

Abstract
Small ubiquitin-like modifiers (SUMOs) are covalently conjugated to target proteins to regulate numerous biological processes, including subcellular localization, protein-protein interactions, and transactivational activities. While the majority of identified SUMO targets are cellular proteins, SUMO modified viral proteins have also been identified. In addition, there are a growing number of examples where viruses alter the sumoylation status of host cell proteins. Work from our laboratory has previously demonstrated that the human cytomegalovirus (HCMV) virion tegument protein pp71 binds to Daxx, a cellular transcriptional co-repressor, and promotes its sumoylation. Here we describe the in vivo techniques used to detect pp71-induced sumoylation of Daxx in a cotransfection system as well as the endogenous SUMO modified form of Daxx in HCMV-infected cells. The approaches we describe can be easily adapted to infections with other viruses and for the detection of sumoylation of other proteins.
1. Introduction
Since its discovery in 1996, the small ubiquitin-like (Ubl) modifier (SUMO), a ~10 kDa protein, has received great attention because of its intriguing functions [1–5]. Sumoylation, a post-translational modification, is a major regulator of protein function that plays an important role in a wide range of cellular processes, such as transcriptional regulation, DNA repair and chromatin remodeling (Reviewed in [6]). In the fission yeast Schizosaccharomyces pombe, deletion of the SUMO/Pmt3 gene results in severe growth impairment [7]. In the budding yeast Saccharomyces cerevisiae, SUMO/Smt3 is essential for viability [8], suggesting that SUMO is critical for the normal function of eukaryotic cells. The importance of sumoylation in mammals has also been confirmed through the knockout of Ubc9, a SUMO conjugating enzyme; transgenic mice lacking the gene for Ubc9 display embryonic lethality [9]. Hundreds of SUMO substrates have been previously described (Reviewed in [6]), and several known targets among them are proteins of great interest (e.g., p53, PML, PCNA and IκBα), drawing further attention to this field. Although the biological consequences of SUMO modification are difficult to illustrate overall, when proteins are modified by SUMO, their functional properties, such as transcriptional activity, intracellular localization, stability, and protein-protein interactions, are usually modified [6].
The biochemical mechanism of sumoylation, in contrast, is well understood (Reviewed in [10]). Sumoylation is an enzymatic process that is biochemically analogous to, but functionally distinct from, ubiquitination. Similar to ubiquitination, sumoylation occurs in a multi-step reaction that covalently conjugates SUMO to a target protein by a single E1 activating enzyme (SAE1/2), a unique E2 conjugating enzyme (Ubc9), and an array of different E3 ligases. SUMO is most often conjugated to lysine residues in ψKxE consensus sequences in target proteins, where ψ corresponds to hydrophobic amino acids, K is a lysine residue, x is any amino acid, and E is a glutamic acid residue. Sumoylation, however, can also occur at lysine residues outside of this motif, and not all ψKxE motifs are sumoylated.
In mammalian cells, there are four SUMO isoforms, SUMO-1 to SUMO-4 [10]. Of these, SUMO-1, SUMO-2, and SUMO-3 are ubiquitously expressed, while SUMO-4 expression patterns remain to be determined, although there is one study illustrating SUMO-4 mRNA expression in the kidney [11]. Although some substrates have preferences for specific SUMO isoform modification, many substrates can be modified by different SUMO proteins. This is somewhat surprising because SUMO-2 and SUMO-3 only share 50% similarity with SUMO-1, while SUMO-2 and SUMO-3 share 95% identity with each other. SUMO-2/3 have the ability to form polySUMO chains in a similar manner to polyubiquitin chain formation, but SUMO-1 lacks this ability, and is often used as a polySUMO chain terminator [12]. Sumoylation can also occur at several lysine residues in a substrate (multisumoylation) [10]. In addition to covalent attachment of SUMO to target proteins, specific non-covalent SUMO interaction motifs (SIMs) have been identified [13, 14]. SIM-SUMO interactions have emerged as important mechanisms in the downstream effect of protein sumoylation. For example, Daxx, a cellular transcriptional co-repressor, relies on its SIM to localize to PML nuclear bodies (PML-NBs) and to associate with sumoylated transcription factors to function in transcriptional regulation [15].
Viruses have evolved strategies to manipulate and usurp the host sumoylation pathway (Reviewed in [16]). Examples of viral proteins modified by SUMO as well as viral proteins that have effects on host sumoylation are constantly increasing. CELO adenoviral protein Gam1 targets the host E1 SUMO activating enzyme for proteasomal degradation, thereby inhibiting the host sumoylation system [17]. Kaposi’s sarcoma associated herpesvirus encodes K-bZip, which acts as an E3 SUMO ligase to increase the sumoylation of p53 and Rb [18]. Human cytomegalovirus (HCMV) infection also affects host sumoylation. The viral IE1 protein disrupts PML-NBs through the modulation of sumoylation [19], which is an important step for the virus in overcoming host antiviral responses. Work from our lab has also demonstrated that the HCMV pp71 protein enhances the sumoylation of Daxx, and this process requires its ability to interact with Daxx [20].
