

Abstract
Streptococcus pneumoniae expresses more than 90 capsule types, and currently available pneumococcal vaccines are designed to provide serotype-specific protection. Consequently, serotyping of pneumococcal isolates is important for determining the serotypes to be included in pneumococcal vaccines and to monitor their efficacy. Yet serotyping of pneumococcal isolates has remained a significant technical challenge. By multiplexing many assays, we have now developed a simple yet comprehensive serotyping assay system that can not only identify all known pneumococcal serotypes but also subdivide nontypeable (NT) isolates into those with or without the conventional capsule locus. We have developed this assay system to require only six key reagents: two are used in one multiplex inhibition-type immunoassay, and four are required in two multiplex PCR-based assays. The assay system is largely automated by a seamless combination of monoclonal antibody-based and PCR-based multiplex assays using the flow cytometric bead array technology from Luminex. The assay system has been validated with a panel of pneumococci expressing all known pneumococcal serotypes and was found to be easily transferable to another laboratory.
INTRODUCTION
Streptococcus pneumoniae is an important human pathogen expressing more than 90 serologically distinct polysaccharide (PS) capsules (5, 12, 21). Currently available pneumococcal vaccines provide serotype-specific protection (1, 23). Consequently, in the wake of the unqualified success of the 7-valent conjugate vaccine, the serotypes covered by the vaccine have become uncommon, whereas nonvaccine serotypes have become more common than before (13, 28). Also, the prevalence of a pneumococcal serotype differs for different locations and times (9, 11). Consequently, it is important to survey the serotypes of pneumococcal isolates circulating in many populations in order to monitor the efficacy of current vaccines and to select serotypes for future vaccines.
Classically, pneumococci are serotyped by the quellung reaction (12, 17). However, the quellung reaction is very labor-intensive, and several new approaches have appeared. One approach utilizes PCR-based systems that analyze DNA sequences of the capsular polysaccharide synthesis gene locus (cps), which contains the genes for capsule biosynthesis (3, 4, 15, 16, 19, 24). Another approach is a multiplex immunoassay for capsular polysaccharides using a set of monoclonal antibodies (MAbs) to identify the vaccine-related serotypes (30). While these two approaches overcome some of the limitations associated with the quellung reaction, they still have significant limitations. For instance, PCR products have commonly been identified by manual electrophoresis (20) or line blot hybridization (15). Also, due to vaccine-caused shifts in serotype prevalence, the multiplex immunoassay with limited coverage can no longer identify the serotypes of a large fraction of contemporary isolates. Consequently, pneumococcal serotyping still remains a challenge to most laboratories.
An ideal serotyping system should not only be simple to perform but should also cover all pneumococcal serotypes. The ability to test for all known serotypes is important, because the serotype prevalence not only differs due to vaccine-induced serotype shifts but also differs among different regions (11) or over time (9). Also, vaccine usage may increase the prevalence of nontypeable (NT) pneumococci (25), which, in turn, are heterogeneous. A study (10) divided them into two groups: group I isolates have conventional but defective cps, whereas group II has nonconventional cps with novel genes (10). Within NT group II isolates, clade 1 has pspK (encoding pneumococcal surface protein K); clade 2 has aliB-like ORF1 (referred to here as aliC, for simplicity) and aliB-like ORF2 (named aliD); and clade 3 has aliC (10; also I. H. Park et al. unpublished data). To develop a simple yet comprehensive pneumococcal serotyping system, we have seamlessly combined the PCR- and MAb-based approaches to produce an automated and multiplexed typing system that is simple to perform and can easily be transferred to another laboratory.
MATERIALS AND METHODS
Bacterial strains.
Our study used a panel of bacteria that consisted of 290 isolates. The panel included 90 isolates purchased from the Statens Serum Institute (Copenhagen, Denmark), representing the 90 different pneumococcal serotypes described previously (12). In addition, pneumococci expressing the three newly described serotypes (6C, 6D, and 11E), a laboratory strain with no capsule (R36A), and three clades of NT clinical isolates with null capsule genes, recently described by us (Park et al., unpublished), were also included. The panel had five clinical isolates for each of serotypes 1, 3, 4, 5, 6A, 6B, 6C, 7F, 8, 9N, 9V, 10A, 11A, 11E, 12F, 14, 15B, 17F, 18C, 19A, 19F, 20, 22F, 23F, and 33F, four isolates expressing serotype 2, and three 6D isolates. The panel included 53 clinical isolates of pneumococci expressing the serotypes not included in the 23-valent PS vaccine. The panel also included 3 NT pneumococci obtained from the Centers for Disease Control and Prevention (CDC) (see Table 4). In addition to pneumococci, the panel also contained isolates from several closely related species: one Streptococcus mutans, three Streptococcus mitis, and three Streptococcus oralis isolates. It also had one isolate each from S. pyogenes and S. agalactiae.
Table 4.
Testing of clinical samples with the PCR-based multibead assay
| Assay | Serotype | No. of isolates detected |
|---|---|---|
| Quellung | 13 | 3 |
| 29 | 1 | |
| 31 | 3 | |
| 34 | 4 | |
| 15A | 3 | |
| 15C | 8 | |
| 16F | 3 | |
| 18A | 2 | |
| 23A | 3 | |
| 23B | 2 | |
| 38 | 3 | |
| 28A/28F | 3 | |
| 33A | 2 | |
| 35A/35C | 2 | |
| 35B | 4 | |
| 35F | 1 | |
| 7C | 3 | |
| NTa | 3 | |
| PCR based | 13 | 3 |
| 29 | 1 | |
| 31 | 3 | |
| 34 | 4 | |
| 15A/15F | 3 | |
| 15B/15C | 8 | |
| 16F | 3 | |
| 18A/18B/18C/18F | 2 | |
| 23A | 3 | |
| 23B | 2 | |
| 25F/25A/38 | 3 | |
| 28A/28F | 3 | |
| 33F/33A/37 | 2 | |
| 35A/35C/42 | 2 | |
| 35B | 4 | |
| 35F/47F | 1 | |
| 7B/7C/40 | 3 | |
| NT (cpsA+) | 3 |
NT, nontypeable.
All bacterial isolates were recovered from frozen vials on blood agar plates, and isolated colonies were cultured in Todd-Hewitt broth with 0.5 yeast extract (THY) medium overnight. The pneumococci in the liquid cultures were lysed as described previously (30), and the lysates were stored at −20°C until needed. These lysates were used for our serotyping system with 3 reactions: reaction A consists of 26 PSs detected with MAbs, and reactions B and C are PCR based as described below.
Coupling of polysaccharides to microspheres.
