

Abstract
Pediatric brain tumors have always been challenging as well as intriguing in their anatomical, surgical, and postsurgical management-related issues. They are a heterogeneous set of pathologies involving different age groups in childhood and also differ widely from their adult counterparts as far as adjuvant therapies are concerned. Though neurosurgeons across the world are radical in surgery for most of the pediatric tumors, it can often be at the cost of future quality of life in suprasellar tumors. As the time has gone by, the pendulum has swung toward rather conservative and maximal safe surgical resections with adjuvant therapies coming to the forefront. Hence, the aim is to achieve a good quality of life for these children along with a control of tumor growth (rather than cure) and to again tackle the tumors, if required, once these children reach adolescence or adulthood. We have reviewed the literature for different pediatric suprasellar tumors and discussed their current management giving our perspective with illustrative cases.
Keywords: Chemotherapy, pediatric brain tumors, radiotherapy, suprasellar
Introduction
Tumors of central nervous system (CNS) are the most common childhood malignancy and leading cause of cancer related morbidity and mortality with incidence of 2.5–4 per 100,000 children.[1] Pediatric sellar and suprasellar tumors form a separate entity as far as incidence, histology and responsiveness to therapy is concerned. They also differ in their surgical approaches and their adjuvant treatment from the adults. While primary surgical management is the key in most of the adult tumors, chemo- and radiotherapy are the chief modality in some of these pediatric tumors like germinomas. Therefore, the epidemiology of these tumors is different and requires a dedicated multidisciplinary team approach. We have reviewed the literature and tried to focus on common tumors in this region in children and their diagnosis and management related issues.
These pediatric suprasellar tumors may be classified differently.
Grossly, they can be classified as follows:
Sellar tumors: Pituitary adenoma (0.5–2.5%), arachnoid cyst;
Sellar–suprasellar tumors and cysts: Craniopharyngiomas (6–9%), Rathke's cleft cyst, arachnoid cysts;
Suprasellar tumors: Chiasmatic/hypothalamic glioma (4–8%), arachnoid cyst;
Lesions of the stalk: Germ cell tumors (1–2%), histiocytosis X;
Miscellaneous lesions: Hypothalamic hamartoma, metastatic lesions, PNET.
Pituitary adenomas
Pituitary adenomas are commonly seen tumors in adults while they rarely present in children and are often detected in adolescents.[2] They comprise about 3% of all supratentorial tumors in children.[3] Prolactinomas are most common in children followed by ACTH (Adrenal Corticotrophin Hormone) and HGH (human growth hormone) secreting tumors.
Clinical presentation and preoperative work up
Nonfunctioning tumors are rare in childhood and children mainly present with endocrinological symptoms. Pituitary hyperplasia in growing age can rarely become symptomatic with headaches and/or visual symptoms. This may occasionally be associated with hypothyroidism and needs to be carefully evaluated and medically treated with good results.
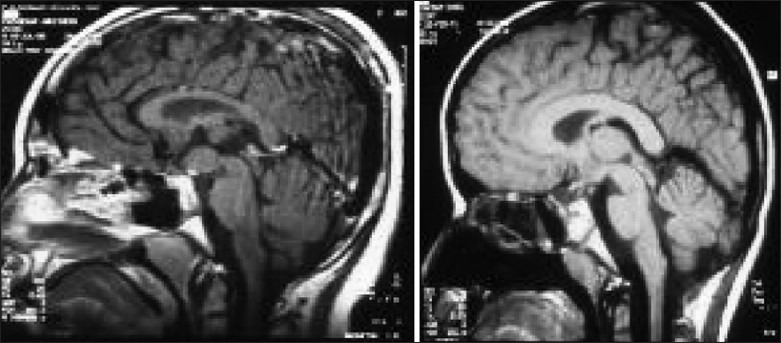
Illustrative case 1: A 16-year-old girl presented with headaches and diminution of vision. Bitemporal hemianopia was observed. Hormonal assay suggested hypothyroidism. Pituitary hyperplasia with optic compression was seen on MRI [Figure 1].
Figure 1.
Pituitary hyperplasia in a growing child responding to thyroid supplements
Treatment with thyroid supplements resulted in relief from headaches and reversal of visual compromise and a scan at 6 weeks showed normal pituitary size.
Prolactinomas are the most common tumors and present with primary or secondary amenorrhoea and galactorrhoea. This may go unnoticed in many. Prolactin (PRL) levels will establish the diagnosis of prolactinoma. Children present with gigantism as opposed to acromegaly in adults although adolescents may have both. Insulin growth factor-I (IGF-I) and serum GH levels will establish the diagnosis of a GH secreting adenoma. Cushing's disease presents similarly as in adults and thorough hormonal work-up remains the key to diagnosis. Serum cortisol, ACTH levels and 24-hour urinary cortisol estimation should be done for Cushing's disease. Failure of the low-dose dexamethasone test to suppress the cortisol secretion is more suggestive. Contrast MRI clinches the diagnosis and localizes the lesion usually, but rarely inferior petrosal sinus sampling will be necessary to diagnose and localize, after imaging fails to diagnose the microadenoma. A 2:1 lateralization is suggestive as in adults.
Management strategy and surgical approaches
The management is similar in the pediatric population as for adults.[4] Cabergoline and Bromocriptine are the drugs of choice for pediatric prolactinoma as in adults. Surgery is indicated in rare cases of rapidly deteriorating vision, CSF leaks or gross hydrocephalus. Trans-sphenoidal resection should be done for ACTH and GH secreting adenomas as initial remission rates up to 60–80% are seen. All signs of hypercortisolism and excess GH secretion usually revert back and significant clinical improvement is usually seen. Nonfunctioning tumors need to be excised for their mass effect and usually visual recovery is expected. Radiotherapy is considered for multiple recurrences and usually avoided in children.
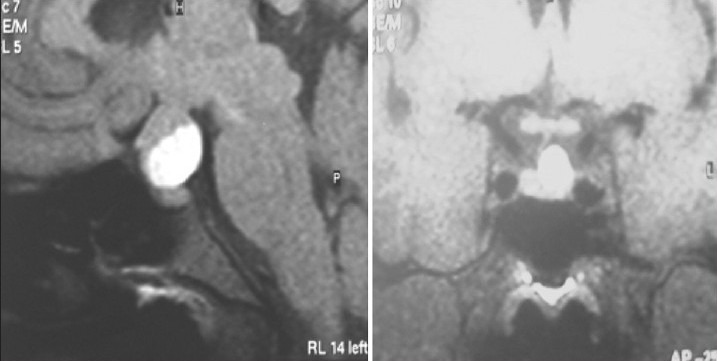
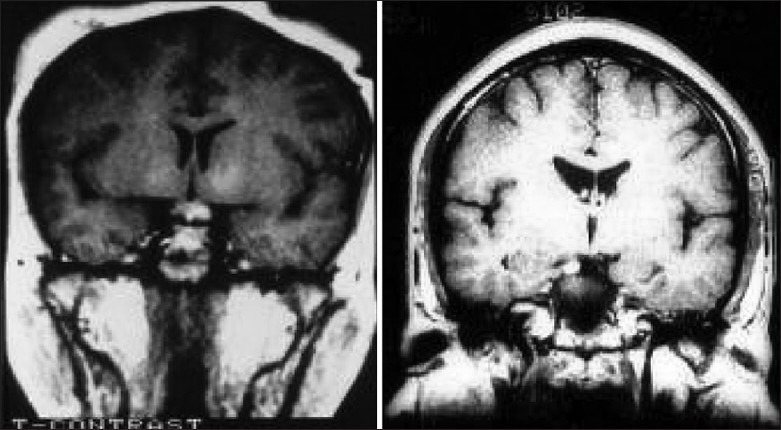
Illustrative case 2: Cushing's disease: A 16-year-old male patient presented with symptoms of increased weight and striae over abdomen. He had signs suggestive of Cushing's disease with increased serum ACTH and serum cortisol levels. Two adenomas were seen on MRI. Transsphenoidal surgery for excision of both adenomas resulted in clinical recovery as well as return of hormone levels to normal [Figure 2a and b].
