

Abstract
Motivation: Homology search for RNAs can use secondary structure information to increase power by modeling base pairs, as in covariance models, but the resulting computational costs are high. Typical acceleration strategies rely on at least one filtering stage using sequence-only search.
Results: Here we present the multi-segment CYK (MSCYK) filter, which implements a heuristic of ungapped structural alignment for RNA homology search. Compared to gapped alignment, this approximation has lower computation time requirements (O(N4) reduced to O(N3)), and space requirements (O(N3) reduced to O(N2)). A vector-parallel implementation of this method gives up to 100-fold speed-up; vector-parallel implementations of standard gapped alignment at two levels of precision give 3- and 6-fold speed-ups. These approaches are combined to create a filtering pipeline that scores RNA secondary structure at all stages, with results that are synergistic with existing methods.
Availability: http://selab.janelia.org/publications.html#KolbeEddy11
Contact: eddys@janelia.hhmi.org
1 INTRODUCTION
A covariance model (CM) is a probabilistic profile model of one or more structural RNAs with a consensus secondary structure. CMs are used for database search and for multiple alignment, and form the basis for the Rfam database of known structural RNAs (Gardner et al., 2011). Explicitly modeling the secondary structure allows the model to capture information about an RNA when the base pairs are preserved even if the sequence changes. A CM includes states that can describe a base pair as a single unit, so that the score can favor good combinations rather than only single residue conservation; the states are arranged in a branching tree structure that reflects the shared consensus fold of the RNA. Inclusion of secondary structure increases the sensitivity and specificity of homology searches for RNA compared to sequence profile methods (Freyhult et al., 2007). CMs do neglect some higher order features of RNA structure, such as pseudoknots, but this is generally acceptable for homology search. A large obstacle to the application of CMs is one of the computational costs: both memory and time requirements have worse scaling behavior than their sequence-only counterparts.
Many different approaches have been used for increasing the speed of CM search and alignment. Acceleration methods generally fall into three classes: direct hardware acceleration of the base method (Liu and Schmidt, 2005), heuristic filters that reduce the number of sequences to be searched and heuristic bands that reduce the number of possible alignments per sequence. Filtering and banding techniques for RNA search can be based on sequence-only or sequence-and-structure models, but one common approach is a sequence-only filter such as the BLAST filter used in the Rfam database pipeline (Gardner et al., 2011). Recent work in filtering methods has focused on the automatic design of profile HMMs and on models that include small amounts of structural information (Weinberg and Ruzzo, 2004a, b, 2006). Structure description languages and heuristic RNA structural search methods stand on their own, but can also be used as structure-based filters for CM search (Bafna and Zhang, 2004; Lowe and Eddy, 1997; Macke et al., 2001). A recent example in this area also focuses on automatic rather than manual creation of filters (Sun and Buhler, 2008). Banding approaches for CM search include HMM bands (Brown, 2000) and structure-derived bands (Nawrocki and Eddy, 2007), and similar methods are used in other types of RNA algorithms such as combined alignment and folding (Havgaard et al., 2005; Mathews and Turner, 2002).
The Infernal software package includes an accelerated search pipeline that incorporates several of these ideas (Nawrocki et al., 2009). This pipeline begins with maximum-likelihood HMM filters derived from the CM (Weinberg and Ruzzo, 2006), followed by HMM-banded structural alignment (Brown, 2000). These results are further passed to alignment using structure-based query-dependent banding (QDB) (Nawrocki and Eddy, 2007). The final scoring is done by the Inside algorithm, which integrates over all possible alignments rather than only considering the single best alignment. The use of filters does decrease sensitivity somewhat, but it provides ~30-fold speed improvements (Nawrocki et al., 2009).
The 3.0 version of the HMMER software package for profile hidden Markov models (http://hmmer.janelia.org) also includes an acceleration pipeline for searches, but its approach is conceptually quite different. HMMER3 begins with ungapped alignment, under a model that produces short alignment segments with position-specific scores and no indels, and also includes a loop that allows the detection and combined scoring of a set of these hits interspersed with unaligned sequence; this is called the multi-segment Viterbi (MSV) algorithm. A second stage performs more typical Viterbi alignment including gaps, before a final filter that calculates the Forward score, the total likelihood of the sequence, integrating over all possible alignments. Sequences that pass all three filter stages are subjected to some additional processing before being displayed in the results. In addition to this tiered approach moving from simpler to more complex models, HMMER3 uses SIMD vector parallelization at each filter stage to decrease the time requirements. The first stage MSV calculation runs with 16-fold vector parallelization, and the successive stages gradually reduce the number of simultaneous operations in order to increase the precision of the calculations. This acceleration strategy has been quite effective, with internal benchmarks indicating that HMMER now has speed comparable to BLAST for many typical searches.