The detection of sumoylated proteins in vivo can be challenging because only a small fraction of target proteins are sumoylated [6]. Sumoylation is a very dynamic process involving both SUMO conjugating enzymes and deconjugating enzymes (Reviewed in [21]). Deconjugating enzymes, a family of isopeptidases known as SENPs (Sentrin-specific proteases) function in cleaving SUMO molecules from target proteins. This may explain the low level of detection for sumoylated proteins in vivo. Furthermore, some sumoylated proteins may not be isolated very well due to limited solubility or their tight interaction with organelles and structures of the cell. Because of these obstacles during the detection of sumoylation in vivo, many methods have been developed to investigate substrate sumoylation in vitro using the purified SUMO, SUMO conjugating enzymes, and substrates produced in bacteria [21, 22]. Although this is a powerful tool, the non-physiological conditions limit their biological relevance.
In this article, we describe protocols to detect protein sumoylation in virus-infected cells in vivo. We focus on techniques that have been used to detect in vivo HCMV pp71-mediated Daxx sumoylation as an example by describing:
Protocols for detecting Ubl (such as SUMO)-mediated covalent modifications of Daxx in HCMV-infected cells (Sections 2.1 and 2.2)
A protocol for detecting and verifying the SUMO modification of Daxx with a cotransfection-coimmunoprecipitation assay (Section 2.3)
A method for detecting endogenous sumoylation of Daxx in HCMV-infected cells by immunoprecipitation assay (Section 2.4)
These methods would be applicable to other proteins and infections with other viruses with little adjustment.
2. Detection of Daxx sumoylation in vivo
2.1. Preparation of protein extracts from HCMV-infected cell populations
To detect endogenous Daxx covalently modified by ubiquitin or Ubls during HCMV infection, HCMV permissive cells, either human foreskin fibroblasts (HFs) or telomerase immortalized HFs (Tert-HFs) are commonly used. Different types of appropriate cell lines can be used for other viral infections.
The protocol uses SDS as denaturant and it provides a reliable means to quickly generate protein extracts for Western blot analysis of covalent modification of Daxx. Maintaining denaturing conditions throughout the procedure has advantages in detecting ubiquitin or Ubl modifications. It not only improves protein solubilization, but also reduces false positives as non-covalent interactions are completely eliminated. To preserve native modifications by ubiquitin or Ubls, cysteine protease inhibitors such as N-ethylmaleimide (NEM) and iodoacetate (IAA) are often required. Lysis of cells in the presence of NEM alone or in combination with IAA has been shown to result in higher levels of conjugates compared to extract prepared without inhibitors (see ref. [23] for example). In our experiment [20], this treatment also resulted in the appearance of higher molecular weight conjugates of Daxx (Figure 1). Because the basic biochemical mechanisms of conjugation and deconjugation are common to ubiquitin and Ubls [6], the method described here is widely applicable with little modifications to detect other protein Ub/Ubl modifications. The protocol is described for cultured cells grown in a single well of a 6-well plate, containing approximately 5×105 cells.
Figure 1. Isopeptidase inhibitors enhance detection of SUMO-modified Daxx.
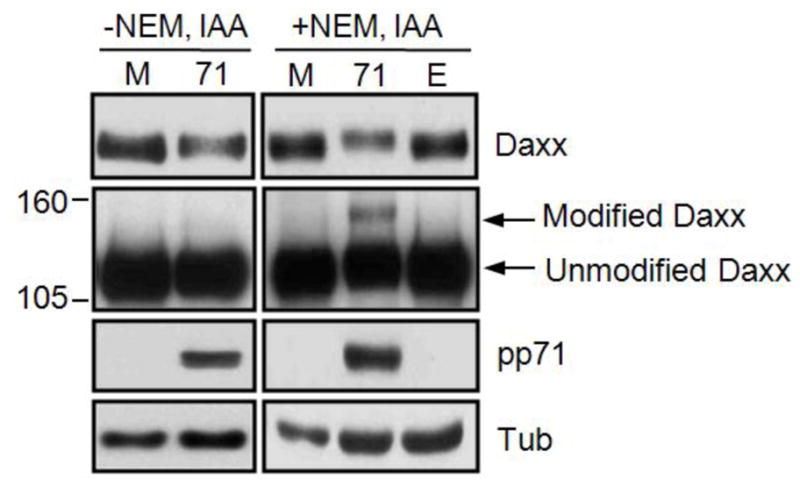
HFs were mock transduced (M) or transduced with an rAd the expresses pp71 (71) or an empty (E) rAd that does not encode a transgene. Lysates were harvested at 24 hours post transduction with or without NEM and IAA as indicated and analyzed by Western blot. Short (top) and long (middle) exposures are shown. Numbers on the left represent molecular weight standards in kilodaltons. Tubulin (Tub) serves as a loading control. Image from reference 20 used with permission.
PROCEDURE: Cell lysate preparation for Ub/Ubl conjugate detection
Set up cultures
-
1
Two days prior to infection with HCMV, split cells (either HFs or Tert-HFs) seeding 3×105 cells per well of a 6-well plate into 2ml of 10% FBS-containing DMEM medium.
-
2
Optional: Change the medium the next day.
Viral infection
-
3
For HCMV infection, remove the medium from confluent cells, then incubate with HCMV by adding virus at a multiplicity of infection of 3 in a 1 ml total volume at 37°C. For mock-infected control culture, expose cells to a 1 ml of mock-infecting 10% FBS-containing DMEM medium. After an hour, remove the virus inoculum and return the old medium to the cells.
Alternative method: Infect cells with recombinant adenovirus (rAd) expressing pp71.
Tip: For negative controls, mock infection, or pp71-null mutant HCMV or rAd expressing mutant pp71 unable to bind to Daxx could be used.
