30]
Obtained as white solid (0.29 g, 85%) using 15 (0.38 g, 1.3 mmol) as the starting material; m.p. 182–186°C; Rf = 0.40 (n-hexane:EtOAc 60:40); 1H NMR (400 MHz; DMSO-d6; TMS) δ 15.25 (s, 1H), 14.25 (s, 1H), 8.18 (d, J = 7.9 Hz, 2H), 7.90 (d, J = 7.8 Hz, 2H), 7.81 (d, J = 7.8 Hz, 2H), 7.60-7.40 (m, 3H), 7.17 (s, 1H); 13C NMR (100 MHz; DMSO-d6; TMS) δ 98.3, 127.5 (2), 127.7 (2), 129.1 (2), 129.6 (2), 133.9, 139.1, 145.8, 163.6, 170.8, 190.2 (2). Anal. Calcd. for C16H12O4. 1/6 H2O: C, 67.36; H, 4.59. Found: C, 67.20; H, 4.51.
Ethyl 2-hydroxy-4-(2-nitrophenyl)-4-oxobut-2-enoate (16)
Obtained as white solid (0.54 g, 65%) using 2-nitroacetophenone (0.51 g, 3.12 mmol) as the starting material; m.p. 178–182°C; Rf = 0.59 (n-hexane:EtOAc 80:20); 1H NMR (400 MHz; DMSO-d6; TMS) δ 10.81 (bs, 1H), 8.10 (d, J = 8.0 Hz, 1H), 7.84 (t, J = 7.2 Hz, 1H), 7.75 (t, J = 7.6 Hz, 1H), 7.69 (d, J = 6.6 Hz, 1H), 6.49 (s, 1H), 4.25 (q, J = 6.9 Hz, 2H), 1.26 (t, J = 7.0 Hz, 3H).
2-Hydroxy-4-(2-nitrophenyl)-4-oxobut-2-enoic acid (16a) [30]
Obtained as pink solid (0.23 g, 75%) using 16 (0.34 g, 1.3 mmol) as the starting material; m.p. 195–198°C; Rf = 0.47 (EtOAc : n-hexane 60:40); 1H NMR (400 MHz; DMSO-d6; TMS) δ 12.99 (bs, 2H), 8.11 (d, J = 8.1 Hz, 1H), 7.84 (t, J = 7.5 Hz, 1H), 7.75 (t, J = 7.2 Hz, 1H), 7.69 (d, J = 7.5 Hz, 1H), 6.40 (s, 1H); 13C NMR (100 MHz; DMSO-d6; TMS) δ 103.40, 124.67 (2), 129.36, 132.15, 134.53, 135.06, 147.11, 164.17 (2). Anal. Calcd. for C10H7NO6: C, 50.63; H, 2.98; N, 5.91. Found: C, 50.57; H, 2.98; N, 5.73.
Ethyl 4-(2-fluorophenyl)-2-hydroxy-4-oxobut-2-enoate (17)
Obtained as white solid (0.56 g, 76%) using 2-fluoroacetophenone (0.43 g, 3.12 mmol) as the starting material; m.p. 176–178°C, Rf = 0.65 (n-hexane:EtOAc 60:40); 1H NMR (400 MHz; DMSO-d6; TMS) δ 14.34 (bs, 1H), 7.89 (t, J = 7.2 Hz, 1H), 7.71 (q, J = 6.6 Hz, 1H), 7.45 – 7.34 (m, 2H), 6.85 (s, 1H), 4.29 (q, J = 7.0 Hz, 2H), 1.29 (t, J = 7.0 Hz, 3H). Anal. Calcd. for C12H11FO4.1/5H2O: C, 59.60; H, 4.75. Found: C, 59.59; H, 4.47.