Because of the similarity between the underlying models of HMMs and CMs, we reasoned that Infernal might also benefit from an acceleration pipeline modeled after the one in HMMER. We knew that the HMM-based filters are less effective for RNA families that have diverse members and little primary sequence conservation, so we wanted to design an approach that would use structural information at all stages. This necessitated a somewhat different design of each pipeline stage, but we sought to retain the core ideas: short, ungapped, multi-hit alignment for initial screening; gradually increasing alignment complexity through multiple filter stages; and parallelization to increase the base speed of each alignment stage.
2 ALGORITHM
Before moving to a description of the method, we give a brief overview of an established notation for covariance models; a more complete description is available in earlier works (Durbin et al., 1998; Eddy, 2002). Sequence positions are indexed by i, j, and k; xi indicates the residue of sequence x at position i. A subsequence xi..xj has length d=j−i+1. A covariance model is represented as M states of various types—Start (S), Pair emit (P), Left emit (L), Right emit (R), Delete (D), Bifurcation (B), End (E), with the type of state v represented by 𝒮v—related by transition probabilities tv(y) for moving from state v to state y. For those states which model residues, there is also a set of emission probabilities ev(xi) for single bases and ev(xi, xj) for pairs.
The states are conceptually ordered into larger groups called nodes, where one state in each node represents consensus, and the remainder provide alternatives of insertions and deletions. The nodes are organized in a branching tree structure that represents the consensus fold of the RNA: beginning from a single Start state, proceeding through various states to produce residues, and branching at bifurcation states to produce new stems, with each branch finally terminating at an End state. An overview of the relationship between a multiple alignment, a structure and a CM is given in Figure 1.
Fig. 1.

Structure of a CM from a multiple sequence alignment. (A) A toy multiple sequence alignment, with conserved base pairs annotated by angle brackets. (B) Corresponding structure for the human sequence. (C) Model construction of a CM. On the left is the ‘guide tree’, corresponding to the consensus structure, with node types and numbers in boxes, and sequence residues in circles (the human sequence is shown). On the right is a small section of a full CM with insert and delete states. Converting the CM to be consensus-only produces a model identical to the guide tree, except that the consensus state now also includes residue probabilities, where the guide tree does not.
2.1 Ungapped single-hit consensus matches
The first step in our heuristic filter considers ungapped consensus hits. By removing the possibility of gaps, we simplify the model considerably, removing all insertion and deletion states and leaving each consensus state as the only representative of its node. This also eliminates the need for transition probabilities, with a deterministic path describing the complete model. We can then define a consensus hit to be a match to an unbranched continuous range of these states. The unbranched nature of matches is a key step in model simplification: as long as matches are unbranched, extending an alignment that currently includes (v,j,d) is a fixed time operation, since the next state is uniquely defined and it corresponds to known changes in the values of j and d. In contrast, extending through a bifurcation requires time proportional to d, since each position could split the sequence between the two halves of the branch. Although match extension is ungapped, we do not constrain the initial value of d, allowing significant variation in the length of sequence outside the match, unlike the otherwise conceptually similar SSP filters (Sun and Buhler, 2008). The scoring of a match is still based on the emission probabilities of its states, so a hit need not contain the exact consensus sequence.
We can represent each possible hit as a state range w..y in our simplified consensus model. The state which ‘begins’ the hit (closer to the root of the model), w, will always be a state that represents at least one residue. The hit may extend through an arbitrary number of model states, matching 5′ and 3′ residues as described by the consensus model. The final state of the match y may be another emitting state or it may be an end state (ϵ). In the first case, the total match consists of two (possibly distant) subsequences aligned to this range of states, xw..y and 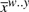 corresponding to the 5′ and 3′ portions, respectively (one but not both of these may be empty). In the second case, reaching a model end connects the two portions of sequence, leaving only a single uninterrupted subsequence xw..ϵ. These cases are similar to the V-type and wedge-type subproblems described in Eddy (2002).
corresponding to the 5′ and 3′ portions, respectively (one but not both of these may be empty). In the second case, reaching a model end connects the two portions of sequence, leaving only a single uninterrupted subsequence xw..ϵ. These cases are similar to the V-type and wedge-type subproblems described in Eddy (2002).