Each of 26 pneumococcal capsular PSs (serotypes 1, 2, 3, 4, 5, 6A, 6B, 6C, 7F, 8, 9N, 9V, 10A, 11A, 11E, 12F, 14, 15B, 17F, 18C, 19A, 19F, 20, 22F, 23F, and 33F) was conjugated to one of 26 types of MicroPlex microspheres (Luminex, San Antonio, TX). All of the PSs were purchased from the ATCC (Manassas, VA) except for those of serotypes 6A, 6C, and 11E. Serotype 6A PS was obtained from G. Schiffman (Brooklyn, NY), but serotype 6C and 11E PSs were purified in our laboratory (21, 32). For each of the PSs, 12 million carboxyl MicroPlex microspheres (Luminex, San Antonio, TX) were washed twice in morpholineethanesulfonic acid (MES) buffer (100 mM MES in water [pH 6]), resuspended in 1 ml of adipic acid dihydrazide (ADH) solution (35 mg/ml of ADH in MES buffer), and mixed with 200 μl of 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC) solution (200 mg/ml of EDC in MES buffer). The reaction mixture was briefly sonicated using a Bransonic 2200 sonicator (Branson Ultrasonics Corporation, Danbury, CT) and was vortexed gently before being incubated for 1 h at room temperature (RT) in the dark. The beads were washed with water and were resuspended in 400 μl of water. The resuspended beads were first mixed with 320 μl of a PS solution and 80 μl of an EDC solution (100 mg/ml of EDC in water) and then incubated with shaking at 37°C for various periods. PS concentrations (10 to 250 μg/ml) and incubation periods (1 to 18 h) differed for different serotypes. After the incubation, 80 μl of 500 mM glucose and 80 μl of 500 mM glycine (pH 7) were added to the reaction mixture. After a 30-min incubation at RT with shaking, the beads were washed with the wash buffer (phosphate-buffered saline [PBS] with 0.1% Tween 20 and 0.02% sodium azide) and were resuspended in 1 ml of storage buffer (PBS with 0.01% Tween 20, 1% bovine serum albumin [BSA], 0.1% glucose, and 0.02% sodium azide). After the bead was aged for 10 days at 4°C, the stock bead mixture was made by mixing all 26 bead suspensions together with 54 ml of storage buffer, and this mixture was stored frozen in 1-ml aliquots.
Multibead serotyping assay with MAbs (reaction A).
One frozen vial of PS-coated stock bead mixture was thawed, diluted 3-fold with 2 ml of blocking buffer (PBS with 0.1% Tween 20, 0.02% sodium azide, and 1% BSA), and resuspended by sonicating. Twenty-five microliters of the bead mixture was added to each well of a 96-well plate with a filter bottom (Thermo Fisher, Waltham, MA), and the beads were washed twice with wash buffer using vacuum suctioning. After resuspension of the beads with 25 μl of blocking buffer, 25 μl of one diluted bacterial lysate and 25 μl of the diluted MAb pool were added to each well. The diluted bacterial lysates were prepared by diluting the bacterial lysates 10- or 30-fold in blocking buffer. The MAb pool was prepared by mixing 26 different hybridoma culture supernatants (see Table 1), stored frozen in 1-ml aliquots, and then diluted 3-fold with blocking buffer before use. After a 30-min incubation at RT in the dark with shaking (700 rpm), the plate was washed four times with the wash buffer. To detect bound MAb, 50 μl of phycoerythrin (PE)-conjugated goat anti-mouse immunoglobulin (BD Pharmingen, Franklin Lakes, NJ) was diluted 1:200 in blocking buffer and was added to each well. The plates were incubated for 30 min in the dark with shaking. After 4 washes, the beads were resuspended in 75 μl of wash buffer, and the 96-well plate was then incubated for 2 h in the dark at RT before being placed in a Bio-Plex 200 analyzer (Bio-Rad, Hercules, CA) for the acquisition of fluorescence data. The fluorescence data of each sample for each serotype were converted to a normalized signal by the following equation: normalized signal = 100 × (sample signal − background signal)/(blank-control signal − background signal). For an unknown sample, the serotype with a normalized signal of <33% was selected. To facilitate data analysis, a computer program generated a table containing the normalized signals of all serotypes for each sample. The normalized signals were displayed in red when they were less than 33%, in blue when they were between 33 and 67%, and in black when they were above 67%.
Table 1.
Specificity and sensitivity of the MAb-based serotyping assay
| MAb no. | MAb designation | Serotype specificitya | IC50 (ng/ml)b |
|---|---|---|---|
| 1 | Hyp1G4 | 1 | 8 |
| 2 | Hyp2M2 | 2 | 10 |
| 3 | Hyp3M6 | 3 | 6 |
| 4 | Hyp4M3 | 4 | 6 |
| 5 | Hyp5M3 | 5 | 12 |
| 6 | Hyp6AM3 | 6A | 3 |
| 7 | Hyp6BM1 | 6B | 77 |
| 8 | Hyp6DM5 | 6C/6D | 14 |
| 9 | Hyp7FM15 | 7F/7A | 52 |
| 10 | Hyp8M2 | 8 | 10 |
| 11 | Hyp9NM3 | 9N | 8 |
| 12 | Hyp9VM7 | 9V/9A | 1 |
| 13 | Hyp10AG3 | 10A/39 | 58 |
| 14 | Hyp11AM2 | 11A/11D/11F | 13 |
| 15 | Hyp11AM1 | 11E/(11A/11D/11F) | 15 |
| 16 | Hyp12FM3 | 12F | 26 |
| 17 | Hyp14M11 | 14 | 5 |
| 18 | Hyp15BG5 | 15B/(15C) | 68 |
| 19 | Hyp17FM6 | 17F/17A | 6 |
| 20 | Hyp18CM1 | 18C | 5 |
| 21 | Hyp19AG1 | 19A | 5 |
| 22 | Hyp19FM3 | 19F | 5 |
| 23 | Hyp20G5 | 20 | 29 |
| 24 | Hyp22FM2 | 22F/22A | 4 |
| 25 | Hyp23FG3 | 23F | 16 |
| 26 | Hyp33FG1 | 33F/33A | 18 |
Serotypes in parentheses are those with partial cross-reaction. Hyp10AG3 does not cross-react with 10B, unlike Danish factor serum 10d (12).
The PS concentration required to inhibit 50% of MAb binding to the enzyme-linked immunosorbent assay plate.
Coupling of DNA probes to microspheres.
