

Abstract
Background
β-thalassemic syndromes are inherited red cell disorders characterized by severe ineffective erythropoiesis and increased levels of reactive oxygen species whose contribution to β-thalassemic anemia is only partially understood.
Design and Methods
We studied erythroid precursors from normal and β-thalassemic peripheral CD34+ cells in two-phase liquid culture by proteomic, reverse transcriptase polymerase chain reaction and immunoblot analyses. We measured intracellular reactive oxygen species, heme levels and the activity of δ-aminolevulinate-synthase-2. We exposed normal cells and K562 cells with silenced peroxiredoxin-2 to H2O2 and generated a recombinant peroxiredoxin-2 for kinetic measurements in the presence of H2O2 or hemin.
Results
In β-thalassemia the increased production of reactive oxygen species was associated with down-regulation of heme oxygenase-1 and biliverdin reductase and up-regulation of peroxiredoxin-2. In agreement with these observations in β-thalassemic cells we found decreased heme levels related to significantly reduced activity of the first enzyme of the heme pathway, δ-aminolevulinate synthase-2 without differences in its expression. We demonstrated that the activity of recombinant δ-aminolevulinate synthase-2 is inhibited by both reactive oxygen species and hemin as a protective mechanism in β-thalassemic cells. We then addressed the question of the protective role of peroxiredoxin-2 in erythropoiesis by exposing normal cells to oxidative stress and silencing peroxiredoxin-2 in human erythroleukemia K562 cells. We found that peroxiredoxin-2 expression is up-regulated in response to oxidative stress and required for K562 cells to survive oxidative stress. We then showed that peroxiredoxin-2 binds heme in erythroid precursors with high affinity, suggesting a possible multifunctional cytoprotective role of peroxiredoxin-2 in β-thalassemia.
Conclusions
In β-thalassemic erythroid cells the reduction of δ-aminolevulinate synthase-2 activity and the increased expression of peroxiredoxin-2 might represent two novel stress-response protective systems.
Keywords: β-thalassemia, heme biosynthesis, oxidative stress, ROS, peroxiredoxin-2
Introduction
β-thalassemias are common inherited red cell disorders characterized by absent or reduced synthesis of β globin chains. Despite extensive knowledge of the molecular defects causing β-thalassemia, less is known about the mechanisms responsible for the associated ineffective erythropoiesis and reduced red cell survival.1,2 β-thalassemic erythropoiesis is characterized by an imbalance of α/β globin chain synthesis, mostly evident in the homozygous forms, leading to the accumulation of excess free α globin chains associated with reduced heme production. How the globin chain imbalance might affect the rate of heme synthesis is still a matter of investigation.3,4 Increased levels of reactive oxygen species (ROS) have been reported to contribute to the anemia of β-thalassemia; the mechanisms protecting against ROS have been only partially investigated.1,5,6
Previous studies identified a small protein that stabilizes the α chains (AHSP, α hemoglobin-stabilizing protein) and that partially protects the erythroid precursors from the α chain excess. Indeed, anemia in β-thalassemic mice is more severe in those animals which are AHSP-deficient.7–9 However, the impact of AHSP deficiency in β thalassemia patients is still under evaluation and the link between decreased AHSP expression and severity of β-thalassemic syndromes remains speculative.9 Another protective factor in β-thalassemic erythropoiesis is the heme-regulated inhibitor of protein translation, which represses globin translation in heme-deficient erythroid precursors.10 Heme-regulated inhibitor of protein translation plays a role in murine β-thalassemia, since anemia is more severe in β-thalassemic mice genetically lacking this protein.10,11 While the heme-mediated mechanisms of globin synthesis control are partially understood, less is known about factors that modulate heme synthesis in erythroid cells.
δ-aminolevulinate synthase-2 (ALAS-2) is the first and rate-limiting enzyme of the heme biosynthetic pathway in erythroid precursors12–15 Heme regulates the non-erythroid ALAS-1 at multiple levels including reduction of transcription and translation, destabilization of mRNA, inhibition of mitochondrial transport of precursor protein and direct heme-dependent inhibition of the enzyme activity.15 It is well known that heme binding may inhibit mitochondrial translocation of ALAS-2 precursor, and that the post-transcriptional control of the enzyme’s synthesis is iron-dependent through iron-regulatory proteins.16 However, regulatory mechanisms of ALAS-2 activity remain unknown.12–14 Heme turnover is controlled by heme-oxygenases: HO-1, the inducible form, and HO-2, the constitutive isoform.17 Both isoforms catalyze the rate-limiting step in the oxidative degradation of heme to biliverdin. Subsequently biliverdin reductase (BVR) degrades biliverdin to bilirubin, which has strong cytoprotective properties.18
Design and Methods
Cell culture of erythroid precursors from CD34+ and K562 cells
Peripheral blood from adult normal volunteers and from transfusion-independent β-thalassemia patients (i.e., patients with β-thalassemia intermedia) was collected, after informed consent had been obtained according to the guidelines established by the Ethics Committee for human subject studies of the University of Milan. We analyzed 20 erythroid cultures obtained from peripheral blood of different normal subjects and 20 erythroid cultures obtained from ten homozygous β-thalassemic patients (β0cod39) (see Online Supplementary Design and Methods). We evaluated the morphology and the expression of the surface markers, CD71 and glycophorin A, in erythroid cells at days 7 and 14 of culture, corresponding to early and late erythroid precursors, as previously described19,20 (Online Supplemental Design and Methods and Online Supplementary Figure S1A-C).
The K562 human erythroleukemia cell line was obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA). K562 cells were maintained in Iscove’s medium (Sigma Aldrich, Milan, Italy), supplemented with 10% fetal bovine serum (Celbio Pero, Milan, Italy), 10 U/mL penicillin and 0.1 mg/mL streptomycin (Pen/Step) (Celbio Pero). The cell line was grown in a humidified, 5% CO2 atmosphere, at 37 °C.
Measurements of hydrogen peroxide and superoxide anion as reactive oxygen species in erythroid precursors
ROS production in erythroid precursors was measured as reported in the Online Supplementary Design and Methods.
Proteomic analysis of cultured cells
Two-dimensional cell culture analysis
Cells for two-dimensional (2D) electrophoresis were lysed (in medium containing bicine 25 mM, NaF 25 mM, NaCl 0.4 M, EDTA 20 mM, Triton 1.5%, Na3VO4 1 mM, benzamidine 3 mM and a tablet with a cocktail of protease inhibitors) and delipidated as previously described.21,22 Details on the generation of 2D maps are reported in the Online Supplementary Design and Methods.