In outline, an optimal dynamic programming (DP) algorithm matching only these single-hit consensus segments would look like this: for each model state v in our consensus model, and for each sequence position j from 1 to L, and for each subsequence length d from 1 to j, the score α(v,j,d) is the better of either starting a new alignment at that triple, or continuing an existing alignment from the next state in the consensus path, if any. The best alignment then corresponds to the set of (v,j,d) that maximize α. Because these hits are by definition unbranched, there is no point where we need to consider subdividing the sequence in order to find the best hit. This reduces the time complexity of the algorithm from O(N4) to O(N3). Further, since we can calculate α(v,j,d) in one slice of the DP cube, analogous to sequence alignment scores in one row of a DP matrix, as long as traceback is not required memory usage can be reduced from O(N3) to O(N2).
2.2 Multi-hit consensus matches
We expect that a single unbranched consensus match will be short, being limited both by the presence of insertions and deletions in real sequences, and by the branch divisions in the complete structure. Therefore, we believe single-hit matches will be insufficient to reliably discriminate between homologous sequences and random similarities. Our approach was designed to be multi-hit, finding local groups of hits which can provide stronger signal than a single match. Requiring these sets of hits to be fully consistent with the CM would re-introduce the complexity that was eliminated in the unbranched match stage, bringing the running time back up to O(N4). Instead, we derive a simpler probabilistic model from the CM that allows these short hits to form arbitrary nested structures, possibly—but not necessarily—consistent with the original consensus fold, as shown in Figure 2. It is possible at this stage to have high-scoring regions that are significantly rearranged compared with the CM, but because this is designed only as a filter, these sequences will be eliminated at later, more stringent alignment stages.
Fig. 2.

Multi-hit ungapped consensus matches. (A) Example of non-coding RNA, from Figure 1. Each shaded block represents one of many potential consensus match segments. (B) Two consensus match segments in a database sequence. (C) Two consensus match segments to the same portions of the model, but in a rearranged configuration.
A simple grammar provides the connections between the ungapped segments in order to allow nested and sequential hits, potentially with intervening unaligned sequence. This grammar has two non-terminals: H, which produces an ungapped homologous model segment, and J, which produces unaligned sequence and connects model segments under the following rules:
where a is any unaligned residue, and w..ϵ and w..y can be any valid ranges in the model. This model has the effect of replacing all model bifurcations with a single bifurcation capable of joining any two segments.
Like the CM it is derived from, this model is intended to be probabilistic, and so each of the possible rules described above has an associated probability. The first rule chooses between these three potential productions with probabilities t1, t2 and t3, respectively. The second rule potentially has a very large number of different productions, depending on the number of valid state ranges w..y in the model. We intend for all possible hits to be equally likely [similar to HMMER3′s uniform segment length distribution (Eddy, 2008)], so this transition is given a uniform value of tH for all possibilities of w..y. The relative weight of V-type (y≠ϵ) to wedge-type cases (y=ϵ) is determined by the numbers of each type that are possible in the model. The choice of these parameters will be described in more detail below.
Given a consensus model with M states, a window length W, and a sequence of length L to search, we calculate a semi-HMM with two states, alternating between segments of non-homologous sequence and segments matching to the grammar non-terminal J, similar to models used in gene finders (Burge and Karlin, 1997; Durbin et al., 1998). We then use the score γ to track all positive-scoring alignments as given in Figure 3. For hit detection, γ is traced back, and each usage of J(j, d) is checked against a minimum reporting threshold. This algorithm has the same general time complexity as other variants of the CYK algorithm adapted for scanning long sequences, namely O(MLW+BLW2), where B is the number of bifurcation states (B<M, and generally B ~ log(M)). The major changes for MSCYK are a large reduction of M, and B fixed at 1, rather than being a variable defined by the model, reducing time complexity to O(LW2) ~ O(N3). As full traceback of which model segments were used is not required, memory requirements can also be reduced from O(MW2) to O(W2).