Figure 2.
(a) MRI with double micro adenomas (noncontrast enhancing). (b) Postoperative MRI after complete excision of both adenomas
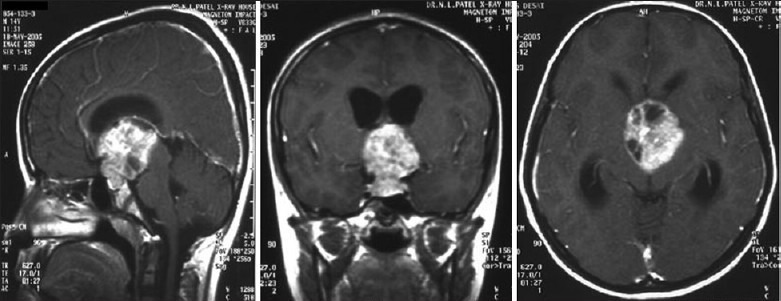
Illustrative case 3- acromegaly: A 16-year-old male presented with a rapid increase in height with intermittent headaches for 8 months. On examination he was found to have features suggestive of gigantism with no visual, motor, and sensory deficit. His hormonal profile was GH level 106 ng/ml, S.IGF-1 level 1113 ng/ml, and s. Prolactin 70.5 ng/ml [Figure 3a and b].
Figure 3a.
Contrast MR images showing a pituitary adenoma
Figure 3b.
Post-op contrast sagittal and coronal MRI images at 3-month follow-up, GH level 3.5ng/ml
Transsphenoidal excision of the adenoma resulted in normalization of hormonal levels and relief from headaches.
Hypophysitis is a rare entity found in children in the suprasellar area. Hypophysitis usually presents with hypocortisolism and hypothyroidism; this is managed with steroids and hormonal supplementation.
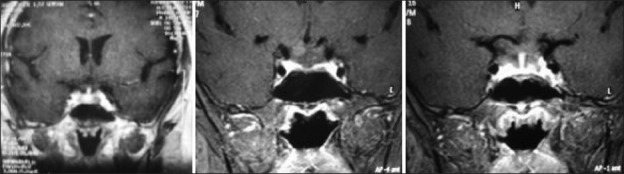
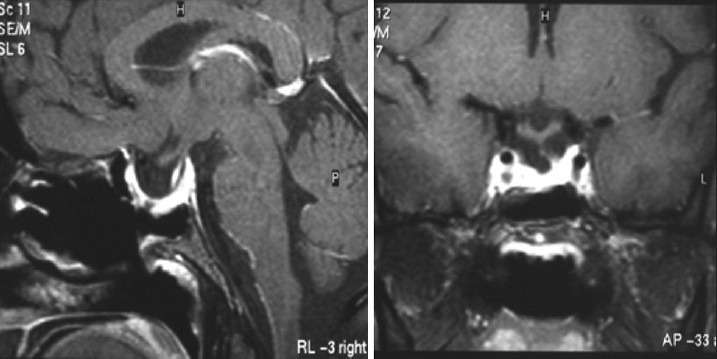
Illustrative case 4: Hypophysitis – We had a case of a 17-year-old female with hypopituitarism with enlarged stalk suggestive of lymphoid hypophysitis [Figure 4a and b].
Figure 4.
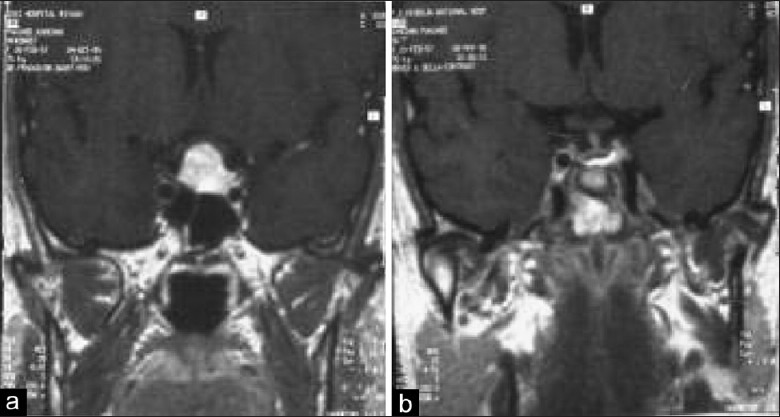
(a and b) Preoperative MRI of lymphoid hypophysitis
Other sellar tumors
Arachnoid cysts and Rathke's cleft cysts may rarely occur in the suprasellar region in children. Arachnoid cysts may present with visual symptoms, hydrocephalus, macrocephaly, pyramidal tract signs, and endocrinopathies (precocious puberty). Bobble head doll syndrome is usually rare.
Two types may be seen:
Suprasellar arachnoid cyst.
Sellar (Infra diaphragmatic) arachnoid cyst.
Suprasellar cysts are usually diverticula from Lilequist's membrane and may present with precocious puberty, gelastic seizures, and hydrocephalous in children; sellar cysts present at a later age, with visual symptoms and hypopituitarism and are usually infradiaphragmatic.
For suprasellar arachnoid cysts, various treatment options include cysto-peritoneal shunting or percutaneous ventriculocystostomy. We have been practicing Endoscopic double fenestration for these kinds of cysts which help in avoiding shunts completely. Lastly, subfrontal or pterional craniotomies have been done by some in the past; the less invasive modalities have made those approaches a rare practice today.
Illustrative case 5: Adolescent boy presenting with hydrocephalic symptoms [Figure 5a and b].
Figure 5a.
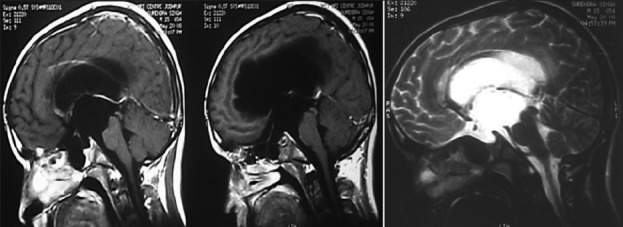
MRI scan of suprasellar arachnoid cyst
Figure 5b.
Postoperative MR scan after double fenestration with good flow across the stoma and resolution of hydrocephalous and clinical symptoms
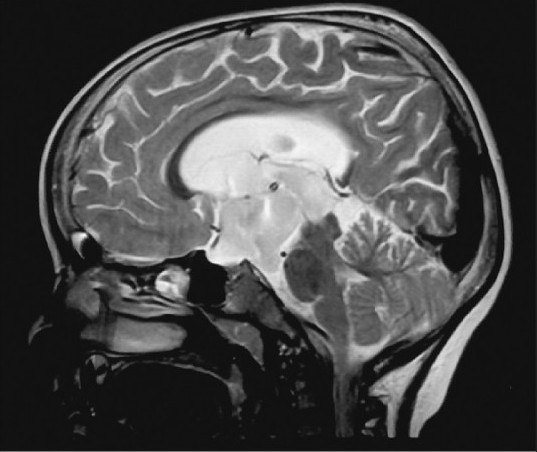
Illustrative case 6: Young lady presenting with progressive visual deficit and hypopituitarism [Figure 6a and b].