Image analysis and statistical analytic strategy
Spots differently expressed were identified by Progenesis SameSpots software (Non Linear Dynamics, Newcastle-Upon-Tyne, UK).22 Details are reported in the Online Supplementary Design and Methods. The selected spots were excised from colloidal Coomassie-stained gels and identified by MALDI-TOF MS/MS analysis. Details are reported in the Online Supplementary Design and Methods.
Immunoblot analysis
Cells for one-dimensional electrophoresis were solubilized as described by Karur et al.,23 with a few modifications. The details are reported in the Online Supplementary Design and Methods. Whenever indicated culture media underwent a purification step using a Qproteome Albumin/IgG Depletion column, according to the manufacturer’s protocol (Qiagen, Vancouver, Canada) and 80 μg of proteins were studied by immunoblot analysis.
Peroxiredoxin-2: cytofluorimetric and immunoprecipitation assay
To evaluate the effects of oxidative damage on peroxiredoxin-2 (PRDX2) expression, we exposed normal erythroid precursors to hydrogen peroxide (H2O2) (16 μM), as previously reported.24 Details on cytoflourimetric analysis are reporetd in the Online Supplementary Design and Methods. Whenever indicated, PRDX2 was immunoprecipitated from cell lysates (11×106 cells). Details are given in the Online Supplementary Design and Methods.
Heme concentration measurement
Heme concentration in erythroid precursors was determined by a spectrophotometric method that exploits the heme peroxidative activity on chlorpromazine dye; for details and related references see the Online Supplementary Design and Methods.
Quantitative real-time polymerase chain reaction
For quantitative real-time polymerase chain reaction (qRT-PCR) total RNA was isolated from cell pellets on days 7 and 14 of culture by the method of Chomczynski and Sacchi;25 for details see the Online Supplementary Design and Methods.
δ-aminolevulinate synthase-2 activity assay
ALAS-2 was a generous gift of Dr. Gloria C. Ferreira (University of Tampa, FL; USA). ALAS-2 activity was determined as reported by Hunter et al.26 on the cytoplasmic fraction of erythroid precursors from controls, control-treated H2O2 cells and β-thalassemic cells on days 7 and 14 of culture; for details see the Online Supplementary Design and Methods. Analyses of the tertiary and secondary structure of ALAS-2 are reported in the Online Supplementary Design and Methods.
Silencing of peroxiredoxin-2 in K562 cells and exposure to oxidative stress
Details on silencing of PRDX2, the treatment of K562 cells and measurement of cell growth are provided in the Online Supplementary Design and Methods.27
Kinetic and inhibition assays of peroxidase activity of peroxiredoxin-2 and measurements of hemin binding to peroxiredoxin-2
Human PRDX2 was cloned, expressed and purified to homogeneity as reported in the Online Supplementary Design and Methods. The measurement of PRDX2 kinetic parameters, using H2O2 in substrate and inhibition assays in the presence of hemin, were performed as reported in the Online Supplementary Design and Methods. The equilibrium dissociation binding constant of PRDX2 with hemin was evaluated by exploiting protein tryptophan intrinsic fluorescence quenching, following addition of increasing hemin concentration, as a sensor of hemin binding. Details on the analysis of kinetics and inhibition of PRDX2 and equilibrium binding studies are reported in the Online Supplementary Design and Methods.
Results
Increased oxidative stress in β-thalassemic erythroid precursors showing different proteomic two-dimensional maps
We studied erythroid precursors from normal and β-thalassemic peripheral CD34+ cells in two-phase liquid culture in early (7 days of culture) and late (14 days of culture) phases of maturation (Online Supplementary Figure S1A,B). We previously validated this in vitro model of β-thalassemic ineffective erythropoiesis by both morphological and cytofluorimetric analyses.19 Since the percentage of cells positive for the surface markers CD71 and glycophorin A were similar in normal and β-thalassemic cells at 7 and 14 days of culture, we studied the cells at these time points (Online Supplementary Figure S1C).
We first measured the amount of ROS as markers of oxidative stress during maturation of both normal and β-thalassemic erythroid precursors. We observed a significant increase of ROS production in both early and late β-thalassemic erythroid precursors (Online Supplementary Figure S1D). No significant differences were observed within β-thalassemic erythroid cells in different stages of maturation, whereas normal controls showed a slight increase in ROS production only in late erythroblasts (Online Supplementary Figure S1D).
We then evaluated the proteomes of normal and β-thalassemic erythroid precursors. Comparing normal and β thalassemic cells, at 7 and 14 days of culture 55 spots were differently expressed, of which 25 were identified by mass spectrometry (Figure 1A, Table 1). The proteins differently expressed were grouped into eight functional clusters: (i) cell structure-related proteins; (ii) metabolic enzymes; (iii) stress-response and chaperones; (iv) ras-related system; (v) intracellular signaling cascade; (vi) ubiquitin-proteasome system; (vii) hemoglobin complex and (viii) not functionally classified protein (Table 1). As expected, β-globin chains were detected in normal mature (day 14 of culture) but not in normal early erythroid precursors nor in β-thalassemia cells (Figure 1A, Table 1).
Figure 1.

β-thalassemic erythroid precursors show increased oxidative stress, reduced biliverdin reductase/ heme-oxygenase 1 expression and heme content. (A) Two-dimensional (2D) gel analysis of erythroid precursors from normal (control) and β-thalassemic (β thal cells) subjects at 7 and 14 days of culture. The images show representative gels of ten others with similar results. The differently expressed spots were identified (black circle) by image analysis, excised from the colloidal Coomassie-stained gels and analyzed by mass spectrometry (see also Design and Methods and Online Supplementary Design and Methods). Spot numbers correspond to those reported in Table 1. (B) Immunoblot analysis of BVR, HO-1, HO-2 and ALAS-2 in normal (C7, C14) and β-thalassemic (T7 and T14) cells. We showed on the colloidal Coomassie-stained gel the band at ~ 37 kDa used as a loading control and identified by mass spectrometry as the glycerol 3P-dehydrogenase cytoplasmic domain (Acc. n: P21695, 12% sequence coverage). One representative gel of ten others with similar results is presented. (C) Intracellular heme levels in normal (white bars: C7, C14) and β-thalassemic (gray bars: T7 and T14) erythroid cells. Experiments were repeated in triplicate, the error bars represent the S.E.M.
Table 1.
Identification of proteins from normal and β-thassemic erythroid progenitors.

In control cells the following proteins were found in a more acid form compared to their theoretical pI: BVR (spots 14, 15), carbonic anhydrase 1 (spots 9, 12), Ras-GEF domain containing family member 1A (spot 4) and inorganic pyrophosphase 2 mitochondrial precursors (spot 2) (Table 1; Figure 1A). In β-thalassemia cells the following proteins were in a more acid form compared to their theoretical pI: PRDX2 (spots 43, 48) and phospho 2, pyridoxal 5′-phosphate phosphatase (spot 30), while stathmin was detected at higher molecular weight than the theoretical one (Table 1, Figure 1A).