4-(2-Fluorophenyl)-2-hydroxy-4-oxobut-2-enoic acid (17a) [30]
Obtained as white solid (0.22 g, 82%) using 17 (0.31 g, 1.3 mmol) as the starting material; m.p. 192–195°C, Rf = 0.59 (n-hexane:EtOAc 70:30); 1H NMR (400 MHz; DMSO-d6; TMS) δ 14.29 (bs, 2H), 7.91 (t, J = 7.3 Hz, 1H), 7.71 (q, J = 6.7 Hz, 1H), 7.39 (dd, J = 6.7 Hz, 5.6 Hz, 2H), 6.90 (s, 1H); 13C NMR (100 MHz, DMSO-d6, TMS) δ 102.28, 117.47, 123.83, 125.69, 130.70, 136.12, 159.94, 163.48, 170.16, 187.44. Anal. Calcd. for C10H7FO4: C, 57.13; H, 3.36. Found: C, 56.95; H, 3.45.
2-Hydroxy-4-oxo-4-o-tolylbut-2-enoic acid (18a) [30]
Obtained as white solid (0.19 g, 71%) using 2-methylacetophenone (0.42 g, 3.12 mmol) as the starting material followed by subsequent hydrolysis of the ester; m.p. 167–171°C; Rf = 0.68 (n-hexane:EtOAc 60:40); 1H NMR (400 MHz; DMSO-d6; TMS) δ 14.12 (bs, 2H), 7.69 (d, J = 7.2 Hz, 1H), 7.48 (t, J = 7.4 Hz, 1H), 7.37-7.32 (m, 2H), 6.72 (s, 1H), 2.47 (s, 3H); 13C NMR (100 MHz, DMSO-d6, TMS) δ 21.00, 102.04, 126.67, 129.40, 132.20, 132.53, 136.15, 138.46, 161.88 (2), 163.66. Anal. Calcd. for C11H10O4: C, 64.07; H, 4.89. Found: C, 63.95; H, 4.67.
Ethyl 4-(3-fluorophenyl)-2-hydroxy-4-oxobut-2-enoate (19)
Obtained as white solid (0.54 g, 73%) using 3-fluoroacetophenone (0.43 g, 3.12 mmol) as the starting material; m.p. 165–168°C; Rf = 0.61 (n-hexane:EtOAc 60:40); 1H NMR (400 MHz; CDCl3; TMS ) δ 15.23 (s, 1H), 7.81 (d, J = 7.8 Hz, 1H), 7.72-7.69 (m, 1H), 7.52 (q, J = 7.2 Hz, 1H), 7.34 (t, J = 7.2 Hz, 1H), 7.09 (s, 1H), 4.42 (q, J = 7.1 Hz, 2H), 1.43 (t, J = 7.2 Hz, 3H).
4-(3-Fluorophenyl)-2-hydroxy-4-oxobut-2-enoic acid (19a)
Obtained as white solid (0.21 g, 77%) using 19 (0.31 g, 1.3 mmol) as the starting material; m.p. 187–189°C; Rf = 0.47 (EtOAc : n-hexane 60:40); 1H NMR (400 MHz; DMSO-d6; TMS) δ 14.26 (bs, 2H), 7.92 (d, J = 7.7 Hz, 1H), 7.86 (d, J = 7.6 Hz, 1H), 7.62 (q, J = 7.2 Hz, 1H), 7.54 (m, 1H), 7.10 (s, 1H); 13C NMR (100 MHz, DMSO-d6, TMS) δ 98.60, 114.88, 121.32, 124.52, 131.73, 137.45, 161.56, 163.49, 170.88, 189.26. Anal. Calcd. for C10H7FO4. 1/5 H2O: C, 56.19; H, 3.49. Found: C, 56.51; H, 3.32.
Ethyl 4-(3-cyanophenyl)-2-hydroxy-4-oxobut-2-enoate (20)
Obtained as white solid (0.58 g, 76%) using 3-cyanoacetophenone (0.45 g, 3.12 mmol) as the starting material; m.p. 178–181°C, Rf = 0.67 (n-hexane:EtOAc 70:30); 1H NMR (400 MHz; DMSO-d6; TMS) δ 14.49 (bs, 1H), 8.56 (s, 1H), 8.34 (d, J = 7.8 Hz, 1H), 8.14 (d, J = 7.7 Hz, 1H), 7.77 (t, J = 7.9 Hz, 1H), 7.19 (s, 1H), 4.32 (q, J = 7.1 Hz, 2H), 1.32 (t, J = 7.0 Hz, 3H).