To prepare beads for conjugation, 2 million carboxyl MicroPlex microspheres (Luminex, San Antonio, TX) were washed with water before being suspended in 50 μl of 0.1 M MES (pH 4.5) by sonication. Two microliters of the oligonucleotide probe stock (100 μM in water, kept at −20°C) and 2.5 μl of fresh EDC solution (10 mg/ml in MES) were added to the bead suspension, and the reaction mixture was mixed well and was incubated for 30 min at RT in the dark with shaking. After the first incubation, 2.5 μl of fresh EDC solution (10 mg/ml) was added to the reaction mixture before another 30-min incubation at RT in the dark with shaking. The beads were washed first with 1 ml of 0.02% Tween 20 and then with 1 ml of 0.1% sodium dodecyl sulfate (SDS) and were resuspended in 400 μl of TE buffer (10 mM Tris–1 mM EDTA [pH 8.0]) by vortexing and sonication. All of the beads were mixed together, and the resulting bead pool (stock bead mixture) was stored frozen in 0.4-ml aliquots.
Multibead serotyping assay with multiplex PCR (reactions B and C).
For serotypes not identified by MAbs, two multiplex PCRs were performed in 25-μl volumes containing 5 μl of the bacterial lysate (diluted 1:50) and 20 μl of the PCR mixture. The PCR mixture was made by mixing 990 μl of 2× primer pool, 990 μl of water, and 20 μl (100 U) of Takara Ex Taq DNA polymerase (Takara Bio, Madison, WI). One aliquot (990 μl) of 2× primer pool is made by mixing 250 μl of 10× Ex Taq buffer (Takara Bio, Madison, WI), 200 μl of 2.5 mM deoxynucleoside triphosphates (dNTP), and 6.25 μl of each stock primer (except the lytA primers) in Table 2, followed by the addition of enough water to make the final volume 990 μl. Instead of 6.25 μl, only 5.0 μl of the lytA primer stocks was added. One of each primer pair has a biotin label at the 5′ end for detection by streptavidin. Thermal cycling was performed in a Mastercycler gradient system (Eppendorf, Westbury, NY) using the following conditions: 94°C for 15 min; 35 amplification cycles of 94°C for 30 s, 55°C for 30 s, and 72°C for 1 min; and finally 72°C for 10 min for extension. The PCR products were kept at 4°C until further analysis.
Table 2.
Sequences of primers and probes for reaction B
| Specificity | Oligonucleotide codea | Sequence (5′–3′)b | Source or reference |
|---|---|---|---|
| lytA | 5322 | CAA CCG TAC AGA ATG AAG CGG | 33 |
| 1006 | GTC TTT CCG CCA GTG ATA AT | ||
| 3322 | TTA TTC GTG CAA TAC TCG TGC G | ||
| cpsA (except 25A, 25F, 38) | 5202 | GCA GTA CAG CAG TTT GTT GGA CTG ACC | 20 |
| 1039 | GG TGC TGT CAC ACT CGT CAG | This study | |
| 3202 | GAA TAT TTT CAT TAT CAG TCC CAG TC | 20 | |
| 7B, 7C, 40 | 5317 | AAA ACT CAA GTA TCT GTG C(T)CA CCT T | 33 |
| 1001 | TCC AAA TTT SAG TAA ACC AAC CTA A | ||
| 3317 | CAT CTC TAT TCG ACC TTG CGT TA | ||
| 21 | 5352 | CAA TTC TAC TGA GTC CAT ATT ATG AAA | 33 |
| 1017 | GAT AGT TTC TCT GTA TCA AAT AGC GA | ||
| 3352 | ACC ATC GTA CCT GCA CCA TAA | ||
| 33F, 33A, 37 | 5351 | TCA ACT AGT CAA GGA TTT GAT GG | 14 |
| 1016 | TGA TAC CAC AAG TAA CAG AGT CG | ||
| 3351 | CGT ATC AGA TTT GCG ATT TC | ||
| 15B, 15C | 5346 | TAA TAA GCG GAT GAT TGT AGC G | 14 |
| 1011 | GAG CAG GAA TCA GAA CAC AAT C | ||
| 3346 | TAT ACT GAT TAA CTT TCC AGA TGG G | ||
| 16A | 5361 | CCG CTC ACG GTA TGG ACT A | 33 |
| 1026 | GGA GTA AAT GAT GTG TAG TGA AAA CC | ||
| 3361 | CCA GCA ATA TAC TCA GGA AAT AAT TC | ||
| 16F | 5342 | TTG TTC TTA CAT TTA GCC GTA GTG | 33 |
| 1007 | GTT GAA AGA ATA CGA TTC CTA CAA G | ||
| 3342 | TCG TCG TTG AAA ACA ATT TCT TAC | ||
| 18 | 5607 | AAT TGT TCT TTT CCT GTA CTC AGT C | 14 |
| 1039 | GGA GGA CTT AGT CAA TTT ATC TTG | ||
| 3607 | CGA ACC ATT GAA ACT ATC ATC TG | ||
| 23A | 5518 | TTT ACT TTA ATT TAT AGC TTT TTG GCT AA | 33 |
| 1002 | TGC CTT TTT TAA CGA GGT TG | ||
| 3318 | GGT GCA TGA GTT AGG AGA AAG TG | ||
| 23B | 5319 | GGA TCG TTG TTC ATA GCG G | 33 |
| 1003 | ATA ATT ACT GGT CTG TGA TTT TTC TTT | ||
| 3319 | GAT AAT AAA GAA ATT ACT AAC CAT GTC GT | ||
| 25F, 25A, 38 | 5343 | GAC TAC AAA CTG CGG TAG TAG AAA TG | 33 |
| 1008 | ATA GGA ACT CTA GGG TTT AGT TTT TTC | ||
| 3343 | TGG AAC AAT TCT AAT CGT TAA TAC G | ||
| 10A, 10B | 5373 | TGA GCT ATT TAA GGA CCT GGG | 14 |
| 1024 | GTT TAG AAA CCT TGC CCA GG | ||
| 3373 | GCA AGC GTC ACT TTC TTG A | ||
| 43 | 5369 | AGA TCA AAT GGT GGT ATT AGG AA | 33 |
| 1034 | TCG GGT GTA CAA ATC CTA AAC TA | ||
| 3369 | GGA ATA GAT CAT TAA CCC TAA TGA AT | ||
| 36 | 5368 | CAA TTT CCC CTT ATT CTG TAG TTC | 33 |
| 1033 | AGA TAA ATA CAT CAT TAT TGA CGA ACA | ||
| 3368 | TGG AGA TCC CCA AGA GAA AAT A | ||
| 48 | 5372 | GCA TTT GGA GTT ATT GCC CTA C | 33 |
| 1037 | CCT ATA AAC ACA CTC AAA ACT AGC A | ||
| 3372 | CGA CGG AAT CAA TAT AAA TAA GTG ATA | ||
| 34 | 5608 | TTA AAA GTA TTA TTG GTA GTG ATT CTT TTG | This study |
| 1040 | AA TCC ATT GGT ACT CTT CAC AAA | ||
| 3608 | TGT AAA GAC ATT CCC TGT AGG C | ||
| 35A, 35C, 42 | 5367 | GGA GAC TA(/G)T TAA AAC TTT TTT CGT TC | 33 |
| 1032 | CCT ACT TTA TTA ATG CCT GTT TGA G | ||
| 3367 | TTA AGT AAG TCT TCG CAA TCC AG | ||
| 35F, 47F | 5521 | ATA AAA AGA AAG TCT TTG CCA GAG | 33 |
| 1005 | AAA GTC ACA TC(T)T AAA ATT GAC ACA AC | ||
| 3321 | CAA CTT TTG GAA GAT ACT GAA CAT AA | ||
| 35B | 5345 | CTA ATT TGG CTA TGA AGC TAA TCC C | 33 |
| 1010 | AAG CGT GAA AAA TTT TAA TAA AAG AC | ||
| 3345 | TAA CTT AAA TAG GCA TTA ACA AAA TAG GT | ||
| 6C, 6D | 5325 | CATTTTAGTGAAGTTGGCGGTGGAGTT | This study |
| 1042 | TTTGCAGGTGTAGTAGGCAGG | ||
| 3645 | ACTTTACACGACAAACCCATAGAAGC |
Oligonucleotide codes beginning with 1, 3, or 5 designate probes, reverse primers, or forward primers, respectively.