Fig. 3.

Pseudo-code for the MSCYK algorithm. State types 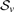 are Pair, Left, or Right emitting, or End.
are Pair, Left, or Right emitting, or End.
3 IMPLEMENTATION
3.1 Parameterization
In the above description, we have used the values t1, t2, t3 and tH without discussion of how these parameters are set or determined. Because they represent probabilities, they are constrained to the [0, 1] interval, and we introduce an additional constraint, which is the expected sequence length generated by the model. We set the expected length to be equal to the consensus length of the CM, a proxy for the expected length of that CM. Given the expected sequence length 〈n〉, the expected length of a single consensus hit 〈nH〉 and the fraction of all model fragments that are V-type r (those that do not reach an End state),
and
leaving t1. Intuitively, t1 describes the proportion of unaligned residues—higher values imply longer stretches of unaligned residues and fewer model segments overall, and the converse, lower values favor fewer unaligned residues and more frequent matches. Based on anecdotal testing of discrimination between real and random sequences, the default for t1 is set to 0.25.
The parameter tH is designed to provide a uniform distribution across all model fragments w..y that non-terminal H could match, defined as 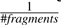 . The number of fragments, expected length of fragments 〈nH〉 and proportion of V-type fragments r are entirely dependent on the original CM, and are calculated when building the consensus-only model. (Implementation details are available in the function cm_consensus_Convert().)
. The number of fragments, expected length of fragments 〈nH〉 and proportion of V-type fragments r are entirely dependent on the original CM, and are calculated when building the consensus-only model. (Implementation details are available in the function cm_consensus_Convert().)
3.2 P-value determination
In order to effectively use this short match alignment method as a filter, we must be able to determine a score threshold for each model that will pass a certain percentage of all input sequences. Although this model is unusual in terms of sequence alignment, it is still a probabilistic model with log-likelihood scoring, so we expect that maximum scores will follow an Extreme Value Distribution (EVD, specifically a type I EVD or Gumbel distribution). An EVD had parameters μ for location, and λ for scale, here corresponding to the rate of decrease in the frequency of increasingly higher scores. With the accurate parameters for the EVD, we can easily convert between scoring thresholds, P-values and expected filter pass rates. Ideally, these parameters would conform to the conjectures of Eddy (2008), with λ=log(2). Otherwise, if λ values for the full CM and MSCYK models were near-equal, this value could be derived from the EVD already fit for the CM.
To test the fit of an EVD, and the behavior of λ if so, we performed simulations of the scores for alignment of random sequences under MSCYK alignment. Figure 4A gives a typical example, with some divergence from an EVD for low scoring sequences, but quickly converging to an exponential tail for higher scoring sequences. Testing across all models in Rfam confirmed that scores fit an exponential tail reasonably well. Unfortunately, the parameter values μ and λ show no particular trends. Although centered near ln(2), λ has too much variation to be approximated as constant, neither μ or λ is correlated with the corresponding parameters from the full CM (Fig. 4B). We concluded that individual EVD fitting of each model is required. By default, the score simulation is performed with 10 000 random sequences of 1 kb each, fitting an exponential tail to the 20% of sequences with the highest maximum scores.
Fig. 4.

Extreme value distribution fits for MSCYK scores. (A) An example EVD (in red) fit to the scores for simulated random sequences against a model of tRNAs (RF00005; 1052 sequences, average length 73). The default score simulation is shown in green (10 000 sequences); a larger simulation of 300 000 random sequences is in black. Dotted lines at the end of each simulation indicate uncertainty due to small numbers of sequences observed. (B) Estimated lambda values for both CM scores and MSCYK scores, across all Rfam families. Tolerances of ±3% are shown for approximating with a constant lambda of ln(2), or for setting lambda for MSYCK equal to the lambda for the (already-simulated) EVD for the CM.
3.3 Vector parallelization
Dynamic programming algorithms are highly amenable to acceleration by vector parallelization (Farrar, 2007; Rognes and Seeberg, 2000; Wozniak, 1997); the HMMER3 pipeline applied these techniques to its filter stages with good synergistic effect (http://hmmer.janelia.org). We have implemented three versions of vector parallel alignment; two of them score standard CYK alignment (at 4× and 8× parallelism) and one implements the MSCYK heuristic at 16×.
