

Abstract
Efficient cofermentation of d-glucose, d-xylose, and l-arabinose, three major sugars present in lignocellulose, is a fundamental requirement for cost-effective utilization of lignocellulosic biomass. The Gram-positive anaerobic bacterium Clostridium acetobutylicum, known for its excellent capability of producing ABE (acetone, butanol, and ethanol) solvent, is limited in using lignocellulose because of inefficient pentose consumption when fermenting sugar mixtures. To overcome this substrate utilization defect, a predicted glcG gene, encoding enzyme II of the d-glucose phosphoenolpyruvate-dependent phosphotransferase system (PTS), was first disrupted in the ABE-producing model strain Clostridium acetobutylicum ATCC 824, resulting in greatly improved d-xylose and l-arabinose consumption in the presence of d-glucose. Interestingly, despite the loss of GlcG, the resulting mutant strain 824glcG fermented d-glucose as efficiently as did the parent strain. This could be attributed to residual glucose PTS activity, although an increased activity of glucose kinase suggested that non-PTS glucose uptake might also be elevated as a result of glcG disruption. Furthermore, the inherent rate-limiting steps of the d-xylose metabolic pathway were observed prior to the pentose phosphate pathway (PPP) in strain ATCC 824 and then overcome by co-overexpression of the d-xylose proton-symporter (cac1345), d-xylose isomerase (cac2610), and xylulokinase (cac2612). As a result, an engineered strain (824glcG-TBA), obtained by integrating glcG disruption and genetic overexpression of the xylose pathway, was able to efficiently coferment mixtures of d-glucose, d-xylose, and l-arabinose, reaching a 24% higher ABE solvent titer (16.06 g/liter) and a 5% higher yield (0.28 g/g) compared to those of the wild-type strain. This strain will be a promising platform host toward commercial exploitation of lignocellulose to produce solvents and biofuels.
INTRODUCTION
The production of ABE (acetone, butanol, and ethanol) solvent through biological processes has a long history (3, 8, 12). Among the three fermentation products, butanol is not only an important bulk industrial chemical but also a high-quality transportation fuel (11). To address an economic bottleneck, namely, the excessively high feedstock cost in ABE bioproduction, traditional cereal substrates (e.g., maize and wheat) are gradually being abandoned, whereas lignocellulose, the most abundant renewable biomass, is arousing worldwide interest (18). Therefore, Clostridium acetobutylicum, an important solvent-producing bacterium, once often used in corn-based fermentation, has demonstrated potential value in lignocellulose-based ABE solvent production (9, 25).
C. acetobutylicum is capable of utilizing a variety of carbohydrates, including hexoses and pentoses (50). Among these sugars, d-xylose and l-arabinose are the major pentoses contained in lignocellulose (2). Although C. acetobutylicum is able to use these two pentoses as carbon sources, this process is inhibited when d-glucose, the most abundant monosaccharide contained in lignocellulose, is present (14, 22). This is caused by the effect of carbon catabolite repression (CCR), a common phenomenon observed in many microbes (28). The CCR effect can limit the efficient utilization of d-xylose and l-arabinose in fermenting lignocellulosic hydrolysates, because d-glucose and the two pentoses are formed simultaneously when lignocellulose is depolymerized to fermentable sugars (2).
The phosphoenolpyruvate (PEP)-dependent phosphotransferase system (PTS) often plays an important role in carbohydrate transport in bacteria (37). In the model firmicute Bacillus subtilis, the PTS has been proven to be crucial for CCR (10, 43, 44). A typical PTS contains enzyme I (EI), enzyme II (EII) and a histidine-containing protein (HPr). The glycolytic intermediate glucose-6-phosphate, which is derived from EII-mediated phosphorylation during d-glucose uptake, as well as the subsequent intermediate product, fructose-1,6-bisphosphate, play key roles in CCR (16). Therefore, the PTS was chosen as a target for engineering to reduce the CCR caused by d-glucose, the so called “glucose repression,” in some microbes; however, this has always resulted in impaired or suspended d-glucose consumption (13, 32). Besides “glucose repression,” the insufficient ability of C. acetobutylicum to ferment d-xylose posed another problem in the fermentation of sugar mixtures. Compared to d-glucose, several more steps are required to catalyze conversion of d-xylose to glyceraldehyde-3-P, a key intermediate located at the intersection of the glycolysis and d-xylose pathways (20, 47), and thus some rate-limiting steps likely occur during this process (21, 24). In addition, C. acetobutylicum showed lower butanol tolerance in fermenting d-xylose compared to d-glucose (33), which also resulted in incomplete d-xylose consumption. Given the importance of simultaneous or rapid sequential fermentation of all major sugars in biomass hydrolysates, especially d-glucose and d-xylose, metabolic engineering strategies that can overcome “glucose repression” on d-xylose and l-arabinose and the low-efficiency of d-xylose consumption are required.
We previously identified and inactivated CcpA, a pleiotropic regulator, to analyze the cofermentation of d-glucose and d-xylose by C. acetobutylicum (38). Disruption of the ccpA gene eliminated CCR, however, the engineered strain showed an unexpected and severe accumulation of butyrate, resulting in a defective growth. Therefore, pH had to be controlled during the fermentation process to neutralize excess acids in the broth, which is obviously uneconomical for industrial-scale ABE fermentation. In the present study, a new strategy that enabled C. acetobutylicum to efficiently coferment d-glucose, d-xylose, and l-arabinose is presented. We first inactivated the glcG gene (45), which greatly reduced “glucose repression” of d-xylose and l-arabinose metabolism. Next, the rate-limiting steps of the d-xylose pathway were confirmed, and the bottleneck was relieved. The resulting recombinant strain, obtained through these combined strategies, exhibited a significantly improved capacity to coferment d-glucose, d-xylose, and l-arabinose, which should prove valuable in industrial ABE production from lignocellulosic hydrolysates.
MATERIALS AND METHODS
Strains, plasmids, and cultivation conditions.
The bacterial strains and plasmids used in the present study are listed in Table 1. Escherichia coli cells were grown at 37°C in Luria-Bertani medium or on Luria-Bertani agar (Luria-Bertani plus 1.5% [wt/vol] Difco agar). C. acetobutylicum and C. butyricum cells were grown anaerobically (Thermo Forma, Inc., Waltham, MA) at 37°C in liquid Clostridium growth medium (CGM) or on solid CGM (plus 2% [wt/vol] Difco agar) (48). The P2 medium was also used for C. acetobutylicum growth (4). For E. coli, ampicillin and spectinomycin were used at concentrations of 100 and 50 μg/ml, respectively, when needed. For C. acetobutylicum, 25 μg of erythromycin/ml was used when needed. C. acetobutylicum strains were maintained in 20% (vol/vol) glycerol and stored at −20°C.