Among the differently expressed proteins in β-thalassemia we found decreased expression of BVR and increased expression of three proteins involved in oxidative defense: glutathione peroxidase 1, in the early phase of β-thalassemic erythropoiesis, PRDX2 throughout all β-thalassemic erythropoiesis and heat shock protein 27 (HSP27) in the late phase of β-thalassemic erythropoiesis (Table 1). We validated the proteomic results by both qRT-PCR for the mRNA expression and immunoblot analysis with specific antibodies for the following genes/proteins: BVR, PRDX2 and HSP27.
β-thalassemic erythroid precursors show reduced biliverdin reductase/ heme oxygenase-1 expression and decreased intracellular heme levels
In β-thalassemic erythropoiesis mRNA levels of BVR were almost unmodified during erythroid maturation and were markedly lower than those in controls (Online Supplementary Figure S2B). BVR protein expression was modulated during erythropoiesis in both cell types but markedly reduced in β-thalassemic erythroid precursors compared to in control cells, in agreement with the proteomic data (Figure 1A,B, Online Supplementary Figure S2C).
Since BVR is a part of the protective system involved in heme metabolism through heme-oxygenases, we then evaluated heme-oxygenase expression and heme content in both normal and β-thalassemic erythroid precursors. We observed increased expression of HO-1 gene and protein in both normal and β thalassemic cultures during maturation. However, both mRNA and protein HO-1 were markedly lower in β-thalassemic cells than in controls (Online Supplementary Figures S1B and S2C, Figure 1B), in agreement with the reduced BVR expression observed in β-thalassemic erythroid precursors. HO-2 gene expression was up-regulated in late normal erythroid precursors and down-regulated in β-thalassemic erythroid precursors. The expression of HO-2 protein was similar in both cell types with a reduction at day 14 of culture (Online Supplementary Figures S1B and S2C, Figure 1B). The discrepancy between HO-2 gene-protein levels in normal cells may be related either to a regulation of HO-2 transcriptional levels as shown in other models28 or to HO-2 protein stability as recently reported by Ding et al.29
We then asked whether the reduction of the elements of the BVR/HO-1 system was paralleled by changes in intra-cellular heme levels in β-thalassemic erythropoiesis. As shown in Figure 1C, the intracellular heme content was significantly increased in early β-thalassemic precursors but strongly reduced in late β thalassemic precursors, compared to the levels in controls. Since ALAS-2 catalyzes the rate-limiting step in heme biosynthesis in erythroid cells, we evaluated ALAS-2 expression and activity during erythropoiesis.
δ-aminolevulinate synthase-2 activity is reduced in β-thalassemic precursors and is down-regulated by reactive oxygen species and heme
We found low expression of ALAS-2 gene and almost undetectable protein during early erythroid erythropoiesis but markedly and similarly increased ALAS-2 gene and protein expression in late erythroid precursors from both cell types (Figure 1B, Online Supplementary Figure S1B,C).
We then measured the activity of ALAS-2 during erythropoiesis and found that it was increased in normal late erythropoiesis, as expected, but significantly reduced in β-thalassemic erythropoiesis (Figure 2A). This result is in line with the lower heme levels in β-thalassemic late erythropoiesis than in normal late erythropoiesis. In addition we treated the control cells with 16 μM H2O2, as previously reported by Uchida et al.,24 and found a marked reduction in ALAS-2 activity and in heme content compared to the activity of normal controls at day 14 of culture (data not shown).
Figure 2.

ALAS-2 activity is reduced in β-thalassemic erythroid precursors and is affected by oxidative stress. (A) ALAS-2 activity in normal and β-thalassemic erythroid cells at day 7 ( white bars: C7, T7) and day 14 of culture (gray bars: C14, T14). An aliquot of 20 μL of the cytoplasmic fractions of day 7 and 14 erythroid precursors from normal controls or β-thalassemic patients was diluted into the assay reaction mixture and activity was measured in the presence of 10 μM pyridoxal 5′-phosphate (see Design and Methods and Online Supplementary Design and Methods). The activity determined at 14 days in control cells was set as 100%. Experiments were repeated in triplicate, the error bars represent the S.E.M.; *P< 0.05 compared to control cells; °P<0.05 comparing 14 vs. 7 days. (B) Recombinant 5-δ aminolevulinate synthase (ALAS-2) activity in the presence of saturating concentrations of H2O2 or hemin. Error bars represent the means ± SD, *P<0.05 compared to control cells; °P<0.05 compared to baseline. Inset: The IC50 value measured was then used to determine the Ki value. 5-δ aminolevulinate synthase (ALAS-2) activity in the presence of increasing concentrations of H2O2 or hemin. The IC50 value measured was then used to determine the Ki value. Errors were less than 10%.
To address whether ROS or heme levels might affect ALAS-2 activity, we used recombinant ALAS-2. As shown in Figure 2B, ALAS-2 activity was reduced in the presence of increasing concentrations of either hemin, (a commonly used heme analog) or H2O2 (ROS agent). The Ki values (inset) calculated from the IC50 were 5±2 μm and 2±0.5 μM for H2O2 and hemin, respectively. These Ki values are in the range of the ROS and heme concentrations measured (Figure 1C and Online Supplementary Figure S1D), supporting the observation of reduced ALAS-2 activity as a protective mechanism in β-thalassemic cells characterized by high ROS production. We also determined that the secondary structure of ALAS-2 is unaffected by the presence of heme or H2O2 (Online Supplementary Figure S2D), thus excluding the possibility that these ligands damage the structural integrity of the protein. Interestingly, subtle pyridoxal 5′-phosphate active site changes could be detected by visible CD analysis, thus corroborating heme and/or hydrogen peroxide binding (Online Supplementary Figure S2D, inset). Based on all these data, we propose that heme biosynthesis in β-thalassemic erythropoiesis might be regulated by an interplay between itself and ROS levels, resulting in ALAS-2 inhibition likely with the aim of protecting the developing cells from the cytotoxic effects of newly synthesized heme.
Peroxiredoxin-2 expression is increased in β-thalassemic erythropoiesis
We then evaluated expression of PRDX2, CATALASE, and HSP27 genes and protein during erythropoiesis in normal and β-thalassemic cells.
