

Abstract
Proper assembly of viruses must occur through specific interactions between capsid proteins. Many double-stranded DNA viruses and bacteriophages require internal scaffolding proteins to assemble their coat proteins into icosahedral capsids. The 303 amino acid bacteriophage P22 scaffolding protein is mostly helical, and its C-terminal helix-turn-helix (HTH) domain binds to the coat protein during virion assembly, directing the formation of an intermediate structure called the procapsid. The interaction between coat and scaffolding protein HTH domain is electrostatic, but the amino acids that form the protein-protein interface have yet to be described. In the present study, we used alanine scanning mutagenesis of charged surface residues of the C-terminal HTH domain of scaffolding protein. We have determined that P22 scaffolding protein residues R293 and K296 are crucial for binding to coat protein and that the neighboring charges are not essential but do modulate the affinity between the two proteins.
Keywords: virus assembly, ankyrin repeat, electrostatic interactions, recombineering, scaffolding protein, procapsid
Introduction
Viruses assemble through a highly concerted process with accuracy and reproducibility. Many icosahedral viruses form their capsid shells from a single major capsid or coat protein that adopts quasi-equivalent conformations. In order to accurately adopt such icosahedral symmetry, the coat proteins of many double-stranded (ds) DNA animal viruses (e.g., Herpesviruses) and bacteriophages (e.g., lambda, φ29, T4, T7, P2 and P22) require the assistance of internal scaffolding proteins, which are only transiently present during virion assembly. Scaffolding protein and coat protein co-assemble into stable, virion precursor structures called procapsids, and scaffold is removed before or during DNA packaging (reviewed by Dokland, 1999; Fane and Prevelige, 2003).
In the case of bacteriophage P22, which infects Salmonella enterica serovar Typhimurium, the absence of scaffolding protein results in inefficient coat protein assembly in vitro (Prevelige et al., 1988) and assembly of coat protein into aberrant structures in vivo (King and Casjens, 1974). When scaffolding protein is present, assembly of procapsids occurs through a nucleation-limited process (Prevelige et al., 1988; Parent et al., 2005). Completed procapsids contain 415 molecules of coat protein, 60 to 300 molecules of scaffolding protein, a single dodecameric portal protein ring, and several molecules of auxiliary ejection proteins (Botstein et al., 1973; Casjens and King, 1974; Parker et al., 2001). DNA then packaged into procapsids and the capsid shell undergoes an expansion that results in a 10% increase in radius (Earnshaw et al., 1976; Prasad et al., 1993). In many dsDNA bacteriophages and Herpesviridae, scaffolding protein is cleaved by proteases (reviewed in Fane and Prevelige, 2003). However, in other viruses the scaffolding protein is not cleaved, and this is the case for bacteriophage P22. Its scaffolding protein exits the procapsid intact before or during DNA packaging, probably through channels in the center of the coat protein hexons (Prasad et al., 1993; Chen et al., 2011; Parent et al., 2010; Teschke and Parent. 2010). P22’s scaffolding protein autoregulates its own synthesis (King et al., 1978; Casjens and Adams, 1985; Casjens et al., 1985; Wyckoff and Casjens, 1985), and in lysis-inhibited infections, is recycled through approximately five rounds of procapsid assembly (King and Casjens, 1974; Casjens and King, 1974). In addition to its role in coat shell assembly, P22 scaffolding protein is required for incorporation of the portal protein (Weigele et al., 2005). The P22 virion is completed by the addition of tails to the portal vertex of the DNA-containing particle (reviewed by Casjens and Thuman-Commike, 2011).
The P22 scaffolding protein is mostly helical, 303 amino acids in length, with a highly charged helix-turn-helix (HTH) structure at its C-terminus (Figure 1; Tuma et al., 1998; Sun et al., 2000). Maximum fidelity of assembly is dependent upon the full length scaffolding protein; however, the C-terminal fragment of scaffolding protein that contains amino acids 238–303 is able to support assembly of procapsid-like particles, albeit with increased formation of aberrant structures (Parker et al., 1998). Deletion analysis has shown that residues 280–294 of the HTH comprise the minimal coat-binding region (MCBR) (Weigele et al., 2005; Greene and King, 1996; Greene and King, 1999), which binds coat protein mainly through electrostatic interactions (Parker and Prevelige, 1998; Parent et al., 2005). More specifically, 25 mM sodium chloride allows proper assembly of procapsids in vitro, but 1 M NaCl inhibits this reaction. Inhibition is likely due to destabilization of the interaction between coat protein monomers and scaffolding protein (Parker and Prevelige, 1998). Furthermore, in the absence of sodium chloride in vitro assembly reactions result in kinetically-trapped, bowl-shaped ‘half procapsid’ structures (Parent et al., 2005). These incomplete structures result from an increased affinity between scaffolding protein and coat protein in low salt, which leads to the formation of too many nuclei and ultimately causes complete depletion of coat protein monomers. These partially assembled procapsid structures will be called ‘halves’ herein (Parent et al., 2005). In the present study, we investigated the importance of charged residues on the surface of scaffolding protein’s HTH domain in binding to coat protein and stimulating procapsid assembly. We identified two residues that are most important for binding to coat protein and show that other charged residues serve as modulators of this interaction.
Figure 1.

The P22 scaffolding protein helix-turn-helix domain (HTH). The NMR structure of the HTH domain, residues 264–303, is shown. Helix 1 comprises amino acids 269 through 283, the turn region is amino acids 284 through 288, and helix 2 is amino acids 289 through 300. The minimal coat-binding region is comprised of amino acids 280–294. All charged residues are depicted as stick diagrams. The structure (PDB ID: 2gp8) was manipulated using PyMol (San Carlos, CA).
Results
Scaffolding protein binding to coat protein in vivo is refractory to single amino acid changes in its HTH domain
Mutant scaffolding proteins expressed from the phage P22 genome
In order to begin to dissect the details of scaffolding protein-coat protein contacts, scaffolding protein variants with changes that alter different surface exposed residues of the HTH domain were tested for in vivo function. Mutations were recombineered into a P22 prophage lysogen (see Materials and Methods), the mutant prophages were induced to lytic growth with mitomycin C and the phage yield was measured by titering (Table 1). This assay provides a simple means to assess the ability of each scaffolding variant to fully support procapsid and virion assembly in vivo. Prophage mutants with charges reversed or neutralized at positions D281, K286, D288, E290, K294, K296, K298, K300 or R303 did not block scaffolding protein function in vivo. Only a charge reversal of arginine 293 to glutamic acid completely blocked scaffolding protein function. Interestingly, when amino acid R293 is replaced by alanine or glycine the resulting phages produced normal or nearly normal virion yields, indicating that even this positive charge is not absolutely required for functionality.
Table 1.
In vivo functionality of P22 phages with altered scaffolding proteins
| Scaffolding protein variant change | Induction #1a | Induction #2a |
|---|---|---|
| Wild-type c | 9.5×109 | 3×1010 |
| K278A | 6×109 | 7×109 |
| D281K | 8×109 | 1×1010 |
| A282Dc | 1×1011 | 8×1010 |
| K286Eb | 6×1010 | 5×1010 |
| D288Rc | 2×1010 | 4×1010 |
| E290Ac | 6×109 | 7×109 |
| E290K | 1×1010 | 2×1010 |
| T291D | 2×1010 | 6×1010 |
| R293E | <104 | 2×102 |
| R293Ab,c | 6×1010 | 5×1010 |
| R293G | 1×109 (nt) | 9×109 |
| K294E | 9×1010 | 5×1010 |
| R293A/K294A | 2×1010 | 1×1010 |
| K296A | 8×109 | 1×1010 |
| A297D | 8×1010 | 1×1011 |
| K298A | 5×109 | 5×1010 |
| 5-Alab | 6×1010 | 2×1010 |
| 6-Ala | <105 | <105 |
| SM1c | 1×1010 | 1×1010 |
| SM2 | <105 | <105 |
| SM3 | <105 | <105 |
| SM4 | 1×109 | 2×108 |
| D299–303 | <104 | <104 |
| C-terminal his-tag c | 1×1010 | nd |
| N-terminal his-tag c | 1×1011 | nd |
Number of phage/mL produced upon induction in two independent experiments (titered on host strain DB7004; see Materials and Methods).
Prophage does not carry the 13amH101 mutation and is in sup+ strain DB7004; all others are in supo strain UB-20. This does not cause any interpretational difference except that the yield could be somewhat lower because of earlier lysis.
Scaffolding protein has P at position 259 (the others have H in this position; this difference does not affect its function, see text).
nt No exogenous tails added.
nd Not done
We also tested the effect of the HTH substitutions on assembly of procapsid-like particles in vivo by expressing scaffolding protein N-terminal deletion mutant (Δ1–140) from a plasmid during an infection by a scaffolding protein and DNA packaging defective P22 as described by Weigele et al. (2005). None of the substitutions at eleven different surface exposed residues blocked the truncated scaffolding protein’s ability to bind coat protein by this test (Supplementary Material Table S1). The failure of these many changes to inactivate scaffolding protein indicates that the interactions between scaffolding and coat proteins are distributed and/or redundant. Since these in vivo assays were so insensitive to these amino acid changes, in vitro assembly studies were pursued where the reaction conditions could be readily controlled.