Table 1.
Bacterial strains and plasmids
| Strain or plasmid | Relevant characteristicsa | Source or referenceb |
|---|---|---|
| Strains | ||
| C. butyricum DSM 10702 | Wild type | DSMZ |
| C. acetobutylicum | ||
| ATCC 824 | Wild type | ATCC |
| 824glcG | glcG::intron/pWJ1-glcG | This study |
| 824glcG-P | 824glcG/pIMP1-Pthl | This study |
| 824glcG-xylT | 824glcG/pIMP1-xylTthl | This study |
| 824glcG-xylA | 824glcG/pIMP1-xylAthl | This study |
| 824glcG-xylB | 824glcG/pIMP1-xylBthl | This study |
| 824glcG-TBA | 824glcG/pIMP1-xylTthl-xylBAthl | This study |
| E. coli | ||
| ER2275 | hsdR mcr recA1 endA1 | NEB |
| DH5α | General cloning host strain | Takara |
| Plasmids | ||
| pANS1 | Φ3TI, p15A origin; Sper | E. T. Papoutsakis (27) |
| pSY6 | Group II intron, ltrA | 40 |
| pWJ1 | Derived from pSY6 with pCB102 ORI instead of pIM13 ORI | This study |
| pWJ1-glcG | Derived from pWJ1 for intron insertion in glcG at 269/270 nt | This study |
| pIMP1-Pptb | ColE1 ORI; Ampr; pIM13 ORI; MLSr; ptb (cac3076) promoter region of C. acetobutylicum ATCC 824 | E. T. Papoutsakis |
| pIMP1-Pthl | Derived from pIMP1-Pptb, with thl (cac2873) promoter instead of ptb promoter | This study |
| pIMP1-xylTthl | Derived from pIMP1-Pthl, with xylT gene (cac1345) expressing cassette added | This study |
| pIMP1-xylAthl | Derived from pIMP1-Pthl, with xylA gene (cac2610) expressing cassette added | This study |
| pIMP1-xylBthl | Derived from pIMP1-Pthl, with xylB gene (cac2612) expressing cassette added | This study |
| pIMP1-xylTthl-Pthl | Derived from pIMP1-xylTthl, with thl (cac2873) promoter added. | This study |
| pIMP1-xylTthl-xylBAthl | Derived from pIMP1-xylTthl, with xylBA operon (from cac2612 to cac2610) added | This study |
glcG, glucose-specific PTS permease; xylT, xylose transporter; xylA, xylose isomerase; xylB, xylulokinase; hsdR, host-specific restriction deficient; mcr, methylcytosine-specific restriction abolished; recA1, homologous recombination abolished; endA1, endonucleases abolished; Sper, spectinomycin resistance; ltrA, LtrA protein, required for trans-splicing; ColE1 ORI, ColE1 origin of replication; Ampr, ampicillin resistance; pIM13 ORI, Gram-positive origin of replication; pCB102 ORI, Gram-positive origin of replication, which was unstable in C. acetobutylicum (15); MLSr, macrolide-lincosamide-streptogramin resistance; ptb, phosphotransbutyrylase; thl, thiolase; mls, gene encoding macrolide-lincosamide-streptogramin.
DSMZ, Deutsche Sammlung von Mikroorganismen und Zellkulturen, Braunschweig, Germany; ATCC, American Type Culture Collection, Manassas, VA; NEB, New England Biolabs, Beverly, MA.
Construction of plasmids pWJ1, pWJ1-glcG, pIMP1-xylTthl, pIMP1-xylAthl, pIMP1-xylBthl, and pIMP1-xylTthl-xylBAthl.
The replicon pCB102 was obtained by PCR using C. butyricum DSM 10702 genomic DNA as a template and pCB102-up/pCB102-dn as primers (see Table S1 in the supplemental material). The PCR fragment of pCB102 was digested with ClaI and SmaI and then inserted into plasmid pSY6, which was digested with the same restriction enzymes, yielding plasmid pWJ1. The pIM13 replicon of pWJ1 was then replaced by pCB102. The glcG-TargeTron target sequence of 350 bp was amplified by using primers glcG269|270a-IBS, glcG269|270a-EBS1d, and glcG269|270a-EBS2 to retarget the RNA portion of the intron according to the protocol of a TargeTron gene knockout system kit (Sigma-Aldrich, St. Louis, MO). Plasmid pWJ1-glcG was then obtained by inserting the glcG-TargeTron fragment into the XhoI and BsrGI sites of plasmid pWJ1. All primers involved in constructing the pWJ1-glcG plasmid are listed in Table S1 in the supplemental material. Primers IBS, EBS1d, and EBS2 were designed using the Clostron tool (www.clostron.com).
The C. acetobutylicum thl (thiolase, cac2873) promoter was obtained by PCR using thl-up/thl-dn as primers (see Table S1 in the supplemental material). The PCR fragment containing the thl promoter was digested with PstI and EcoRI and then inserted into plasmid pIMP1-Pptb, which was digested with the same restriction enzymes, yielding plasmid pIMP1-Pthl. The xylT (cac1345), xylA (cac2610), and xylB (cac2612) genes were amplified via PCR using the primer pairs of xylT-up/xylT-dn, xylA-up/xylA-dn, and xylB-up/xylB-dn, respectively (see Table S1 in the supplemental material). The obtained xylT, xylA, and xylB fragments were digested with SalI/XbaI, BamHI/SmaI, and BamHI/EcoRI, respectively, and then cloned into plasmid pIMP1-Pthl digested with the same restriction enzymes, yielding the plasmids pIMP1-xylTthl, pIMP1-xylAthl, and pIMP1-xylBthl, respectively (Fig. 1). Using plasmid pIMP1-Pthl as a template and Thl2-up/Thl2-dn as primers (see Table S1 in the supplemental material), the thl promoter fragment was obtained, digested with XbaI and BamHI, and then inserted into plasmid pIMP1-xylTthl to yield plasmid pIMP1-xylTthl-Pthl. The xylBA genes (cac2610 to cac2612) were amplified via PCR using primer pairs of xylBA-up/xylBA-dn (see Table S1 in the supplemental material). The obtained xylBA fragment was digested with BamHI and SmaI and then cloned into plasmid of pIMP1-xylTthl-Pthl, which was digested with the same restriction enzymes, yielding plasmid pIMP1-xylTthl-xylBAthl (Fig. 1).