In control erythroid precursors PRDX2, CATALASE, and HSP27 genes were up-regulated at day 14 of culture corresponding to late erythropoiesis (Online Supplementary Figure S3A). In β thalassemic cells mRNA levels of PRDX2 and CATALASE genes were almost unmodified during erythroid maturation, but the expression of PRDX2 and CATALASE genes was 5-fold increased and 2-fold decreased, respectively, compared to the expression in controls (Online Supplementary Figure 3SA). HSP27 mRNA levels were similarly up-regulated in late erythropoiesis in both cell models (Online Supplementary Figure 3SA).
PRDX2 protein expression was strongly increased in β thalassemic cells, especially at day 7 when it was present as a double band (Figure 3). This finding may be related to post-translational modifications of the protein, which would be consistent with the proteomic data and similar to what has been described in other cell models.30–33 In more mature (day 14) cells PRDX2 expression was slightly increased in β-thalassemia, but was abundant in culture media, further strengthening the increased PRDX2 expression at this stage (Figure 3 and Online Supplementary Figure S3C). In contrast the expression of PRDX1 and six proteins was similar in both types of cell and almost exclusively limited to the early phase of maturation (Online Supplementary Figure S3D). The abundance of catalase protein was similar in both normal and β-thalassemic early erythroid cells, but was slightly increased in late normal cells (Figure 3, Online Supplementary Figure S3B). HSP27 protein expression was significantly higher in β-thalassemic cells in late erythropoiesis than in normal cells (Figure 3, Online Supplementary Figure S3B). The discrepancy between HSP27 mRNA and protein levels observed in normal cells might be related to mRNA translational events, as reported for other types of cells.34 Collectively these data indicate up-regulation of PRDX2 and HSP27, likely as a response to the high pro-oxidant status of β-thalassemic cells. In addition, the finding of PRDX2 in the culture media seems to reflect modulation rather than non-specific release, since there were no significant changes in catalase release into the culture media during early or late erythropoiesis in both cell types.
Figure 3.
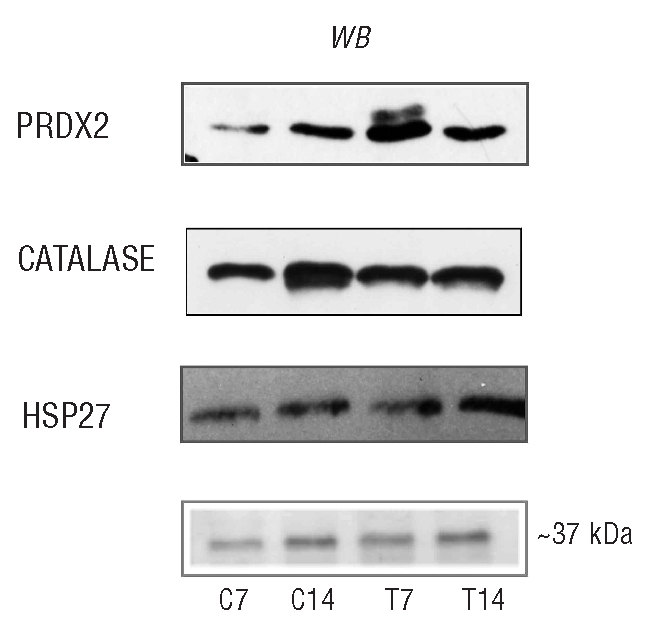
Peroxiredoxin-2 expression is increased in β-thalassemic erythroid precursors. Immunoblot analysis of PRDX2, CATALASE and HSP27 protein in normal and β thalassemic erythroid cells. PRDX2, CATALASE and HSP27 protein expression in normal (C7, C14) and β thalassemic (T7 and T14) cells. We showed on the colloidal Coomassie stained gel the band at ~ 37 kDa used as loading control and identified by mass spectrometry as the glycerol 3P-dehydrogenase cytoplasmic domain (Accession number: P21695, 12% sequence coverage). One representative gel of ten others with similar results is presented.
Since the anti-oxidant system was sensitive to H2O2, as catalase was similarly expressed in both normal and β-thalassemic cells, while PRDX2 was up-regulated during all β-thalassemic erythropoiesis, we considered PRDX2 an interesting candidate as a new cytoprotective system in stress erythropoiesis such as that occurring in β-thalassemia.
Peroxiredoxin-2 is up-regulated in response to oxidative stress and protects erythroid precursor
PRDX2 is a typical 2-cysteine (Cys-51 and Cys-172) peroxiredoxin. Previous studies have shown that oxidative stress can up-regulate the expression of PRDX2 in other cell types and that genetically modified cells over-expressing PRDX2 are generally more protected from severe oxidative stress.32,35
To evaluate the possible protective role of PRDX2 against oxidative stress in erythropoiesis, we added H2O2 to normal erythroid precursors (Figure 4A,B). We observed a delay in cell growth between days 7 to 10 and increased PRDX2 protein expression on day 10 followed by cell recovery on day 14 of culture (Figure 4A,B, Online Supplementary Figure S3E-G).
Figure 4.

Effects of oxidative stress on PRDX2 protein expression in normal erythroid precursors and in K562 cells with PRDX2 depletion. (A) Effect of H2O2 (16 μM) on cell proliferation of control erythroid precursors. Data are presented as mean ± SD (n=3). (B) Flow-cytometric analysis of PRDX2 in normal erythroid precursors exposed to H2O2 (16 μM). Each culture was divided into two separate cultures with and without H2O2 (16 μM). The ordinate represents the number of cells displaying the fluorescent intensity given by the abscissa (see also Online Supplementary Data for cell gating strategy). The figure shows the results from one typical erythroid culture representative of three studied. (C) Effect of PRDX2 depletion during erythroid differentiation of K562 cells. Human K562 cells were induced to differentiate with hemin (50 μM) after 24 h of transfection with K562 wild type and non-silencing vector as control (Crtl), and shRNAmir plasmid against PRDX2. Samples were collected at specific time points (after transfection): before hemin addition, and at days 4 and 6 after hemin addition. Erythroid differentiation was assessed by FACS analysis for glycophorin A (CD235A). Analyses were performed using a FACSCalibur (Becton Dickinson, San Jose, CA, USA) with CELL Quest software, version 3.3, after gating for viable cells. The upper panel shows the immunoblot analysis with specific anti-PRDX2 antibody in silenced cells (shPRDX2), non-silencing vector as control cells (Crtl) and K562 cells. One representative gel of three other with similar results is presented. (D) Effect of H2O2 on cell viability of parental (upper panel) and hemin-induced (lower panel) k562 cells after silencing. Data are presented as mean ± SD (n=3). Cells were treated by 30 min incubation with 50 μM H2O2 (
 K562 wild type, □Ctrl, non-silencing vector, and ■shPRDX2 after transfection; ▵K562 wild type,
K562 wild type, □Ctrl, non-silencing vector, and ■shPRDX2 after transfection; ▵K562 wild type,
 Ctrl, non-silencing vector, and ▴ shPRDX2, hemin-induced cells). Cell growth was evaluated by seeding the cells, after repeated washing, in Iscove’s complete medium (1.5x105 cells/mL); the number of viable cells was evaluated at 24 and 48 h by the trypan blue dye exclusion test;47 Data are presented as ratio of trypan blue-positive cells; n=3 for all points.