Analysis of scaffolding protein-coat protein interactions in vitro
Charge-removal amino acid substitutions in the scaffolding protein MCBR do not significantly affect its stability or overall secondary structure
To avoid concerns that the structure of scaffolding protein would be affected by more drastic changes, we studied charge removal (alanine) substitutions, since alanine has a high propensity for helix formation (Creighton, 1993) For ease of purification, all scaffolding proteins carried the pET-15b N-terminal hexa-histidine containing tag MGSHHHHHHSSGLVPRGSH- (referred to as ‘his-tag’ herein), which does not affect scaffolding protein function in vivo (Table 1). To determine whether the charge-removal alanine substitutions affect the overall structure and stability of full-length scaffolding protein, the melting temperatures (Tm) of the mutant proteins were analyzed by circular dichroism (Figure S1). The Tm of the his-tagged wild type (WT) scaffolding protein was within error (≤1 °C) of authentic untagged WT scaffolding protein (Table S2). The Tms of the single mutant proteins were all within 2.3 °C of his-tagged WT scaffolding protein, and there was no change in the cooperativity of the unfolding reaction (data not shown). Partial chymotrypsin digestion of the mutant proteins also showed no evidence for any changes in overall structure (data not shown). Mass spectrometry analysis indicated a chymotrypsin cleavage site at amino acid 164 in the his-tagged protein that is not present in the untagged WT protein, but no differences in the rates of proteolysis were observed. These findings suggest that the various amino acid substitutions caused no gross structural changes to scaffolding protein.
Coat binding activity of scaffolding proteins with altered MCBRs
Since electrostatic interactions are critical for the scaffolding protein:coat protein interaction (Parker and Prevelige, 1998; Parent et al., 2005), in this mutational study we initially focused on surface charge removal alanine substitutions in the MCBR (amino acids 280 through 294, above). First we characterized the ability of mutationally altered scaffolding proteins to re-enter through holes in the procapsid lattice and bind to preassembled empty coat protein shells (procapsids chemically stripped of scaffolding protein; Greene and King, 1994). This assay allows examination of coat binding without the requirement that the scaffolding protein be able to nucleate assembly. Scaffolding protein’s ability to bind the inside of shells is correlated with its to bind to coat protein monomers (Parker et al., 1988; Suhanovsky et al., 2010; Teschke and Fong, 1996). Purified scaffolding proteins were incubated with empty procapsid shells, and the scaffolding protein bound to shells was pelleted by ultracentrifugation. The amount of bound scaffolding protein was quantified by densitometry of SDS-PAGE gels of the resuspended pellet (Figure 2). The his-tagged WT protein reproducibly bound slightly less efficiently than the untagged wild-type scaffolding protein. This observation was reproduced in both the salt titration and kinetic experiments below, suggesting that the his-tag has a small negative affect on the activity of the protein. Nonetheless, the his-tagged WT scaffolding protein is fully functional for procapsid assembly in vitro. The K286A, D288A, E290A and K294A scaffolding proteins bound shells with similar efficiency to his-tagged WT scaffolding protein, and the R293A and R293E proteins were deficient in shell binding. The importance of R293 is in agreement with observations that fragment 141–292 is unable to bind shells (Parker et al., 1998), and that R293E protein is not able to support phage assembly in vivo (above). To further investigate the ability of scaffolding protein variants to support procapsid assembly, in vitro procapsid assembly reactions were examined in the following sections.
Figure 2.

R293A, R293E and K296A scaffolding proteins are deficient in binding to empty procapsid shells. Purified shells were incubated with each scaffolding protein variant overnight at room temperature. The resulting samples were pelleted through a 20% sucrose cushion and analyzed in 12.5% SDS-PAGE. Shown in the top panel: c, coat protein; s, scaffolding protein; sh, histidine-tagged scaffolding protein. The bands were quantified by densitometry and bars represent the ratio of scaffolding protein to coat protein (S:C) incorporated into shells for each scaffolding protein variant relative to that of authentic WT scaffolding protein. The minor band that overlaps the s-h position in the wild type lane is the P22 encoded ejection protein gp20 that is present in our non-histidine tagged WT scaffolding protein preparation. It is not present in the reactions with his-tagged scaffolding proteins and therefore has no effect on the quantification of the gel bands.
In vitro procapsid assembly activity of scaffolding proteins with altered MCBRs
When purified P22 scaffolding protein and coat protein monomers are mixed under appropriate conditions, procapsid-like particles spontaneously assemble (Fuller and King, 1982; Prevelige et al., 1988; Parent et al., 2005). For ease of discussion, we refer to this process as in vitro procapsid assembly. P22 procapsid assembly is a nucleation-dependent process, which follows sigmoidal reaction kinetics (Prevelige et al., 1993). Analysis of the nucleation (lag) and elongation (~linear growth) phases can dissect the proteins’ function during procapsid assembly. In such experiments the procapsid assembly is observed either by visualizing procapsids, partially assembled states, and monomers as bands in agarose electrophoresis gels (Teschke et al., 2003), or by following light scattering at 500 nm (Prevelige et al., 1988). Under the conditions of our experiments, coat protein undergoes some uncontrolled assembly without scaffolding protein, which accounts for approximately 6% of the final products (M. Suhanovsky and C. Teschke, unpublished).
Proper assembly of procapsids in vitro is highly dependent on the anion concentration, which is consistent with the electrostatic interaction between coat and scaffolding proteins (Parent et al., 2005; Parker and Prevelige, 1998). In the absence of salt, the affinity between scaffolding protein and coat protein is high, giving rise to bowl-like ‘halves’ as the product of the in vitro assembly reaction at the scaffolding/coat proteins ratio used in the experiments below; however, scaffolding and coat proteins properly co-assemble into complete procapsids at 60 mM NaCl (Parent et al., 2005). The NaCl lowers the affinity between coat and scaffolding monomers so that fewer nuclei are formed and there is sufficient coat protein to complete assembly of procapsids. Here we use the observation of the assembly of complete procapsids as NaCl concentrations are increased as an indirect means to monitor coat protein:scaffolding protein affinity; the lower the NaCl concentration at which assembly products switch from ‘halves’ to whole procapsids, the weaker the binding between coat and scaffolding proteins during nucleation (Parent et al., 2005).
Coat binding affinity determined by salt titration of in vitro assembly reactions showed that the E290A and K294A scaffolding proteins have moderately reduced affinity (procapsids form at >15 mM NaCl), the D288A protein’s affinity is more reduced (procapsid assembly at >5 mM NaCl), and the K286A protein is most severely impacted (no NaCl required for procapsid assembly). Scaffolding proteins carrying the R293A or R293E substitution showed no significant assembly product at any NaCl concentration (Figure 3; Table 2). Kinetic analysis of assembly by light scattering measurements at 45 mM NaCl gave similar results with a few quantitative differences; the K286A, D288A, and E290A proteins had half-times for assembly completion of about 5 minutes (the T1/2 of his-tagged WT protein is 3 minutes), while the K294A protein had a significantly longer lag phase and a half time for its elongation phase of approximately 8 minutes (Figure 4B, C). These kinetics suggest that K294A may have a specific effect on the nucleation step, perhaps by affecting scaffolding protein dimerization (Padilla-Meier and Teschke, 2011). Finally, no assembly was observed by light scattering measurements with the R293A or R293E proteins (Figure 4D). Electron micrographs of representative assembly reactions with WT his-tagged and E290A scaffolding proteins are shown in Figure 5. The presence of some aberrant structures in the E290A sample may explain why the light scattering was slightly higher than WT for this protein (Figure 4B). These in vitro assembly experiments confirmed the in vivo results that showed that R293 is important for scaffolding and coat protein interaction.
Figure 3.
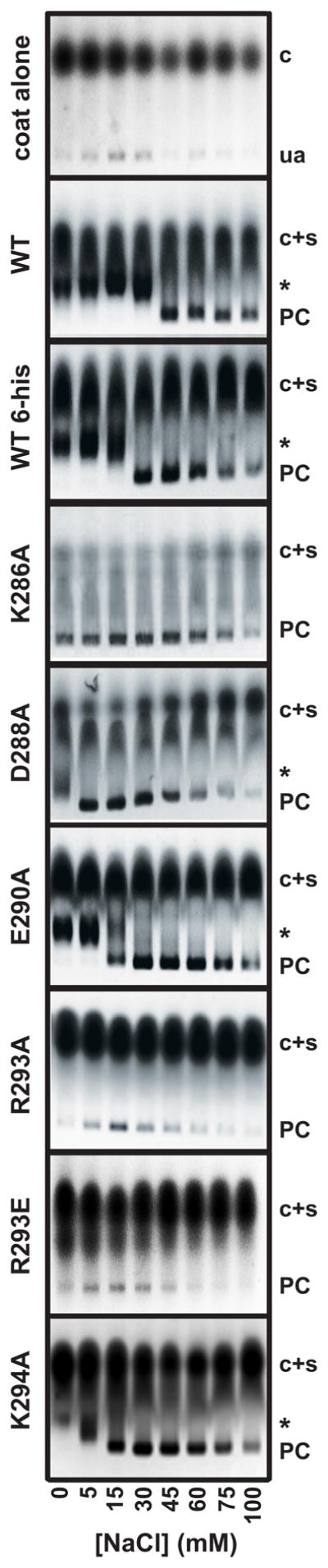
Single amino acid substitutions in the minimal coat-binding region of scaffolding protein affect affinity for coat protein. Each scaffolding protein variant was incubated overnight with coat protein in the presence of increasing final concentrations of NaCl. The products of the assembly reactions were analyzed by electrophoresis through 1.2% agarose gels. Scaffolding protein amino acid substitutions are indicated to the left of each agarose gel. Uncontrolled assembly (ua) of coat protein alone is shown in the panel labeled ‘coat alone’. Gel positions are indicated as follows: coat monomers, ‘c’; coat and scaffolding proteins monomers, labeled ‘c+s’; whole procapsids, ‘PC’; and ‘halves’, an asterisk. It is important to note that coat protein and scaffolding protein co-migrate in the agarose gels. The salt titration gels are representative of at least three experiments.