Fig. 1.

Maps of plasmids pIMP1-xylTthl (A), pIMP1-xylAthl (B), pIMP1-xylBthl (C), and pIMP1-xylTthl-xylBAthl (D). bla, β-lactamase resistance gene; mlsR, macrolide-lincosamide-streptogramin resistance gene; ColE1 ori, replicon functional in E. coli; rep, replicon functional in C. acetobutylicum; thl, thl promoter; xylT, d-xylose transporter; xylA, d-xylose isomerase; xylB, d-xylulokinase; xylBA, xylBA operon (cac2610 to cac2612).
Electroporation of C. acetobutylicum and identification of mutants.
The electroporation of C. acetobutylicum ATCC 824 was performed as follows. All plasmids were first methylated in E. coli ER2275(pANS1) (27) and then electroporated into cells. The cells were plated on CGM agar containing 25 μg of erythromycin/ml and incubated anaerobically at 37°C for 48 h. Identification of positive transformants containing inserts was performed by colony PCR using primers glcG_126-145/glcG_473-492 (see Table S1 in the supplemental material). The PCR fragments were sequenced to confirm the insertion of intron. To eliminate erythromycin resistance for the next step of plasmid electroporation, the glcG-disrupted strain bearing plasmid pWJ1-glcG was successively transferred (once every 12 h) in liquid CGM without antibiotics at 37°C for 2 to 3 days. The cells were then plated on CGM agar and incubated to obtain individual colonies. Those losing plasmids were confirmed by colony PCR using primers glcG_126-145/glcG_473-492 (see Table S1 in the supplemental material). The resulting mutant strain was named 824glcG.
Plasmids pIMP1-xylTthl, pIMP1-xylAthl, pIMP1-xylBthl, and pIMP1-xylTthl-xylBAthl were then introduced into strain 824glcG by electroporation. Positive transformants were identified via colony PCR using the primer pairs dxylT-up/dpIMP1-dn, dxylA-up/dpIMP1-dn, dpIMP1-up/dxylB-dn, and dxylT-overlap-up/dxylBA-overlap-dn (see Table S1 in the supplemental material), yielding strains 824glcG-xylT, 824glcG-xylA, 824glcG-xylB, and 824glcG-TBA, respectively. The empty plasmid pIMP1-Pthl was also introduced into strain 824glcG, and the resulting strain (824glcG-P) was used as a control in the fermentation process. Positive transformants of 824glcG-P were identified by using the primer pair dpIMP1-up/dthl-dn (see Table S1 in the supplemental material).
Fermentations.
Batch fermentations were performed anaerobically in P2 medium at 37°C using d-glucose, d-xylose, and l-arabinose as carbon sources.
Fermentations were performed in 250-ml serum bottles as follows. First, 150 μl of frozen stock was inoculated into 5 ml of liquid CGM, followed by anaerobic incubation at 37°C for 24 h. When the optical density at 600 nm (OD600) of the cells reached 0.8 to 1.0, 2.5-ml portions of the grown cells were inoculated into 50 ml of CGM without antibiotics for the secondary preparation. When the OD600 of cells reached 0.8 to 1.0, ca. 5% (vol/vol) of the inoculum was transferred into 95 ml of P2 medium for fermentation.
Fermentation with 1.5-liter working volumes were performed using a BioFlo 110 bioreactor (New Brunswick Scientific, Edison, NJ). A portion (100 ml) of the secondary culture preparation was inoculated into 1.4 liters of P2 medium. The anaerobic conditions of fermentors were maintained through filtered nitrogen.
Analytical methods.
The cell density (OD600) was determined at A600 using a DU730 spectrophotometer (Beckman Coulter). Solvents (acetone, butanol, and ethanol) were determined by gas chromatography (7890A; Agilent, Wilmington, DE). The concentrations of d-glucose, d-xylose, and l-arabinose were determined by using a high-pressure liquid chromatography system (1200 series; Agilent). Conditions of gas and liquid chromatographs were as described previously (38).
PTS assays in cell extracts.
Cultures were grown for 18 h in P2 medium containing 20 g of glucose/liter as a carbon source, and extracts were prepared as previously described (29, 49). Cells were harvested by centrifugation, washed, and resuspended in 50 mM potassium phosphate buffer (pH 7) containing 5 mM MgCl2 and 1 mM dithiothreitol and broken by two passages through a French press at 20,000 lb/in2. PTS activities in cell extracts were assayed by following the PEP-dependent phosphorylation of d-[U-14C]glucose and methyl α-d-[U-14C]glucoside under conditions described previously (49).
Glucose kinase assays.
C. acetobutylicum and its derivative strains were grown in P2 medium containing 40 g of d-glucose/liter as a carbon source. Cells were collected by centrifugation at 4°C and frozen immediately using liquid nitrogen. The frozen cells were then resuspended in 6 ml of Tris-HCl buffer (50 mM, pH 7.4) containing 10% (vol/vol) glycerol and disrupted (30KPSI, two times) by two passages through a French press at 20,000 lb/in2. Cell debris was separated from the soluble fraction by centrifugation (4°C, 13,400 × g, 30 min), and the cell extract was used for glucokinase assay. The glucokinase activity was assayed using enzyme-linked reactions to detect the reduction of NADP (40).
RNA preparation and real-time PCR analysis.
Culture samples for real-time PCR analysis were collected from 1 liter of P2 medium using 40 g of d-glucose/liter and 20 g of d-xylose/liter as carbon sources. RNA preparation and generation of cDNA were performed as described previously (38).
Each real-time PCR contained 10 μl of iQ SYBR green Supermix (Bio-Rad), 200 nM concentrations of each primer, 1 μl of DNA template, and a sufficient volume of water to reach a final reaction volume of 20 μl. Real-time PCR was performed in a CFX96 real-time PCR detection system (Bio-Rad) as follows: 1 cycle at 95°C for 3 min, followed by 40 cycles at 95°C for 20 s, 55°C for 20 s, and 72°C for 20 s. Three PCRs were performed in parallel for each transcript. The gene cac2679 (encoding pullulanase) was used as an internal control. The relative fold change of RNA transcript (mutant/wild type) was determined according to the 2−ΔΔCT method (36), in which ΔΔCT = (CT tested gene − CT cac2679)mutant − (CT tested gene − CT cac2679)wild type.
RESULTS
Disruption of the glcG gene reduces “glucose repression” of d-xylose and l-arabinose utilization in C. acetobutylicum.