Cell harvest
-
4
Harvest cells using 0.25% trypsin or scrapping at desired time points after infection.
-
5
Wash cell pellets with cold PBS containing 20 mM NEM and 5 mM IAA (Acros Organics).
-
6
Resuspend cell pellets thoroughly in 5 μl of cold PBS containing 20 mM NEM and 5 mM IAA.
Cell lysis
-
7
Lyse cells by adding 35 μl of SDS lysis buffer (20 mM Tris [pH 8.0], 250 mM NaCl, 3 mM EDTA, 10% glycerol, 1% SDS, 0.5% NP-40, 20 mM NEM and 5 mM IAA, and fluoride, protease inhibitors [1 mM Na3VO4, 25 mM NaF, 1 mM phenylmethylsulfonyl 10 μg of pepstatin A/ml, 10 μg of aprotinin/ml, 25 μg of leupeptin/ml, 25 μg of trypsin inhibitor/ml]), then immediately boil samples at 90–95°C in heat block for 20–30 minutes. To help lysis, mix by vortexing samples for 5–10 seconds every 3 minutes during the boiling step.
Tip: Continue procedure until viscosity is eliminated.
-
8
Centrifuge the samples (13K rpm, 10 minutes at room temperature in a standard table-top microfuge), and transfer supernatants to new tubes.
Pause point: Cell lysates from step 8 can be stored at −20°C before further analysis.
2.2. Detection of protein modifications
To detect a Ub/Ubl post-translational modification of the protein of interest, Western blot analysis can be used as protein modification by Ub/Ubl causes a molecular weight shift of about 10 kDa or larger on SDS-PAGE. Although this method would not indicate which modification occurs, Western blot analysis using total cell lysates is a good tool to readily examine if the test protein is modified and thus determine if further investigation of such modification is warranted.
PROCEDURE: Detection of protein modification by Western blot
Determination of protein concentration
-
1
Measure protein concentration using a Bradford assay. Prepare a Bradford reagent: dissolve 100 mg of Coomassie Brilliant Blue G-250 in 50ml of 95% ethanol. Add 100 ml of phosphoric acid, and bring to 1L with distilled water. Filter through Wattman paper.
-
2
Prepare a standard curve using 0, 0.05, 0.1, 0.2, and 0.3 mg/ml BSA. Pipet each BSA dilution into a Bradford reagent, mix well, and incubate for 5 minutes at room temperature. Prepare dilutions of the protein sample of interest along with BSA standard samples. The end volume of samples should be 2ml. Transfer Bradford mixture to plastic cuvette, measure the OD at 595 nm for each sample, and plot the standard curve. Calculate the protein concentration of the test samples from a standard curve.
Tip: SDS can interfere with Bradford assay. Alternative protein assays, such as Lowry protein assay [24], may be required for more accurate protein quantification.
Western blot analysis
-
3
Analyze 50 μg of the protein lysates on Western blots to test for protein modification. Add a loading buffer (standard loading buffer is Laemmli buffer [25]) to sample, and boil the mixture at 95–100°C in a heat block for 5 minutes.
-
4
Prepare a polyacrylamide gel (10 × 8.2 cm). When separated on a polyacrylamide gel, the procedure is abbreviated as SDS-PAGE (SDS polyacrylamide gel electrophoresis). Depending on test protein size, the appropriate percentage of SDS-PAGE gel should be prepared in order to efficiently separate the modified and unmodified forms of the protein (e.g., 15% gel for <20 kDa protein, 12.5% gel for 10–40 kDa protein, 10% gel for 30–70 kDa protein, 7.5% for 50–120 kDa protein, 6% gel for 80–200 kDa protein). 6% or 7.5% gel can be used to separate Daxx, for instance, because the primary species of Daxx (unmodified Daxx) is detected as a ~120 kDa band by SDS-PAGE.
-
5
Load each sample in wells. To determine protein size, an appropriate molecular weight marker should be loaded in a separate lane of the gel. A range of molecular markers are commercially available. Run SDS-PAGE gel for the recommended time depending on the voltage. For Daxx detection, run 7.5% gel at 60V for 5 hours or 6% gel at 60V for 4 hours. A standard running buffer for SDS-PAGE is 1x Tris-glycine buffer: 25 mM Tris base, 190 mM glycine, 0.1% SDS.
-
6
Transfer proteins to a nitrocellulose or PVDF (polyvinyl-difluoride) membrane. Detailed instructions for the transfer process can be found on the websites of the manufacturers of transfer apparatus.
-
7
Perform Western blot analysis. A detailed Western blot procedure can be found in [26]. Use an appropriate primary antibody that specifically detects the protein of interest. We recommend HRP (horseradish peroxidase)-conjugated secondary antibodies for high sensitivity detection. To develop, use X-ray films to control the incubation time. Depending on the abundance of modified species of test protein, a few minutes to an overnight exposure is required.
TROUBLESHOOTING: Troubleshooting advice can be found in Table 1.
Table 1.