Table 2.
In vitro assembly properties of altered scaffolding proteins
| Scaffolding protein variant a | In vitro assembly at 45 mM NaCl | mM NaCl required to achieve procapsid assembly | Relative affinity b |
|---|---|---|---|
| WT | 100% | 60 | +++ |
| K286A | 50% | 0 | ++ |
| D288A | 75% | 5 | ++ |
| E290A | 125% | 15 | ++ |
| R293A | <10% | − | − |
| R293E | <10% | − | − |
| R293G | <10% | 0 | + |
| K294A | 50% | 15 | + |
| K296A | <10% | 0c | − |
| K298A | 44% | 15 | ++ |
| K300A | 27% | 0 | + |
| R303A | 37% | 0 | + |
| 5-Ala | 50% | 0 | + |
| SM1 | 75% | 15 | ++ |
| SM2 | <10% | − | − |
| SM3 | <10% | − | − |
| SM4 | 50% | 0c | + |
All scaffolding proteins carry an N-terminal his-tag (see text).
Qualitative analysis of the relative affinity of scaffolding protein mutants for coat protein monitored by the production of ‘halves’ relative to WT his-tagged scaffolding protein in in vitro assembly reactions, determined by analysis of agarose gels. ‘+++’ denotes presence of ‘halves’ at NaCl concentrations of ≤ 30 mM (affinity similar to WT scaffolding protein); ‘++’ denotes formation of ‘halves’ at NaCl concentrations ≤ 15 mM (mildly decreased affinity); ‘+’ denotes the absence of ‘halves’ at any given NaCl concentration (moderately decreased affinity), and ‘−’ is the absence of assembly products at any NaCl concentration.
Extent of assembly was less than with his-tagged WT protein.
Figure 4.
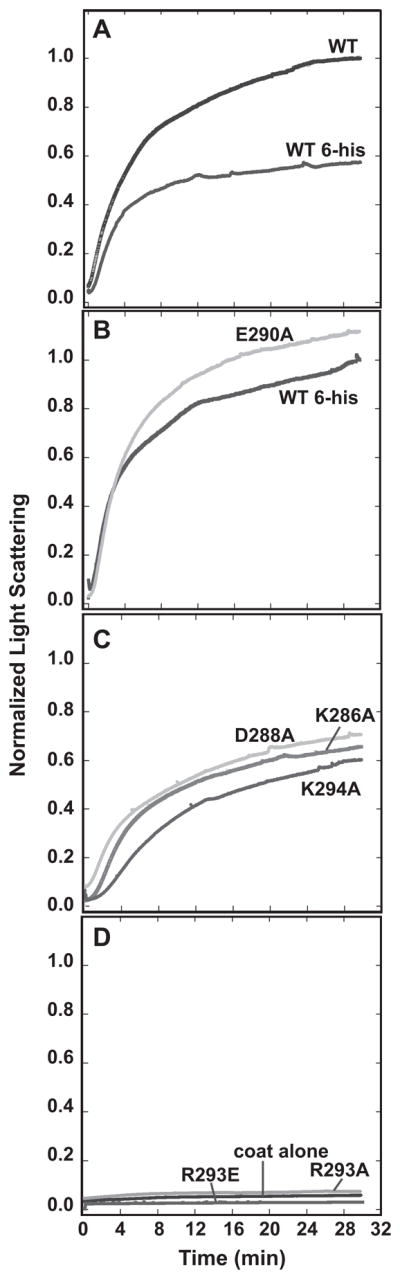
Kinetics of procapsid assembly with mutant scaffolding proteins. Scaffolding variants and coat monomers were rapidly mixed at 45 mM NaCl and the assembly kinetics followed by light scattering at 500 nm. In (A) wild-type with and without the N-terminal histidine-tag are compared. The scaffolding variant assembly reactions were classified as: (B) similar to his-tagged WT, (C) mildly defective, or (D) inactive. Coat protein alone was used to monitor uncontrolled assembly. All traces were normalized relative to wild-type protein without the his-tag as described in Materials and Methods.
Figure 5.
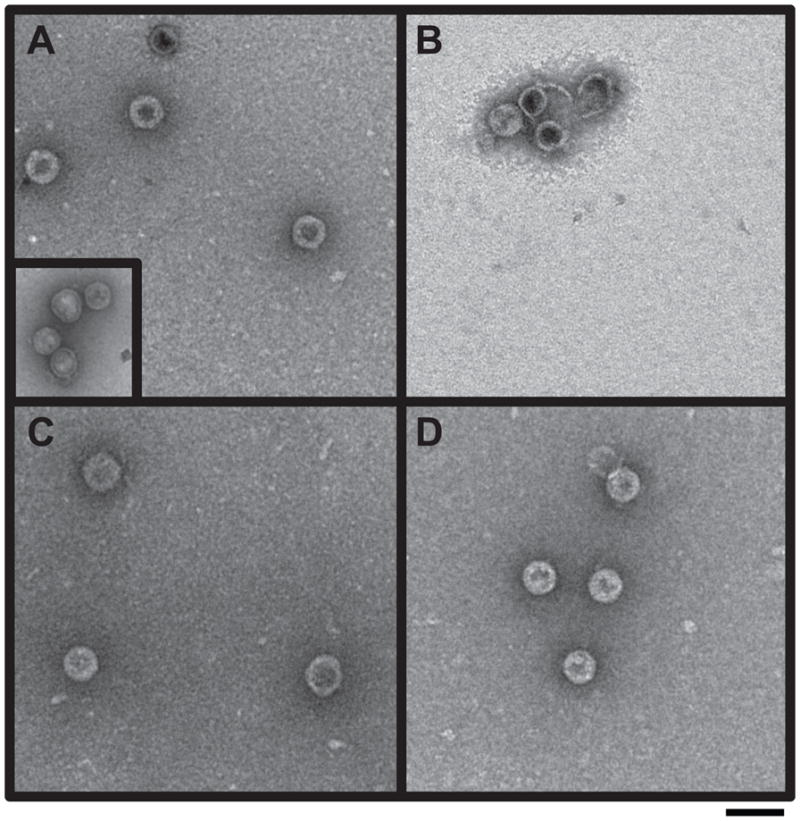
Electron micrographs of in vitro procapsid assembly products. Procapsid assembly reaction products with the following scaffolding proteins were observed by negative stain electron microscopy: (A) WT his-tag; procapsids generated with authentic WT scaffolding protein are shown in the inset, (B) E290A, (C) 5-Ala and (D) SM1. The magnification was 43,000X. Scale bar represents 100 nm.
Individually removing the surface charge of each amino acid in the MCBR resulted in reduced affinity for coat protein; however, these variants still function well enough in vivo to allow phage production. Therefore, multiple charge removal variants 5-Ala and 6-Ala (Figure 6) were generated to test the additivity of these modest changes. The 5-Ala protein has five of the six charged MCBR residues (D281, K286, D288, E290 and K294) changed to alanine. The removal of these charges affected the protein’s melting temperature only slightly, indicating that its overall structure was not grossly different from WT scaffolding protein (Figure S1). The 5-Ala prophage produced normal numbers of progeny upon induction indicating full function in vivo (Table 1). The 6-Ala protein carries R293A in addition to the 5-Ala changes and was not active in in vivo prophage induction experiments (Table 1). Since R293 is crucial for in vitro assembly activity, we did not pursue the in vitro study of the 6-Ala protein. Salt titration assembly experiments showed that no NaCl was required to form whole procapsids with the 5-Ala protein (Figure S2), and light scattering values for assembly at 45 mM NaCl were reduced by ~50% relative to the WT control (Figure 7A). Electron micrographs of the in vitro assembly products showed apparently normal procapsids (Figure 5). We also performed experiments to test whether the precise positioning of the arginine is important by testing mutant SM3 (Figure 6) in which all the charges in the MCBR are removed except K294, which was changed to arginine. It was not active in either procapsid assembly or the prophage induction assays, indicating the positioning of R293 is important (Figure 7B).
Figure 6.