PEP-dependent glucose PTS activity has been detected in C. acetobutylicum ATCC 824 (45), indicating that this activity may play a leading role for d-glucose metabolism in this anaerobe. To attenuate glucose PTS activity in cells, gene glcG (cac0570), which was suggested by bioinformatics analysis to encode the PTS enzyme II (45), was disrupted using TargeTron technology (42). As expected, colony PCR and sequencing data showed that an intron was inserted into the glcG gene at nucleotide positions 269 and 270 (see Fig. S1 in the supplemental material).
Strain 824glcG and wild-type strain 824WT were then cultured to see whether they differed in fermenting mixtures of d-glucose–d-xylose or d-glucose–l-arabinose. The medium contained 40 g of d-glucose/liter with 20 g of d-xylose/liter or 40 g of d-glucose/liter with 20 g of l-arabinose/liter. The pH curve obtained for strain 824glcG was consistent with that of strain 824WT throughout the whole fermentation process (Fig. 2C and F). However, when fermented in the mixture of d-glucose and l-arabinose, strain 824glcG consumed 79% of the total amount of l-arabinose, behaving more efficiently than 824WT (Fig. 2A). When l-arabinose was substituted with d-xylose, strain 824glcG showed an advantage only in the late fermentation stages (after 40 h) and eventually consumed 9.7 g more d-xylose/liter than strain 824WT (Fig. 2D). These results suggested that the disruption of gene glcG relieved “glucose repression,” but some rate-limiting steps still existed in the d-xylose pathway of C. acetobutylicum.
Fig. 2.

Growth and metabolite profiles in batch fermentations of strain 824WT and strain 824glcG in P2 medium containing a mixture of 40 g of d-glucose/liter and 20 g of l-arabinose/liter (A to C) and P2 medium containing a mixture of 40 g of d-glucose/liter and 20 g of d-xylose/liter (D to F). (A and D) Sugar consumption; (B and E) growth; (C and F) pH values. Fermentations were performed in triplicate.
Interestingly, despite inactivation of the glcG gene, the rate of glucose consumption by the 824glcG strain was almost identical to that observed for 824WT (Fig. 2A and D). Assays of PEP-dependent glucose phosphorylation indicated that extracts of the 824glcG strain had a glucose PTS activity similar to that present in extracts of the parental strain 824WT (Fig. 3A). However, PEP-dependent phosphorylation of the glucose analogue methyl α-glucoside was lost in the mutant, an observation consistent with inactivation of a PTS for which the analogue is a substrate (Fig. 3B). In addition, we also considered whether an alternative, non-PTS-mediated route of glucose transport and metabolism might play a greater role in glucose uptake in 824glcG than in 824WT. The ATP-dependent glucose kinase activity of these two strains was therefore measured, since this enzyme would be required to phosphorylate accumulated glucose. The glucose kinase specific activities of 824WT were 0.16 ± 0.02 and 0.23 ± 0.06 U/mg in acidogenic and solventogenic phases, respectively, while those of 824glcG were 0.31± 0.13 and 1.05 ± 0.36 U/mg, respectively. (The OD600 values of the samples taken in the acidogenic phase and solventogenic phase were ca. 1.8 and 4.0, respectively.) Thus, the glucose kinase activities of strain 824glcG were 1.9- and 4.6-fold greater than those of strain 824WT for the acidogenic and solventogenic periods, respectively.
Fig. 3.

Phosphotransferase activities in cell extracts. Phosphorylation of glucose (A) and methyl α-glucoside (B) was monitored in the presence or absence of PEP. Values are the average of duplicate determinations and are presented as nmol of sugar phosphorylated per mg of extract protein.
Rate-limiting steps of d-xylose metabolism occur before PPP.
As mentioned above, only after d-glucose was nearly exhausted did strain 824glcG begin to show an advantage in d-xylose consumption (Fig. 2D), whereas in fermenting the mixture of d-glucose and l-arabinose, 824glcG exhibited faster l-arabinose consumption at an earlier stage (Fig. 2A). Therefore, we speculated that there still existed rate-limiting steps in the initiation of d-xylose metabolism despite relief of the “glucose repression” by glcG gene disruption. Given the fact that d-xylose and l-arabinose normally share the same pathway after metabolic flux enters into PPP, which is followed by glycolysis (Fig. 4), the rate-limiting steps are likely to involve d-xylose transport, d-xylose isomerization, and xylulose phosphorylation (Fig. 4). Comparisons between separate fermentations of d-xylose and l-arabinose (∼16 g/liter) by strain 824WT support this hypothesis. As shown in Fig. 5A, strain 824WT was able to exhaust all l-arabinose within 38 h, at which time only 46% of the total d-xylose was consumed. Moreover, 4.94 g of d-xylose/liter still remained even after 72 h of fermentation. Besides, faster growth was also observed on l-arabinose compared to d-xylose (Fig. 5B). These results strongly suggest that in addition to “glucose repression,” inherently inefficient d-xylose metabolism also needs to be addressed when fermenting mixed sugars by C. acetobutylicum.
Fig. 4.
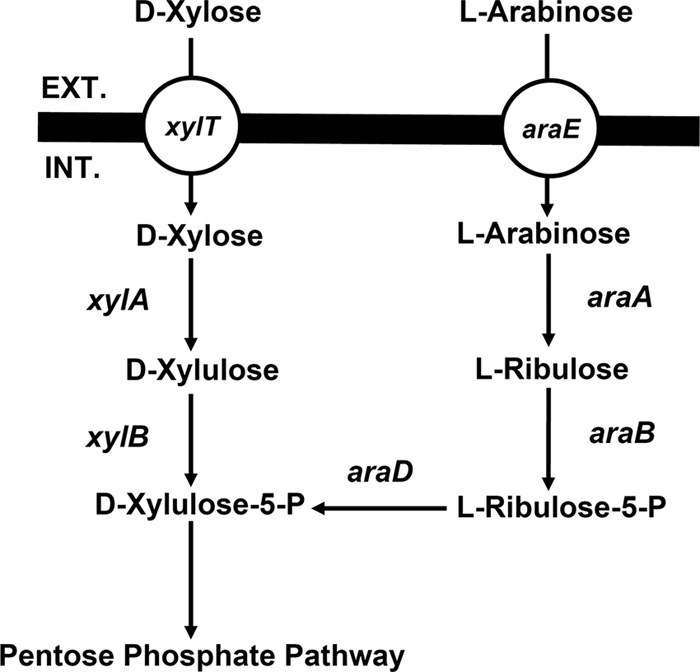
Schematic representation of possible d-xylose and l-arabinose catabolic pathways in C. acetobutylicum. xylT (cac1345), d-xylose transporter; xylA (cac2610), d-xylose isomerase; xylB (cac2612), xylulokinase; araE (cac1339), l-arabinose transporter; araA (cac1342, cac1346), l-arabinose isomerase; araB (cac1344?), l-ribulokinase; araD (cac1341), l-ribulose-5-phosphate 4-epimerase.