4-(3-Cyanophenyl)-2-hydroxy-4-oxobut-2-enoic acid (20a)
Obtained as white solid (0.22 g, 78%) using 20 (0.32 g, 1.3 mmol) as the starting material; m.p. 201–205°C, Rf = 0.47 (EtOAc : n-hexane 60:40); 1H NMR (400 MHz; DMSO-d6; TMS) δ 14.24 (bs, 2H), 8.55 (s, 1H), 8.34 (d, J = 7.9 Hz, 1H), 8.13 (d, J = 7.3 Hz, 1H), 7.77 (t, J = 7.7 Hz, 1H), 7.17 (s, 1H). 13C NMR (100 MHz, DMSO-d6, TMS) δ 98.81, 112.83, 118.43, 130.79, 132.16, 132.63, 136.21, 137.31, 163.40, 170.95, 188.59. Anal. Calcd. for C11H7NO4.1/5 H2O: C, 59.84; H, 3.38; N, 6.34. Found: C, 60.16; H, 3.34; N, 6.23.
4-(3-(1H-Tetrazol-5-yl)phenyl)-2-hydroxy-4-oxobut-2-enoic acid (21a)
According to reported procedure [27], NaN3 (0.16 g, 2.51 mmol) was added to a well stirred reaction mixture of 20a (0.3 g, 1.38 mmol) and ammonium chloride (0.1 g, 1.88 mmol) in anhydrous DMF. The reaction was then heated to 110°C and intermittently monitored by TLC. Upon completion, the reaction mixture was concentrated under vacuum. Water was added and the aqueous layer was acidified with HCl to pH 1. The resulting precipitates were filtered off, washed with water and dried under vacuum. The crude product was purified by precipitating it using hexane: ethyl acetate system to obtain the tetrazole derivative as buff solid (0.27 g, 75%); m.p. 210–213°C, Rf = 0.31 (EtOAc : n-hexane 70 : 30); 1H NMR (400 MHz; DMSO-d6; TMS) δ 15.45 (bs, 2H), 8.67 (s, 1H), 8.35 (d, J = 7.6 Hz, 1H), 8.26 (d, J = 7.6 Hz, 1H), 7.81 (t, J = 7.7 Hz, 1H), 7.17 (s, 1H). [Note: The absence of the tetrazole ring -NH proton was attributed to the protopic tautomerism. Moreover, the existence of the proton was confirmed by alkylation of the tetrazole ring by treating it with benzyl chloride under basic conditions (data not shown)]. Anal. Calcd. for C11H8N4O4: C, 50.77; H, 3.10; N, 21.53. Found: C, 50.63; H, 3.24; N, 21.66.
Ethyl 4-(2,6-difluorophenyl)- 2-hydroxy-4-oxobut-2-enoate (22)
Obtained as white powder (0.68 g, 85%) using 2,6-difluoroacetophenone (0.49 g, 3.12 mmol) as the starting material; m.p. 156–158°C; Rf = 0.60 (n-hexane:EtOAc 70:30); 1H NMR (400 MHz; CDCl3; TMS) δ 14.38 (s, 1H), 7.52 - 7.46 (m, 1H), 7.01 (t, J = 8.6 Hz, 2H), 6.79 (s, 1H), 4.40 (q, J = 7.1 Hz, 2H), 1.40 (t, J = 7.1 Hz, 3H).