Troubleshooting table
| Step | Problem | Possible reasons | Possible solutions |
|---|---|---|---|
| Section 2.2 Step 2 |
The amount of total protein cell lysates using SDS lysis- based protein extraction method is low | Large volume of SDS buffer was used in Section 2.1. Small volume of SDS buffer was used in Section 2.1 Inaccurate Bradford assay |
To avoid loss of protein, extracts should not be too dilute. If a high volume of lysis buffer was used to avoid sample viscosity, try DNase digestion or sonication for genomic DNA fragmentation. Using insufficient volume of lysis buffer prevents cell lysis. 40ul of final volume per 5×105 cells is recommended. SDS could interfere with Bradford assay. Alternative protein assays, such as Lowry protein assay, may be required for accurate protein quantification. |
| Section 2.2 Step 7 |
The SUMO-modified form of Daxx is not detected by Western blotting | Western blot exposure too short Modified and unmodified forms not sufficiently separated |
Because only a small fraction of endogenous Daxx is sumoylated, at least 50ug cell lysates are required for Western blot analysis. Make sure blots are exposed overnight in order to detect SUMO modification. The unmodified form of Daxx is detected as an approximately 120kDa band by SDS-PAGE while the SUMO-modified Daxx is detected as ~140kDa. Run6% or 7.5% PAGE gel for 4–5 hours (or longer if necessary) at 60V to nicely separate the two Daxx species. This will place unmodified Daxx very near the bottom of the gel. |
| Section 2.3 Step 3 |
Unequal protein expression levels | Used CMV promoter-driven plasmids | pp71 transactivates the CMV promoter. Co- expression of pp71 would increase the expression of other co-transfected plasmids that are under control of CMV promoter. Use other promoters (e.g., SV40 promoter). |
| Section 2.4 | IP using SUMO-1 antibody did not show Daxx by Western blotting | Low antibody binding efficiency Lost the sumoylated Daxx during IP |
Use antibody up to ~20ug/IP reaction. Small IP volume (~500ul) is favorable. Incubate antibody overnight (>12 hours) at 4°C. Because IP is performed in RIPA condition (non- denaturing condition), SUMO proteases may be partially active even in the presence of NEM and IAA, or become active during long IP procedure. Crosslinking may be helpful to solve the problem if this is the case. |
| Section 2.4 | Difficult detection of endogenous protein sumoylation | Low sumoylation due to combination of low levels of free pool of SUMO and substrate protein in cells | Use cells that constitutively express SUMO proteins or SUMO-Ubc9 fusion proteins. Despite its weakness of artificial system, increased SUMO machinery can be a useful way to detect endogenous substrate sumoylation. |
Using this method [20], we were able to detect the covalently modified form of Daxx induced by HCMV infection or by rAd-pp71 transduction with an apparent molecular weight that was approximately 20 kDa larger than the unmodified form of Daxx (Figure 1 and 2). We hypothesized that the pp71-induced Daxx modification may be sumoylation.
Figure 2. Tegument-delivered pp71 induces Daxx sumoylation at the start of HCMV infections.
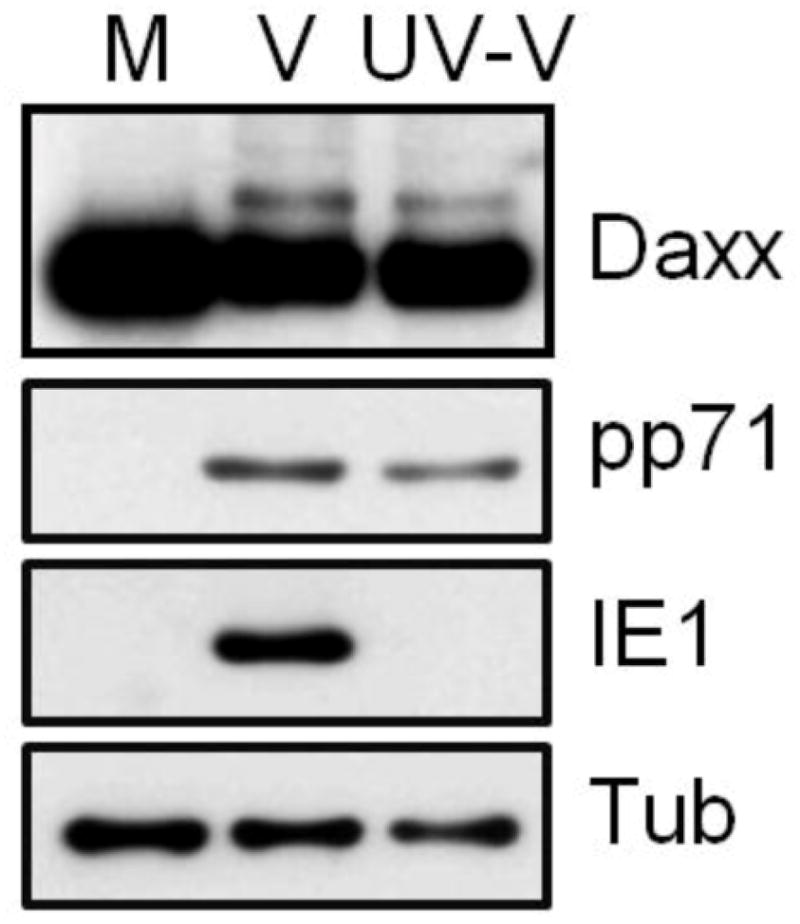
HFs were mock infected (M) or infected wit either wild-type HCMV (V) or UV-inactivated wild-type HCMV (UV-V) at a multiplicity of infection of 3. Lysates harvested at 2 hours post infection were analyzed by Western blot with the indicated antibodies. Image from reference 20 used with permission.