One primer in each primer pair (in boldface) has a biotin label at the 5′ end for detection with avidin. All the probes have an amine group at the 5′ end. Stock primer solutions have 100 μM oligonucleotides in water.
To identify the PCR amplicons, 20 μl of the 1:100-diluted PCR mixture was transferred to a 96-well PCR plate and was heated for 10 min at 95°C. One aliquot of the DNA-coupled stock bead mixture corresponding to the PCR was thawed and washed with 1× tetramethylammonium chloride (TMAC) buffer (3 M TMAC, 1 g/liter SDS, 50 mM Tris-HCl, 4 mM EDTA [pH 8.0]) by centrifugation. The beads were then suspended in 4 ml of 1.5× TMAC at 48°C and were briefly sonicated. Forty microliters of the bead mixture was quickly transferred to each well and was mixed with the PCR product by brief vortexing. The plate was incubated at 48°C for 30 min in the dark. A 135-μl volume of 1× TMAC (48°C) was added to each well of the PCR plate and was mixed before the entire contents of each well were transferred to a filter plate (Millipore, Billerica, MA). After the plate was washed with 1× TMAC, 50 μl of PE-streptavidin (BD Pharmingen, Franklin Lakes, NJ) was added to each well of the plate. After a 20-min incubation at 48°C, the plate was washed once with 1× TMAC and once with PBS–0.1% Tween 20. The beads were resuspended in 75 μl of PBS–0.1% Tween 20, and the filter plate was inserted into a Bio-Plex 200 analyzer (Bio-Rad, Hercules, CA) for the acquisition of fluorescence data. Fluorescence signals were converted to signal-to-noise (S/N) ratios by dividing the fluorescence signal of a sample by the background signal for each serotype. All the control samples produced S/N ratios less than 4 for incorrect serotypes and greater than 30 for the correct serotypes. For an unknown sample, the serotype with an S/N ratio greater than 10 was chosen. To facilitate data analysis, a computer program generated a table containing the S/N ratios of all serotypes for each sample. The S/N ratios were displayed in red when they were greater than 10, in blue when they were between 3 and 10, and in black when they were below 3.
RESULTS
Characterization of MAb-based multibead serotyping assays (reaction A).
Although we have previously described a multiplexed immunoassay for 26 vaccine-related serotypes using MAbs (30), it had a low degree of multiplexing and required several separate assays. We have now overcome these limitations by developing a 26-fold multiplexed assay with a new set of 26 different MAbs (Table 1). While 16 MAbs were monospecific, 10 MAbs were multispecific: they reacted with related serotypes (Table 1). The multispecificity of cross-reactive MAbs would not cause difficulties in serotype surveillance, however, since the cross-reactive serotypes, such as 7A or 9A, are rare and epidemiologically insignificant.
In the assay configuration used here, the multispecific antibodies were fully cross-reactive, as illustrated with a 9A/9V-specific antibody (Fig. 1B), except for MAbs Hyp11AM1 and Hyp15BG5. Hyp11AM1 preferentially bound to serotype 11E and reacted weakly with serotypes 11A, 11D, and 11F. Since Hyp11AM1 is used to identify 11E isolates only, and another MAb (Hyp11AM2) is used to identify serotypes 11A, 11D, and 11F, the unequal binding pattern of Hyp11AM1 caused no difficulties. In the case of serotypes 15B and 15C, Hyp15BG5 reacted with 15C less efficiently, but the unequal cross-reactivity is not relevant, since serotypes 15B and 15C interconvert in vitro (26, 27).
Fig. 1.

(A and B) Normalized fluorescence signals of the beads coated with serotype 14 PS (A) or serotype 9V PS (B) obtained with the panel of test samples (x axis) by use of the multiplexed inhibition type immunoassay with MAbs. The numbers on the X axes represent the numbers assigned to the isolates in the test panel that includes all pneumococcal serotypes and NT isolates. (C and D) Signal-to-noise ratios of the beads coated with probes for serotype 21 (C) and serotype 33F (D) obtained with the panel of test samples by use of the PCR-based multiplexed assay. The panel includes all 93 pneumococcal serotypes that have been identified so far and 4 isolates of NT pneumococci representing both group I and group II. Dashed lines indicate cutoffs and separate the positively reacting serotypes from the nonreacting serotypes.
The specificity of the tests included in this MAb-based multiplex assay, which is named reaction A, was then examined with a panel of pneumococcal isolates representing all 93 known pneumococcal serotypes, as well as both groups of NT pneumococci and some nonpneumococcal streptococcal species (Fig. 1A and B). Nonpneumococcal isolates produced normalized (fluorescence) signals of >67%. All pneumococcal isolates produced normalized signals less than 33% for the tests of their own or cross-reactive serotypes but more than 67% for other, unrelated serotypes. Figure 1 shows the normalized signals obtained by two tests (serotypes 14 and 9V) with all the isolates in the test panel, and Table 1 shows the summary of the specificity for all the tests. A normalized signal of 33% was used as the cutoff for unknown samples. The sensitivities of the tests included in reaction A were determined using purified capsular PS. Although the sensitivities differed more than 100-fold among the tests, all the tests in reaction A showed significant inhibition (i.e., <50% of the normalized signal) at 80 ng/ml of capsular PS (Fig. 2; Table 1); the concentrations of PS inhibiting 50% of MAb binding (50% inhibitory concentrations [IC50]) are shown in Table 1. These sensitivities were sufficient to identify serotypes even when a pneumococcal lysate was diluted 100-fold (data not shown).