At 4× parallelism, CYK is implemented in single-precision floating-point log-odds probabilities, fully replicating the calculation of the non-parallel reference algorithm. The code release provides both SSE_CYKInsideScore(), a score-only version and SSE_CYKDivideAndConquer(), a version with memory-efficient alignment traceback. Empirically, they give ~ 3× speed-up compared with the serial reference versions (without and with traceback, respectively).
The 8× parallel version of CYK uses 16-bit numbers rather than 32-bit; this necessitates a switch from floating-point representation to scaled and rounded integers. Internally, all log-likelihood score values are scaled up by 500 and rounded, with the maximum possible score corresponding to a score of approximately 65 under the CM. We use saturated arithmetic to ensure that extending an alignment with the maximum score retains that score. High-scoring sequences will have multiple alternative alignments that should exceed the cap by variable amounts, so arbitrarily choosing one of those alignments no longer makes sense. Full details for this score-only implementation can be found in SSE_CYKFilter_epi16() in the code. Observed speed-up is ~6-fold (Fig. 5A).
Fig. 5.

Speed-up for vector CYK and MSCYK methods. (A) Time for 4x (red) and 8x (blue) vector CYK implementations, compared with time for scalar implementation, log–log scale. Times measured are for alignment of a sequence of the window length of the model, median of five trials, for all Rfam families. (B) Time for MSCYK search compared to time for scalar CYK search. Red dots are the subset of Rfam families with no bifurcations (simple stems), green dots are for families with a single branch point and blue dots represent more complex families with multiple branches. Times measured are for search of a 10 kb randomly generated sequence.
The MSCYK short match algorithm described above is implemented at even higher parallelism, using 16 simultaneous calculations. This allows only 8 physical bits per score, with the extremely limited range of 0–255. To accommodate this, the floating point values are scaled scaled by 3 and offset for an effective range of −65 to 20 bits of information; again, saturated arithmetic prevents overflow. This implementation also switches from vectorization of adjacent cells in the matrix to using the striped method introduced by Farrar (2007). The full implementation is in function SSE_MSCYK(). The empirical speed-up of this stage depends both on the parallelization and on the MSCYK heuristic; we compare MSCYK to its closest equivalent, a CYK algorithm adapted for scanning long sequences. Total speed-up is model-dependent and ranges from 5- to 100-fold, with larger, more complicated models generally having better relative performance (Fig. 5B).
3.4 Pipeline
The methods described above yield three main components for assembling a search pipeline. The MSCYK scanning algorithm considers a long input sequence, and identifies regions containing ungapped hits to the consensus model. The medium-precision alignment filter examines a sequence window and returns an approximate score for the best gapped alignment within that window. The high-precision alignment function not only determines the best gapped alignment, but also provides an exact score, the boundary coordinates of the alignment, and optionally can give the complete alignment.
To connect these pieces, we also need some post-processing of the initial MSCYK hits to create appropriately sized sequence windows for the later stages. For each CM, there is a W such that no true hits are expected to have length greater than W. The value of W is derived from the query-dependent bands, which calculate the probability distribution over the lengths of hits, meaning we can select a maximum length that sacrifices only a minimal amount of the total probability. Each hit from MSCYK—potentially smaller than the true bounds of the alignment—is expanded from initial bounds (i,j) to (j−W, i+W) so that all sequences of length W that include (i,j) are part of the final window. Separate MSCYK hits which are within W of each other are also merged to reduce redundancy in the list of sequence windows that pass this first filter. The resulting list of sequence windows is appropriate for scoring under the medium-precision filter; those sequences that pass a pre-defined threshold are further passed to the high-precision final alignment.
Each of these main stages has a tunable threshold parameter to control the amount of the search database that reaches the later, more computationally intensive steps. These thresholds are expressed in terms of P-values, requiring calibrated models in order to convert between bit scores and P-values. The MSCYK consensus-hit model is calibrated by score simulation at the beginning of each search, as described above; the medium- and high-precision alignment steps rely on a required CM calibration step as part of the model creation process. By default, the threshold for MSCYK is set such that the hit windows are expected to amount to 2% of the total database size. In general, for a filter pass rate of x, we want one hit (approximate size after padding is 2W) per every 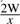 bases. The simulation is set per-kilobase of sequence, so the necessary P-value is
bases. The simulation is set per-kilobase of sequence, so the necessary P-value is 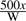 . Since the subsequent stages are markedly slower, this parameter has the largest tunable effect on the speed of the pipeline. The medium-precision filter stage requires individual windows to pass a P-value cutoff, by default 10−5. Finally, the results reported from the final alignment stage are ranked by E-value, with the default showing all hits with E < 1.