2.3. Cotransfection-Coimmunoprecipitation assay to verify SUMO modification of Daxx
Because Daxx is found at PML-NBs where many proteins, including Daxx [27, 28], are found to be covalently modified by SUMO, we asked if pp71 induced Daxx sumoylation. In this section, we describe a method to identify Daxx sumoylation by cotransfection-coimmunoprecipitation. Despite its potential limitation resulting from expression of exogenous gene products, cotransfection assays are widely used to investigate SUMO conjugation due to the following reasons: (a) epitope-tagging of transfected SUMO or the target protein makes it easy to immunoprecipitate the test protein, (b) enrichment of SUMO and/or the target protein allows the easy detection of target protein sumoylation, especially in cases where the endogenous sumoylated form is not abundant in cells, (c) expression levels of conjugating (i.e., SUMO E1 and E2) and/or deconjugating (i.e., SENP family proteins) enzymes are easily manipulated, (d) it is readily applicable for the identification of SUMO conjugation sites on target protein by mutating the potential SUMO acceptor lysine residues, and (e) SUMO paralog specificity (SUMO-1, SUMO-2, and SUMO-3) for target protein can be determined.
We have used this technique [20] for the identification of Daxx modification by pp71 (Figure 3). For transfection efficiency, we chose the human papillomavirus-negative cervical carcinoma cell line C33A instead of HCMV permissive cells. Other transfectable cell types, such as HeLa and 293, are applicable as well. The protocol here is described for cultured cells grown in a 10 cm dish, containing 6–7 × 106 cells. NEM and IAA are included in all buffers for this method.
Figure 3. Co-transfection/immunoprecipitation assay detects sumoylated Daxx.
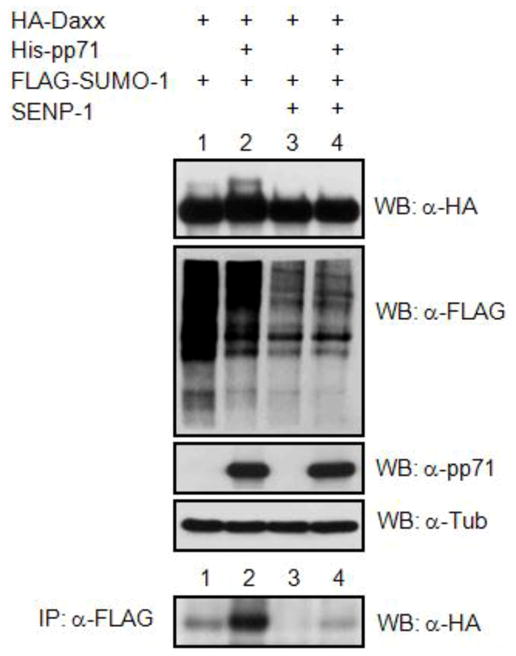
C33A cells were transfected with expression plasmids for the indicated proteins. Lysates were subject to direct Western blotting (WB) or to IP with an anti-FLAG antibody followed by Western blotting with an anti-HA antibody. Image from reference 20 used with permission.
PROCEDURE: Cotranfection-coimmunoprecipitation
Set up cultures
-
1
The day before transfection, split C33A cells so that they will be 70–80% confluent on the day of transfection. Plate the cells at 3×106 cells per 10 cm culture dish with 10ml of 10% FBS-containing DMEM medium.
-
2
Optional: Change the medium for fresh 10% FBS-containing DMEM the next morning.
Transfection
-
3
Cotransfect cells with the desired plasmids using the calcium phosphate method [29] (the total amount of DNA should be 20 μg) or any other applicable transfection methods, such as lipofectamine transfection, following manufacturers’ instructions. An example of transfection combinations for the Daxx sumoylation assay is:
Sample no.1: HA-Daxx+FLAG-SUMO-1
Sample no.2: HA-Daxx+FLAG-SUMO-1+His-pp71
Sample no.3: HA-Daxx+FLAG-SUMO-1+SENP-1
Sample no.4: HA-Daxx+FLAG-SUMO-1+His-pp71+SENP-1
Tip: The tag choice can be variable.
Tip: A nonconjugatable SUMO-1 mutant (i.e., SUMO-1ΔGG, in which the C-terminal diglycine residues are deleted) can be used as another control.
-
4
Remove the DNA precipitates 12–16 hours later. Rinse cells two times with PBS and culture in 10% FBS-containing DMEM for an additional 24 hours before harvesting cells.
Cell harvest and lysis
-
5
Harvest cells as described in Section 2.1. Make sure that all buffers from this point on, including PBS, lysis buffer, and IP buffer, contain NEM and IAA.
-
6
Lyse cells in 200 μl of 1% SDS lysis buffer as described in Section 2.1.
Tip: Sonication can be done prior to the centrifugation if the lysates are highly viscous. Alternatively, DNase I can be used to degrade genomic DNA by adding 2 units to samples and incubating for an hour at 37°C.
Pause point: Cell lysates from step 6 can be stored at −20°C.
Immunoprecipitation
-
7
Subject equal amounts of protein lysates (typically 500–1000 μg) to immunoprecipitation (IP) assay. For IP, dilute lysates into 800 μl of IP buffer (20 mM Tris [pH 8.0], 250 mM NaCl, 3 mM EDTA, 10% glycerol, 0.1% SDS, 0.5% NP-40, 20 mM NEM and 5 mM IAA, and protease inhibitors [1 mM Na3VO4, 25 mM NaF, 1 mM phenylmethylsulfonyl fluoride, 10 μg of pepstatin A/ml, 10 μg of aprotinin/ml, 25 μg of leupeptin/ml, 25 μg of trypsin inhibitor/ml]) and perform all subsequent steps at 4°C. Before the dilution, save 10 μl of cell lysates for use as the pre-IP samples.