Fig. 2.

Normalized fluorescence signals of the PS-coated latex particles in the presence of varying concentrations of homologous free inhibitory PS in solution. The serotypes of inhibitory PS are given in the key. Two horizontal dashed lines indicate 33% and 67% of normalized signals, the values that were used as cutoffs.
Characterization of the PCR-based multibead serotyping assays (reactions B and C).
Although multiplexed PCR methods for typing pneumococci are reported in the literature (15, 20, 33), generally, cumbersome methods are used to identify PCR products (15, 20), and the degree of multiplexing is low (20). We used high degrees (21- and 22-fold) of multiplexing and simplified the identification of PCR products by using Luminex technology. Two PCR-based assays, designated reactions B and C, include tests for the serotypes that were not covered by reaction A, except for tests for 6C/6D and 15B/15C. There are tests for 6C/6D in both reactions A and B, since the 6C/6D test was developed for reaction B before a suitable MAb was developed for reaction A. The test for 15B/15C was included in order to independently monitor the performance of the 15B-specific MAb, which is unequally cross-reactive for 15C.
In addition to serotype-specific tests, tests for lytA and cpsA were added to reactions B and C to confirm the species identification as S. pneumoniae (Tables 2 and 3). Two cpsA tests were included, because the first cpsA test does not detect the cpsA of serotypes 25A, 25F, and 38 (20), but the second cpsA test does. Reaction C has tests for genes such as aliC, aliD, and pspK to classify group II NT pneumococci, which lack cpsA. In contrast, group I NT pneumococci are cpsA+ (10). Even with these additional and overlapping tests, all 43 target genes could be detected easily and quickly with only two PCRs and the Luminex detection assays.
Table 3.
Sequences of primers and probes for reaction C
| Specificity | Oligonucleotide codea | Sequence (5′–3′)b | Source or reference |
|---|---|---|---|
| cpsA for 25A, 25F, 38 | 5694 | GGGAACGACTATCCTGTTGGAAATC | This study |
| 1046 | GTCAGCACTTGGTTTCGTTCGAG | ||
| 3694 | CTTCTGTTGATTCCGTCCTCGATC | ||
| 9N, 9L | 5358 | TCA ATG GCG ACT TTA TTT GC | 14 |
| 1023 | GAA CTT TGG GAA TAT AAT CAA AAG | ||
| 3358 | AGT CTA TTA TCT CCT GTA GGG TGC | ||
| 28F, 28A | 5354 | CAG AGT TTG GTC GAG GTT CCT A | 33 |
| 1019 | AGA ACT AAA TAC AGT GCA ATA ATT GG | ||
| 3354 | GCT CAA CTT TAT TTC TCT AGA ATA AAC A | ||
| 10F, 10C | 5355 | TAG TTT TGG TTA CGT AGT TGT TGA CT | 33 |
| 1020 | GAA AAC TTG CCC AAA TCC TT | ||
| 3355 | GCA ATA(/G) AAT ACT GTA GCA TAC GAT AGT T | ||
| 11B, 11C | 5356 | TCT GGT GCT AAG GGG ATC AA | 33 |
| 1021 | TGC ATA AGC TGA TTA TGA GCA TAG | ||
| 3356 | CCA ATT ACT CCA TTA TCT ATT GCT AAT | ||
| 13 | 5348 | GAT GGG AAA ATA CGA TAT GCT C | 33 |
| 1013 | TGA GCT AAA TGT TGA ATA TTT ATA CCC | ||
| 3348 | GAA AAT CGT AAC ATG GAA AAA GTA A | ||
| 24F, 24A, 24B | 5363 | TCA ACA CTT ATG ATG G(A)TG CCT G | 33 |
| 1028 | CAC AAT CCA AAA CTT AAG TTG TTT C | ||
| 3363 | GCA GAA ACA AAA(/G) GTA AGA ATT ATA GAT ATC | ||
| 12, 44, 46 | 5374 | TGA ATA TGG ACG GTG GAG | 14 |
| 1025 | GAA GAA GTT CAA CAA TCG CT | ||
| 3374 | AGC AAA GAA AGC CGA AAG | ||
| 19B, 19C | 5362 | AGA ATT CGG AGA TTT GTG GTA T | 33 |
| 1027 | TTC GTA CTG AAA ATT CAT TTC G | ||
| 3362 | CAA TCC ACC TCC ATA AAC GA | ||
| 27 | 5364 | GCA GCC ACC TCT TCT CAT TC | 33 |
| 1029 | CGC CAA ATT CTA TAC CAA CTA GTA T | ||
| 3364 | GGA AGG AAC AAC CCA ACA AT | ||
| 32F, 32A | 5365 | GGT ATG CTT ACA ATG AGA CGC | 33 |
| 1030 | CCA CTT CCC AGA GGA AAA TA | ||
| 3365 | AAT TCG TTC CCG GAT AAG ATG | ||
| 15F, 15A | 5347 | TAT TTC CTT CCT ATG GGA CAA C | 33 |
| 1012 | AGT CCT TTC CCA AAT ATA GCA CT | ||
| 3347 | GCA AGC ATT TTA CCA AGT TCA TAA A | ||
| 33B, 33D | 5366 | TCG TTG GAT GAC AAA ACT CTT AC | 33 |
| 1031 | GCT CAA TGT GAC AGG GAG AA | ||
| 3366 | CCT CCC TGA GCC AAA ATA AC | ||
| 31 | 5344 | TGA AAA TCC CTT AGT GAC ATC TG | |
| 1009 | GAG CCT TCT CAA TAG TCA TAA AAA A | 33 | |
| 3344 | GCC ATA ATC AAA AAT AAG TTA GAC ATA A | ||
| 41F, 41A | 5357 | TGA CAC TAT TTA TAA TTG CTT TAT CCT T | 33 |
| 1022 | GGG TGC AAG GTG ATT ATG TAT | ||
| 3357 | TAG CGA GAA ACT ATC TGC ATC TTG | ||
| 29 | 5353 | CCG AAA ATT GTT CAC AGG ATA C | 33 |
| 1018 | TAA CAA GCA GAA TAA GCA AAA TAG C | ||
| 3353 | AGC TTT CTT TTG TAC GAC TCT TTT A | ||
| 45 | 5370 | TAT GCA GGA AAT ATC CGA GAA GG | 33 |
| 1035 | CAG CAT ATC TTG CAC GAT AAT GAA | ||
| 3370 | CCG TGA AAC AGA AAC GCT ATG | ||
| 47A | 5371 | TAT TTG CCA TAA CGG ACT CTA GAA C | 33 |
| 1036 | TTT TTA ACA ACC TTG TAT AGA ATA CCT C | ||
| 3371 | GCT AAA ATA ATA AAT AGC GAA CTT ACT ACA | ||
| 33C | 5375 | CGA ATC GTC ATA AGG CAA AA | 33 |
| 1038 | ACC TAC TGT GAC AGG GAA TAG TAA A | ||
| 3375 | AAT AGG AGT AAC AAA GAG AAG CCT AA | ||
| NCC2 | 5433 | AACACTTGGAACGGAGAATG | This study |
| 1044 | GACCAGATTACCAAGATCCAGCAAC | ||
| 3433 | GCCCTTTGTTATACCTAGATGTTTC | ||
| NCC2 and NCC3 | 5666 | ATGCCAAATGGTTCACGGCTG | This study |
| 1045 | GTGACAGGTATCAAGTACGCAGTG | ||
| 3666 | CTGGTCGTCGATTGCTTTCACAC | ||
| NCC1 | 5658 | GCAAATCAGCCAGTAACTGTGA | This study |
| 1043 | CTGTAGAACAGGCAAAGAGCAAGG | ||
| 3658 | CAAGATAAGCTTTCTGCACCTCT |
Oligonucleotide codes beginning with 1, 3, or 5 designate probes, reverse primers, or forward primers, respectively.