. Since the subsequent stages are markedly slower, this parameter has the largest tunable effect on the speed of the pipeline. The medium-precision filter stage requires individual windows to pass a P-value cutoff, by default 10−5. Finally, the results reported from the final alignment stage are ranked by E-value, with the default showing all hits with E < 1.
4 RESULTS
MSCYK is a heuristic designed for search acceleration; we expect that its speed comes with a loss of sensitivity, but we want the loss to be minimal. To quantify this performance trade-off, we evaluated the MSCYK pipeline using the rmark-2 benchmark used in earlier development of the Infernal package (Nawrocki and Eddy, 2007; Nawrocki et al., 2009). It features 450 test sequences, from 51 known ncRNA gene families, embedded in a 10 Mb ‘pseudogenome’. The test sequences are separated from their corresponding alignments in such a way that no test sequence is >60% identical to any sequence that remains in the set of training alignments. The 51 training alignments contain from 5 to 1080 sequences (median 20); each is used to create a single CM as a search query. The pseudogenome itself is randomly generated from an HMM that produces sequence modeled from a variety of species, such that it includes regions of biased nucleic acid composition.
Homology search methods are evaluated based on their ability to detect the embedded test sequences, given the known members of the family in the training set, while also minimizing the number of false positive hits. A reported alignment is considered a hit if the overlap between the reported sequence and the embedded sequence corresponds to at least half of the shorter of the two. Hits against a true sequence of a different family are considered to be neither true positives or false positives, because there are known to be evolutionary relationships between at least some of the ncRNA families, but these are not all well characterized. (For example, Rfam operationally classifies RNase P into four families.)
We use this benchmark to make two comparisons about the performance of the filtering strategy described here. First, to address whether MSCYK is an effective filtering strategy, we compare the MSCYK pipeline to the reference (unaccelerated) implementation of CM search in Infernal. In addition, Infernal has an existing acceleration pipeline; we compare the results between the two heuristics for their relative performance. The MSCYK filter's model and parameters were not optimized directly against the rmark-2 benchmark; in development, they were trained with Rfam seed alignments and tested for score discrimination between IID background sequence and either sequences from the Rfam full alignments or sequences randomly generated from the training CM.
The existing acceleration pipeline operates in four stages: an HMM filter, an HMM-banded structural filter, a structural filter using query-dependent banding (QDB) and finally alignment with the Inside algorithm. The first HMM-based filter is a sequence-only method, while the second uses an HMM alignment to constrain a structural alignment. QDB is structural alignment constrained by subsequence lengths; these constraints are complementary to the sequence-based HMM constraints. The final alignment stage uses the total log-likelihood calculation (Inside algorithm), rather than the maximum log-likelihood calculation that we have referred to throughout this work (CYK algorithm).
The performance of different filtering approaches on the rmark-2 benchmark are shown in Figure 6. Full search without any filtering stages (black line) has the best sensitivity and specificity trade-offs, and also has by far the longest running time of 648 CPU hours. (All times are aggregate results for the benchmark, with each search being run on a single 2.66 GHz Intel X5550 processor.) At slightly lower sensitivity for a given false positive rate is the existing filter strategy (green). Although these filters do miss some sequences that would be detected without using any filters, the run time of only 23.5 CPU hours makes the trade-off generally worthwhile.
Fig. 6.

ROC curves for the rmark-2 benchmark. Performance plots are shown for Infernal version 1.0, with and without the standard filtering strategy, and the new structure-based parallel filtering strategy both alone and in combination with the existing filters. All methods use default parameters for their filter cut-off values; the curve reflects only the E-value-based sort order of the final output.