-
8
Preclear the samples by the addition of goat serum to a final concentration of 5% for 30 minutes prior to incubation with 40 μl of a 50% slurry of heat-killed protein A-positive Staphylococcus aureus (Calbiochem, Catalog number: 507862) for 30 minutes. To prepare the 50% slurry, resuspend 0.1 g of heat-killed protein A-positive Staphylococcus aureus cells in 1 ml of IP buffer, centrifuge at 13K rpm for 1 minute at 4°C. Discard supernatant and repeat wash. Resuspend in one volume IP buffer to make 50% slurry.
-
9
Spin down samples (13K rpm, 10 minutes at 4°C), and transfer supernatants to new tubes.
-
10
Combine the appropriate antibody (e.g., anti-FLAG) with the cell lysates for one to four hours at 4°C to form the immune complexes. Depending on the antibody, different amounts of antibody could be used. Typically, 3–5 μg of antibody is used for each IP. This could be increased to 10 μg if no signal is observed.
-
11
To prepare Protein A agarose/sepharose, wash the beads twice with distilled water and restore to a 50% slurry with IP wash buffer. Add 30 μl of the 50% resin slurry to the immune complexes, incubate at 4°C for an hour, and wash the resin six times with 1 ml of IP wash buffer. Discard the buffer after each wash.
-
12
To elute the immune complexes, add 30 μl of Laemmli loading buffer and incubate at 95°C for 5–10 minutes.
-
13
Centrifuge to collect eluates, and apply samples for Western blot analysis using appropriate antibodies as described in Section 2.2.
TROUBLESHOOTING: Troubleshooting advice can be found in Table 1.
Denaturing conditions for this experiment are often used to ensure that any detected SUMO proteins represent covalent modifications and not simply a non-covalent interaction through a SIM. Using this approach with FLAG antibodies for the IP and HA antibodies for Western blotting, our lab [20] was able to observe that co-expression of pp71 enhances the modified form of HA-Daxx by FLAG-SUMO-1 (Figure 3, lanes 1 and 2). Importantly, co-expression of the SUMO-specific protease SENP-1, which has ability to deconjugate all three SUMO paralogues from modified substrates [21], prevents this modification of Daxx, validating that Daxx is specifically modified by SUMO (Figure 3, lanes 3 and 4). Mutant substrate that is predicted to not be sumoylated (e.g., mutant with lysine residues substituted by arginine) can also be used as a control to further confirm target protein sumoylation. Likewise, the same method can be applied to identify other Ubl modifications using plasmids expressing the desired Ubls in place of SUMO. Although this technique is powerful, the non-physiological condition (i.e., expression of exogenous gene products) is considered a limitation to this approach. Therefore, this single method alone cannot be regarded as solid proof of sumoylation.
2.4. Immunoprecipitation assay to detect endogenous sumoylated proteins
To complement cotransfection assays, a number of additional methods can be used to further investigate the sumoylation of a target protein, such as mass spectrometry (MS) analysis and IP of an endogenous protein. The mass spectrometric method generally involves affinity purification (e.g., Ni-NTA [30]), proteolytic digestion, and analysis by MS. This approach is useful not only for identifying the proteins modified by SUMO, but also for mapping the residues that are covalently linked to SUMO. Although MS analysis is clearly powerful, there are drawbacks resulting from numerous false positives (nonspecific binding) and false negatives (loss of true SUMO conjugates). IP of the protein followed by Western blot analysis using endogenous proteins is one of the best approaches to detect true SUMO-modified substrates, although low abundance of the endogenous sumoylated target proteins often makes this approach challenging. In this section, we discuss the method used to detect endogenous Daxx sumoylation using the IP method (see ref. [31, 32] for discussion of MS studies).
Two major technical challenges in this type of experiment are the relatively low levels of endogenous sumoylated protein and the quality of the antibody used to capture the sumoylated form of protein. In addition to specificity and affinity, the antibody used for IP should be clean with minimal background (high purity) because overexposure of the Western blot is often required to detect the low levels of modified protein species. IP of endogenous Daxx using the rabbit polyclonal Daxx antibody from Sigma Aldrich (Catalog number: D7810) seems to generate high background in our hands, though the Daxx antibody produces clean Western blots. Mouse monoclonal SUMO-1 antibody from Zymed Laboratories Inc. (Catalog number: 33–2400, now Invitrogen) specifically recognizes both the free form of SUMO-1 and the covalently attached SUMO-1 to target proteins with negligible background after IP followed by Western blotting. For this reason, we [20] chose the SUMO-1 antibody for the IP in order to detect endogenous SUMO modification of Daxx by pp71 (Figure 4).
Figure 4. Co-immunoprecipitation assay detects endogenous sumoylation of Daxx.