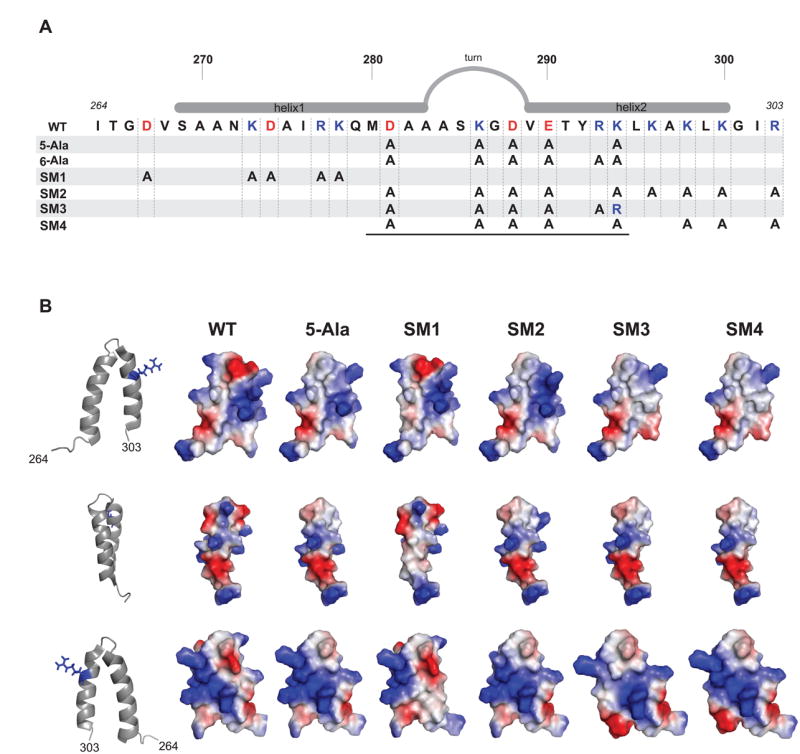
Multi-substituted scaffolding proteins. (A) Schematic of the scaffolding protein helix-turn-helix domain. Positively charged residues are colored blue and negatively charged residues are colored red. The alanine substitutions are shown for each scaffolding mutant with multiple substitutions. The black line below the diagram delimits the minimal coat-binding region, which spans residues M280-K294. (B) The surface charge distribution, generated with PyMol, resulting from each of the scaffolding variants depicted in (A), with exception of 6-Ala, are shown to the right. The orientation of each row is represented by the ribbon structure to the left, in which the residue R293 is shown in blue stick format as a landmark.
Figure 7.

Procapsid assembly kinetics of multi-substituted scaffolding proteins. Scaffolding variants (at 0.5 mg/mL unless otherwise indicated) and coat monomers were rapidly mixed at 45 mM NaCl and the assembly kinetics followed by light scattering at 500 nm. (A) multi-substituted scaffolding proteins (Figure 6) are compared. (B) scaffolding proteins with alterations outside of the MCBR in helix 2 are shown. (C) the concentrations of scaffolding proteins are varied. All traces were normalized relative to his-tagged WT at 0.5 mg/mL. Scaffolding protein amino acid changes are labeled on the kinetic traces. In panel C, the * indicates the overlapping kinetic traces of K296A (0.5 mg/mL), K293A (0.5 mg/mL), and R293E (both 2.0 mg/mL and 0.5 mg/mL) scaffolding proteins.
In summary, scaffolding protein-dependent procapsid assembly is affected greatly by changes at R293, and moderately affected by substitutions, either alone or together, that remove the other five charges in the MCBR. The effects of the non-R293 charge removals do not appear to be strictly additive; if they were, the 5-Ala scaffolding protein would be expected to be inactive. The fact that removal of five of the six charges in the MCBR did not abolish the interaction with coat protein was unforeseen, given the previous evidence that electrostatic interactions are critical for the scaffolding:coat interaction. However, the importance of R293 (and K296, below) is consistent with an electrostatic interaction.
Scaffolding protein surface charges outside the MCBR affect its ability to assemble procapsids
The rather small and non-additive effects on scaffolding protein function generated by changes within the MCBR caused us to consider the possibility that residues outside this region might have similar “accessory” roles in procapsid assembly that could have been missed by our previous deletion analysis (Weigele et al., 2005). In support of this idea, we found that deletion of five C-terminal scaffolding protein amino acids outside the MCBR blocked in vivo virion production (Table 1), even though they are not required for coat binding in vivo (Weigele et al., 2005). Therefore, we changed charged residues in the HTH domain outside the MCBR and examined their function. Mutant SM1 changed five charged amino acids (D267A, K273A, D274A, R277A and K278A) in the N-terminal portion of helix 1 to alanine (Figure 6A). This protein was fully functional for phage production in vivo (Table 1), was mildly affected for in vitro assembly (Figure 7A), and electron micrographs of the in vitro assembly products showed normal procapsids (Figure 5), indicating that the charged residues of helix 1 outside of the MCBR are not very important for assembly.
Mutant SM2 changed the four charged residues (K296, K298, K300 and K303) in the C-terminal half of helix 2 to alanine, in addition to carrying the 5-Ala substitutions (above; Figure 6A). SM2 protein did not support assembly at any NaCl concentration (Figures 7A and S2) and was nonfunctional in vivo (Table 1), showing that charged amino acids outside the MCBR in helix 2 are required for assembly, at least in the absence of the five 5-Ala MCBR charges. To test which of these charged residues is responsible for the loss of function in SM2, we analyzed the single substitution mutants, K296A, K298A, K300A and R303A. The K296A and K298A proteins each retain sufficient activity to allow approximately normal virion production in vivo (Table 1; K300 and R303A were not tested in vivo). However, the K296A protein binds coat protein shells poorly (Figure 2) and assembles into procapsids in vitro very slowly (Figure 7B); after overnight incubation procapsids do accumulate to a much lower yield and only at lower NaCl concentration than the WT control. On the other hand, the K298A, K300A and R303A proteins all bound coat shells with intermediate efficiency (Figure 2) and had moderately reduced activity by light scattering (44%, 27% and 37%, respectively; Figure 7B). In addition, the SM4 scaffolding protein (SM2 with the WT lysine restored at position 296; Figure 6A) functioned in vivo to produce an only moderately reduced phage yield (Table 1).
Thus, we conclude that K296 plays an important role, but is less critical than that of R293, in scaffolding protein function. The K296 charge is important in vitro when present as a single substitution, and is critical in vivo when other nonessential HTH surface charges are also removed. In the absence of all other charged residues of helix 2 and the loop, R293 and K296 alone were sufficient to allow phage production and in vitro procapsid assembly.
High scaffolding protein concentrations rescue assembly of R293A and K296A scaffolding proteins
Scaffolding proteins bearing substitutions R293A or K296A are able to support phage production in vivo, but each has a deleterious effect on in vitro assembly. In lysis-inhibited infected cells, scaffolding protein is estimated to be present at 1 – 5 mg/mL (Parent et al., 2006; Wyckoff and Casjens, 1985), which is higher than the 0.5 mg/mL used in the above in vitro assembly reactions. Therefore, increasing concentrations of the R293A, R293E and K296A scaffolding proteins were mixed with coat protein and assembly was monitored by light scattering. The highest concentration (2.0 mg/mL) of R293A and K296A proteins was able to promote procapsid assembly, albeit at lower substantially lower levels than WT for R293A (Figure 7C). On the other hand, R293E was unable to support assembly even at 2 mg/mL. These findings resolve the apparent difference between in vitro and in vivo measurements of the activity of these two mutant proteins.
Discussion
Virus assembly intermediate structures often contain proteins in metastable conformations. This allows robust assembly into organized capsids, but also allows changes in protein:protein interactions that are needed later in the virus life cycle. During nucleation-dependent assembly of phage P22 procapsids, coat protein must interact with its internal scaffolding protein in order to give rise to procapsids with the correct size and shape; yet these interactions must readily be disrupted so that scaffolding protein can be released from the procapsid and DNA can be packaged.
Two charged surface residues of P22 scaffolding protein’s HTH are crucial to its function
Biophysical analyses have shown that P22 scaffolding protein:coat protein interactions are mainly electrostatic (Parent et al., 2005; Parker and Prevelige, 1998), and the surface of scaffolding protein’s coat binding site is highly charged (Sun et al., 2000) (Figure 6B). We show here that the coat binding function of the C-terminal HTH domain of scaffolding protein is refractory to amino acid changes, and many radical changes such as individual charge reversals or multiple charge removals in the HTH domain do not block its function. Analysis of scaffolding protein alanine substitution mutants in the HTH domain showed that none of the charged surface residues, when changed individually are essential for in vivo assembly. We note that in vivo scaffolding protein is controlled in such a manner that it fills the available coat protein shells to form procapsids (King et al., 1978; Casjens and Adams, 1985; Wyckoff and Casjens, 1985). It is possible that when the scaffolding protein affinity for coat protein is lowered by a substitution, the mutant gene is up-regulated to compensate for poor binding. Such an effect would tend to minimize the in vivo phenotype of such mutants. Nonetheless, disruption of the charge distribution by alanine mutagenesis of residues R293A and K296A caused scaffolding protein to be inactive in in vitro assembly reactions at concentrations of 0.5 mg/mL, and both residues are essential in vivo when other non-essential nearby charged residues are substituted. This requirement for other changes in vivo was shown to be almost certainly due to the fact that the concentration in vivo is higher than 0.5 mg/mL and these two mutant proteins are able to function at higher concentrations. Since circular dichroism measurements showed no significant destabilization of either R293A or the K296A protein, we attribute the changes in the assembly properties of these variants to the lack of crucial charges at the scaffolding protein:coat protein interaction interface. We conclude that, of the fifteen charged residues on the HTH domain surface, only R293 and K296 play crucial roles in scaffolding protein’s ability to bind coat protein and assemble into procapsids. A number of the other charged surface residues appear to play modulatory roles, in that alanine substitutions at these positions do not eliminate function, but rather cause less efficient scaffolding function.