Fig. 5.

Growth and metabolite profiles in batch fermentations of strain 824WT in P2 medium containing 16.26 g of l-arabinose/liter or 16.80 g of d-xylose/liter. (A) Sugar consumption; (B) growth. Fermentations were performed in triplicate.
Overexpression of XylT, XylA, and XylB in strain 824glcG improves d-xylose consumption in the presence of d-glucose.
To alleviate the bottleneck of the d-xylose metabolic pathway, we introduced the genes cac1345, cac2610, and cac2612, which had been identified in our previous study as encoding a d-xylose transporter XylT, d-xylose isomerase XylA, and xylulokinase XylB of C. acetobutylicum (17), separately or together into strain 824glcG. These genes were cloned into the plasmid pIMP1 (27) and overexpressed under the constitutive thiolase (thl) promoter (5). All of the engineered strains (824glcG-xylT, 824glcG-xylA, 824glcG-xylB, and 824glcG-TBA) showed improved d-xylose consumption in the presence of d-glucose compared to strain 824glcG (Fig. 6). Strain 824glcG-TBA exhibited most efficient consumption, being able to use 16.82 g of d-xylose/liter when fermented the mixed sugars (40 g of d-glucose and 20 g of d-xylose/liter), whereas the 824glcG, 824glcG-xylT, 824glcG-xylA, and 824glcG-xylB strains could use only 10.48, 12.41, 14.60, and 11.53 g of d-xylose/liter, respectively. The transcript levels of the xylT, xylA, and xylB genes were then compared in strains 824glcG-TBA and 824glcG-P (control strain bearing the empty plasmid pIMP1). The results showed that during the acidogenic phase, xylT, xylA, and xylB transcripts were increased by 1.17-, 132-, and 119-fold in strain 824glcG-TBA compared to those in 824glcG-P, respectively, and while entering into solventogenic phase, the upregulation of these three genes in 824glcG-TBA became 6.60-, 30-, and 62-fold, respectively. This enhancement of the transcriptional levels suggests an efficient pIMP1-based expression of these genes in strain 824glcG-TBA (Table 2). It should be noted that the fold changes in xylT expression are much lower than those for xylA and xylB. This could be attributed to the much higher transcriptional level of xylT compared to xylA and xylB in the control strain 824glcG-P (see Table S2 in the supplemental material), which made the transcriptional increases of xylT appear less conspicuous than those of xylA and xylB when plasmid-based gene overexpression was introduced into the 824glcG strain.
Fig. 6.

Sugar consumption of strains 824WT, 824glcG, 824glcG-P, 824glcG-xylT, 824glcG-xylA, 824glcG-xylB, and 824glcG-TBA in P2 medium containing a mixture of 40 g of d-glucose/liter and 20 g of d-xylose/liter during batch fermentations. Samples were taken after 96 h. Fermentations were performed in triplicate.
Table 2.
Transcriptional fold changes of xylT, xylA, and xylB of strain 824glcG-TBA compared to stain 824glcG-P
| Gene | Transcriptional level (mean fold change ± SD)a |
|
|---|---|---|
| Acidogenic phase | Solventogenic phase | |
| xylT | 1.17 ± 0.08 | 6.60 ± 0.56 |
| xylA | 132 ± 27 | 30 ± 3 |
| xylB | 119 ± 5 | 62 ± 4 |
The OD600 values for samples taken in acidogenic and solventogenic phases were 3.8 and 7.0, respectively. Fermentations were performed in P2 medium containing 40 g of glucose and 20 g of xylose/liter.
Efficient cofermentation of d-glucose, d-xylose, and l-arabinose achieved by C. acetobutylicum 824glcG-TBA.
To further test the performance of the engineered strain, 824glcG-TBA was compared to 824WT in fermenting a simulated lignocellulosic hydrolysate based on a general ratio of mixed sugars (37.63 g of d-glucose/liter to 14.47 g of d-xylose/liter to 2.89 g of l-arabinose/liter) (2). The fermentation was performed using a bioreactor without pH regulation. As shown in Fig. 7A, strain 824glcG-TBA rapidly consumed the three sugars. A total of 56.02 g of the sugars/liter was consumed within 52 h despite d-xylose being consumed more slowly than the other sugars. In contrast, the wild-type strain could not complete the consumption of d-xylose within the same time under identical conditions (Fig. 7A) and, moreover, it showed a slower l-arabinose consumption rate compared to strain 824glcG-TBA. Therefore, strain 824glcG-TBA was able to produce 9.11 g of butanol/liter and 16.06 g of total solvents/liter within 52 h while fermenting the mixture of these three sugars. During the same time frame, the wild-type strain (824WT) only yielded 7.85 g of butanol/liter and 12.99 g of total solvents/liter (Fig. 7B), with 8.05 g of d-xylose/liter remaining at the end of fermentation (Table 3). As a result, yields of 0.16 g/g for butanol and 0.28 g/g for total solvents were achieved in 824glcG-TBA (Table 3), which was higher than obtained from 824WT (0.14 g/g [butanol yield]; 0.23 g/g [total solvents]). This capacity to ferment sugar mixtures enables strain 824glcG-TBA to meet the requirements for using cellulose hydrolysates as feedstock.
Fig. 7.

Growth and metabolite profiles of strain 824WT and strain 824glcG-TBA in P2 medium containing a mixture of 38 g of d-glucose/liter, 14 g of d-xylose/liter, and 3 g of l-arabinose/liter in batch fermentations. (A) Sugar consumption; (B) butanol and ABE concentration; (C) growth; (D) pH values.
Table 3.