Figure 6a.
Infra-diaphragmatic arachnoid cyst
Figure 6b.
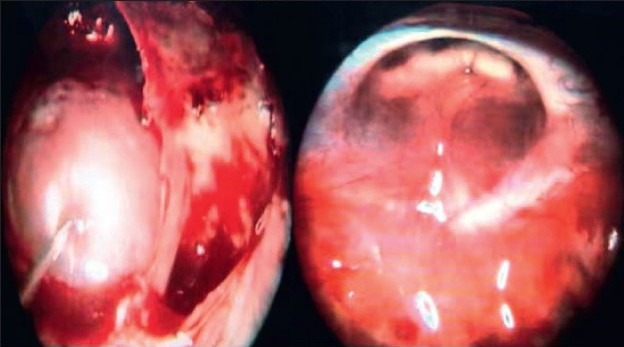
Endoscopic trans-sphenoidal approach for repair of diaphragmatic defect: Intraoperative images
Figure 6c.
Postoperative images showing collapsed cyst
Rathke's cleft cyst is the most common developmental sellar, suprasellar pathology but rarely presents in childhood. Rathke's cleft cysts are epithelium (cuboidal or columnar) lined cystic benign collections thought to arise embryologically from Rathkes cleft, resemble craniopharyngiomas on imaging but usually do not contain calcium. Like other lesions, they can also present with mass effect on surrounding neural structures and endocrinopathies. MRI shows a hypointense lesion on T1 and hyperintense lesion on T2-weighted images primarily within sella resulting in its widening. Ophthalmological and endocrinological work up is mandatory and only after this, patients undergo trans-sphenoidal excision of the cyst wall and drainage of the protein rich fluid. Visual symptoms reverse quickly while endocrinopathies may/may not reverse.
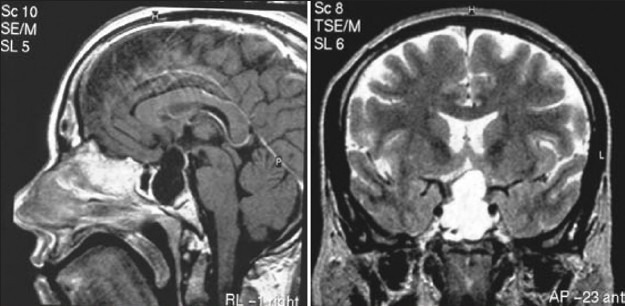
Illustrative case 7: Rathke's cleft cyst [Figure 7a and b].
Figure 7a.
MR Images showing hyperintense cyst along the stalk
Figure 7b.
MR Images after transsphenoidal drainage and partial excision of cyst
Craniopharyngioma
Craniopharyngiomas are the most common tumors arising from the pituitary region apart from pituitary adenomas.[5] These comprise about 6% of all expanding lesions in childhood.[6] These tumors have a bimodal age of presentation, firstly in the 1st or 2nd decade and again in the 5th decade. These tumors have been hypothesized to arise from ectoblastic remnants of Rathke's duct and therefore are commonly found along the path of development of Rathke's pouch from pharynx to the floor of sella as well as above and within the sella.[7] These tumors are partly solid and cystic and can have dense areas of calcification. The cystic fluid is typically like “machine oil.” The microscopic appearance has an external layer of columnar epithelium and a central network of epithelial cells. Keratin deposition is also seen in the cellular stroma. Craniopharyngiomas spark intense reaction from the adjacent brain due to their papillary projections, especially around hypothalamus. Even adherence to arteries is encountered although it is uncommon.
Clinical presentation and preoperative work up
They often present with features of raised intracranial tension (ICT) and endocrine dysfunction (delayed puberty, short stature) in children apart from visual impairment which is more common presentation in adults. Endocrine dysfunction is seen in 80% patients and growth hormone (GH) deficiency is the most commonly found (75% cases).[8] Diabetes insipidus is a relatively uncommon presentation and hyperprolactinemia is found in few due to the stalk effect. Computed tomography (CT) and magnetic resonance Imaging (MRI) are the investigation modalities of choice with coronal and sagittal reconstructions emphasizing on certain key points like solid and cystic areas of tumor, displacement of neurovascular structures and the degree of hydrocephalus; useful also to see the pneumatization of the sphenoid sinus, pre- or postfixed chiasm. High protein content can be visualized as hyperintense signals on T1 weighted images. Similarly fluid attenuated inversion recovery (FLAIR) sequences can help detecting the cystic portions more effectively.
Ophthalmological and endocrinological assessments must be made in detail and especially hypothyroidism and reduced cortisol levels need correction preoperatively.
Management and surgical approaches
The pendulum of management of craniopharyngiomas has once again swung from “radical resections” to “safe resections.” The enthusiasm of radical excision in children at the cost of quality of life issues has been replaced by more logical near-total or subtotal safe resections followed by adjuvant therapy in the form of radiotherapy once the child grows.[9–10] The residual/recurrence can be observed till radiotherapy can be given to them (hyper-fractionated with dose per fraction kept below 2 Gy or radiosurgery). Other adjuvant therapies including intracystic bleomycin, interferon or radioactive yttrium, phosphorus-32 have also emerged as viable options for retreating cystic recurrences. Preoperative shunts may be done if hydrocephalus is causing significant acute symptoms.
Surgical approaches may be
Transsphenoidal (microsurgical or endoscopic).
Transcranial.
Combined endonasal and cranial.
Endoscopic intraventricular.
Stereotactic aspiration and injections.
Pure endonasal endoscopic excision should be attempted in those with smaller chiefly “intrasellar” tumors or with those having large sella with suprasellar extensions (type II, III). Calcified chunks within the tumor and relation with nearby vascular structures must be studied thoroughly before planning any approach.
In comparison, trans-cranial approaches are best suitable for tumors with extensive suprasellar components which are multi-lobulated and more solid with little or no sellar component, nonenlargement of sella or nonpneumatized sphenoid. Pterional approaches are commonly used with following corridors to the suprasellar region (depending on the pre- or postfixation of optic chiasm):
A. Subchiasmatic, B. Optico-carotid, C. Trans-lamina terminalis and D. Occulomotor-carotid.
We now have experience with several cases where children have presented with features of raised ICT due to large suprasellar (type III-ventricular) cystic component obstructing the foramen of Monroe and the solid part remaining intrasellar. In these cases, burr hole endoscopic fenestration and placement of Ommaya reservoir as first stage to reduce ICT followed by endonasal endoscopic excision of the solid component as second stage surgery have been a rewarding option.