Scaffolding protein is in monomer-dimer equilibrium at physiological concentrations (Parker et al., 1997b), and dimers have been shown to be more effective than monomers in triggering procapsid assembly (Parker et al., 1997b; Parker et al., 1998). The oligomeric state of the scaffolding protein variants was not determined in these studies, but there is no evidence that amino acid changes in this region of the protein affect oligomerization of scaffolding protein, since the dimerization site is between residues 1–245 (Sun et al., 2000). Also, previous studies show that increasing the NaCl concentration from 25 mM to 1 M increased the dimerization Kd from 78 μM to 170 μM (Parker and Prevelige, 1998), so the NaCl concentrations used for the in vitro assembly reactions presented here most likely had little effect on the oligomerization of the scaffolding protein variants. Therefore, we believe that it is unlikely that changes in the properties of the mutant scaffolding proteins studied here are due to different oligomeric states.
We also observed that attachment of the 19 amino acid histidine tag to the N-terminus had a small negative affect on the affinity of scaffolding protein for coat protein when compared to native (untagged) scaffolding protein purified from phage infected cells. Biochemical and genetic data indicate that scaffolding protein may be folded upon itself as a U-shaped molecule, allowing the N-terminal region to be in contact with the C-terminal region (Padilla-Meier and Teschke, 2011; Tuma et al., 1998; Weigele et al., 2005). Addition of positive N-terminal charges may create an imbalance in charge distribution on the protein surface. This idea is supported by the observation that the R293E mutation does not block coat protein binding in vivo when present in the C-terminal amino acid 141–303 fragment (Table S1), but does so in full-length scaffolding protein (Table 2). Thus, it appears that the N-terminal part of scaffolding protein has a negative effect on coat binding (Parker et al., 1998; Suhanovsky and Teschke, 2011; P. Weigele and S. Casjens, unpublished data).
Based on our mutagenesis data, a model for the interaction of scaffolding protein with coat protein was generated (Figure 8). According to the salt titration assay, the relative affinity of scaffolding protein for coat protein is least affected by substitutions at D288, E290, K298, K294 (Figure 8, green residues), suggesting that these positions are the least involved in scaffolding protein:coat protein interface. Our data also indicate that residues R293 and K296 are in closest contact with coat protein, and that K286, K300 and K303 have only moderate roles in binding. Figure 8 shows that the side chains of D288, E290, K294, and K298 protrude from the opposite side of the helix as those of R293 and K296. K300 and K303, which cause intermediated effects on coat binding, are clearly accessible from the same side of the HTH as R293 and K296. We hypothesize that it is this side of the HTH that contacts coat protein. The decrease in affinity mediated by K286 may be due to changes in the turn of the HTH. Consistent with this idea, the mutant G287P, which is also in the turn, has a mild effect on assembly (data not shown).
Figure 8.

Model for scaffolding protein interaction with coat protein. A ribbon diagram of scaffolding protein C-terminal HTH domain is shown with stick format residues color coded according to their relative affinity for coat protein: red is inactive (R293), orange very defective (K296), yellow moderately defective (K286, K300 and R303) and green mildly defective (D288, E290, K294 and K298). The gray area symbolizes coat protein with negatively charged residues (denoted by ‘–‘) that interact with scaffolding protein R293 and K296 upon assembly. The N-terminus of the HTH domain is labeled ‘N’ and residue arginine 303 is labeled ‘C’.
Scaffolding protein has been suggested to bind to coat protein at the capsid lattice 3-fold axis of symmetry (‘trimer tips’), represented by the gray region in Figure 8 (Chen et al., 2011; Padilla-Meier and Teschke, 2011; Parent et al., 2010; Thuman-Commike et al., 2000; Thuman-Commike et al., 1996). To date, our results cannot determine whether scaffolding protein interacts at the 3-fold axis of symmetry as a monomer or a dimer. Therefore, in the present model we show the binding of a single scaffolding protein molecule to coat protein even though the dimer is likely the most active form during in vitro assembly (Padilla-Meier and Teschke, 2011; Parker et al., 1998; Tuma et al., 2008). The electrostatic nature of the scaffolding protein and coat protein interaction and the observation that residues R293 and K296 are critical for coat protein binding suggest that acidic residues in coat protein are likely to be involved in this interaction. Indeed, the 3-fold axis of coat protein symmetry presents a prominent acidic patch, comprised of residues D14, E15, E18, E97, and D385 of the three adjacent subunits. Interestingly, upon expansion/maturation, the polypeptide in this region is rearranged so that these residues are no longer surface accessible (Chen et al., 2011; Parent et al., 2010). This change seems likely to be responsible of release of scaffolding protein from the procapsid during expansion/maturation. The search for the region of coat protein interaction with scaffolding protein residues R293 and K296 will require further investigation, presently ongoing in our laboratories.
P22 scaffolding protein as a model for the study of protein:protein interaction surfaces
Protein function and activity are often linked to their ability to bind other protein partners. Virus assembly provides a paradigm of protein:protein interactions in a highly organized process: individual subunits interact to form accurately assembled protein cages that are responsible for protecting the viral genome. Protein-protein interfaces are formed mainly by hydrophobic surfaces that often contain charged residues, especially arginine (Janin et al., 2008). Using a database of more than 2000 proteins, Bogan and Thorn (1998) showed that arginine is one of the most common charged residues present at protein interfaces at critical ‘hot’ spots. The coat protein binding domain of scaffolding protein is structurally homologous to the tetratricopeptide repeat 1 protein (TRP1) (Sun et al., 2000), which is involved in eukaryotic cellular metabolism, and, most importantly, in protein:protein interactions. TRP1 proteins contain ankyrin repeats, which have a HTH motif and are one of the most abundant folds deposited in the PDB (Li at al., 2006; Sedgwick and Smerdon, 1999). Ankyrin repeats can be quite variable in terms of amino acid sequence; however, several residues are conserved. One such residue is an arginine in a designed ankyrin repeat (PDB 2xee; Kramer et al., 2010) that aligns with R293 in scaffolding protein by amino acid sequence, using the program Top Match (Sippl, 2008; Sippl and Wiederstein, 2008). This arginine site in the 2xee ankyrin repeat is also in the second helix and shows structural homology to the scaffolding protein HTH. Interestingly, there is no sequence similarity between φ29’s scaffolding protein and ankyrin repeats even though it has partial structural homology to P22 scaffolding protein, suggesting that these two scaffolding proteins may interact with their respective coat proteins in different ways. We note, however, that the scaffolding proteins of phages φ29 and P2, as well as herpesviruses HSV-1 and cytomegalovirus, all interact with their coat proteins through amino acid sequences near their C-termini (Chang et al., 2009; Fu et al., 2007; Hong et al., 1996; Kennard et al., 1995; Matusick-Kumar et al., 1995; Oien et al., 1997), suggesting that there could be similarities in their mechanisms of action.
Materials and Methods
Bacterial and bacteriophage strains
Salmonella enterica serovar Typhimurium LT2 strain UB-20 (leuA−414, ΔFels2, r−, sup°; the kind gift of Dr. Miriam Susskind) was lysogenized with P22 15− ΔSC302::KanR 13− amH101 to create strain UB-1757. Phages were titered on S. enterica strain amber mutant suppressing strain DB7004 (Winston et al., 1979). Plasmid constructions were transformed into E. coli NF1829 (Schultz et al., 1982).
The prophage used to carry scaffolding protein mutations (P22 15− ΔSC302::KanR 13− amH101 in Salmonella strain UB-1757) carries a kanamycin resistance gene to ensure prophage presence and a nonsense mutation in gene 13 to allow control of lysis by chloroform addition (Botstein et al., 1973). Strain UB-1757 was constructed as follows: the 1100 bp BamHI kanamycin resistance gene carrying fragment from plasmid JM22 (Mekalanos et al., 1983) was cloned into the unique BglII site of plasmid pTP397 (Casjens et al., 1989). The resulting plasmid pSC302 carries P22 DNA between P22 bp 38548 and bp 41134 (GenBank accession number BK000583), except the kanamycin resistance cassette replaces P22 DNA between bp 39139 and 40789; these P22 sequences contain part of phage gene 15 and all of the rha/orf201 gene. The gene 15 product is required for lysis under high divalent cation conditions, and the Rha protein blocks phage growth only in infections of IHF defective hosts (Henthorn and Friedman, 1995). Bacteriophage P22 c+ 13− amH101 was used to infect Salmonella strain UB-20 carrying pSC302. The resulting lysate was used to infect naive UB-20 cells, and a lysogen (strain UB-1757) was isolated by selecting for kanamycin resistance. Sequencing of the insertion junctions in the resulting prophage confirmed that the KanR cassette is located in the phage at the position indicated above; this replacement allele was named 15−ΔSC302::KanR.