2-Hydroxy-4-(2,6-difluorophenyl)-4-oxobut-2-enoic acid (22a) [30]
Obtained as white powder (0.23 g, 79%) using 22 (0.33 g, 1.3 mmol) as the starting material; m.p. 179–181°C; Rf = 0.53 (n-hexane:EtOAc 70:30); 1H NMR (400 MHz; DMSO-d6; TMS) δ 13.09 (bs, 2H), 7.62-7.60 (m, 1H), 7.23 (t, J = 7.9 Hz, 2H), 6.41 (s, 1H); 13C NMR (100 MHz; DMSO-d6; TMS) δ 104.66, 112.66, 112.90, 116.54, 133.80, 158.35, 158.42, 160.85, 160.92, 164.10. Anal. Calcd. for C10H6F2O4. 2/3 H2O: C, 49.99; H, 2.97. Found: C, 49.85; H, 2.60.
Ethyl 4-(2,4-dichlorophenyl)-2-hydroxy-4-oxobut-2-enoate (23)
Obtained as white solid (0.64 g, 71%) using 2,4-dichloroacetophenone (0.58 g, 3.12 mmol) as the starting material; m.p. 134–136°C; Rf = 0.62 (n-hexane:EtOAc 80:20); 1H NMR (400 MHz; CDCl3; TMS) δ 14.57 (s, 1H), 7.61 (d, J = 8.4 Hz, 1H), 7.50 (s, 1H), 7.36 (d, J = 8.5 Hz, 1H), 6.95 (s, 1H), 4.39 (q, J = 7.1 Hz, 2H), 1.39 (t, J = 7.1 Hz, 3H).
4-(2,4-Dichlorophenyl)-2-hydroxy-4-oxobut-2-enoic acid (23a) [30]
Obtained as white solid (0.26 g, 78%) using 23 (0.37 g, 1.3 mmol) as the starting material; m.p. 165–168°C; Rf = 0.62 (DCM:MeOH 98:2); 1H NMR (400 MHz; DMSO-d6; TMS) δ 13.97 (bs, 2H), 7.78 (s, 1H), 7.67 (d, J = 7.7 Hz, 1H), 7.57 (dd, J = 7.9 Hz, 7.7 Hz, 1H), 6.55 (s, 1H); 13C NMR (100 MHz, DMSO-d6, TMS) δ 103.19, 128.28 (2), 130.43, 131.77 (2), 132.11, 135.90, 136.98, 163.83. Anal. Calcd. for C10H6Cl2O4: C, 46.16; H, 2.33. Found: C, 46.19; H, 2.39.
Ethyl 4-(2,4-difluorophenyl)-2-hydroxy-4-oxobut-2-enoate (24)
Obtained as white powder (0.58 g, 72%) using 2,4-difluoroacetophenone (0.49 g, 3.12 mmol) as the starting material; m.p. 190–192°C; Rf = 0.73 (n-hexane:EtOAc 60:40); 1H NMR (400 MHz; DMSO-d6; TMS) δ 15.26 (bs, 1H), 7.99 (q, J = 7.9 Hz, 1H), 7.48 (t, J = 7.1 Hz, 1H), 7.29 (t, J = 7.1 Hz, 1H), 6.87 (s, 1H), 4.29 (q, J = 7.1 Hz, 2H), 1.29 (t, J = 7.0 Hz, 3H).
4-(2,4-Difluorophenyl)-2-hydroxy-4-oxobut-2-enoic acid (24a) [30]
Obtained as white solid (0.21 g, 70%) using 24 (0.33 g, 1.3 mmol) as the starting material; m.p. 210–214°C, Rf = 0.51 (EtOAc : n-hexane 60:40); 1H NMR (400 MHz; DMSO-d6; TMS) δ 14.29 (bs, 2H), 8.00 (q, J = 8.0 Hz, 1H), 7.48 (dd, J = 7.7 Hz, 7.4 Hz, 1H), 7.29 (dt, J = 7.6 Hz, 7.3 Hz, 1H), 6.86 (s, 1H); 13C NMR (100 MHz, DMSO-d6, TMS) δ 102.18, 105.94, 113.27, 120.96, 132.88, 141.99, 163.46, 167.06, 169.43, 186.78. Anal. Calcd. for C10H6F2O4: C, 52.64; H, 2.65. Found: C, 52.90; H, 2.81.