The MSCYK pipeline (dashed blue line), has notably worse sensitivity than either unfiltered search or the previous filtering methods. The time required to run the MSCYK pipeline is only a little slower than the existing filter strategies. The first-stage structural filter alone here requires 25.4 CPU hours, with the second and third stages combining to require about half as much time again, for a total run time in the range of 37 CPU hours. Although this is a significant speed-up over the reference implementation, it is not an improvement over the existing acceleration pipeline.
The major loss in sensitivity for the new filtering approach occurs at the very beginning of the pipeline, at the ungapped consensus alignment stage. We looked at the sequences that fail to pass this stage, and they broadly share two general features. First, they have lower than average information content per position (i.e. less conservation), and require longer alignments in order to produce a significant hit, but the requirement of ungapped, unbranched alignment limits the possible length of hits. Second, they have insertions or deletions at all (or nearly all) of the stem–loops in the model. This is not inherently surprising; the ends of stem–loops commonly show less conservation and more changes in length. However, the model has a significant score preference for alignments that reach the end of a hairpin with no intervening unaligned sequence; without at least one well-conserved stem–loop to anchor or seed the potential hit, the scores of these sequences are well down into the noise of scores from random sequences.
Despite its lower sensitivity when evaluated alone, MSCYK should be complementary to the existing Infernal pipeline, because it implements structure-based rather than sequence-based filters. To test this, we examined the overlap in hits between the acceleration strategies, and found that MSCYK identified a significant number of hits that had been missed by the sequence-based approach. To leverage this complementarity, we designed a small driver script which runs both pipelines and determines a union set of their results. Because the two pipelines use different scoring methods for their final stages (CYK versus Inside), all hits are re-scored under the Inside algorithm to provide unified output. This combined approach has sensitivity and false positive results which are competitive with running Infernal without any filters at all (Fig. 6, dotted red line).
The running time for the combined approach is the sum of the time required for each pipeline, plus some overhead. In the proof-of-concept runs, the benchmark run took 61.2 CPU hours, still 10-fold better than the time required when running the search without any filters. This gives only a rough upper bound on the time required if a pipeline was designed from the ground up to use both methods; since the majority of sequences are identified in common by both approaches, many alignments are unnecessarily performed multiple times, in addition to the simple overhead of running two searches and combining the results.
5 DISCUSSION
Although MSCYK does not have the sensitivity required to stand on its own, this structure-based heuristic is successful in identifying true hits that are missed by sequence-based methods. The combined set of hits from both pipeline methods has sensitivity approaching that of unfiltered search; although there are some sequences which are not found by either method, they are few in number. These performance gains in sensitivity relative to the existing filters are large enough to suggest that this method or a similar structure-based approach will be an important component of improving the performance of CM-based RNA search in the future.
In this work, we have not implemented a production-quality filter pipeline in Infernal; the results shown here are from a simple proof-of-concept script. A more efficient production pipeline would take steps to reduce the amount of time spent in the structure-based portion of the analysis. This could be done both toward the end of the pipeline—combining the sets of hits earlier to reduce redundant alignment of sequences found by both methods—and at the beginning of the pipeline, reducing the number of sequences that are subjected to the structure-based pipeline at all. One approach to this might be to start with an HMM-based sequence-only filter, with sequences that clearly pass being moved directly to the main processing pipeline, but with a second group of sequences with lower but potentially interesting ‘twilight-zone’ scores being analyzed under a structure-based pipeline optimized for alignments with lower sequence identity. These adjustments will be increasingly important with an expected upcoming move of Infernal from HMMER2 to HMMER3 for its underlying engine for HMM filters. The dramatically increased speed of HMMER3 should provide large automatic speed gains for the existing HMM filters, rendering the structure-based filters increasingly slow by comparison.
Funding: Howard Hughes Medical Institute.
Conflict of Interest: none declared.