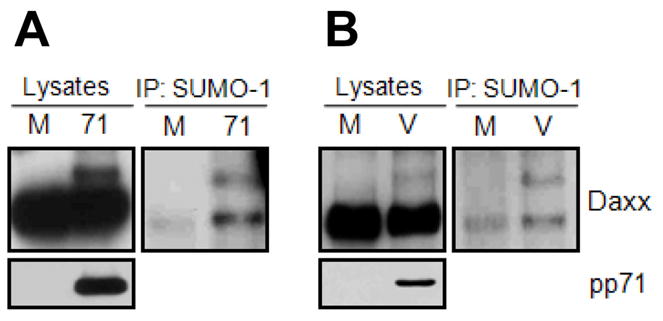
A. HFs were mock transduced (M) or transduced with an rAd that expresses pp71 (71). Lysates were subject to direct Western blotting (WB) or to IP with an anti-SUMO-1 antibody followed by Western blotting with an anti-Daxx antibody. B. HFs were mock infected (M) or infected with HCMV (V). Lysates were subject to direct Western blotting (WB) or to IP with an anti-SUMO-1 antibody followed by Western blotting with an anti-Daxx antibody. Image from reference 20 used with permission.
Protein lysates from HFs infected with HCMV (or transduced with rAd-pp71) were analyzed by IP with a SUMO-1 antibody followed by Western blotting with a Daxx antibody. Lysates from mock-infected HFs were used as a control. When IP analysis was performed using 6 × 106 cells in the SDS denaturing condition as described in section 2.3, the level of IP was observed to be very low, making it hard to detect the sumoylated form of Daxx by Western blot analysis. To improve the IP condition, a modified IP protocol was employed. The major differences were as follows:
Use ~6 × 107 cells for endogenous IP assay (approximately 10 times more cells).
Lyse cells in RIPA lysis buffer (50 mM Tris [pH 8.0], 150 mM NaCl, 5 mM EDTA, 10% glycerol, 0.1% SDS, 1% NP-40, 1% sodium deoxycholate, 20 mM NEM and 5 mM IAA, and protease inhibitors), instead of 1% SDS lysis buffer.
Incubate the antibody (e.g., anti-SUMO-1) with cell lysates overnight at 4°C to increase the formation of the immune complexes.
Wash the resin six times with 1ml of RIPA buffer.
Using the modified protocol, we were able to detect endogenous sumoylated Daxx in HCMV-infected or rAd-pp71-infected cells. What makes the IP level different between the two methods, aside from the increased pre-IP input protein amounts, is not clear. It is possible that the different buffer compositions between SDS and RIPA IP buffers (i.e., differences in SDS or salt concentrations) cause altered affinity of the SUMO-1 antibody. Importantly, using RIPA buffer for the SUMO IP assay may result in the detection of non-covalent interactions in addition to covalent SUMO conjugations. For example, our IP-Western analysis [20] using RIPA buffer reveals a ~120 kDa Daxx band corresponding to the unmodified Daxx in addition to a ~140 kDa band corresponding to sumoylated Daxx (Figure 4). Coimmunoprecipitated unmodified Daxx is detected presumably from interactions with sumoylated proteins that bind to unmodified Daxx through its SIM. Nevertheless, this IP result implies that SUMO-1 can be conjugated to Daxx in physiological conditions, and that this conjugation is mainly detected after HCMV infection. Reciprocal IP/Western blot analysis can be also performed if good quality antibodies are available. The same protocol performed with different antibodies (e.g., anti-SUMO-2) will be applicable in determining which SUMO paralogue preferentially modifies Daxx in HCMV-infected cells.
3. Future directions
Proteomic approaches to identify and map sites of protein modification are becoming more reliable and more common. However, they still must be confirmed by widely accepted protocols such as the ones we present here. The major limitations of these approaches are the lack of quality antibodies, the need for ectopic protein overexpression, and the residual activity of isopeptidase (even in the presence of NEM and IAA) in crude cell lysates. Technologic improvement in these areas will increase the feasibility of these assays. The real future will not be simply in identifying sites of protein modification, but in determining their effects on protein function. This will be especially challenging for protein modifications like Daxx sumoylation, where only a vanishingly small fraction of the total protein is modified.
Research highlights.
Sumoylation of viral and cellular proteins modulates their functions.
Extracting or identifying sumoylated proteins from cellular lysates is challenging.
Enhancing solubility and inhibiting isopeptidases solves most technical difficulties.
Immunoprecipitation/Western blot can be used to detect endogenous sumoylation.
Epitope tagged proteins can increase the sensitivity of detecting sumoylated antigens.