NS5B inhibition assay
For evaluating the biological activity of the compounds, the enzymatic reaction mixtures containing 20 mM Tris–HCl (pH 7.0), 100 mM NaCl, 100 mM sodium glutamate, 0.5 mM DTT, 0.01% BSA, 0.01% Tween-20, 5% glycerol, 20 U/mL of RNase Out, 0.5 μM of poly rA/U12, 25 μM UTP, 2–5 μCi [α-32P]UTP, 300–500 ng of NS5BCΔ21 and 1.0 mM MnCl2 with or without inhibitors in a total volume of 25 μL were incubated for 1 h at 30°C. The concentration of DMSO in all reactions was maintained constant at 10%. Reactions were terminated by the addition of ice-cold 5% (v/v) trichloroacetic acid (TCA) containing 0.5 mM pyrophosphate. The quenched reaction mixtures were incubated at −20°C for 1 h to precipitate out denatured polymeric RNA products, transferred to GF-B filters, washed twice with 5% (v/v) TCA containing 0.5 mM pyrophosphate to remove unincorporated UTP, and rinsed three times with water and once with ethanol before vacuum drying. The amount of radioactive UMP incorporated into RNA products was quantified on a liquid scintillation counter (Packard). Activity of NS5B in the absence of the inhibitor but containing an equivalent amount of DMSO (control reaction) was set at 100% and that in the presence of the inhibitor was quantified relative to this control. The concentration of inhibitors inhibiting 50% of NS5B RdRp activity (IC50) was calculated from the inhibition curves as a function of inhibitor concentration and values obtained represent an average of at least two independent measurements in duplicate.
Molecular modeling
Molecular docking computations were carried out on a Dell Precision 470n workstation with the RHEL 4.0 operating system using Glide 5.5 (Schrodinger, LLC, New York, NY). 3D Structures of the entire target DKAs were constructed using the fragment dictionary of Maestro 9.0 (Schrodinger, LLC, New York, NY) and geometry was optimized by Macromodel program v9.7 using the OPLS-AA force field with the steepest descent followed by truncated Newton conjugate gradient protocol. Each ligand structure was subsequently processed with the LigPrep v2.3 program (Schrodinger, LLC, New York, NY) to assign the protonation states appropriate for physiological pH, using the “ionizer” subprogram. A maximum of 100 conformers were generated for each ligand using a combination of Monte Carlo Multiple Minimum and low mode conformational searching in conjugation with OPLS_2005 force field (Schrodinger, LLC, New York, NY). Minimized structures were filtered with a maximum relative energy difference of 5 kcal/mol to exclude redundant conformers. The X-ray crystal structure of NS5B polymerase in complex with rUTP (PDB ID: 1GX6), [33] obtained from the RCSB Protein Data Bank (PDB), was used to dock conformational library of each ligand. The protein was optimized for docking using the “Protein Preparation Wizard” and “Prime-Refinement Utility” of Maestro 9.0. The grids were generated using bound rUTP with the default parameters. The Glide docking program v5.5 (Schrodinger, LLC, New York, NY) was used to dock the conformational library of each ligand as per the details described in our previous studies [24, 34].
Acknowledgments
This work was supported by the National Institute of Health Research Grant CA153147 to N.K-B., and St. John’s University Seed Grant No. 579-1110 to T. T. T.