Scaffolding gene mutations in the P22 15− ΔSC302::KanR 13− amH101 prophage of Salmonella strain UB-1757 were created by lambda Red-mediated recombineering as described by Karlinsey (2007). The TetRA tetracycline resistance cassette was PCR amplified from Tn10dTc in the genome of Salmonella enterica strain TH2788 using primers that have 3′-end sequences of the tetA and tetR primers with 46 bp of the desired P22 sequence at their 5′-ends. Subsequently, this fragment was inserted into the P22 prophage of UB-1757 by electroporation using a BIORAD Gene Pulser (25 μF, 2.4 kV, 200 Ω through tetracycline resistant transformants (Karlinsey, 2007). Two recipient strains were constructed in which TetRA replaces P22 gene 8 codons 279 to 294 (UB-1751) or codons 266 to 303 (UB-1838). Correct placement of the cassette was confirmed by PCR amplification of the inserted cassettes using tetOUT and T1test oligonucleotides paired with suitable oligonucleotides that prime 400–900 bp away from the TetRA insertion site in the P22 genome, followed by sequencing of the cassette and insertion boundaries within the amplicon (Karlinsey, 2007). Insertion of the TetRA cassette and its replacements (below), were done in the presence of the ampicillin resistance-encoding plasmid pKD46, which supplies the necessary lambda Red recombination proteins (Datsenko and Wanner, 2000; Karlinsey, 2007). This plasmid was removed by growth overnight on plates at 40 °C when it was no longer needed. Replacement of the TetRA cassette with a mutant scaffolding protein gene sequence was performed as follows: a portion of the plasmid-borne mutant scaffolding protein gene (below) was PCR amplified with primers that amplify 400–800 bp that span the site at which the TetRA cassette is inserted into the prophage. This amplified DNA was used to electroporate UB-1751 or UB-1838 cells, and transformants were selected for loss of the TetRA cassette on plates containing 24 mg/ml fusaric acid (Sigma, St. Louis, MO), 20 mM ZnCl2 and 0.25 mg/ml anhydrotetracycline hydrochloride (Thermo Fisher Scientific, Waltham, MA) while retaining kanamycin resistance. The loss of tetracycline resistance and presence of prophage mutations were confirmed by sequencing using either whole phage DNA (when the phage was fully functional) or relevant PCR products amplified from nonfunctional prophages as template; the entire scaffolding protein gene was sequenced in each of the mutant phages to ensure no additional scaffolding protein mutations were present.
Prophages were induced to lytic growth as follows: cells were grown in LB at 37 °C to 2 × 108 cells/ml, induced by addition 0.5 μg/ml Mitomycin C (Sigma, St. Louis, MO). The cultures were then shaken overnight at 37°C and lysed by addition of chloroform. Since P22 prophage induction results in virions that are deficient in tailspikes (Adams et al., 1985; Israel et al., 1967; Schwarz and Berget, 1989b), and these phages lack a functional gene 15, titers were determined with S. enterica indicator strain DB7004 on LB plates containing 10 mM citrate after addition of saturating amounts of tailspike protein as previously described (Schwarz and Berget, 1989a; Schwarz and Berget, 1989b; Wu et al., 2002).
Plasmids
The mutant scaffolding protein genes were constructed for expression in plasmid PP073. This plasmid was created by PCR amplification of the scaffolding protein gene from P22 c1-7 13− amH101 (Botstein et al., 1973) using primers whose 5′-tails create an NdeI site overlapping the ATG start codon and a BamHI site immediately after the TAA stop codon. After cleavage with NdeI and BamHI, the amplified DNA was cloned into plasmid pET-15b (Novagen, EMD Biosciences, Darmstadt, Germany), which places a hexa-histidine tag at the N-terminus of the expressed protein. The complete scaffolding protein genes of all the mutant plasmids were sequenced to confirm that only the directed mutations were present. This P22 strain and the resulting plasmid PP073 carry a P259H change from wild type P22 in its scaffolding protein gene that does not affect scaffolding function (Parker et al., 1997a), and which is present in some of the mutant prophage constructs (see footnote c of Table 1). For in vitro experiments, all constructs were modified to have a proline at position 259. Mutations in PP073 were made using the QUICKCHANGE® site directed mutagenesis and were transformed into DH5αNF1829 or XL-1 blue competent E. coli cells.
Protein purification
The plasmids with mutant scaffolding protein genes described above were transformed into E. coli strain BL21(DE3)/pLysS (Studier, 1991), cells were grown in Luria-Bertani media at 37°C to an O.D.600 nm of ~ 0.2, protein expression was induced by the addition of 0.4 mM (final concentration) isopropyl β-D-1-thiogalactopyranoside, and the culture was aerated for an additional 4 hours. Cells were harvested by centrifugation and resulting pellets were frozen and lysed in the presence of protease inhibitor cocktail (Sigma-Aldrich, St. Louis, MO). Cell debris was removed by centrifugation, the scaffolding protein-containing soluble material was loaded onto a cobalt-NTA affinity Talon column (Clontech, Mountain View, CA), and the protein was purified by imidazole step-gradient elution. The his-tagged scaffolding proteins eluted in the presence of 250 mM imidazole. Pooled fractions containing purified scaffolding protein were concentrated to 5–10 mg/mL by transferring the protein to a 3.5-kDa cut-off dialysis bag, which was then placed on a bed of PEG 20,000. The protein concentration was determined using the Pierce 660 nm Protein Assay as instructed by the manufacturer (Pierce, Rockford, IL).
Circular dichroism
The secondary structure and stability of the mutant scaffolding proteins were analyzed using circular dichroism. Protein concentrations were 0.5 mg/mL in 10 mM sodium phosphate, pH 7.6. Spectra were obtained between 200–250 nm, with 0.5 nm scan step and 15 sec averaging time per point. Melting curves were obtained for each mutant in 10 mM sodium phosphate, pH 7.6 at 0.5 mg/mL. The temperature was ramped at 0.3 °C/min from 20 to 80 °C and data collected at 222 nm every 1 °C. The resultant melting curve data was converted to percentage folded (a) by using the formula:
Where θ is the observed ellipticity, θi is the ellipticity at 20 °C and θf is the ellipticity at the highest temperature. Melting temperatures were obtained by averaging the temperature values of two data points nearest the 50% unfolded point, i.e., where a values were 0.5. The data were not fit to any folding model.
Shell refilling assay
Empty procapsid shells were generated as previously described (Prevelige et al., 1988; Teschke, 1999). Shells at 0.25 mg/mL were incubated with scaffolding protein at 0.33 mg/mL in 10 mM sodium phosphate, pH 7.6, for 12–18 hours at room temperature. This sample was layered on a 7 ml 20 % sucrose cushion and spun at 40,000 rpm for 1.5 hours in a Sorvall S50-ST rotor. The free scaffolding protein remained at the top of the tube and so was separated from that which had refilled the shells. The pellet was resuspended in 50 μL of 10 mM sodium phosphate, pH 7.6 by shaking at 4 °C overnight. Twenty microliters of 5X SDS-PAGE reducing sample loading buffer was added to 50 μL of both pellet and samples were analyzed by 12.5% SDS-PAGE. The resulting gel was then quantified by densitometry and the ratio of scaffolding protein to coat protein in each lane was calculated.
Coat monomer purification
For the assembly reactions, P22 coat monomer protein was obtained by denaturing purified P22 coat protein shells as previously described (Anderson and Teschke, 2003; Parent et al., 2007). Briefly, 8 mg/mL shell stocks were diluted 1:4 into buffered 10 M urea and incubated for 30 minutes. An equal volume of 10 mM sodium phosphate, pH 7.6, was added to the unfolded coat sample, to give a coat monomer final concentration of 1 mg/mL, followed by extensive dialysis against 10 mM sodium phosphate, pH 7.6. During dialysis, some coat protein undergoes uncontrolled shell assembly, which is removed from soluble monomeric coat by ultracentrifugation.
Assembly reactions
The kinetics of assembly
Assembly-competent coat monomers were diluted to 0.5 mg/mL in 10 mM sodium phosphate, pH 7.6. The desired scaffolding protein was placed in a quartz cuvette, to give a final 0.5 mg/mL concentration. Assembly reactions were triggered by the rapid mixing of the coat protein solution with the scaffolding protein solution, with a final NaCl concentration of 45 mM. The reactions were then followed by light scattering measured by a SLM Aminco Bowman 2 spectrofluorometer, with emission and excitation wavelengths set to 500 nm, bandpass of 4 nm in both emission and excitation pathways, with 1 sec increments. Unless otherwise noted, each kinetic trace was followed for 30 minutes and the temperature was kept constant at 20 °C. Resulting kinetic traces were normalized to the wild-type scaffolding protein with or without the N-terminal his-tag by dividing each kinetics light scattering values by the wild-type maximum light scattering value. Assembled samples were further analyzed by electron microscopy (see below). For assembly reactions with extra scaffolding protein, coat protein monomers at 0.5 mg/mL were mixed with scaffolding protein variants at either 0.5 or 2.0 mg/mL in 10 mM phosphate buffer with 45 mM NaCl (final concentration). Assembly product formation was observed by monitoring light scattering at 500 nm over 60 minutes.
Assembly reactions assayed at completion
In order to determine the effect of the scaffolding protein amino acid substitutions on the relative affinity for coat protein, the proteins were mixed at 0.5 mg/mL each and incubated in the presence of final NaCl concentrations of 0, 5, 15, 30, 45, 60, 75 and 100 mM. The samples were incubated overnight at room temperature. Reactions were diluted 2:1 in 50% glycerol, TAE buffer (0.04 M Tris-acetate, 1 mM EDTA and 0.01% bromophenol blue. Samples were loaded onto a 1.2% SeaKem LE agarose (Rockland, ME) gels in TAE buffer and run at 100 V for 60 minutes. Gels were stained in 10% acetic acid, 0.003% Coomassie blue R250.
Electron microscopy
Four microliters of procapsids assembled in vitro were spotted onto carbon coated, copper grids and negatively stained with 1% uranyl acetate. Grids were visualized in an FEI 493 Technai Biotwin transmission electron microscope with nominal magnification of 43,000 x operated at 80 kV.