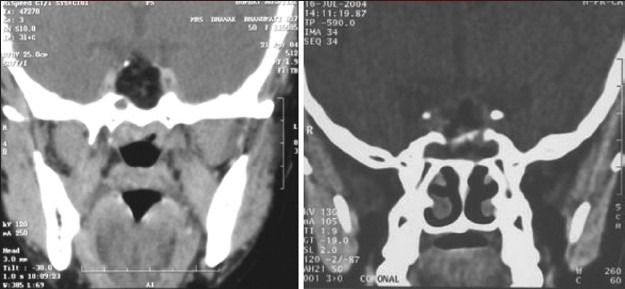
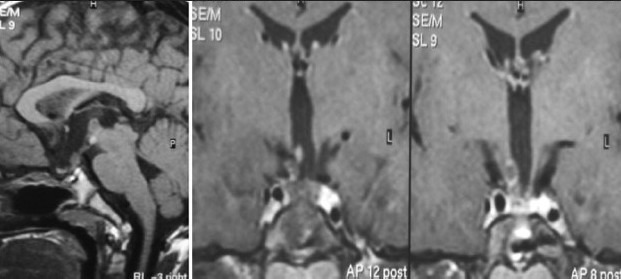
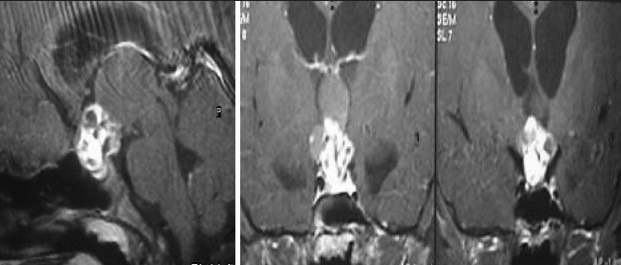
Illustrative case 8: A 6-year-old male came with headache and vomiting for 15 days. On examination, no focal neurological deficit was observed with a normal visual examination. Hormonal evaluation showed only hypothyroidism. He underwent endoscopic fenestration and insertion of reservoir as an emergency procedure on the same day. Three days later, he underwent transnasal endoscopic extended transsphenoidal surgery for near-total excision of the tumor [Figure 8a–c].
Figure 8a.
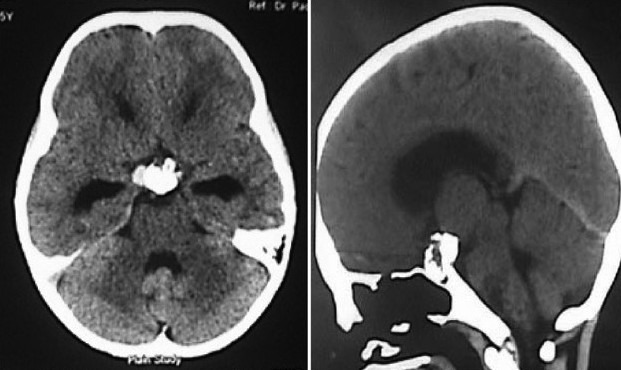
CT scan axial and sagittal images showing calcification
Figure 8c.
Postoperative MRI follow-up at 1 year shows a small nongrowing residue. The child needed thyroid and cortisol supplements
Figure 8b.
MR axial T1 and contrast with coronal images showing solid cystic tumor
Postoperatively, these children should be monitored closely for endocrinological dysfunctions (if not present earlier) due to damage to the stalk. Cortisol replacement is crucial in the immediate postoperative period. These patients are at a high risk for developing diabetes insipidus due to antidiuretic hormone (ADH) deficiency and measuring urine and serum osmolality, electrolytes, urine-specific gravity and bodyweight on daily basis is essential. Those with accidental stalk sectioning will require long-term ADH replacements. We have found the Desmopressin nasal spray quite effective as far as compliance is concerned.
Radiotherapy as adjuvant therapy must be used with caution as it may result in pan-hypopituitarism later in life or may lead to radiation necrosis, optic neuritis, and dementia.[11] Intracystic therapies may also be useful in certain cases where an Ommaya reservoir can be placed stereotactically or with navigation guidance.
Chiasmatic-hypothalamic gliomas
Optic pathway/chiasmatic gliomas comprise 4–8% of childhood intracranial tumors occurring most commonly in the first decade.[12–13] They affect 20–30% of children with neurofibromatosis-1 (NF-1).[14] NF-1-associated tumors are bilateral and multifocal and can involve anything between optic nerve to visual cortex.[15] Chiasmatic gliomas progress rapidly and involve the hypothalamus rapidly. Almost 25% optic pathway gliomas are confined to optic disK and nerve whereas 40-75% involve the chiasm. Of those involving chiasm, 33–60% also involve hypothalamus[16–18] (presenting as diencephalic syndrome). Dutton et al., in a meta-analysis of optic nerve gliomas, have emphasized that these patients with hypothalamic gliomas are common in young age (<1 year).[16] They occur equally in males and females and rarely can occur in adults.
Clinical presentation and preoperative work up
The child usually presents with painless proptosis with some degree of visual impairment and spasmus nutans. Precocious puberty and diencephalic syndrome may be one form of presentation. On fundoscopy, optic pallor or disk oedema may be encountered. Visual acuity might be reduced. They can also present as hypothalamic mass causing obstructive hydrocephalus at the level of foramen of Monroe. CT (with bone windows) forms an important diagnostic modality along with fat suppressed contrast MRI images of the orbit and suprasellar region.[19] A classic J-shaped sella turcica and enlarged optic canals can be seen on plain X-rays. Histologically they are low-grade gliomas[20] (pilocytic astrocytoma/fibrillary astrocytoma/pilomyxoid astrocytoma).
Management strategy and surgical approaches
Astrup advocated observation for newly diagnosed OPG, resection for progressive intraorbital tumors, radiotherapy for progressive chiasmatic tumors in older children, and chemotherapy in children younger than 5 years.[21] The role of surgery is very limited in this subset of tumors. Surgical excision can be only achieved in unilateral tumors with no visual acuity. Therefore, contemporary indications for surgery include single nerve involvement causing progressive, disfiguring proptosis, blindness, or both, or exophytic chiasm tumors causing mass effect or hydrocephalus. Chemotherapy is the chief modality of treatment in children younger than 5 years. Concurrent carboplatin and vincristine in a 10-week induction phase, followed by 48 weeks of maintenance carboplatin/vincristine, is the protocol given by Packer et al.[22,23] Temozolamide can also be used as a second line therapy.[24] Older children may receive radiotherapy (5000 rads) in addition to above.
Surgical approaches used are
transfrontal–transorbital for optic nerve gliomas and
pterional for chiasmatic/hypothalamic gliomas.
There is clear consensus on the surgical excision of solitary optic nerve glioma presenting with visual loss/proptosis. Tumors with neurofibromatosis usually do not progress and are managed conservatively. Whether to go for complete excision of chiasmatic gliomas (leading to complete blindness) to relieve mass effect and hydrocephalus or to biopsy followed by radiotherapy/chemotherapy (as per the age of the child) is still controversial. Optico chiasmatic gliomas in the suprasellar area are common in infants presenting with visual deficits; the commonest histopatholgy being pilocytic astrocytoma followed by diffuse astrocytoma.
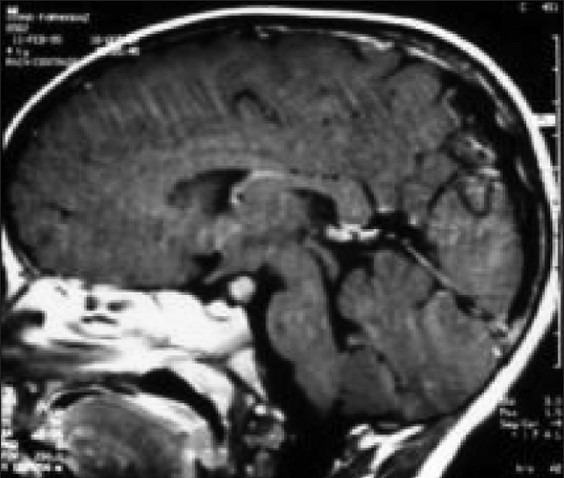
Illustrative case 9: 1 and 1/2 years old child presented with difficult;ty in walking. On examination, no focal neurological deficits found. Developemental history was normal [Figure 9a and b].