One primer in each primer pair (in boldface) has a biotin label at the 5′ end for detection with avidin. All the probes have an amine group at the 5′ end. Stock primer solutions have 100 μM oligonucleotides in water.
The specificities of reactions B and C were examined with a panel of pneumococci, including all 93 pneumococcal serotypes and both groups of NT pneumococci (Fig. 1C and D). All pneumococcal isolates produced S/N ratios greater than 30 for the tests of their own serotypes but less than 4 for other serotypes. Nonpneumococcal isolates produced S/N ratios less than 2. Figure 1 shows the results obtained by a monospecific test (serotype 21) and a multispecific test (serotypes 33F, 33A, and 37) with the bacterial isolates in the test panel. An S/N ratio of 10 was chosen as the cutoff for unknown samples, and the specificities of all assays are summarized in Tables 2 and 3. The assay is sensitive enough to detect 5 pg of DNA, which corresponds to ∼2,000 copies of pneumococcal DNA. In practical terms, most of the lysates could be diluted an additional 50-fold, and therefore, the assay has sufficient sensitivity for practical uses.
Analysis of archived clinical isolates with MAb and PCR tests.
Although all the tests in our assay system were tested with a test panel, including all known serotypes, reaction A was reevaluated with a panel of clinical isolates that included three 6D isolates, four serotype 2 isolates, and five isolates each of serotypes 1, 3, 4, 5, 6A, 6B, 6C, 7F, 8, 9N, 9V, 10A, 11A, 11E, 12F, 14, 15B, 17F, 18C, 19A, 19F, 20, 22F, 23F, and 33F. All clinical isolates were correctly identified (see Table S1 in the supplemental material).
Testing reactions B and C with clinical isolates was difficult, because the serotypes detected by the two reactions are not common. Nevertheless, we tested our assay with 53 clinical isolates expressing the serotypes relevant to reactions B and C. Concordant results were obtained with all isolates (Table 4). Three samples that were originally identified as NT were cpsA+ and lacked pspK, aliC, and aliD. These 3 isolates, therefore, belong to NT group I, with defective cps loci (10).
Testing of mixtures of two different serotypes.
Although our assay was designed for individual pneumococcal isolates, it is inherently capable of detecting multiple serotypes in one sample. To investigate this capability, we prepared three pneumococcal lysates spiked with a secondary serotype at a 1% or 10% level. One was a type 3 lysate spiked with serotype 6B; another was a type 5 lysate spiked with type 12F; and the third was type 19F mixed with 33F. The serotypes used for spiking were chosen because the test was least sensitive of all serotypes (serotype 6B) or the lysates had a low titer, and therefore, these spiked samples provided stringent testing conditions. When the samples were tested with reaction A, the primary serotype was identified without difficulty in all samples. The secondary serotypes could be identified easily (<33%) in all 3 samples containing 10% (Fig. 3), but the samples containing the secondary serotype at 1% produced normalized signals that were substantially reduced (<67%) but not below 33%. Although additional studies are needed, reaction A should identify all secondary serotypes present at a 10% abundance level but may not identify some secondary serotypes at a 1% abundance level. Analogous studies of reactions B and C suggest that they should detect the secondary serotypes present at a 0.1 to 1% abundance.
Fig. 3.
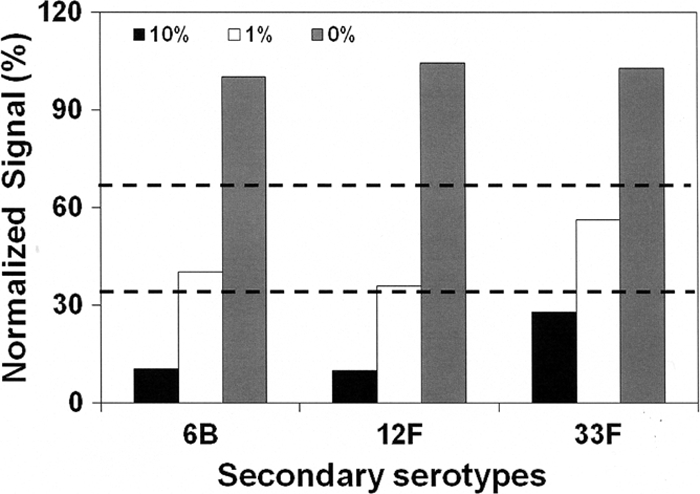
Normalized signals for the secondary serotypes in three samples. The abundances of the secondary serotypes are 10%, 1%, and 0%. Two dashed horizontal lines at 33% and 67% are used for decision making.
Portability of the assay system.
We found that the six key reagents (1 MAb pool, 2 primer pools, and 3 bead mixtures) have long shelf lives. To investigate whether our system is simple and robust enough to be transferred to another laboratory with minimal experience in serotyping, we sent the six key reagents, our assay protocol, and 75 test samples to K. H. Kim's laboratory in Seoul, South Korea, which has equivalent equipment (Bio-Plex). The test samples were blinded and included 72 samples representing all 69 serotyping reactions performed by the assay system. Seven samples were mixtures of two serotypes. The laboratory was able to set up all three reactions with ease and correctly identified all of the samples.