REFERENCES
- Bafna V., Zhang S. FastR: fast database search tool for non-coding RNA. Proc. IEEE Comput. Syst. Bioinform. Conf. 2004:52–61. doi: 10.1109/csb.2004.1332417. [DOI] [PubMed] [Google Scholar]
- Brown M.P. Small subunit ribosomal RNA modeling using stochastic context-free grammars. Proc. Int. Conf. Intell. Syst. Mol. Biol. 2000;8:57–66. [PubMed] [Google Scholar]
- Burge C., Karlin S. Prediction of complete gene structures in human genomic DNA. J. Mol. Biol. 1997;268:78–94. doi: 10.1006/jmbi.1997.0951. [DOI] [PubMed] [Google Scholar]
- Durbin R., et al. Biological Sequence Analysis: Probabilistic Models of Proteins and Nucleic Acids. Cambridge, UK: Cambridge University Press; 1998. [Google Scholar]
- Eddy S.R. A memory-efficient dynamic programming algorithm for optimal alignment of a sequence to an RNA secondary structure. BMC Bioinformatics. 2002;3:18. doi: 10.1186/1471-2105-3-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eddy S.R. A probabilistic model of local sequence alignment that simplifies statistical significance estimation. PLoS Comput. Biol. 2008;4:e1000069. doi: 10.1371/journal.pcbi.1000069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrar M. Striped Smith-Waterman speeds database searches six times over other SIMD implementations. Bioinformatics. 2007;23:156–161. doi: 10.1093/bioinformatics/btl582. [DOI] [PubMed] [Google Scholar]
- Freyhult E.K., et al. Exploring genomic dark matter: A critical assessment of the performance of homology search methods on noncoding RNA. Genome Res. 2007;17:117–125. doi: 10.1101/gr.5890907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner P.P., et al. Rfam: Wikipedia, clans and the “decimal” release. Nucleic Acids Res. 2011;39:D141–D145. doi: 10.1093/nar/gkq1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Havgaard J.H., et al. Pairwise local structural alignment of RNA sequences with sequence similarity less than 40% Bioinformatics. 2005;21:1815–1824. doi: 10.1093/bioinformatics/bti279. [DOI] [PubMed] [Google Scholar]
- Liu T., Schmidt B. Parallel RNA secondary structure prediction using stochastic context-free grammars. Concurr. Comput. Pract. Exp. 2005;17:1669–1685. [Google Scholar]
- Lowe T.M., Eddy S.R. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25:955–964. doi: 10.1093/nar/25.5.955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macke T.J., et al. RNAMotif, an RNA secondary structure definition and search algorithm. Nucleic Acids Res. 2001;29:4724–4735. doi: 10.1093/nar/29.22.4724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathews D.H., Turner D.H. Dynalign: an algorithm for finding the secondary structure common to two RNA sequences. J. Mol. Biol. 2002;317:191–203. doi: 10.1006/jmbi.2001.5351. [DOI] [PubMed] [Google Scholar]
- Nawrocki E.P., Eddy S.R. Query-dependent banding (QDB) for faster RNA similarity searches. PLoS Comput. Biol. 2007;3:e56. doi: 10.1371/journal.pcbi.0030056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nawrocki E.P., et al. Infernal 1.0: Inference of RNA alignments. Bioinformatics. 2009;25:1335–1337. doi: 10.1093/bioinformatics/btp157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rognes T., Seeberg E. Six-fold speed-up of Smith-Waterman sequence database searches using parallel processing on common microprocessors. Bioinformatics. 2000;16:699–706. doi: 10.1093/bioinformatics/16.8.699. [DOI] [PubMed] [Google Scholar]
- Sun Y., Buhler J. Designing secondary structure profiles for fast ncRNA identification. Comput. Syst. Bioinformatics Conf. 2008;7:145–156. [PubMed] [Google Scholar]
- Weinberg Z., Ruzzo W.L. Exploiting conserved structure for faster annotation of non-coding RNAs without loss of accuracy. Bioinformatics. 2004a;20(Suppl. 1):I334–I341. doi: 10.1093/bioinformatics/bth925. [DOI] [PubMed] [Google Scholar]
- Weinberg Z., Ruzzo W.L. RECOMB '04. New York, NY: ACM; 2004b. Faster genome annotation of non-coding RNA families without loss of accuracy; pp. 243–251. [Google Scholar]
- Weinberg Z., Ruzzo W.L. Sequence-based heuristics for faster annotation of non-coding RNA families. Bioinformatics. 2006;22:35–39. doi: 10.1093/bioinformatics/bti743. [DOI] [PubMed] [Google Scholar]
- Wozniak A. Using video-oriented instructions to speed up sequence comparison. Comput. Appl. Biosci. 1997;13:145–150. doi: 10.1093/bioinformatics/13.2.145. [DOI] [PubMed] [Google Scholar]