Ctrl, non-silencing vector, and ▴ shPRDX2, hemin-induced cells). Cell growth was evaluated by seeding the cells, after repeated washing, in Iscove’s complete medium (1.5x105 cells/mL); the number of viable cells was evaluated at 24 and 48 h by the trypan blue dye exclusion test;47 Data are presented as ratio of trypan blue-positive cells; n=3 for all points.
We then silenced PRDX2 in K562 and observed decreased cell viability and a decreased capacity of shK562 cells to differentiate (Figure 4C,D). When shK562 cells were exposed to oxidative stress (H2O2), we observed a marked reduction of their viability compared to that of cells expressing PRDX2 (Figure 4D). These data support a protective role of PRDX2 in stress erythropoiesis.
Peroxiredoxin-2 binds heme in erythroid precursors and shows high affinity heme-binding features
We then hypothesized that PRDX2 might be involved in additional protective mechanism(s) in stress erythropoiesis as observed with some bacterial and eukaryotic peroxiredoxin isoforms.36,37 As shown in Figure 5A, we observed the presence of heme-specific colorimetric reactivity in immunoprecipitated PRDX2 from both normal and β thalassemic cells, suggesting that heme is bound to PRDX2. This observation supports the hypothesis that PRDX2 interacts with heme during erythropoiesis. It is interesting to note that PRDX2-heme complexes were more abundant in β thalassemic cells on day 7 than in control cells and in normal cells in late erythropoiesis than in either β thalassemic cells at day 14 of culture or early normal erythropoiesis, reflecting the differences in heme levels observed during erythropoiesis (Figure 1A).
Figure 5.

Recovery of heme bound to immunoprecipitated PRDX2 and functional features of recombinant PRDX2 in the presence or absence of heme. (A) Heme bound to immunoprecipitated PRDX2 is expressed as a ratio between the heme levels measured as reported in the Design and Methods section and the amount of PRDX2 immunoprecipitated and evaluated by densitometric analyses. The heme levels were measured in the following samples: column 1: anti-peroxiredoxin-2 antibody (Ab) incubated with heme; column 2: recombinant PRDX2 alone; column 3: recombinant PRDX2 previously bound to heme and then immunoprecipitated (Ab) with specific anti-PRDX2 antibody (black); columns 4–7 immunoprecipitated PRDX2 from normal controls (C) and β-thalassemic (T) erythroid precursors at the different time points studied C7 and T7 (white bars, 7 days of culture), C14 and T14 (gray bars, 14 days of culture). Errors were reported as the S.E.M. as bars. *compared to control cells; °comparison of 14 versus 7 days. (B) Kinetic parameters of recombinant PRDX2 in the presence of increasing concentrations of H2O2. The data points were fitted to equation 1 which accounts for substrate inhibition. Errors were less than 10%. (C) Inhibitory effect of increasing concentrations of hemin on recombinant PRDX2 activity in the presence of saturating concentrations of H2O2. The IC50 was determined by fitting inhibition data to a dose-response sigmoidal curve. The inset shows the binding of hemin to PRDX2 determined by following tryptophan emission quenching after the addition of increasing concentrations of hemin. Data were fitted to equation 2. (D) The absorption spectra of the PRDX2-heme complex measured in the presence of: oxidized (Fe3+) (−), reduced (Fe2+) (…) and 1 mM DTT (-.-.). Equimolar amounts of PRDX2 and hemin were mixed and UV-visual spectra recorded in 50 mM Hepes pH 7.5 at room temperature. Heme bound to recombinant PRDX2 was reduced with a 1000-fold molar excess of sodium dithionite. The visible spectrum of unbound PRDX2 is reported (- - -).
In order to characterize the features of PRDX2 better, we generated a recombinant human PRDX2 as a useful instrument for studying PRDX2 in the presence of ROS and heme and for evaluating whether heme binding might affect PRDX2 peroxidase activity. The kinetic parameters of the purified PRDX2 using H2O2 as a substrate (see also the Online Supplementary Design and Methods) showed the dependence of the initial rate of PRDX2 on H2O2 concentration, displaying substrate inhibition behavior. Fitting to equation (1) gave the values, kcat= 13.2±0.4 s−1, Km= 0.34±0.04 μM and Ki= 33±3 μM, this last value probably being due to overoxidation of the catalytic cysteine (Cys-51) in the recombinant enzyme (Figure 5B). We then evaluated the binding of PRDX2 to hemin by fluorescence. The addition of hemin to PRDX2 caused a concentration-dependent quenching of tryptophan emission allowing the calculation of a Kd value of 46±7 nM (inset of Figure 5C). The high affinity binding of hemin to PRDX2 induced a decrease in the peroxidase activity of PRDX2 (Figure 5C).The inhibition assays carried out at a saturating concentration of H2O2 (8 μM) in the presence of various concentrations of hemin led to the determination of an IC50 value of 57±6 nM, a value in good agreement with the Kd determined (Figure 5C). In order to gain insight into the mode of heme binding, the spectrophotometric features of PRDX2 were measured in the absence or presence of hemin (Figure 5D). While the native PRDX2 did not display absorbance in the 400–700 nm region, the addition of hemin caused the appearance of a band at 417 nm (Soret band) and a large broad peak in the 550 nm region. This spectrum is different from that of free hemin whose absorbance maxima were centered at 385 and 650 nm (data not shown). Following the addition of PRDX2, the heme spectrum showed significant changes when reduced with a 1000-fold molar excess of sodium dithionite (a common iron-reducing agent). As shown in Figure 5D the Soret band became slightly sharper and red-shifted to 428 nm, whereas in the 550 nm region the absorbance split into two well-resolved 530 and 560 nm bands. The addition of 1 mM DTT (a cysteine-reducing agent) determined a red-shift of the absorbance band of the PRDX2-hemin complex to 419 nm and broad absorbance in the 550 nm region. Overall, the spectral data, displaying features typical of coordination of iron in a low spin and hexacoordinated state, indicate that hemin (heme) binds specifically to PRDX2.
Discussion
Among the defense strategies used by β-thalassemic erythroid precursors against oxidative stress2,7,10,38 we identified two novel cytoprotective mechanisms. The first is the negative feedback of ROS/ heme levels on ALAS-2 activity; the second is the up-regulation of PRDX2 as both an anti-oxidant and a heme-binding protein (Figure 6).