Acknowledgments
This work was supported by NIH grant AI074984 to RFK. RFK is a Burroughs Welcome Fund Investigator in the Pathogenesis of Infectious Disease.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Matunis MJ, Coutavas E, Blobel G. J Cell Biol. 1996;135:1457–70. doi: 10.1083/jcb.135.6.1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mahajan R, Delphin C, Guan T, Gerace L, Melchior F. Cell. 1997;88:97–107. doi: 10.1016/s0092-8674(00)81862-0. [DOI] [PubMed] [Google Scholar]
- 3.Shen Z, Pardington-Purtymun PE, Comeaux JC, Moyzis RK, Chen DJ. Genomics. 1996;36:271–9. doi: 10.1006/geno.1996.0462. [DOI] [PubMed] [Google Scholar]
- 4.Boddy MN, Howe K, Etkin LD, Solomon E, Freemont PS. Oncogene. 1996;13:971–82. [PubMed] [Google Scholar]
- 5.Okura T, Gong L, Kamitani T, Wada T, Okura I, Wei CF, Chang HM, Yeh ET. J Immunol. 1996;157:4277–81. [PubMed] [Google Scholar]
- 6.Geiss-Friedlander R, Melchior F. Nat Rev Mol Cell Biol. 2007;8:947–56. doi: 10.1038/nrm2293. [DOI] [PubMed] [Google Scholar]
- 7.Tanaka K, Nishide J, Okazaki K, Kato H, Niwa O, Nakagawa T, Matsuda H, Kawamukai M, Murakami Y. Mol Cell Biol. 1999;19:8660–72. doi: 10.1128/mcb.19.12.8660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Meluh PB, Koshland D. Mol Biol Cell. 1995;6:793–807. doi: 10.1091/mbc.6.7.793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nacerddine K, Lehembre F, Bhaumik M, Artus J, Cohen-Tannoudji M, Babinet C, Pandolfi PP, Dejean A. Dev Cell. 2005;9:769–79. doi: 10.1016/j.devcel.2005.10.007. [DOI] [PubMed] [Google Scholar]
- 10.Gareau JR, Lima CD. Nat Rev Mol Cell Biol. 2010;11:861–71. doi: 10.1038/nrm3011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bohren KM, Nadkarni V, Song JH, Gabbay KH, Owerbach D. J Biol Chem. 2004;279:27233–8. doi: 10.1074/jbc.M402273200. [DOI] [PubMed] [Google Scholar]
- 12.Ulrich HD. Mol Cell. 2008;32:301–5. doi: 10.1016/j.molcel.2008.10.010. [DOI] [PubMed] [Google Scholar]
- 13.Song J, Durrin LK, Wilkinson TA, Krontiris TG, Chen Y. Proc Natl Acad Sci U S A. 2004;101:14373–8. doi: 10.1073/pnas.0403498101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Minty A, Dumont X, Kaghad M, Caput D. J Biol Chem. 2000;275:36316–23. doi: 10.1074/jbc.M004293200. [DOI] [PubMed] [Google Scholar]
- 15.Lin DY, Huang YS, Jeng JC, Kuo HY, Chang CC, Chao TT, Ho CC, Chen YC, Lin TP, Fang HI, Hung CC, Suen CS, Hwang MJ, Chang KS, Maul GG, Shih HM. Mol Cell. 2006;24:341–54. doi: 10.1016/j.molcel.2006.10.019. [DOI] [PubMed] [Google Scholar]
- 16.Boggio R, Chiocca S. Curr Opin Microbiol. 2006;9:430–6. doi: 10.1016/j.mib.2006.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Boggio R, Colombo R, Hay RT, Draetta GF, Chiocca S. Mol Cell. 2004;16:549–61. doi: 10.1016/j.molcel.2004.11.007. [DOI] [PubMed] [Google Scholar]
- 18.Chang PC, Izumiya Y, Wu CY, Fitzgerald LD, Campbell M, Ellison TJ, Lam KS, Luciw PA, Kung HJ. J Biol Chem. 2010;285:5266–73. doi: 10.1074/jbc.M109.088088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ahn JH, Brignole EJ, 3rd, Hayward GS. Mol Cell Biol. 1998;18:4899–913. doi: 10.1128/mcb.18.8.4899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hwang J, Kalejta RF. J Virol. 2009;83:6591–8. doi: 10.1128/JVI.02639-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim JH, Baek SH. Biochim Biophys Acta. 2009;1792:155–62. doi: 10.1016/j.bbadis.2008.12.008. [DOI] [PubMed] [Google Scholar]
- 22.Werner A, Moutty MC, Moller U, Melchior F. Methods Mol Biol. 2009;497:187–99. doi: 10.1007/978-1-59745-566-4_12. [DOI] [PubMed] [Google Scholar]
- 23.Eloranta JJ, Hurst HC. J Biol Chem. 2002;277:30798–804. doi: 10.1074/jbc.M202780200. [DOI] [PubMed] [Google Scholar]
- 24.Kirazov LP, Venkov LG, Kirazov EP. Anal Biochem. 1993;208:44–8. doi: 10.1006/abio.1993.1006. [DOI] [PubMed] [Google Scholar]
- 25.Laemmli UK. Nature. 1970;227:680–5. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 26.Fido RJ, Tatham AS, Shewry PR. Methods Mol Biol. 1995;49:423–37. doi: 10.1385/0-89603-321-X:423. [DOI] [PubMed] [Google Scholar]
- 27.Bernardi R, Pandolfi PP. Nat Rev Mol Cell Biol. 2007;8:1006–16. doi: 10.1038/nrm2277. [DOI] [PubMed] [Google Scholar]
- 28.Lindsay CR, Morozov VM, Ishov AM. Front Biosci. 2008;13:7132–42. doi: 10.2741/3216. [DOI] [PubMed] [Google Scholar]
- 29.Kingston RE, Chen CA, Okayama H. Curr Protoc Immunol. Unit 10. Chapter 10. 2001. p. 13. [DOI] [PubMed] [Google Scholar]
- 30.Tatham MH, Rodriguez MS, Xirodimas DP, Hay RT. Nat Protoc. 2009;4:1363–71. doi: 10.1038/nprot.2009.128. [DOI] [PubMed] [Google Scholar]
- 31.Wohlschlegel JA. Methods Mol Biol. 2009;497:33–49. doi: 10.1007/978-1-59745-566-4_3. [DOI] [PubMed] [Google Scholar]
- 32.Blomster HA, Hietakangas V, Wu J, Kouvonen P, Hautaniemi S, Sistonen L. Mol Cell Proteomics. 2009;8:1382–90. doi: 10.1074/mcp.M800551-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]