Supplementary Material
Acknowledgments
We thank G. Pauline Padilla-Meier for the scaffolding protein R293G data, Peter E. Prevelige, Jr. for the use of his mass spectrometer, Kelly Hughes for Salmonella strains, and members of the Teschke lab for critical reading of this manuscript. This work was supported by NIH grants R01 GM076661 to CMT and R01 AI074825 to SRC.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adams MB, Brown HR, Casjens S. Bacteriophage P22 tail protein gene expression. J Virol. 1985;53:180–184. doi: 10.1128/jvi.53.1.180-184.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson E, Teschke CM. Folding of Phage P22 Coat Protein Monomers: kinetic and thermodynamic properties. Virology. 2003;313:184–197. doi: 10.1016/s0042-6822(03)00240-x. [DOI] [PubMed] [Google Scholar]
- Bogan AA, Thorn KS. Anatomy of hot spots in protein interfaces. J Mol Biol. 1998;280:1–9. doi: 10.1006/jmbi.1998.1843. [DOI] [PubMed] [Google Scholar]
- Botstein D, Waddell CH, King J. Mechanism of head assembly and DNA encapsulation in Salmonella phage P22. I. Genes, proteins, structures and DNA maturation. J Mol Biol. 1973;80:669–695. doi: 10.1016/0022-2836(73)90204-0. [DOI] [PubMed] [Google Scholar]
- Casjens S, King J. P22 morphogenesis. I. Catalytic scaffolding protein in capsid assembly. J Supramol Struct. 1974;2:202–224. doi: 10.1002/jss.400020215. [DOI] [PubMed] [Google Scholar]
- Casjens S, Adams MB. Post-transcriptional modulation of bacteriophage P22 scaffolding protein gene expression. J Virol. 1985;53:185–191. doi: 10.1128/jvi.53.1.185-191.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casjens S, Adams MB, Hall C, King J. Assembly-controlled autogenous modulation of bacteriophage P22 scaffolding protein gene expression. J Virol. 1985;53:174–179. doi: 10.1128/jvi.53.1.174-179.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casjens S, Eppler K, Parr R, Poteete AR. Nucleotide sequence of the bacteriophage P22 gene 19 to 3 region: identification of a new gene required for lysis. Virology. 1989;171:588–598. doi: 10.1016/0042-6822(89)90628-4. [DOI] [PubMed] [Google Scholar]
- Casjens SR, Thuman-Commike PA. Evolution of mosaically related tailed bacteriophage genomes seen through the lens of phage P22 virion assembly. Virology. 2011;411:393–415. doi: 10.1016/j.virol.2010.12.046. [DOI] [PubMed] [Google Scholar]
- Chang JR, Spilman MS, Rodenburg CM, Dokland T. Functional domains of the bacteriophage P2 scaffolding protein: identification of residues involved in assembly and protease activity. Virology. 2009;384:144–150. doi: 10.1016/j.virol.2008.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen DH, Baker ML, Hryc CF, Dimaio F, Jakana J, Wu W, Dougherty M, Haase-Pettingell C, Schmid MF, Jiang W, Baker D, King JA, Chiu W. Structural basis for scaffolding-mediated assembly and maturation of a dsDNA virus. Proc Natl Acad Sci USA. 2011;108:1355–1360. doi: 10.1073/pnas.1015739108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creighton TE. Proteins: structures and molecular properties. 2. W. H. Freeman and Company; New York: 1993. [Google Scholar]
- Datsenko KA, Wanner BL. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci USA. 2000;97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dokland T. Scaffolding proteins and their role in viral assembly. Cel Mol Life Sci. 1999;56:580–603. doi: 10.1007/s000180050455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Earnshaw W, Casjens S, Harrison SC. Assembly of the head of bacteriophage P22: x-ray diffraction from heads, proheads and related structures. J Mol Biol. 1976;104:387–410. doi: 10.1016/0022-2836(76)90278-3. [DOI] [PubMed] [Google Scholar]
- Fane BA, Prevelige PE., Jr Mechanism of scaffolding-assisted viral assembly. Adv Protein Chem. 2003;64:259–299. doi: 10.1016/s0065-3233(03)01007-6. [DOI] [PubMed] [Google Scholar]
- Fu CY, Morais MC, Battisti AJ, Rossmann MG, Prevelige PE., Jr Molecular dissection of o29 scaffolding protein function in an in vitro assembly system. J Mol Biol. 2007;366:1161–1173. doi: 10.1016/j.jmb.2006.11.091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuller MT, King J. Assembly in vitro of bacteriophage P22 procapsids from purified coat and scaffolding subunits. J Mol Biol. 1982;156:633–665. doi: 10.1016/0022-2836(82)90270-4. [DOI] [PubMed] [Google Scholar]
- Greene B, King J. Binding of scaffolding subunits within the P22 procapsid lattice. Virology. 1994;205:188–197. doi: 10.1006/viro.1994.1634. [DOI] [PubMed] [Google Scholar]
- Greene B, King J. Scaffolding mutants identifying domains required for P22 procapsid assembly and maturation. Virology. 1996;225:82–96. doi: 10.1006/viro.1996.0577. [DOI] [PubMed] [Google Scholar]
- Greene B, King J. In vitro unfolding/refolding of wild type phage P22 scaffolding protein reveals capsid-binding domain. J Biol Chem. 1999;274:16135–16140. doi: 10.1074/jbc.274.23.16135. [DOI] [PubMed] [Google Scholar]
- Henthorn KS, Friedman DI. Identification of related genes in phages phi 80 and P22 whose products are inhibitory for phage growth in Escherichia coli IHF mutants. J Bact. 1995;177:3185–3190. doi: 10.1128/jb.177.11.3185-3190.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong Z, Beaudet-Miller M, Durkin J, Zhang R, Kwong AD. Identification of a minimal hydrophobic domain in the herpes simplex virus type 1 scaffolding protein which is required for interaction with the major capsid protein. J Virol. 1996;70:533–540. doi: 10.1128/jvi.70.1.533-540.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Israel JV, Anderson TF, Levine M. In vitro morphogenesis of phage P22 from heads and base-plate parts. Proc Natl Acad Sci USA. 1967;57:284–291. doi: 10.1073/pnas.57.2.284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janin J, Bahadur RP, Chakrabarti P. Protein-protein interaction and quaternary structure. Quarterly Reviews of Biophysics. 2008;41:133 –180. doi: 10.1017/S0033583508004708. [DOI] [PubMed] [Google Scholar]
- Karlinsey JE. lambda-Red genetic engineering in Salmonella enterica serovar Typhimurium. Methods Enzymol. 2007;421:199–209. doi: 10.1016/S0076-6879(06)21016-4. [DOI] [PubMed] [Google Scholar]
- Kennard J, Rixon FJ, McDougall IM, Tatman JD, Preston VG. The 25 amino acid residues at the carboxy terminus of the herpes simplex virus type 1 UL26.5 protein are required for the formation of the capsid shell around the scaffold. J Gen Virol. 1995;76:1611–1621. doi: 10.1099/0022-1317-76-7-1611. [DOI] [PubMed] [Google Scholar]
- King J, Casjens S. Catalytic head assembly protein in virus morphogenesis. Nature. 1974;251:112–119. doi: 10.1038/251112a0. [DOI] [PubMed] [Google Scholar]
- King J, Hall C, Casjens S. Control of the synthesis of phage P22 scaffolding protein is coupled to capsid assembly. Cell. 1978;15:551–560. doi: 10.1016/0092-8674(78)90023-5. [DOI] [PubMed] [Google Scholar]
- Kramer MA, Wetzel SK, Pluckthun A, Mittl PR, Grutter MG. Structural determinants for improved stability of designed ankyrin repeat proteins with a redesigned C-capping module. J Mol Biol. 2010;404:381–91. doi: 10.1016/j.jmb.2010.09.023. [DOI] [PubMed] [Google Scholar]
- Li J, Mahajan A, Tsai MD. Ankyrin Repeat: A unique motif mediating protein-protein interactions. Biochemistry. 2006;45:15168–15178. doi: 10.1021/bi062188q. [DOI] [PubMed] [Google Scholar]
- Matusick-Kumar L, Newcomb WW, Brown JC, McCann PJ, 3rd, Hurlburt W, Weinheimer SP, Gao M. The C-terminal 25 amino acids of the protease and its substrate ICP35 of herpes simplex virus type 1 are involved in the formation of sealed capsids. J Virol. 1995;69:4347–4356. doi: 10.1128/jvi.69.7.4347-4356.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mekalanos JJ, Swartz DJ, Pearson GD, Harford N, Groyne F, de Wilde M. Cholera toxin genes: nucleotide sequence, deletion analysis and vaccine development. Nature. 1983;306:551–557. doi: 10.1038/306551a0. [DOI] [PubMed] [Google Scholar]
- Oien NL, Thomsen DR, Wathen MW, Newcomb WW, Brown JC, Homa FL. Assembly of herpes simplex virus capsids using the human cytomegalovirus scaffold protein: critical role of the C terminus. J Virol. 1997;71:1281–1291. doi: 10.1128/jvi.71.2.1281-1291.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padilla-Meier GP, Teschke CM. Conformational changes in bacteriophage p22 scaffolding protein induced by interaction with coat protein. J Mol Biol. 2011;410:226–240. doi: 10.1016/j.jmb.2011.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parent KN, Doyle SM, Anderson E, Teschke CM. Electrostatic interactions govern both nucleation and elongation during phage P22 procapsid assembly. Virology. 2005;340:33–45. doi: 10.1016/j.virol.2005.06.018. [DOI] [PubMed] [Google Scholar]