Figure 6.

Schematic model of cytoprotective mechanisms in response to oxidative stress in β-thalassemic erythroid cells. In β-thalassemic erythropoiesis ROS inhibits 5-δ aminolevulinate synthase (ALAS-2) activity, reducing the neosynthesis of heme, and induces peroxiredoxin-2 (PRDX2) expression. In the early stage of β-thalassemic eythropoiesis, ROS and heme levels are both increased and PRDX2 acts on both targets; in more mature cells, when ROS levels are still high and heme levels are reduced, ROS might become the PRDX2 major target (see text for details).
In β thalassemic erythroid precursors the finding of low intracellular heme levels, confirming results of a previous study,4 is in agreement with the observed down-regulation of the HO-1/BVR system, involved in heme catabolism. This observation led us to determine the activity of ALAS-2 in β-thalassemic erythroid precursors and investigate whether the activity of ALAS-2 was affected by ROS and/or heme. Here, we first demonstrated the reduction of ALAS-2 activity in β-thalassemic erythroid precursors, possibly related to the inhibitory effects of ROS on ALAS-2 function. In fact, recombinant ALAS-2 activity is inhibited by H2O2 and hemin with similar Ki, suggesting a common binding site, possibly the Cys-Pro motifs (Cys70-Pro71) in the mature form of ALAS-2.39 Moreover, since the Ki values are in the range of the ROS concentrations found in β-thalassemic precursors (Figure 1B), we suggest that ALAS-2 is inhibited by ROS in β-thalassemic cells. Similar effects of ROS have been reported for the activity of ALAS-1, the constitutive ubiquitous isoform, through a still unknown molecular mechanism.40 Based on this experimental evidence, we propose that ALAS-2 activity in erythroid precursors is affected by both heme and ROS levels. The resulting reduction of heme biosynthesis observed in late β-thalassemic erythropoiesis, prevents the cytotoxic effect of excess free heme.
The other novel protective mechanism we found in β-thalassemic cells is the up-regulation of PRDX2 expression. PRDX2 is a typical 2-cysteine (Cys) peroxiredoxin, whose catalytic and resolving cysteine residues are Cys 51 and 172, respectively. In other cell types it has been demonstrated that PRDX2 is induced by oxidative stress and that PRDX2 over-expression protects cells against oxidative damage.41,42 In erythroid cells, PRDX2 expression was reported in the mouse erythroleukemia (MEL) cell line, in normal erythroid precursors31,43,44 and in bone marrow and spleen from normal mice.30,45 We recently reported increased PRDX2 expression in the spleen and bone marrow from two β-thalassemia mouse models, compared to the expression in controls.30 Here, we found increased levels of PRDX2 mRNA/protein during β-thalassemic erythropoiesis. In addition, PRDX2 was present in a more acidic form, similarly to that found in Jurkat T cells exposed to oxidative stress as reported by Rabilloud et al.,32 suggesting a possible protective role of Prdx2 in β-thalassemic erythropoiesis, which is characterized by a high oxidative cell environment. In fact, when normal erythroid precursors were exposed to oxidative stress, we observed up-regulation of PRDX2 expression and cell recovery. In addition, after silencing PRDX2, K562 cells showed decreased differentiation and reduced survival in conditions of oxidative stress, supporting the cytoprotective role of PRDX2 during stress erythropoiesis.
The observation that PRDX2 is abundantly expressed during β-thalassemic erythropoiesis and is required for cell survival during oxidative stress together with its high evolutionary conservation suggests a broader role for PRDX2 besides its anti-oxidant function. In fact, we showed that PRDX2 binds heme in erythroid precursors, possibly playing an additional role to protect maturing cells by free heme. Using recombinant PRDX2 we showed that PRDX2 specifically binds heme with decreased PRDX2 peroxidase activity. The heme-binding affinity of PRDX2 is greater than that reported for other members of the peroxiredoxin family. In particular, PRDX2 binds heme with a Kd that is approximately 2- and 10-fold lower than that reported for PRDX136 and the bacterial peroxiredoxin counterpart AhpC,37 respectively. With regards to AhpC, Lechardeur et al. recently showed no involvement of catalytic cysteines in interaction with hemin, indicating that hemin binding is dissociable from catalysis and multimerization.41 In the human PRDX2, we showed that the PRDX2-hemin complex displays characteristics of a hexa-coordinated heme iron, even if it has not yet been possible to establish whether it is bis-His, bis-Met or His-Met coordination (i.e. amino acids whose side chains are known to be favored in cytochromes).46
We propose that in early β thalassemic erythroid precursors, characterized by high levels of ROS and heme, PRDX2 targets both ROS and heme to reduce oxidative stress. In late β-thalassemic erythropoiesis, when ROS levels are still high but heme levels are reduced, ROS might become the major target of PRDX2 (Figure 6). Further studies are needed to characterize these novel cytoprotective systems better in both normal and pathological erythropoiesis.
Acknowledgments
The authors are grateful to Dr. Gloria C Ferreira (University of Tampa, FL, USA) for the kind gift of recombinant ALAS-2. This work was supported by grants from the Italian Ministero dell’Università e della Ricerca (PRIN 2008) to CC, LDF,MDC, AI, by grants from Regione Campania (DGRC2362/07), and by Telethon project GGP09004 to AI. We thank Dr A. Matte’, Dept. of Medicine, University of Verona, for his help with PRDX2 immunoprecipitation experiments.
Footnotes
The online version of this article has a Supplementary Appendix.
Authorship and Disclosures
The information provided by the authors about contributions from persons listed as authors and in acknowledgments is available with the full text of this paper at www.haematologica.org.
Financial and other disclosures provided by the authors using the ICMJE (www.icmje.org) Uniform Format for Disclosure of Competing Interests are also available at www.haematologica.org.