Fermentation parameters of strains 824WT and 824glcG-TBA after 52 h of batch fermentation
| Strain | Content (g/liter) |
Productivity (g/liter · h) | ABE yield (g/g) | Butanol yield (g/g) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Initial sugar |
Residual sugar |
Product |
|||||||||||
| Glucose | Xylose | Arabinose | Glucose | Xylose | Arabinose | Acetone | Butanol | Ethanol | ABE | ||||
| 824WT | 38.50 | 15.70 | 3.46 | 0.00 | 8.05 | 0.00 | 3.99 | 7.85 | 1.15 | 12.99 | 0.25 | 0.23 | 0.14 |
| 824glcG-TBA | 38.54 | 14.92 | 3.51 | 0.00 | 0.95 | 0.00 | 5.07 | 9.11 | 1.88 | 16.06 | 0.31 | 0.28 | 0.16 |
DISCUSSION
For this study, C. acetobutylicum ATCC 824, a model strain of solventogenic clostridia, was genetically engineered to obtain the capability to efficiently coferment mixtures of d-glucose, d-xylose, and l-arabinose in solvent production. Based on the engineering strategy proposed here, the disruption of gene glcG, which encodes a d-glucose PTS enzyme II, was the initial step. This gene is recognized as a key factor that may contribute to “glucose repression” of nonpreferred sugars (1, 6, 44). However, defective and suspended d-glucose consumption had previously been observed in E. coli, B. subtilis, and S. clavuligerus when genes responsible for glucose transport were inactivated or were expressed at low levels (13, 34, 35). Thus, it was possible that glcG would not be an ideal gene target for engineering to relieve “glucose repression.” However, we found that the effect of inactivation of glcG was quite different in C. acetobutylicum compared to the earlier studies in that the mutant could ferment glucose efficiently. The loss of PTS activity for methyl α-glucoside, which has been proposed to be a substrate of GlcG (44), was consistent with inactivation of this system. However, the glcG mutant retained a considerable residual PTS activity for glucose that was comparable to the activity in the wild type. Including GlcG, the C. acetobutylicum genome encodes 13 phosphotransferases (30). Several of these systems belong to the glucose-glucoside and mannose-fructose-sorbose PTS families, and some have been shown to be expressed in glucose-grown cells (41). It is therefore likely that one or more of these systems can contribute to glucose uptake and phosphorylation in the mutant. Furthermore, assays of glucose kinase indicated that the 824glcG strain had elevated activity of this enzyme compared to the parental strain. Therefore, as has been demonstrated in Clostridium beijerinckii (26), coupling of glucose uptake to ATP-dependent phosphorylation may provide an alternative route for glucose utilization in C. acetobutylicum, which becomes important under certain physiological conditions. While glucose kinase (EC 2.7.1.2), like other hexokinases, can catalyze phosphorylation of other hexoses under certain conditions (23), it has not been found yet to harbor activities toward pentoses and therefore is unlikely to contribute to the utilization of d-xylose and l-arabinose.
The continued ability of the glcG mutant to take up and phosphorylate d-glucose, by either or both of the indicated mechanisms, was reflected in its ability to utilize d-glucose as efficiently as the parental strain in fermentation experiments. On the other hand, the glcG mutant strain showed a marked deficiency in “glucose repression” with respect to fermentation of d-xylose and l-arabinose. This finding would appear to implicate GlcG as an important determinant of the repression mechanism in C. acetobutylicum.
Other than glucose repression, the inherent inefficiency of d-xylose utilization is another problem to be addressed in fermenting sugar mixtures using C. acetobutylicum. Direct overexpression of C. acetobutylicum genes responsible for d-xylose transport, as well as catalytic enzymes (d-xylose isomerase, xylulokinase, and enzymes of PPP) is undoubtedly the primary strategy to be considered. However, combining all of these genes may yield a DNA fragment estimated to be over 9 kb. Introduction of such a large DNA fragment plus the plasmid pIMP1 skeleton (4.8 kb) into C. acetobutylicum appears to be difficult now (31, 42). Therefore, identifying the rate-limiting steps and reducing the number of genes overexpressed is particularly important, since this would allow the plasmid-based expression to be feasible.
In C. acetobutylicum, d-xylose transport is the initial and perhaps a relatively inefficient step during d-xylose metabolism, because no d-xylose ABC-type transporter, which was considered to be mainly responsible for the d-xylose transport, was found by our previous bioinformatics analysis (17). Only putative d-xylose and undefined sugar symporters (cac1339, cac3422, and cac1345) were observed. In general, symporters exhibit much lower affinity for d-xylose compared to ABC transporters (21), which indicates that d-xylose uptake may be a rate-limiting step in d-xylose metabolism by C. acetobutylicum. The contribution of gene cac1345 overexpression to improved d-xylose consumption also supports this speculation (Fig. 6). d-Xylose isomerization and xylulose phosphorylation, catalyzed by XylA and XylB, respectively, are the first and important two steps in d-xylose metabolism after d-xylose uptake. Therefore, efficient expression of XylA and XylB is essential for the utilization of d-xylose. Our previous study confirmed that C. acetobutylicum genes cac2610 and cac2612 are responsible for encoding XylA and XylB, respectively, and their indispensable roles in d-xylose metabolism were also shown (17). However, just as in some other bacteria (e.g., B. subtilis) (39), a possible d-xylose repressor, XylR (cac2613), was discovered next to the xylB gene in C. acetobutylicum ATCC 824 (19). The disruption of cac2613 resulted in faster d-xylose consumption, which indicated that this gene may play the role of a repressor of cac2610 to cac2613 and be responsible for the initial low d-xylose consumption rate (19). Therefore, to overcome these limitations, genes cac1345, cac2610, and cac2612 were coexpressed via plasmid pIMP1 under the control of the constitutive thl promoter.
The strategy described in the present study presented an engineered strain with significantly improved capabilities of utilizing d-xylose and l-arabinose in the presence of d-glucose. However, we also noticed that consumption of d-xylose still lagged behind d-glucose when a mixture of the three sugars was fermented (Fig. 7A). This may be partly due to the nonspecificity of cac1345 in d-xylose transport. Although cac1345 has been shown to act as a d-xylose transporter in our previous work (17), its primary role in l-arabinose transport was also strongly suggested (41). Therefore, it seems necessary to find a more specific and efficient d-xylose transporter, such as an ABC-type d-xylose transporter, to enhance d-xylose uptake by C. acetobutylicum. Another important factor that can influence d-xylose metabolism is butanol inhibition. It has been shown that only 8 g of butanol/liter could completely inhibit cultures growing on d-xylose (33), and this inhibition likely acted on the cell membrane functionality (7, 46). As illustrated in Fig. 7, at the time of exhaustion d-glucose, the butanol concentration in the medium was high enough to inhibit d-xylose consumption. Therefore, to improve this process, two strategies can probably be used: first, increasing the tolerance of the engineered strain to butanol, and second, accelerating d-xylose consumption so that d-xylose can be exhausted before reaching the inhibitory concentration of butanol. This will be the focus of our future work.