- Parent KN, Khayat R, Tu LH, Suhanovsky MM, Cortines JR, Teschke CM, Johnson JE, Baker TS. P22 coat protein structures reveal a novel mechanism for capsid maturation: Stability without auxiliary proteins or chemical cross-links. Structure. 2010;18:1568–82. doi: 10.1016/j.str.2009.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parent KN, Suhanovsky MM, Teschke CM. Phage P22 procapsids equilibrate with free coat protein subunits. J Mol Biol. 2007;365:513–522. doi: 10.1016/j.jmb.2006.09.088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parent KN, Zlotnick A, Teschke CM. Quantitative analysis of multi-component spherical virus assembly: Scaffolding protein contributes to the global stability of phage P22 procapsids. J Mol Biol. 2006;359:1097–1106. doi: 10.1016/j.jmb.2006.03.068. [DOI] [PubMed] [Google Scholar]
- Parker MH, Brouillette CG, Prevelige PJ. Kinetic and calorimetric evidence for two distinct scaffolding protein binding populations within the bacteriophage P22 procapsid. Biochemistry. 2001;40:8962–70. doi: 10.1021/bi0026167. [DOI] [PubMed] [Google Scholar]
- Parker MH, Jablonsky M, Casjens S, Sampson L, Krishna NR, Prevelige PE., Jr Cloning, purification, and preliminary characterization by circular dichroism and NMR of a carboxyl-terminal domain of the bacteriophage P22 scaffolding protein. Protein Sci. 1997a;6:1583–1586. doi: 10.1002/pro.5560060722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker MH, Stafford WF, III, Prevelige PE., Jr Bacteriophage P22 scaffolding protein forms oligomers in solution. J Mol Biol. 1997b;268:655–665. doi: 10.1006/jmbi.1997.0995. [DOI] [PubMed] [Google Scholar]
- Parker MH, Casjens S, Prevelige PE., Jr Functional Domains of Bacteriophage P22 Scaffolding Protein. J Mol Biol. 1998;281:69–71. doi: 10.1006/jmbi.1998.1917. [DOI] [PubMed] [Google Scholar]
- Parker MH, Prevelige PE., Jr Electrostatic interactions drive scaffolding/coat protein binding and procapsid maturation in bacteriophage P22. Virology. 1998;250:337–349. doi: 10.1006/viro.1998.9386. [DOI] [PubMed] [Google Scholar]
- Prasad BVV, Prevelige PE, Jr, Marieta E, Chen RO, Thomas D, King J, Chiu W. Three-dimensional transformation of capsids associated with genome packaging in a bacterial virus. J Mol Biol. 1993;231:65–74. doi: 10.1006/jmbi.1993.1257. [DOI] [PubMed] [Google Scholar]
- Prevelige PE, Jr, Thomas D, King J. Scaffolding protein regulates the polymerization of P22 coat subunits into icosahedral shells in vitro. J Mol Biol. 1988;202:743–757. doi: 10.1016/0022-2836(88)90555-4. [DOI] [PubMed] [Google Scholar]
- Prevelige PE, Jr, Thomas D, King J. Nucleation and growth phases in the polymerization of coat and scaffolding subunits into icosahedral procapsid shells. Biophys J. 1993;64:824–35. doi: 10.1016/S0006-3495(93)81443-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz J, Silhavy T, Berman M, Fiil N, Emr S. A previously unidentified gene in the spc operon of Escherichia coli K12 specifies a compnent of the protein export machinery. Genetics. 1982;121:635–649. doi: 10.1016/0092-8674(82)90422-6. [DOI] [PubMed] [Google Scholar]
- Schwarz J, Berget P. Characterization of bacteriophage P22 tailspike mutant proteins with altered endorhamnosidase and capsid assembly activities. J Biol Chem. 1989a;264:20112–20119. [PubMed] [Google Scholar]
- Schwarz JJ, Berget PB. The isolation and sequence of missense and nonsense mutations in the cloned bacteriophage P22 tailspike protein gene. Genetics. 1989b;121:635–649. doi: 10.1093/genetics/121.4.635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedgwick SG, Smerdon SJ. The ankyrin repeat: a diversity of interactions on a common structural framework. Trends Biochem Sci. 1999;24:311–316. doi: 10.1016/s0968-0004(99)01426-7. [DOI] [PubMed] [Google Scholar]
- Sippl MJ. On distance and similarity in fold space. Bioinformatics. 2008;24:872–873. doi: 10.1093/bioinformatics/btn040. [DOI] [PubMed] [Google Scholar]
- Sippl MJ, Wiederstein M. A note on difficult structure alignment problems. Bioinformatics. 2008;24:426–427. doi: 10.1093/bioinformatics/btm622. [DOI] [PubMed] [Google Scholar]
- Studier FW. Use of bacteriophage T7 lysozyme to improve an inducible T7 expression system. J Mol Biol. 1991;219:37–44. doi: 10.1016/0022-2836(91)90855-z. [DOI] [PubMed] [Google Scholar]
- Suhanovsky MM, Parent KN, Dunn SE, Baker TS, Teschke CM. Determinants of bacteriophage P22 polyhead formation: the role of coat protein flexibility in conformational switching. Mol Microbiol. 2010;77:1568–82. doi: 10.1111/j.1365-2958.2010.07311.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suhanovsky MM, Teschke CM. Bacteriophage P22 capsid size determination: Roles for the coat protein telokin-like domain and the scaffolding protein amino-terminus. Virology. 2011;417:418–29. doi: 10.1016/j.virol.2011.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Parker MH, Weigele P, Casjens S, Prevelige PEJ, Krishna NR. Structure of the coat protein-binding domain of the scaffolding protein from a double-stranded DNA virus. J Mol Biol. 2000;297:1195–1202. doi: 10.1006/jmbi.2000.3620. [DOI] [PubMed] [Google Scholar]
- Teschke CM. Aggregation and assembly of phage P22 temperature-sensitive coat protein mutants in vitro mimic the in vivo phenotype. Biochemistry. 1999;38:2873–2881. doi: 10.1021/bi982739f. [DOI] [PubMed] [Google Scholar]
- Teschke CM, Fong DG. Interactions between coat and scaffolding proteins of phage P22 are altered in vitro by amino acid substitutions in coat protein that cause a cold-sensitive phenotype. Biochemistry. 1996;35:14831–14840. doi: 10.1021/bi960860l. [DOI] [PubMed] [Google Scholar]
- Teschke CM, McGough A, Thuman-Commike PA. Penton release from P22 heat-expanded capsids suggests importance of stabilizing penton-hexon interactions during capsid maturation. Biophysical Journal. 2003;84:2585–2592. doi: 10.1016/S0006-3495(03)75063-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teschke CM, Parent KN. ‘Let the phage do the work’: using the phage P22 coat protein structures as a framework to understand its folding and assembly mutants. Virology. 2010;401:119–130. doi: 10.1016/j.virol.2010.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thuman-Commike PA, Greene B, Jokana J, Prasad BVV, King J, Prevelige PE, Jr, Chiu W. Three-dimensional structure of scaffolding-containing phage P22 procapsids by electron cryo-microscopy. J Mol Biol. 1996;260:85–98. doi: 10.1006/jmbi.1996.0383. [DOI] [PubMed] [Google Scholar]
- Thuman-Commike PA, Greene B, Jakana J, McGough A, Prevelige PE, Chiu W. Identification of additional coat-scaffolding interactions in a bacteriophage P22 mutant defective in maturation. J Virol. 2000;74:3871–3873. doi: 10.1128/jvi.74.8.3871-3873.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuma R, Parker MH, Weigele P, Sampson L, Sun Y, Krishna NR, Casjens S, Thomas GJJ, Prevelige PEJ. A helical coat protein recognition domain of the bacteriophage P22 scaffolding protein. J Mol Biol. 1998;281:81–94. doi: 10.1006/jmbi.1998.1916. [DOI] [PubMed] [Google Scholar]
- Tuma R, Tsuruta H, French KH, Prevelige PE. Detection of intermediates and kinetic control during assembly of bacteriophage P22 procapsid. J Mol Biol. 2008;381:1395–1406. doi: 10.1016/j.jmb.2008.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weigele PR, Sampson L, Winn-Stapley DA, Casjens SR. Molecular genetics of bacteriophage P22 scaffolding protein’s functional domains. J Mol Biol. 2005;348:831–844. doi: 10.1016/j.jmb.2005.03.004. [DOI] [PubMed] [Google Scholar]
- Winston R, Botstein D, Miller JH. Characterization of amber and ochre suppressors in Salmonella typhimurium. J Bact. 1979;137:433–439. doi: 10.1128/jb.137.1.433-439.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, Sampson L, Parr R, Casjens S. The DNA site utilized by bacteriophage P22 for initiation of DNA packaging. Mol Microbiol. 2002;45:1631–1646. doi: 10.1046/j.1365-2958.2002.03114.x. [DOI] [PubMed] [Google Scholar]
- Wyckoff E, Casjens S. Autoregulation of the bacteriophage P22 scaffolding protein gene. J Virol. 1985;53:192–197. doi: 10.1128/jvi.53.1.192-197.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.