References
- 1.Rachmilewitz E, Schrier SL. Pathophysiology of beta thalassemia. In: Steinberg MH, Forget BG, Higgs DR, Nagel RL, editors. Disorders of Hemoglobin. Cambridge: Cambridge University Press; 2001. pp. 233–51. [Google Scholar]
- 2.Rund D, Rachmilewitz E. Beta-thalassemia. N Engl J Med. 2005;353(11):1135–46. doi: 10.1056/NEJMra050436. [DOI] [PubMed] [Google Scholar]
- 3.Huang SC, Benz EJ. posttranscriptional factors influencing the hemoglobin content of the red cells. In: Steinberg MH, Forget BG, Higgs DR, Nagel RL, editors. Disorders of Hemoglobin. Cambridge: Cambridge University Press; 2001. pp. 146–73. [Google Scholar]
- 4.Forget BG. The thalassemia syndromes. In: Hoffman R, Benz EJ, Shatill SJ, et al., editors. Hematology: Basic Principles and Practice. Philadelphia: Churchill Livingstone; 1999. pp. 485–92. [Google Scholar]
- 5.Olivieri O, De Franceschi L, Capellini MD, Girelli D, Corrocher R, Brugnara C. Oxidative damage and erythrocyte membrane transport abnormalities in thalassemias. Blood. 1994;84(1):315–20. [PubMed] [Google Scholar]
- 6.Amer J, Goldfarb A, Fibach E. Flow cytometric measurement of reactive oxygen species production by normal and thalassaemic red blood cells. Eur J Haematol. 2003;70(2):84–90. doi: 10.1034/j.1600-0609.2003.00011.x. [DOI] [PubMed] [Google Scholar]
- 7.Weiss MJ, dos Santos CO. Chaperoning erythropoiesis. Blood. 2009;113(10):2136–44. doi: 10.1182/blood-2008-09-115238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lai MI, Jiang J, Silver N, Best S, Menzel S, Mijovic A, et al. Alpha-haemoglobin stabilising protein is a quantitative trait gene that modifies the phenotype of beta-thalassaemia. Br J Haematol. 2006;133(6):675–82. doi: 10.1111/j.1365-2141.2006.06075.x. [DOI] [PubMed] [Google Scholar]
- 9.Yu X, Kong Y, Dore LC, Abdulmalik O, Katein AM, Zhou S, et al. An erythroid chaperone that facilitates folding of alpha-globin subunits for hemoglobin synthesis. J Clin Invest. 2007;117(7):1856–65. doi: 10.1172/JCI31664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen JJ. Regulation of protein synthesis by the heme-regulated eIF2alpha kinase: relevance to anemias. Blood. 2007;109(7):2693–9. doi: 10.1182/blood-2006-08-041830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Han AP, Fleming MD, Chen JJ. Heme-regulated eIF2alpha kinase modifies the phenotypic severity of murine models of erythropoietic protoporphyria and beta-thalassemia. J Clin Invest. 2005;115(6):1562–70. doi: 10.1172/JCI24141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schranzhofer M, Schifrer M, Cabrera JA, Kopp S, Chiba P, Beug H, et al. Remodeling the regulation of iron metabolism during erythroid differentiation to ensure efficient heme biosynthesis. Blood. 2006;107(10):4159–67. doi: 10.1182/blood-2005-05-1809. [DOI] [PubMed] [Google Scholar]
- 13.Nakajima O, Takahashi S, Harigae H, Furuyama K, Hayashi N, Sassa S, et al. Heme deficiency in erythroid lineage causes differentiation arrest and cytoplasmic iron overload. EMBO J. 1999;18(22):6282–9. doi: 10.1093/emboj/18.22.6282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sadlon TJ, Dell’Oso T, Surinya KH, May BK. Regulation of erythroid 5-aminolevulinate synthase expression during erythropoiesis. Int J Biochem Cell Biol. 1999;31(10):1153–67. doi: 10.1016/s1357-2725(99)00073-4. [DOI] [PubMed] [Google Scholar]
- 15.Furuyama K, Kaneko K, Vargas PD. Heme as a magnificent molecule with multiple missions: heme determines its own fate and governs cellular homeostasis. Tohoku J Exp Med. 2007;213(1):1–16. doi: 10.1620/tjem.213.1. [DOI] [PubMed] [Google Scholar]
- 16.Wallander ML, Leibold EA, Eisenstein RS. Molecular control of vertebrate iron homeostasis by iron regulatory proteins. Biochim Biophys Acta. 2006;1763(7):668–89. doi: 10.1016/j.bbamcr.2006.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ponka P, Richardson DR. Can ferritin provide iron for hemoglobin synthesis? Blood. 1997;89(7):2611–3. [PubMed] [Google Scholar]
- 18.Baranano DE, Rao M, Ferris CD, Snyder SH. Biliverdin reductase: a major physiologic cytoprotectant. Proc Natl Acad Sci USA. 2002;99(25):16093–8. doi: 10.1073/pnas.252626999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.De Franceschi L, Ronzoni L, Cappellini MD, Cimmino F, Siciliano A, Alper SL, et al. K-CL co-transport plays an important role in normal and beta thalassemic erythropoiesis. Haematologica. 2007;92(10):1319–26. doi: 10.3324/haematol.11556. [DOI] [PubMed] [Google Scholar]
- 20.Ronzoni L, Bonara P, Rusconi D, Frugoni C, Libani I, Cappellini MD. Erythroid differentiation and maturation from peripheral CD34+ cells in liquid culture: cellular and molecular characterization. Blood Cells Mol Dis. 2008;40(2):148–55. doi: 10.1016/j.bcmd.2007.07.006. [DOI] [PubMed] [Google Scholar]
- 21.De Franceschi L, Biondani A, Carta F, Turrini F, Laudanna C, Deana R, et al. PTPepsilon has a critical role in signaling transduction pathways and phosphoprotein network topology in red cells. Proteomics. 2008;8(22):4695–708. doi: 10.1002/pmic.200700596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Siciliano A, Turrini F, Bertoldi M, Matte A, Pantaleo A, Olivieri O, et al. Deoxygenation affects tyrosine phosphoproteome of red cell membrane from patients with sickle cell disease. Blood Cells Mol Dis. 2010;44(4):233–42. doi: 10.1016/j.bcmd.2010.02.007. [DOI] [PubMed] [Google Scholar]
- 23.Karur VG, Lowell CA, Besmer P, Agosti V, Wojchowski DM. Lyn kinase promotes erythroblast expansion and late-stage development. Blood. 2006;108(5):1524–32. doi: 10.1182/blood-2005-09-008243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Uchida E, Morimoto K, Kawasaki N, Izaki Y, Abdu Said A, Hayakawa T. Effect of active oxygen radicals on protein and carbohydrate moieties of recombinant human erythropoietin. Free Radic Res. 1997;27(3):311–23. doi: 10.3109/10715769709065769. [DOI] [PubMed] [Google Scholar]
- 25.Chomczynski P, Sacchi N. The single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction: twenty-something years on. Nat Protoc. 2006;1(2):581–5. doi: 10.1038/nprot.2006.83. [DOI] [PubMed] [Google Scholar]