Footnotes
Author Contributions
Performed design, synthesis and molecular modeling: A.Ba, M.R.P and T.T.T. Carried out NS5B purification, NS5B inhibition assays and data analysis: G.K.R, A.Bb and N.K-B. Wrote the paper: A.Ba, M.R.P, T.T.T and N.K-B.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Choo QL, Kuo G, Weiner AJ, Overby LR, Bradley DW, Houghton M. Isolation of a cDNA clone derived from a blood-borne non-A, non-B viral hepatitis genome. Science. 1989;244:359–362. doi: 10.1126/science.2523562. [DOI] [PubMed] [Google Scholar]
- 2.Alter MJ, Margolis HS, Krawczynski K, Judson FN, Mares A, Alexzender WJ, Hu PY, Miller JK, Gerber MA, Sampliner RE. The Natural History of Community-Acquired Hepatitis C in United States. N Engl J Med. 1992;327:1899–1905. doi: 10.1056/NEJM199212313272702. [DOI] [PubMed] [Google Scholar]
- 3.Alter MJ. The detection, transmission, and the outcome of hepatitis c virus infection. Infect Agents Dis. 1993;2:155–166. [PubMed] [Google Scholar]
- 4.Di Bisceglie AM, Order SE, Klein JL, Waggoner JG, Sjogren MH, Kuo G, Houghton M, Choo QL, Hoofnagle JH. The role of chronic viral hepatitis in hepatocellular carcinoma in the United States. Am J Gastroenterol. 1991;86:335–338. [PubMed] [Google Scholar]
- 5.Alter MJ. Epidemiology of Hepatitis C Virus infection. World J Gastroenterol. 2007;13:2436–2441. doi: 10.3748/wjg.v13.i17.2436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang QM, Heinz BA. Recent advances in prevention and treatment of Hepatitis C Virus infections. Prog Drug Res. 2000;55:1–32. doi: 10.1007/978-3-0348-8385-6_1. [DOI] [PubMed] [Google Scholar]
- 7.Abrignani S, Houghton M, Hsu HH. Prespectives for a vaccine against Hepatitis C Virus. J Hepatol. 1999;31:259–265. doi: 10.1016/s0168-8278(99)80413-9. [DOI] [PubMed] [Google Scholar]
- 8.Huang Z, Murray MG, Secrist JA., 3rd Recent development of therapeutics for chronic HCV infection. Antiviral Res. 2006;71:351–362. doi: 10.1016/j.antiviral.2006.06.001. [DOI] [PubMed] [Google Scholar]
- 9.Ferenci P, Fried MW, Shiffman ML, Smith CL, Marinos G, Goncales FL, Jr, Haussinger D, Daigo M, Carosi G, Dhumeaux D, Craxi A, Chaneac M, Reddy KR. Predicting sustained virological responses in chronic hepatitis C patients treated with peginterferon alfa-2a (40KD)/ribavirin. J Hepatol. 2005;43:425–433. doi: 10.1016/j.jhep.2005.04.009. [DOI] [PubMed] [Google Scholar]
- 10.Arataki K, Kumada H, Toyota K, Ohishi S, Takahashi S, Tazuma S, Chayama K. Evolution of hepatitis C virus quasispecies during ribavirin and interferon-alpha-2b combination therapy and interferon-alpha-2b monotherapy. Intervirology. 2006;49:352–361. doi: 10.1159/000095155. [DOI] [PubMed] [Google Scholar]
- 11.Behrens SE, Tomei L, De Francesco R. Identification of RNA dependent RNA polymerase of Hepatitis C Virus. Embo J. 1996;15:12–22. [PMC free article] [PubMed] [Google Scholar]
- 12.Moradpour D, Penin F, Rice CM. Replication of Hepatitis C Virus. Nat Rev Microbiol. 2007;5:453–463. doi: 10.1038/nrmicro1645. [DOI] [PubMed] [Google Scholar]
- 13.Wang QM, Heinz BA. Recent advances in prevention and treatment of hepatitis C virus infections. Prog Drug Res. 2001;56:79–110. doi: 10.1007/978-3-0348-7784-8_3. [DOI] [PubMed] [Google Scholar]