Figure 9a.
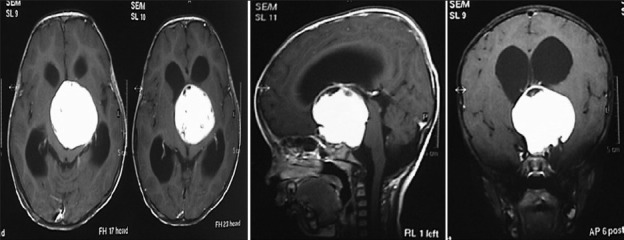
MR contrast axial, sagittal and coronal image with large hypothalamic tumor
Figure 9b.
MR follow-up at 3 years after radical excision of this pilocytic astrocytoma with good recovery
Illustrative case 10: Young child with neurofibromatosis and mild proptosis. No tumor progression observed over 4 years [Figure 10].
Figure 10.
Optic nerve glioma with neurofibromatosis, stable over 4 years
Illustrative case 11: A 10-year-old male presented with diminution of vision for several months. On examination vision in the right eye was hand movements and left eye vision was 3 feet. His MRI scan suggested an optico-chiasmatic pilocytic astrocytoma. Biopsy followed by chemotherapy was performed [Figure 11].
Figure 11.
Preoperative MR scan with cortical dysplastic changes with an optico-chiasmal lesion (pilocytic astrocytoma)
Miscellaneous lesions
Hypothalamic hamartomas
Hypothalamic hamartomas are usually small and asymptomatic presenting in the first two decades.
Rarely, they increase in size and present with symptoms due to mass effect on nearby neural structures and due to endocrinopathies. Le Marquand and Russell described this entity for the first time as normal brain tissue (mixture of normal neurons and glial cells) presenting at an abnormal site.[25] A child may present with hyperactivity syndrome with hyperphagia, behavioural disorders, gelastic seizures, autonomic dysfunction, somnolence, and precocious puberty. Pedunculated hamartomas usually present with precocious puberty which can be managed by GnRH analog. Sessile hamartomas present with gelastic seizures. MRI reveals a lesion which is isointense on T1 and hyperintense on T2 weighted images with poor contrast enhancement and an epicentre in the hypothalamic region (tuber cinereum) with downward extension into the sella.
Medroxyprogesterone acetate is generally prescribed although surgery may also be helpful especially for patients with precocious puberty and gelastic seizures.
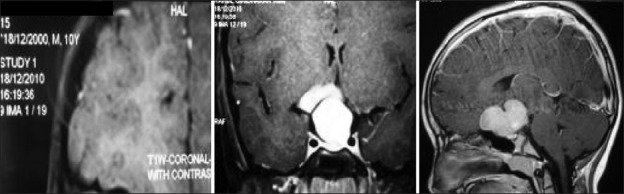
Illustrative case 12: Exophytic hypothalamic hamartoma presenting with precocious puberty. This child was managed conservatively with GnRH analog [Figure 12].
Figure 12.
Pedunculated hypothalamic hamartoma
Illustrative case 13: An 8-year-old boy presenting with precocious puberty, gelastic seizures, and behavioural problems [Figure 13].
Figure 13.
MR images of sessile hamartoma, completely excised with by surgery with improved behavior
Lesions of the infundibulum
Germinoma
Hyphophysitis
Histiocytosis X
Germinomas are tumors of germ cell derivation commonly seen in children and adolescents. They represent less than 1% of all intracranial neoplasms.[26] The pineal region is the most commonly involved site although suprasellar, posterior third ventricular, interpeduncular regions may also be affected. Rare occurrences may also be seen in lateral ventricles, basal ganglia or the posterior fossa. Suprasellar germinomas constitute approximately 20% of all the germ cell tumors.[27]
Types of suprasellar germinomas
There are basically three variants:
Type I: Arising from the pineal region primarily and metastasizing to third ventricular floor, hypothalamus, pituitary stalk, and visual apparatus.
Type II: Same as type I, but the origin is from suprasellar region primarily.
Type III: Develops more inferiorly along the hypothalamo-neurohypophysial axis, optic apparatus, stalk, and third ventricular floor. These are mainly intrasellar and displace the pituitary gland.
Clinical presentation and preoperative work up
Kageyama and Belsky described the classical triad of diabetes insipidus (DI), pan-hypopituitarism and visual disturbances for suprasellar germinomas. DI is a striking presentation of suprasellar germinomas. It is seen in almost all the patients and is a key point in clinching the diagnosis. This happens due to the invasion of the neurohypophysis. Visual diminution and bitemporal hemianopsia is seen in 85–90% patients. They also present with endocrinopathies (hypopituitarism or precocious puberty); features of raised ICT may be present due to hydrocephalus. Therefore, hormonal assays form an integral part of investigations for suprasellar germinomas. Males are commonly affected with pineal region germinomas while no such predilection exists for suprasellar ones.
On imaging, they are hypointense on T1-weighted images, hyperintense on T2-weighted images, and show extremely intense contrast enhancement, and hence, gadolinium-enhanced brain and whole-spine MRI form the imaging of choice. Ectopic deposits may be found anywhere in the ventricular system with metastasis seen in up to 10-57% cases. CSF (cerebrospinal fluid) and serum tumor markers (placental alkaline phosphatase: PLAP, alfa-feto protein: AFP, beta human chorionic gonadotropin: B-HCG) must be done. Histological confirmation remains essential however. Histologically, they are composed of sheets of polygonal cells in lymphocyte rich connective tissue stroma. They can be pure germinomas or may have mixed histologic features of endodermal sinus tumor, choriocarcinoma or teratomas.
Management strategy and surgical approaches
These suprasellar germinomas can create diagnostic dilemmas on imaging. They can be easily confused with optic/chiasmatic gliomas on MRI and if the serum and CSF studies for tumor markers are also negative, surgical exploration for biopsy, and histological confirmation helps differentiate. Patients with hydrocephalus may also need ventriculo-peritoneal shunts before definitive surgery. Today endoscopic third ventriculostomy (ETV) has emerged as a safe and viable option for noncommunicating hydrocephalus in children. Simultaneous ETV for triventricular hydrocephalus and biopsy from the floor or posterior third ventricular region may be performed in these cases. Otherwise, navigation or stereotactic biopsy may be done to achieve the same goal if there is no associated hydrocephalous. CSF dissemination is a potential risk and deposits may be found anywhere along the cranio-spinal subarachnoid space; here cytological examination for malignant cells is helpful. Radical excision is not possible as they are diffusely infiltrating and also, need not be attempted as they are extremely radio and chemosensitive.[28] Etoposide- and Cisplatin-based therapy with cranio-spinal radiation (if malignant cytology, ectopic deposits or more than one intracranial site involvement is detected) forms the basis of treatment.[29] The total dose should be given at the rate of 150–200 rad/day over 5–6 weeks (a total of 5000 rads). Long-term survival can be up to 90% after 10 years of follow-up. However, nongerminomatous tumors fare poorly, and hence, cranio-spinal irradiation should be given in all the cases.