- 26.Hunter GA, Ferreira GC. A continuous spectrophotometric assay for 5-aminolevulinate synthase that utilizes substrate cycling. Anal Biochem. 1995;226(2):221–4. doi: 10.1006/abio.1995.1217. [DOI] [PubMed] [Google Scholar]
- 27.Comelli M, Londero D, Mavelli I. Severe energy impairment consequent to inactivation of mitochondrial ATP synthase as an early event in cell death: a mechanism for the selective sensitivity to H2O2 of differentiating erythroleukemia cells. Free Radic Biol Med. 1998;24(6):924–32. doi: 10.1016/s0891-5849(97)00373-0. [DOI] [PubMed] [Google Scholar]
- 28.He JZ, Ho JJ, Gingerich S, Courtman DW, Marsden PA, Ward ME. Enhanced translation of heme oxygenase-2 preserves human endothelial cell viability during hypoxia. J Biol Chem. 2010;285(13):9452–61. doi: 10.1074/jbc.M109.077230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ding B, Gibbs PE, Brookes PS, Maines MD. The coordinated increased expression of biliverdin reductase and heme oxygenase-2 promotes cardiomyocyte survival: a reductase-based peptide counters beta-adrenergic receptor ligand-mediated cardiac dysfunction. FASEB J. 2011;25(1):301–13. doi: 10.1096/fj.10-166454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Matte A, Low PS, Turrini F, Bertoldi M, Campanella ME, Spano D, et al. Peroxiredoxin-2 expression is increased in beta-thalassemic mouse red cells but is displaced from the membrane as a marker of oxidative stress. Free Radic Biol Med. 2010;49(3):457–66. doi: 10.1016/j.freeradbiomed.2010.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rabilloud T, Berthier R, Vincon M, Ferbus D, Goubin G, Lawrence JJ. Early events in erythroid differentiation: accumulation of the acidic peroxidoxin (PRP/TSA/NKEF-B) Biochem J. 1995;312(Pt 3):699–705. doi: 10.1042/bj3120699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rabilloud T, Heller M, Gasnier F, Luche S, Rey C, Aebersold R, et al. Proteomics analysis of cellular response to oxidative stress. Evidence for in vivo overoxidation of peroxiredoxins at their active site. J Biol Chem. 2002;277(22):19396–401. doi: 10.1074/jbc.M106585200. [DOI] [PubMed] [Google Scholar]
- 33.Low FM, Hampton MB, Peskin AV, Winterbourn CC. Peroxiredoxin 2 functions as a noncatalytic scavenger of low-level hydrogen peroxide in the erythrocyte. Blood. 2007;109(6):2611–7. doi: 10.1182/blood-2006-09-048728. [DOI] [PubMed] [Google Scholar]
- 34.Stetler RA, Gao Y, Signore AP, Cao G, Chen J. HSP27: mechanisms of cellular protection against neuronal injury. Curr Mol Med. 2009;9(7):863–72. doi: 10.2174/156652409789105561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Phalen TJ, Weirather K, Deming PB, Anathy V, Howe AK, van der Vliet A, et al. Oxidation state governs structural transitions in peroxiredoxin II that correlate with cell cycle arrest and recovery. J Cell Biol. 2006;175(5):779–89. doi: 10.1083/jcb.200606005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Iwahara S, Satoh H, Song DX, Webb J, Burlingame AL, Nagae Y, et al. Purification, characterization, and cloning of a heme-binding protein (23 kDa) in rat liver cytosol. Biochemistry. 1995;34(41):13398–406. doi: 10.1021/bi00041a017. [DOI] [PubMed] [Google Scholar]
- 37.Lechardeur D, Fernandez A, Robert B, Gaudu P, Trieu-Cuot P, Lamberet G, et al. The 2-Cys peroxiredoxin alkyl hydroperoxide reductase c binds heme and participates in its intracellular availability in Streptococcus agalactiae. J Biol Chem. 2010;285(21):16032–41. doi: 10.1074/jbc.M109.024505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen C, Lorimore SA, Evans CA, Whetton AD, Wright EG. A proteomic analysis of murine bone marrow and its response to ionizing radiation. Proteomics. 2005;5(16):4254–63. doi: 10.1002/pmic.200401295. [DOI] [PubMed] [Google Scholar]
- 39.Lathrop JT, Timko MP. Regulation by heme of mitochondrial protein transport through a conserved amino acid motif. Science. 1993;259(5094):522–5. doi: 10.1126/science.8424176. [DOI] [PubMed] [Google Scholar]
- 40.Kaliman PA, Barannik TV. Regulation of delta-aminolevulinate synthase activity during the development of oxidative stress. Biochemistry (Mosc) 1999;64(6):699–704. [PubMed] [Google Scholar]
- 41.Zhang P, Liu B, Kang SW, Seo MS, Rhee SG, Obeid LM. Thioredoxin peroxidase is a novel inhibitor of apoptosis with a mechanism distinct from that of Bcl-2. J Biol Chem. 1997;272(49):30615–8. doi: 10.1074/jbc.272.49.30615. [DOI] [PubMed] [Google Scholar]
- 42.Kang SW, Baines IC, Rhee SG. Characterization of a mammalian peroxiredoxin that contains one conserved cysteine. J Biol Chem. 1998;273(11):6303–11. doi: 10.1074/jbc.273.11.6303. [DOI] [PubMed] [Google Scholar]
- 43.Babusiak M, Man P, Sutak R, Petrak J, Vyoral D. Identification of heme binding protein complexes in murine erythroleukemic cells: study by a novel two-dimensional native separation -- liquid chromatography and electrophoresis. Proteomics. 2005;5(2):340–50. doi: 10.1002/pmic.200400935. [DOI] [PubMed] [Google Scholar]
- 44.Petrak J, Myslivcova D, Man P, Cmejlova J, Cmejla R, Vyoral D. Proteomic analysis of erythroid differentiation induced by hexa-methylene bisacetamide in murine erythroleukemia cells. Exp Hematol. 2007;35(2):193–202. doi: 10.1016/j.exphem.2006.10.007. [DOI] [PubMed] [Google Scholar]
- 45.Unwin RD, Smith DL, Blinco D, Wilson CL, Miller CJ, Evans CA, et al. Quantitative proteomics reveals posttranslational control as a regulatory factor in primary hematopoietic stem cells. Blood. 2006;107(12):4687–94. doi: 10.1182/blood-2005-12-4995. [DOI] [PubMed] [Google Scholar]
- 46.Spolaore B, De Filippis V, Fontana A. Heme binding by the N-terminal fragment 1–44 of human growth hormone. Biochemistry. 2005;44(49):16079–89. doi: 10.1021/bi051374d. [DOI] [PubMed] [Google Scholar]
- 47.Cook JA, Mitchell JB. Viability measurements in mammalian cell systems. Anal Biochem. 1989;179(1):1–7. doi: 10.1016/0003-2697(89)90191-7. [DOI] [PubMed] [Google Scholar]