- 14.Di Marco SD, Volpari C, Tomei L, Altamura S, Harper S, Narjes F, Koch U, Rowley M, De Francesco RD, Migliaccio G, Carfi A. Interdomain communication in hepatitis C virus polymerase abolished by small molecule inhibitors bound to a novel allosteric site. J Biol Chem. 2005;280:29675–29700. doi: 10.1074/jbc.M505423200. [DOI] [PubMed] [Google Scholar]
- 15.Love RA, Parge HE, Yu X, Hickey MJ, Diehl W, Gao J, Wriggers H, Ekker A, Wang L, Thomson JA, Dragovich PS, Fuhrman SA. Crystallographic identification of a noncompetitive inhibitor binding site on the Hepatitis C Virus NS5B polymerase enzyme. J Virol. 2003;77:7575–7581. doi: 10.1128/JVI.77.13.7575-7581.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tedesco R, Shaw AN, Bambal R, Chai D, Concha NO, Darcy MG, Dhanak D, Fitch DM, Gates A, Gerhardt WG, Halegoua DL, Han C, Hofmann GA, Johnston VK, Kaura AC, Liu N, Keenan RM, Lin-Goerke J, Sarisky RT, Wiggall KJ, Zimmerman MN, Duffy KJ. 3-(1,1-Dioxo-2H-(1,2,4)-benzothiadiazin-3-yl)-4-hydroxy-2(1H)-quinolinones, Potent Inhibitors of Hepatitis C Virus RNA-Dependent RNA Polymerase. J Med Chem. 2006;49:971–983. doi: 10.1021/jm050855s. [DOI] [PubMed] [Google Scholar]
- 17.Hang JQ, Yang Y, Harris SF, Leveque V, Whittington HJ, Rajyaguru S, Ao-Ieong G, McCown MF, Wong A, Giannetti AM, Le Pogam S, Talamas F, Cammack N, Najera I, Klumpp K. Slow binding inhibition and mechanism of resistance of non-nucleoside polymerase inhibitors of Hepatitis C Virus. J Biol Chem. 2009;284:15517–15529. doi: 10.1074/jbc.M808889200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang M, Ng KK, Cherney MM, Chan L, Yannopoulos CG, Bedard J, Morin N, Nguyen-Ba N, Alaoui-Ismaili MH, Bethell RC, James MN. Non-nucleoside Analogue Inhibitors Bind to an Allosteric Site on HCV NS5B Polymerase Crystal Structures and mechanisms of inhibition. J Biol Chem. 2003;278:9489–9495. doi: 10.1074/jbc.M209397200. [DOI] [PubMed] [Google Scholar]
- 19.Summa V, Petrocchi A, Matassa VG, Taliani M, Laufer R, Francesco R, Altamura S, Pace P. HCV NS5b RNA-Dependent RNA Polymerase Inhibitors: From α,γ-Diketoacids to 4,5-Dihydroxypyrimidine- or 3-Methyl-5-hydroxypyrimidinonecarboxylic Acids. Design and Synthesis. J Med Chem. 2004;47:5336–5339. doi: 10.1021/jm0494669. [DOI] [PubMed] [Google Scholar]
- 20.Hazuda D, Felock P, Witmer M, Wolfe A, Stillmock K, Grobler JA, Espeseth A, Gabryelski L, Schleif W, Blau C, Miller MD. Inhibitors of Strand Transfer That Prevent Integration and Inhibit HIV-1 Replication in Cells. Science. 2000;287:646–650. doi: 10.1126/science.287.5453.646. [DOI] [PubMed] [Google Scholar]
- 21.Di Santo R, Fermeglia M, Ferrone M, Paneni MS, Costi R, Artico M, Roux A, Gabriele M, Tardif KD, Siddiqui A, Pricl S. Simple but highly effective three-dimensional chemical-feature-based pharmacophore model for diketo acid derivatives as hepatitis C virus RNA-dependent RNA polymerase inhibitors. J Med Chem. 2005;48:6304–6307. doi: 10.1021/jm0504454. [DOI] [PubMed] [Google Scholar]
- 22.Powdrill MH, Deval J, Narjes F, De Francesco R, Gotte M. Mechanism of hepatitis C virus RNA polymerase inhibition with dihydroxypyrimidines. Antimicrob Agents Chemother. 2010;54:977–983. doi: 10.1128/AAC.01216-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu Y, Jiang WW, Pratt J, Rockway T, Harris K, Vasavanonda S, Tripathi R, Pithawalla R, Kati WM. Mechanistic study of HCV polymerase inhibitors at individual steps of the polymerization reaction. Biochemistry. 2006;45:11312–11323. doi: 10.1021/bi060511j. [DOI] [PubMed] [Google Scholar]