DISCUSSION
Since currently available pneumococcal vaccines provide serotype-specific protection and alter the distribution of pneumococcal serotypes, continued surveillance of serotypes of pneumococcal isolates is critical in monitoring pneumococcal vaccine efficacy and in improving vaccine formulations. However, the classical typing method used by many laboratories is labor-intensive, and therefore, serotyping surveillance is often limited to a small number of isolates and a few commonly found serotypes. By modifying and combining various approaches developed by us and others (8, 15, 20, 30), we have now developed a simple yet comprehensive serotyping assay system that can test pneumococcal isolates for all the known serotypes with only three multiplexed assays performing 69 different tests with only 6 key reagents (1 MAb pool, 2 primer pools, and 3 bead mixtures). The assay system is rapid and largely automated due to the use of flow cytometric bead array technology. Although the assay uses microtiter plates to accommodate many isolates, it can be used with individual tubes (e.g., Eppendorf tubes) for handling a few samples. In addition, the assay system is robust enough to be transferred to a distant laboratory with a suitable instrument but little experience in serotyping pneumococci. In summary, our assay system has overcome current limitations in serotyping pneumococci and should vastly simplify pneumococcal serotype surveillance.
Our assay system differs from other assays in several ways. First, although there are automated assay systems similar to ours using Luminex technology, these systems, unlike ours, can detect only a limited number of common serotypes (e.g., 40 serotypes). Second, our system is highly flexible due to the seamless integration of immunoassays and DNA assays that share the sample types and the assay platform. Consequently, our system can be readily expanded by adding either an immunoassay or DNA assays, which have complementary strengths and weaknesses (8, 18). This expandability allowed us to add several additional tests for identifying S. pneumoniae species in reactions B and C. Finally, our assay system is easy to standardize because it uses well-defined and extensively characterized reagents, such as oligonucleotides and MAbs.
Despite its strengths, our assay system does have some limitations. Our system does not distinguish some serotypes (e.g., 9A and 9V; serotypes 33A, 33F, and 37), and resolution of such serotyping ambiguities would require conventional manual methods based on polyclonal factor sera or specific DNA tests. Although we anticipate that these serotyping ambiguities should not cause practical difficulties in serotype surveillance, additional tests may be incorporated into our assay system, if necessary. Another limitation may be that our system has been developed for testing individual pneumococcal isolates. This restriction may be undesirable in studies of nasopharyngeal carriage of pneumococci, because the nasopharynx frequently harbors multiple serotypes (2), and identifying all the serotypes in the nasopharynx would require testing many isolates from a single swab. We imposed this restriction because pneumococcal and nonpneumococcal cps genes can be very similar to each other (29), and nonpneumococcal organisms may be the sources of the targets detected by any DNA detection system. Such false-positive detection may also occur with MAb-based immunoassays. However, our assay system is intrinsically capable of detecting multiple serotypes in a sample, and the genetic similarity may not cause practical problems. Consequently, the restriction of using isolates should be reevaluated in the future by studying nasopharyngeal swabs or their broth enrichment cultures (7), along with pneumococcal isolates from the swabs.
The vaccine-related serotypes that were included in reaction A could have been tested by PCR, as we have done for other serotypes. Nevertheless, the use of MAbs for reaction A provides several advantages. First, serotype surveillance often requires testing of pneumococcal isolates only for the vaccine-related serotypes, and the MAb-based reaction A would satisfy this need simply, without PCR. Also, the immunoassay provides more-reliable serotyping information, since it tests the capsule structure directly, unlike DNA tests, which provide indirect information. This was illustrated with a 19F strain that was genetically more similar to 19A (22) and with serotype 11A isolates that could not be genetically distinguished from serotype 11D (6). Also, the ability of the immunoassay to quantitatively measure capsular PS in samples can be valuable. For instance, with the immunoassay, pleural fluids were found to contain enough capsular PS to neutralize the anti-PS antibodies present in the host (31). Reaction A should be useful in vaccine production as well, since it can be used to quantify capsular PS during its purification and conjugation.
Because of difficulties in typing pneumococci, our knowledge of pneumococcal serotype prevalence is limited to select geographical areas and is deficient in a large part of the world. The areas with limited studies may have different serotype distributions or may harbor unusual serotypes. Because our assay can identify all serotypes and can be placed in laboratories with little experience in serotyping, the system should facilitate surveys of pneumococcal serotypes in different parts of the world. Since our assay can serotype pneumococcal isolates completely, including different clades of group II NT pneumococci, we may find new capsule types. Perhaps our assay system may help us to discover new serotypes and new clades among NT pneumococci as well as to accomplish complete surveillance in different parts of the world.
Supplementary Material
ACKNOWLEDGMENTS
We thank C. Steele for allowing us to use his Bio-Plex instrument, B. Beall at the CDC for generous support with bacterial isolates, and K. Honjo for tireless technical assistance.
This work was supported by NIAID-DMID-AI-30021 (to M.H.N.). The University of Alabama at Birmingham has IP rights on monoclonal antibodies and has applied for a patent on the serotypes 6C and 6D. M.H.N., J.Y., J.L., and W.H.B. are employees of the university.
Footnotes
Supplemental material for this article may be found at http://cvi.asm.org/.
Published ahead of print on 7 September 2011.