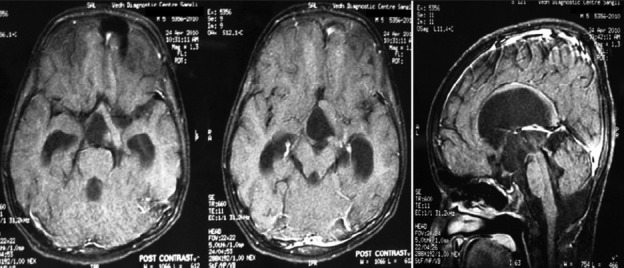
Illustrative case 14: A 14-year-old boy presented with complaints of bilateral diminution of vision (left>right) for 3 months with complaints of polyuria and polydipsia for 2 years. On examination visual acuity was 2 feet on the right side and 3 feet on the left side with fundoscopy showing bilateral optic atrophy. The right pupil showed relative afferent pupillary defect. Serum tumor markers were normal and hormonal profile showed hypopituitarism [Figure 14].
Figure 14a.
MR images sagittal, coronal and axial showing suprasellar germinoma
Figure 14b.
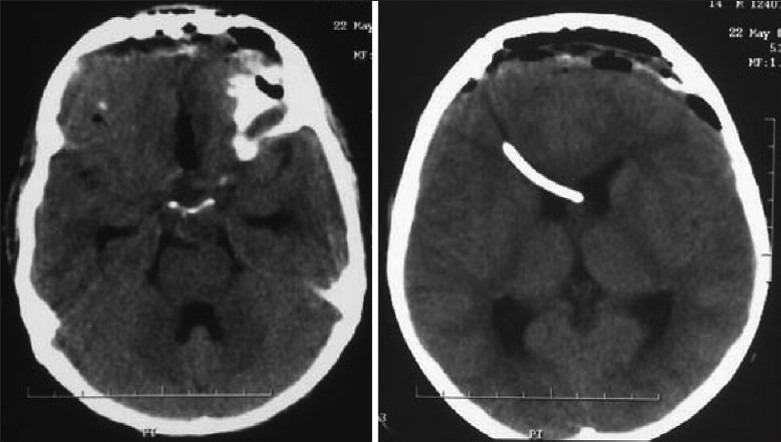
Postexcision CT scan shows subtotal excision. Child had chemoradiation and is tumor free and doing well after 7 years with hormone supplementation
Miscellaneous lesion of the suprasellar area
Suprasellar chordoma
Medulloblastoma metastasis
Clival meningioma
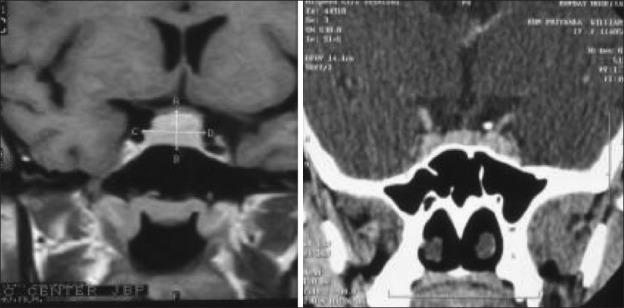
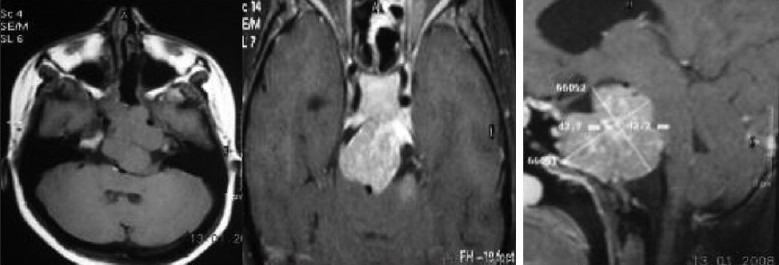
Chordomas are rare tumors found in the suprasellar region in children Illustrative case 15 [Figure 15]. We had a 16-year-old patient who presented with isolated left 6th nerve palsy with no other neurological deficit. The images showed chordoma involving petrous apex extending into suprasellar and cavernous sinus.
Figure 15.
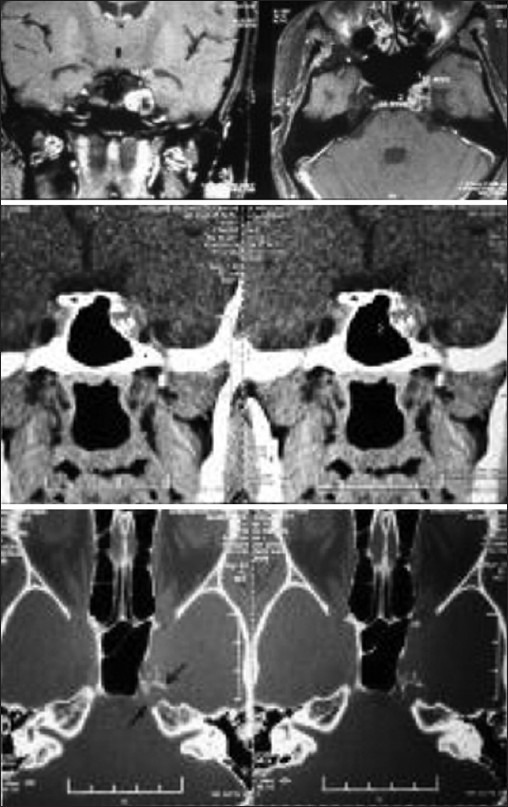
MR and CT images showing heterogeneously enhancing chordoma with erosion of the clinoid process
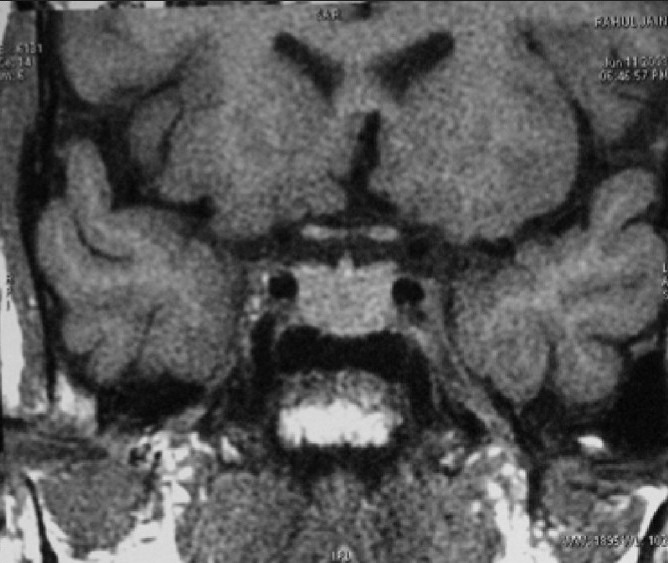
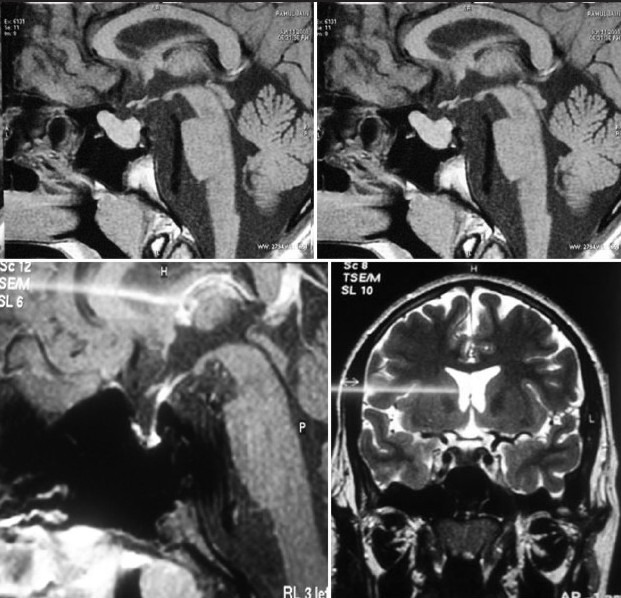
Clival meningiomas with suprasellar extension are rare in children Illustrative case 16 [Figure 16a–c] We had a case of a 17-year-old female who presented with headache with intermittent vomiting and diplopia. On examination, she had 6th nerve palsy with papilledema.