Supplementary Material
ACKNOWLEDGMENTS
This study was supported by the National Basic Research Program of China (973: 2007CB707803, 2011CBA00806), the Knowledge Innovation Program of the Chinese Academy of Sciences (KSCX2-EW-G-1 and KSCX2-EW-J-12), the National Natural Science Foundation of China (31070075), the Knowledge Innovation Program of the Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences (2010KIP204), the Program for S&T Cooperation Project of Jilin Province, Chinese Academy of Sciences (2010SYHZ0048), and the China Partnering Award of the UK Biotechnology and Biological Sciences Research Council (BB/G530341/1). S.Y. was funded by Dupont Young Professor Award.
We thank Eleftherios T. Papoutsakis (University of Delaware, Newark) for plasmids pIMP1, pIMP1-Pptb, and pANS1 and Cong Ren and Shiyuan Hu (Key Laboratory of Synthetic Biology, Institute of Plant Physiology and Ecology, Shanghai Institutes for Biological Sciences, CAS, China) for helpful suggestions and thoughtful discussions.
Footnotes
Supplemental material for this article may be found at http://aem.asm.org/.
Published ahead of print on 16 September 2011.
REFERENCES
- 1. Abranches J., Candella M. M., Wen Z. T., Baker H. V., Burne R. A. 2006. Different roles of EIIABMan and EIIGlc in regulation of energy metabolism, biofilm development, and competence in Streptococcus mutans. J. Bacteriol. 188:3748–3756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Aristidou A., Penttila M. 2000. Metabolic engineering applications to renewable resource utilization. Curr. Opin. Biotechnol. 11:187–198 [DOI] [PubMed] [Google Scholar]
- 3. Awang G. M., Jones G. A., Ingledew W. M. 1988. The acetone-butanol-ethanol fermentation. Crit. Rev. Microbiol. 15(Suppl. 1):S33–S67 [DOI] [PubMed] [Google Scholar]
- 4. Baer S. H., Blaschek H. P., Smith T. L. 1987. Effect of butanol challenge and temperature on lipid composition and membrane fluidity of butanol-tolerant Clostridium acetobutylicum. Appl. Environ. Microbiol. 53:2854–2861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bermejo L. L., Welker N. E., Papoutsakis E. T. 1998. Expression of Clostridium acetobutylicum ATCC 824 genes in Escherichia coli for acetone production and acetate detoxification. Appl. Environ. Microbiol. 64:1079–1085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bettenbrock K., et al. 2006. A quantitative approach to catabolite repression in Escherichia coli. J. Biol. Chem. 281:2578–2584 [DOI] [PubMed] [Google Scholar]
- 7. Bowles L. K., Ellefson W. L. 1985. Effects of butanol on Clostridium acetobutylicum. Appl. Environ. Microbiol. 50:1165–1170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chiao J. S., Sun Z. H. 2007. History of the acetone-butanol-ethanol fermentation industry in China: development of continuous production technology. J. Mol. Microbiol. Biotechnol. 13:12–14 [DOI] [PubMed] [Google Scholar]
- 9. Dellomonaco C., Fava F., Gonzalez R. 2010. The path to next generation biofuels: successes and challenges in the era of synthetic biology. Microb. Cell Fact 9:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Deutscher J., et al. 1994. Loss of protein kinase-catalyzed phosphorylation of HPr, a phosphocarrier protein of the phosphotransferase system, by mutation of the ptsH gene confers catabolite repression resistance to several catabolic genes of Bacillus subtilis. J. Bacteriol. 176:3336–3344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Durre P. 2007. Biobutanol: an attractive biofuel. Biotechnol. J. 2:1525–1534 [DOI] [PubMed] [Google Scholar]
- 12. Durre P. 1998. New insights and novel developments in clostridial acetone/butanol/isopropanol fermentation. Appl. Microbiol. Biotechnol. 49:639–648 [Google Scholar]
- 13. Eiteman M. A., Lee S. A., Altman E. 2008. A co-fermentation strategy to consume sugar mixtures effectively. J. Biol. Eng. 2:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ezeji T., Blaschek H. P. 2008. Fermentation of dried distillers' grains and solubles (DDGS) hydrolysates to solvents and value-added products by solventogenic clostridia. Bioresource Technol. 99:5232–5242 [DOI] [PubMed] [Google Scholar]
- 15. Fox M. E., et al. 1996. Anaerobic bacteria as a delivery system for cancer gene therapy: in vitro activation of 5-fluorocytosine by genetically engineered clostridia. Gene Ther. 3:173–178 [PubMed] [Google Scholar]
- 16. Gorke B., Stulke J. 2008. Carbon catabolite repression in bacteria: many ways to make the most out of nutrients. Nat. Rev. Microbiol. 6:613–624 [DOI] [PubMed] [Google Scholar]
- 17. Gu Y., et al. 2010. Reconstruction of xylose utilization pathway and regulons in Firmicutes. BMC Genomics 11:255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hill J., Nelson E., Tilman D., Polasky S., Tiffany D. 2006. Environmental, economic, and energetic costs and benefits of biodiesel and ethanol biofuels. Proc. Natl. Acad. Sci. U. S. A. 103:11206–11210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hu S., et al. 2011. Comparative genomic and transcriptomic analysis revealed genetic characteristics related to solvent formation and xylose utilization in Clostridium acetobutylicum EA 2018. BMC Genomics 12:93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jeffries T. W. 2006. Engineering yeasts for xylose metabolism. Curr. Opin. Biotechnol. 17:320–326 [DOI] [PubMed] [Google Scholar]
- 21. Jojima T., Omumasaba C. A., Inui M., Yukawa H. 2010. Sugar transporters in efficient utilization of mixed sugar substrates: current knowledge and outlook. Appl. Microbiol. Biotechnol. 85:471–480 [DOI] [PubMed] [Google Scholar]
- 22. Kanouni A. E., et al. 1998. The improvement of glucose/xylose fermentation by Clostridium acetobutylicum using calcium carbonate. World J. Microbiol. Biotechnol. 14:431–435 [Google Scholar]
- 23. Kawai S., Mukai T., Mori S., Mikami B., Murata K. 2005. Hypothesis: structures, evolution, and ancestor of glucose kinases in the hexokinase family. J. Biosci. Bioeng. 99:320–330 [DOI] [PubMed] [Google Scholar]