- 24.Chen Y, Bopda-Waffo A, Basu A, Krishnan R, Silberstein E, Taylor DR, Talele TT, Arora P, Kaushik-Basu N. Characterization of aurintricarboxylic acid as a potent hepatitis C virus replicase inhibitor. Antivir Chem Chemother. 2009;20:19–36. doi: 10.3851/IMP1286. [DOI] [PubMed] [Google Scholar]
- 25.Santo RD, Costi R, Roux A, Miele G, Crucitti GC, Iacovo A, Rosi F, Lavecchia A, Marinelli L, Giovanni CD, Novellieno E, Palmisano L, Andreotti M, Amici R, Galluzzo CM, Palamara AT, Pommier Y, Marchand C. Novel quinolinonyl diketo acid derivatives as HIV-1 Integrase inhibitors: Design, synthesis and biological activities. J Med Chem. 2008;51:4744–4750. doi: 10.1021/jm8001422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Crestey F, Stiebing S, Legay R, Callot V, Rault S. Design and synthesis of a new indazole library: direct conversion of N-methoxy-N-methylamides (Winreb amides) to 3-keto and 3-formylindazoles. Tetrahedron. 2007;63:419–428. [Google Scholar]
- 27.Diana GD, Cutcliffe D, Volkots DL, Mallamo JP, Bailey TR, Vescio N, Oglesby RC, Nitz TJ, Wetzel J, Giranda V, Pevear DC, Dutko FJ. Antipicornavirus activity of tetrazole analogues related to Disoxaril. J Med Chem. 1993;36:3240–3250. doi: 10.1021/jm00074a004. [DOI] [PubMed] [Google Scholar]
- 28.Summa V, Petrocchi A, Pace P, Matassa VG, De Francesco R, Altamura S, Tomei L, Koch U, Neuner P. Discovery of α,γ-Diketo Acids as Potent Selective and Reversible Inhibitors of Hepatitis C Virus NS5b RNA-Dependent RNA Polymerase. J Med Chem. 2004;47:14–17. doi: 10.1021/jm0342109. [DOI] [PubMed] [Google Scholar]
- 29.Lohmann V, Korner F, Herian U, Bartenschlager R. Biochemical properties of hepatitis C virus NS5B RNA-dependent RNA polymerase and identification of amino acid sequence motifs essential for enzymatic activity. J Virol. 1997;71:8416–8428. doi: 10.1128/jvi.71.11.8416-8428.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Freundlich JS, Sacchettini JC, Kriger IV, Loerger TR, Gawandi V. Inhibitors of Mycobacterium tuberculosis malate synthase, methods of making and uses thereof. WO 2010047774. Pct Int Appl. 2010:A2, 20100429.
- 31.Chen B, Yin HF, Wang ZS, Xu JH, Fan LQ, Zhao J. Facile synthesis of enantiopure 4- substituted 2- hydroxyl butyrolactones using Robust Fusarium Lactonase. Adv Synth Catalysis. 2009;35:2959–2966. [Google Scholar]
- 32.Bozdyreva KS, Smirnova IV, Maslivets AN. Five-Membered 2,3-Dioxo Heterocycles: L. Synthesis and thermolysis of 3-Aroyl- and 3-Hetaroyl-5-phenyl-1,2,4,5-tetrahydropyrrolo[1,2-a] quinoxalin 1,2,4-triones. Russian J Org Chem. 2005;41:1081–1088. [Google Scholar]
- 33.Bressanelli S, Tomei L, Rey FA, Francesco RD. Structural analysis of the hepatitis C virus RNA polymerase in complex with ribonucleotides. J Virol. 2002;76:3482–3492. doi: 10.1128/JVI.76.7.3482-3492.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Talele TT, McLaughlin ML. Molecular docking/dynamics studies of Aurora A kinase inhibitors. J Mol Graphics and Model. 2008;26:1213–1222. doi: 10.1016/j.jmgm.2007.11.003. [DOI] [PubMed] [Google Scholar]