Figure 16a.
Preoperative MR images
Figure 16c.
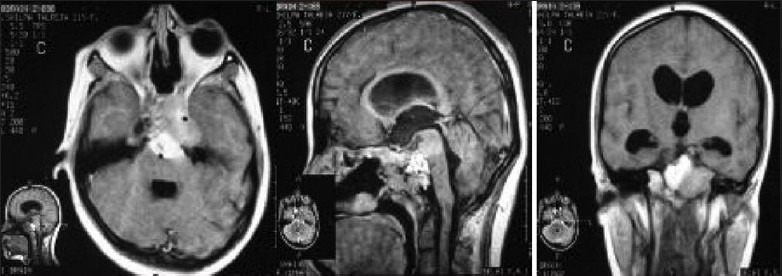
Postoperative MR images
Figure 16b.
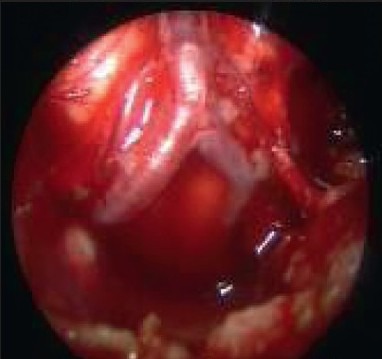
Intraoperative image showing the V-B junction through extended endoscopic skull-base approach
Primitive neuroectodermal tumor (PNET) are midline tumors in children around the age of 1 year. Commonly they are found in posterior fossa infratentorially, but occasionally may occur in the paraventricular or suprasellar region.
Illustrative case 17: A 7-year-old patient presenting with intermittent headache with visual diminution. MRI shows homogenously enhancing lesion in the midline in the suprasellar area [Figure 17].
Figure 17.
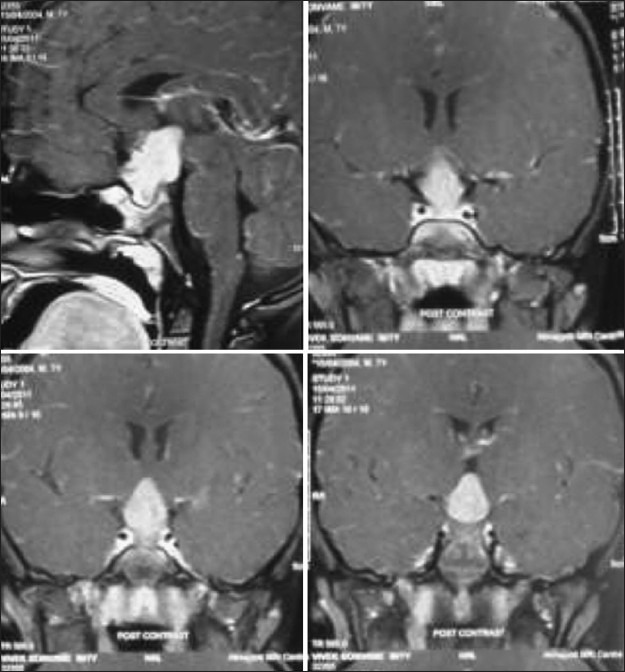
MRI images of suprasellar PNET
Craniotomy for optic decompression and biopsy was carried out with confirmation of PNET. Chemotherapy was instituted with remission of the disease.
Metastasis can occur in the suprasellar area usually through seeding of medulloblastomas, ependymomas or germinomas.
Illustrative case 18: Operated case of medulloblastoma, showed metastasis in suprasellar region on followup imaging. Ths responded well to craniospinal irradiation and chemotherapy [Figure 18a and b].
Figure 18a.
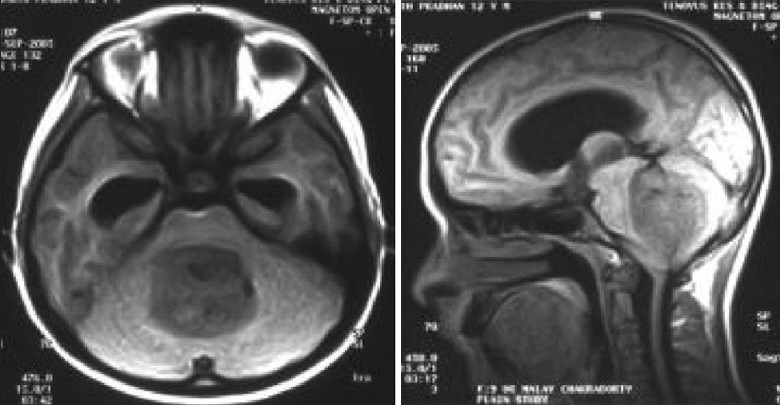
MR images of medulloblastoma
Figure 18b.
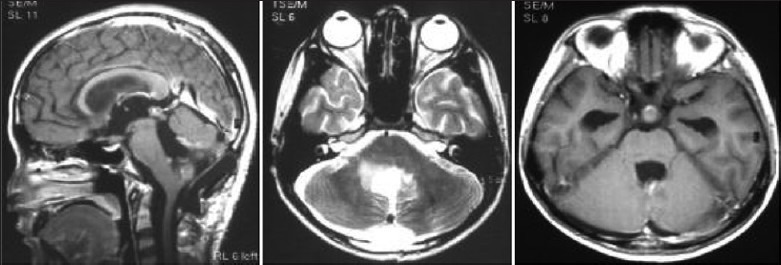
Follow-up MR images after medulloblastoma excision show suprasellar nodule
Summary
Pediatric suprasellar tumors are a separate entity as far as pathologies are concerned. Unlike meningiomas and pituitary adenomas in adults, Craniopharyngiomas, optic gliomas, and Germinomas form the main lesions in the region. While “safe” surgery remains the treatment of choice for Craniopharyngiomas, chemo-radiotherapy after biopsy may be the way to go for Germinomas and diffuse optico-chiasmatic gliomas. Radical surgery for some of these exophytic lesions can be rewarding. Thorough preoperative work-up including hormonal and tumor marker assays and detailed radiological assessment will help in clinching the diagnosis and rational treatment approaches.
Footnotes
Source of Support: Nil.
Conflict of Interest: None declared.