- 24. Kuyper M., Winkler A. A., van Dijken J. P., Pronk J. T. 2004. Minimal metabolic engineering of Saccharomyces cerevisiae for efficient anaerobic xylose fermentation: a proof of principle. FEMS Yeast Res. 4:655–664 [DOI] [PubMed] [Google Scholar]
- 25. la Grange D. C., den Haan R., van Zyl W. H. 2010. Engineering cellulolytic ability into bioprocessing organisms. Appl. Microbiol. Biotechnol. 87:1195–1208 [DOI] [PubMed] [Google Scholar]
- 26. Lee J., Mitchell W. J., Tangney M., Blaschek H. P. 2005. Evidence for the presence of an alternative glucose transport system in Clostridium beijerinckii NCIMB 8052 and the solvent-hyperproducing mutant BA101. Appl. Environ. Microbiol. 71:3384–3387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mermelstein L. D., Papoutsakis E. T. 1993. In vivo methylation in Escherichia coli by the Bacillus subtilis phage φ3T I methyltransferase to protect plasmids from restriction upon transformation of Clostridium acetobutylicum ATCC 824. Appl. Environ. Microbiol. 59:1077–1081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mitchell W. J. 1998. Physiology of carbohydrate to solvent conversion by clostridia. Adv. Microb. Physiol. 39:31–130 [DOI] [PubMed] [Google Scholar]
- 29. Mitchell W. J., Booth I. R. 1984. Characterization of the Clostridium pasteurianum phosphotransferase system. J. Gen. Microbiol. 130:2193–2200 [Google Scholar]
- 30. Mitchell W. J., Tangney M. 2005. Carbohydrate uptake by the phosphotransferase system and other mechanisms, p. 155–175 In Durre P. (ed.), Handbook on clostridia. CRC Press, Boca Raton, FL [Google Scholar]
- 31. Nakotte S., Schaffer S., Bohringer M., Durre P. 1998. Electroporation of, plasmid isolation from and plasmid conservation in Clostridium acetobutylicum DSM 792. Appl. Microbiol. Biotechnol. 50:564–567 [DOI] [PubMed] [Google Scholar]
- 32. Nichols N. N., Dien B. S., Bothast R. J. 2001. Use of catabolite repression mutants for fermentation of sugar mixtures to ethanol. Appl. Microbiol. Biotechnol. 56:120–125 [DOI] [PubMed] [Google Scholar]
- 33. Ounine K., Petitdemange H., Raval G., Gay R. 1985. Regulation and butanol inhibition of d-xylose and d-glucose uptake in Clostridium acetobutylicum. Appl. Environ. Microbiol. 49:874–878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Paulsen I. T., Chauvaux S., Choi P., Saier M. H., Jr 1998. Characterization of glucose-specific catabolite repression-resistant mutants of Bacillus subtilis: identification of a novel hexose:H+ symporter. J. Bacteriol. 180:498–504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Perez-Redondo R., Santamarta I., Bovenberg R., Martin J. F., Liras P. 2010. The enigmatic lack of glucose utilization in Streptomyces clavuligerus is due to inefficient expression of the glucose permease gene. Microbiology 156:1527–1537 [DOI] [PubMed] [Google Scholar]
- 36. Pfaffl M. W. 2001. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 29:e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Postma P. W., Lengeler J. W., Jacobson G. R. 1993. Phosphoenolpyruvate:carbohydrate phosphotransferase systems of bacteria. Microbiol. Rev. 57:543–594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ren C., et al. 2010. Identification and inactivation of pleiotropic regulator CcpA to eliminate glucose repression of xylose utilization in Clostridium acetobutylicum. Metab. Eng. 12:446–454 [DOI] [PubMed] [Google Scholar]
- 39. Rodionov D. A., Mironov A. A., Gelfand M. S. 2001. Transcriptional regulation of pentose utilization systems in the Bacillus/Clostridium group of bacteria. FEMS Microbiol. Lett. 205:305–314 [DOI] [PubMed] [Google Scholar]
- 40. Seno E. T., Chater K. F. 1983. Glycerol catabolic enzymes and their regulation in wild-type and mutant strains of Streptomyces coelicolor A3(2). J. Gen. Microbiol. 129:1403–1413 [DOI] [PubMed] [Google Scholar]
- 41. Servinsky M. D., Kiel J. T., Dupuy N. F., Sund C. J. 2010. Transcriptional analysis of differential carbohydrate utilization by Clostridium acetobutylicum. Microbiology 156:3478–3491 [DOI] [PubMed] [Google Scholar]
- 42. Shao L., et al. 2007. Targeted gene disruption by use of a group II intron (TargeTron) vector in Clostridium acetobutylicum. Cell Res. 17:963–965 [DOI] [PubMed] [Google Scholar]
- 43. Singh K. D., Schmalisch M. H., Stulke J., Gorke B. 2008. Carbon catabolite repression in Bacillus subtilis: quantitative analysis of repression exerted by different carbon sources. J. Bacteriol. 190:7275–7284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Stulke J., Martin-Verstraete I., Glaser P., Rapoport G. 2001. Characterization of glucose-repression-resistant mutants of Bacillus subtilis: identification of the glcR gene. Arch. Microbiol. 175:441–449 [DOI] [PubMed] [Google Scholar]
- 45. Tangney M., Mitchell W. J. 2007. Characterisation of a glucose phosphotransferase system in Clostridium acetobutylicum ATCC 824. Appl. Microbiol. Biotechnol. 74:398–405 [DOI] [PubMed] [Google Scholar]
- 46. Vollherbst-Schneck K., Sands J. A., Montenecourt B. S. 1984. Effect of butanol on lipid composition and fluidity of Clostridium acetobutylicum ATCC 824. Appl. Environ. Microbiol. 47:193–194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wamelink M. M., Struys E. A., Jakobs C. 2008. The biochemistry, metabolism, and inherited defects of the pentose phosphate pathway: a review. J. Inherit. Metab. Dis. 31:703–717 [DOI] [PubMed] [Google Scholar]
- 48. Wiesenborn D. P., Rudolph F. B., Papoutsakis E. T. 1988. Thiolase from Clostridium acetobutylicum ATCC 824 and its role in the synthesis of acids and solvents. Appl. Environ. Microbiol. 54:2717–2722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Yu Y., Tangney M., Aass H. C., Mitchell W. J. 2007. Analysis of the mechanism and regulation of lactose transport and metabolism in Clostridium acetobutylicum ATCC 824. Appl. Environ. Microbiol. 73:1842–1850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zverlov V. V., Berezina O., Velikodvorskaya G. A., Schwarz W. H. 2006. Bacterial acetone and butanol production by industrial fermentation in the Soviet Union: use of hydrolyzed agricultural waste for biorefinery. Appl. Microbiol. Biotechnol. 71:587–597 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.