REFERENCES
- 1. Anonymous. 2010. Licensure of a 13-valent pneumococcal conjugate vaccine (PCV13) and recommendations for use among children—Advisory Committee on Immunization Practices (ACIP), 2010. MMWR Morb. Mortal. Wkly. Rep. 59:258–261 [PubMed] [Google Scholar]
- 2. Austrian R. (ed.), 2000. The enduring pneumococcus: unfinished business and opportunities for the future, p. 3–7. In Tomasz A. (ed.), Streptococcus pneumoniae: molecular biology and mechanisms of disease. Mary Ann Liebert, Inc., Larchmont, NY. [DOI] [PubMed] [Google Scholar]
- 3. Batt S. L., Charalambous B. M., McHugh T. D., Martin S., Gillespie S. H. 2005. Novel PCR-restriction fragment length polymorphism method for determining serotypes or serogroups of Streptococcus pneumoniae isolates. J. Clin. Microbiol. 43:2656–2661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Brito D. A., Ramirez M., de Lencastre H. 2003. Serotyping Streptococcus pneumoniae by multiplex PCR. J. Clin. Microbiol. 41:2378–2384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Calix J. J., Nahm M. H. 2010. A new pneumococcal serotype, 11E, has variably inactivated wcjE gene. . J. Infect. Dis. 202:29–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Camilli R., et al. 2011. Complete genome sequence of a serotype 11A, ST62 Streptococcus pneumoniae invasive isolate. BMC Microbiol. 11:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. da Gloria Carvalho M., et al. 2010. Revisiting pneumococcal carriage by use of broth enrichment and PCR techniques for enhanced detection of carriage and serotypes. J. Clin. Microbiol. 48:1611–1618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Dunbar S. A., Vander Zee C. A., Oliver K. G., Karem K. L., Jacobson J. W. 2003. Quantitative, multiplexed detection of bacterial pathogens: DNA and protein applications of the Luminex LabMAP system. J. Microbiol. Methods 53:245–252 [DOI] [PubMed] [Google Scholar]
- 9. Finland M., Barnes M. W. 1977. Changes in occurrence of capsular serotypes of Streptococcus pneumoniae at Boston City Hospital during selected years between 1935 and 1974. J. Clin. Microbiol. 5:154–166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hathaway L. J., Stutzmann Meier P., Battig P., Aebi S., Muhlemann K. 2004. A homologue of aliB is found in the capsule region of nonencapsulated Streptococcus pneumoniae. J. Bacteriol. 186:3721–3729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hausdorff W. P., Siber G., Paradiso P. R. 2001. Geographical differences in invasive pneumococcal disease rates and serotype frequency in young children. Lancet 357:950–952 [DOI] [PubMed] [Google Scholar]
- 12. Henrichsen J. 1995. Six newly recognized types of Streptococcus pneumoniae. J. Clin. Microbiol. 33:2759–2762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hicks L. A., et al. 2007. Incidence of pneumococcal disease due to non-pneumococcal conjugate vaccine (PCV7) serotypes in the United States during the era of widespread PCV7 vaccination, 1998-2004. . J. Infect. Dis. 196:1346–1354 [DOI] [PubMed] [Google Scholar]
- 14. Kong F., Brown M., Sabananthan A., Zeng X., Gilbert G. L. 2006. Multiplex PCR-based reverse line blot hybridization assay to identify 23 Streptococcus pneumoniae polysaccharide vaccine serotypes. J. Clin. Microbiol. 44:1887–1891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kong F., et al. 2005. A molecular-capsular-type prediction system for 90 Streptococcus pneumoniae serotypes using partial cpsA-cpsB sequencing and wzy- or wzx-specific PCR. J. Med. Microbiol. 54:351–356 [DOI] [PubMed] [Google Scholar]
- 16. Lawrence E. R., Griffiths D. B., Martin S. A., George R. C., Hall L. M. 2003. Evaluation of semiautomated multiplex PCR assay for determination of Streptococcus pneumoniae serotypes and serogroups. J. Clin. Microbiol. 41:601–607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lund E. 1960. Laboratory diagnosis of Pneumococcus infections. Bull. World Health Organ. 23:5–13 [PMC free article] [PubMed] [Google Scholar]
- 18. Miller M. B., Tang Y. W. 2009. Basic concepts of microarrays and potential applications in clinical microbiology. Clin. Microbiol. Rev. 22:611–633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. O'Halloran D. M., Cafferkey M. T. 2005. Multiplex PCR for identification of seven Streptococcus pneumoniae serotypes targeted by a 7-valent conjugate vaccine. J. Clin. Microbiol. 43:3487–3490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pai R., Gertz R. E., Beall B. 2006. Sequential multiplex PCR approach for determining capsular serotypes of Streptococcus pneumoniae isolates. J. Clin. Microbiol. 44:124–131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Park I. H., et al. 2007. Discovery of a new capsular serotype (6C) within serogroup 6 of Streptococcus pneumoniae. J. Clin. Microbiol. 45:1225–1233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Pimenta F. C., et al. 2009. Rarely occurring 19A-like cps locus from a serotype 19F pneumococcal isolate indicates continued need of serology-based quality control for PCR-based serotype determinations. J. Clin. Microbiol. 47:2353–2354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Robbins J. B., et al. 1983. Considerations for formulating the second-generation pneumococcal capsular polysaccharide vaccine with emphasis on the cross-reactive types within groups. . J. Infect. Dis. 148:1136–1159 [DOI] [PubMed] [Google Scholar]
- 24. Rubin L. G., Rizvi A. 2004. PCR-based assays for detection of Streptococcus pneumoniae serotypes 3, 14, 19F and 23F in respiratory specimens. J. Med. Microbiol. 53:595–602 [DOI] [PubMed] [Google Scholar]
- 25. Sa-Leao R., et al. 2009. Changes in pneumococcal serotypes and antibiotypes carried by vaccinated and unvaccinated day-care centre attendees in Portugal, a country with widespread use of the seven-valent pneumococcal conjugate vaccine. Clin. Microbiol. Infect. 15:1002–1007 [DOI] [PubMed] [Google Scholar]
- 26. van Selm S., van Cann L. M., Kolkman M. A., A. van der Zeijst B., van Putten J. P. 2003. Genetic basis for the structural difference between Streptococcus pneumoniae serotype 15B and 15C capsular polysaccharides. Infect. Immun. 71:6192–6198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Venkateswaran P. S., Stanton N., Austrian R. 1983. Type variation of strains of Streptococcus pneumoniae in capsular serogroup 15. . J. Infect. Dis. 147:1041–1054 [DOI] [PubMed] [Google Scholar]
- 28. Whitney C. G., et al. 2003. Decline in invasive pneumococcal disease after the introduction of protein-polysaccharide conjugate vaccine. N. Engl. J. Med. 348:1737–1746 [DOI] [PubMed] [Google Scholar]
- 29. Yang J., Ritchey M., Yoshida Y., Bush C. A., Cisar J. O. 2009. Comparative structural and molecular characterization of ribitol-5-phosphate-containing Streptococcus oralis coaggregation receptor polysaccharides. J. Bacteriol. 191:1891–1900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yu J., da Gloria Carvalho M., Beall B., Nahm M. H. 2008. A rapid pneumococcal serotyping system based on monoclonal antibodies and PCR. J. Med. Microbiol. 57:171–178 [DOI] [PubMed] [Google Scholar]
- 31. Yu J., Salamon D., Marcon M., Nahm M. H. 2011. Pneumococcal serotypes causing pneumonia with pleural effusion in pediatric patients. J. Clin. Microbiol. 49:534–538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zartler E. R., et al. 2009. Structure of the capsular polysaccharide of pneumococcal serotype 11A reveals a novel acetylglycerol that is the structural basis for 11A subtypes. J. Biol. Chem. 284:7318–7329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhou F., Kong F., Tong Z., Gilbert G. L. 2007. Identification of less-common Streptococcus pneumoniae serotypes by a multiplex PCR-based reverse line blot hybridization assay. J. Clin. Microbiol. 45:3411–3415 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.