References
- 1.Packer RJ. Brain tumors in children. Arch Neurol. 1999;56:421–5. doi: 10.1001/archneur.56.4.421. [DOI] [PubMed] [Google Scholar]
- 2.Colao A, Loche S, Cappa M, Di Sarno A, Landi ML, Sarnacchiaro F, et al. Prolactinomas in children and adolescents. Clinical presentation and long-term follow-up. J Clin Endocrinol Metab. 1998;83:2777–80. doi: 10.1210/jcem.83.8.5001. [DOI] [PubMed] [Google Scholar]
- 3.Blackwell RE, Younger BJ. Long-term medical therapy and follow up of pediatric adolescent patients with prolactin-secreting macroadenomas. Fertil Steril. 1986;45:713–6. doi: 10.1016/s0015-0282(16)49346-5. [DOI] [PubMed] [Google Scholar]
- 4.Jagannathan J, Dumont AS, Jane JA, Jr, Laws ER., Jr Pediatric sellar tumors: Diagnostic procedures and management. Neurosurg Focus. 2005;18:1–5. [PubMed] [Google Scholar]
- 5.Bunin GR, Surawicz TS, Witman PA, Preston-Martin S, Davis F, Bruner JM. The descriptive epidemiology of craniopharyngioma. J Neurosurg. 1998;89:547–51. doi: 10.3171/jns.1998.89.4.0547. [DOI] [PubMed] [Google Scholar]
- 6.Lafferty AR, Chrousos GP. Pituitary tumors in children and adolescents. J Clin Endocrinol Metab. 1999;84:4317–23. doi: 10.1210/jcem.84.12.6215. [DOI] [PubMed] [Google Scholar]
- 7.Podoshin L, Rolan L, Altman MM, Peyser E. “Pharyngeal” craniopharyngioma. J Laryngol Otol. 1970;84:93–9. doi: 10.1017/s0022215100071681. [DOI] [PubMed] [Google Scholar]
- 8.Sklar CA. Craniopharyngioma: Endocrine abnormalities at presentation. Pediatr Neurosurg. 1994;21(Suppl):S18–20. doi: 10.1159/000120856. [DOI] [PubMed] [Google Scholar]
- 9.Hayward R. The present and future management of childhood craniopharyngioma. Childs Nerv Syst. 1999;15:764–76. doi: 10.1007/s003810050468. [DOI] [PubMed] [Google Scholar]
- 10.Hetelekidis S, Barnes PD, Tao ML, Fischer EG, Schneider L, Scott RM, et al. 20-year experience in childhood craniopharyngioma. Int J Radiat Oncol Biol Phys. 1993;27:189–95. doi: 10.1016/0360-3016(93)90227-m. [DOI] [PubMed] [Google Scholar]
- 11.Fahlbusch R, Honegger J, Paulus W, Huk W, Buchfelder M. Surgical treatment of craniopharyngiomas: Experience with 168 patients. J Neurosurg. 1999;90:237–50. doi: 10.3171/jns.1999.90.2.0237. [DOI] [PubMed] [Google Scholar]
- 12.Combs SE, Schulz-Ertner D, Moschos D, Thilmann C, Huber PE, Debus J. Fractionated stereotactic radiotherapy of optic pathway gliomas: Tolerance and long-term outcome. Int J Radiat Oncol Biol Phys. 2005;62:814–9. doi: 10.1016/j.ijrobp.2004.12.081. [DOI] [PubMed] [Google Scholar]
- 13.Czyzyk E, Józwiak S, Roszkowski M, Schwartz RA. Optic pathway gliomas in children with and without neurofibromatosis 1. J Child Neurol. 2003;18:471–8. doi: 10.1177/08830738030180070401. [DOI] [PubMed] [Google Scholar]
- 14.Allen JC. Initial management of children with hypothalamic and thalamic tumors and the modifying role of neurofibromatosis-1. Pediatr Neurosurg. 2000;32:154–62. doi: 10.1159/000028922. [DOI] [PubMed] [Google Scholar]
- 15.Medlock MD, Madsen JR, Barnes PD, Anthony DS, Cohen LE, Scott RM. Optic chiasm astrocytomas of childhood. 1. Long-term follow-up. Pediatr Neurosurg. 1997;27:121–8. doi: 10.1159/000121239. [DOI] [PubMed] [Google Scholar]
- 16.Dutton JJ. Gliomas of the anterior visual pathway. Surv Ophthalmol. 1994;38:427–52. doi: 10.1016/0039-6257(94)90173-2. [DOI] [PubMed] [Google Scholar]
- 17.Jenkin D, Angyalfi S, Becker L, Berry M, Buncic R, Chan H, et al. Optic glioma in children: Surveillance, resection, or irradiation? Int J Radiat Oncol Biol Phys. 1993;25:215–25. doi: 10.1016/0360-3016(93)90342-s. [DOI] [PubMed] [Google Scholar]
- 18.Khafaga Y, Hassounah M, Kandil A, Kanaan I, Allam A, El Husseiny G, et al. Optic gliomas: A retrospective analysis of 50 cases. Int J Radiat Oncol Biol Phys. 2003;56:807–12. doi: 10.1016/s0360-3016(02)04512-1. [DOI] [PubMed] [Google Scholar]
- 19.Pepin SM, Lessell S. Anterior visual pathway gliomas: The last 30 years. Semin Ophthalmol. 2006;21:117–24. doi: 10.1080/08820530500350449. [DOI] [PubMed] [Google Scholar]
- 20.Komotar RJ, Burger PC, Carson BS, Brem H, Olivi A, Goldthwaite PT, et al. Pilocytic and pilomyxoid hypothalamic/chiasmatic astrocytomas. Neurosurgery. 2004;54:72–80. doi: 10.1227/01.neu.0000097266.89676.25. [DOI] [PubMed] [Google Scholar]
- 21.Astrup J. Natural history and clinical management of optic pathway glioma. Br J Neurosurg. 2003;17:327–35. doi: 10.1080/02688690310001601216. [DOI] [PubMed] [Google Scholar]
- 22.Packer RJ, Bilaniuk LT, Cohen BH, Braffman BH, Obringer AC, Zimmerman RA, et al. Intracranial visual pathway gliomas in children with neurofibromatosis. Neurofibromatosis. 1988;1:212–22. [PubMed] [Google Scholar]
- 23.Packer RJ, Savino PJ, Bilaniuk LT, Zimmerman RA, Schatz NJ, Rosenstock JG, et al. Chiasmatic gliomas of childhood. A reappraisal of natural history and effectiveness of cranial irradiation. Childs Brain. 1983;10:393–403. [PubMed] [Google Scholar]
- 24.Horwich A, Bloom HJ. Optic gliomas: Radiation therapy and prognosis. Int J Radiat Oncol Biol Phys. 1985;11:1067–79. doi: 10.1016/0360-3016(85)90052-5. [DOI] [PubMed] [Google Scholar]
- 25.Le Marquand HS, Russell DS. A case of pubertas praecox in a boy associated with a tumour in the floor of the third ventricle. Berk Hosp Rep. 1934-1935;3:31–61. [Google Scholar]
- 26.Alberchesten R, Klee JG, Moller JE. Primary intracranial germ cell tumors including five cases of endodermal sinus tumor. Acta Pathol Microbiol Scand. 1972;80(Suppl 233):S32–8. [PubMed] [Google Scholar]
- 27.Jellinger K. Primary intracranial germ cell tumors. Acta Neuropathol (Berl) 1973;25:291–306. doi: 10.1007/BF00691757. [DOI] [PubMed] [Google Scholar]
- 28.Leibel SA, Sheline GE. Radiation therapy for neoplasms of the brain. J Neurosurg. 1987;66:1–22. doi: 10.3171/jns.1987.66.1.0001. [DOI] [PubMed] [Google Scholar]
- 29.Kobayashi T, Yoshida J, Ishiyama J, Noda S, Kito A, Kida Y. Combination chemotherapy with cisplatin and etoposide for malignant intracranial germ cell tumor. An experimental and clinical-study. J Neurosurg. 1989;70:676–81. doi: 10.3171/jns.1989.70.5.0676. [DOI] [PubMed] [Google Scholar]