

Abstract
The Leishmania species cause a variety of human disease syndromes. Methods for diagnosis and species differentiation are insensitive and many require invasive sampling. Although quantitative PCR (qPCR) methods are reported for leishmania detection, no systematic method to quantify parasites and determine the species in clinical specimens is established. We developed a serial qPCR strategy to identify and rapidly differentiate Leishmania species and quantify parasites in clinical or environmental specimens. SYBR green qPCR is mainly employed, with corresponding TaqMan assays for validation. The screening primers recognize kinetoplast minicircle DNA of all Leishmania species. Species identification employs further qPCR set(s) individualized for geographic regions, combining species-discriminating probes with melt curve analysis. The assay was sufficient to detect Leishmania parasites, make species determinations, and quantify Leishmania spp. in sera, cutaneous biopsy specimens, or cultured isolates from subjects from Bangladesh or Brazil with different forms of leishmaniasis. The multicopy kinetoplast DNA (kDNA) probes were the most sensitive and useful for quantification based on promastigote standard curves. To test their validity for quantification, kDNA copy numbers were compared between Leishmania species, isolates, and life stages using qPCR. Maxicircle and minicircle copy numbers differed up to 6-fold between Leishmania species, but the differences were smaller between strains of the same species. Amastigote and promastigote leishmania life stages retained similar numbers of kDNA maxi- or minicircles. Thus, serial qPCR is useful for leishmania detection and species determination and for absolute quantification when compared to a standard curve from the same Leishmania species.
INTRODUCTION
The Leishmania spp. are kinetoplastid protozoa that are transmitted to humans and other mammalian hosts by a sand fly vector. The spectrum of symptomatic human leishmaniasis is wide, and the most important factor determining the clinical outcome of infection seems to be the species of Leishmania. Nonetheless, there are various clinical presentations of disease due to each species (9, 21, 28), and increasing numbers of reports document atypical presentations of leishmaniasis, sometimes but not always in the setting of immunocompromise (15, 50). Differentiation between the Leishmania species is an issue because there are overlapping and dynamic geographic regions of risk and different susceptibilities to treatment (6, 14). Thus, a method of diagnosis that is sensitive enough to detect low levels of the parasite in asymptomatic or early symptomatic infection and can distinguish between the different Leishmania species could be of tremendous utility in regions of endemicity and nonendemicity (37).
The procedures for diagnosis of leishmaniasis are often invasive, and isolates are frequently difficult to grow in vitro. Tests to distinguish between the Leishmania species have traditionally involved separation of isoenzymes in culture-derived parasites, which takes several weeks (11). Leishmania spp. have been detected in and isolated from blood cultures of subjects with all forms of leishmaniasis (19, 38, 39) and in the blood of asymptomatic individuals living in regions of risk (10, 23, 34, 39). The possibility that leishmania may be present in the bloodstream more often than previously recognized led us to hypothesize that amplification-based methods to detect parasite DNA in blood or serum might be a feasible means of diagnosis.
Nucleic acid-based methods avoid the need for parasite cultivation, replacing this with either hybridization or amplification (22, 33, 39, 65). The latter approaches provide the advantage of increased sensitivity. Amplification methods reported for the detection of individual Leishmania species include conventional PCR (3, 20, 61) or quantitative PCR methods, including reverse transcriptase quantitative PCR (RT-qPCR), DNA-based qPCR, quantitative nucleic acid sequence-based amplification (QT-NASBA), and in situ hybridization to quantify Leishmania spp. in blood or tissue samples (61).
Protozoa belonging to the order Kinetoplastida, including Leishmania spp. and Trypanosoma spp., characterized by a prominent kinetoplast structure containing the mitochondrial DNA in the parasites' single mitochondrion. Whereas Leishmania spp. have 35 to 36 chromosomes in their nuclear genomes (49), the kinetoplast contains hundreds of DNA maxicircles encoding genes that are destined for RNA editing and thousands of DNA minicircles, circular molecules with a conserved origin of replication encoding guide RNA sequences for RNA editing (12). Because of their abundance, specificity, and repetitive nature, kinetoplast DNA (kDNA) sequences have frequently been targeted for nucleic acid-based detection (32, 40, 41, 45, 46). A drawback of the use of kDNA for parasite quantification is the uncertainty of whether the kDNA copy number differs between Leishmania species, strains, and growth stages.
The goal of this study was to develop a serial nucleic acid amplification-based method for the diagnosis and species determination of Leishmania spp. parasites in human- or animal-derived tissues. As such, we developed a set of primers and probes for serial qPCR assays. The assays were sensitive enough to detect low levels of parasites and to distinguish between Leishmania species in human specimens. Using non-species-discriminating probes, we quantified the relative differences in kDNA copy numbers between parasite species, among isolates of the same species, and between stages of the same parasite strain. The serial qPCR assays have potential applications for diagnosis and species discrimination, as well as novel approaches to determining parasite load and posttreatment response in infected humans.
MATERIALS AND METHODS
Leishmania species and strains.
Promastigotes were cultivated in modified minimal essential medium (HOMEM) (5) or in Schneider's insect medium with 10% heat-inactivated fetal calf serum (FCS) and 50 μg gentamicin/ml. DNA was extracted from the species and strains listed in Table 1. Leishmania (Leishmania) chagasi MHOM/BR/00/1669 was originally isolated from a Brazilian with visceral leishmaniasis (VL).
Table 1.
Sources of Leishmania spp. isolates used in this project development
| Species (comment) | Source | Name, if any (reference) |
|---|---|---|
| L. (L.) tropica 1 | F. Steurer, CDC | FJ |
| L. (L.) tropica 2 | F. Steurer, CDC | You |
| L. (L.) tropica 3 | F. Steurer, CDC | Sp |
| L. (L.) tropica 4 | F. Steurer, CDC | KiR |
| L. (L.) tropica 5 | F. Steurer, CDC | GH |
| L. (L.) tropica 6 | F. Steurer, CDC | TAM |
| L. (L.) chagasi | S. Jeronimo, UFRN, Natal, Brasil | MHOM/BR/00/1669 |
| L. (L.) chagasi (attenuated) | Above isolate multiply passaged in vitro, M. Wilson | MHOM/BR/00/1669 strain L5 |
| L. (L.) infantum | D. McMahon-Pratt, Yale University | (1) |
| L. (L.) donovani | Buddy Ullman, OHSU | D1700 (27) |
| L. (L.) donovani (reference species) | Shyam Sundar, Banaras Hindu University | LEM 138 (MHOM/IN/DEVI) |
| L. (L.) donovani 1 | Shyam Sundar, Banaras Hindu University | BHU 764 |
| L. (L.) donovani 2 | Shyam Sundar, Banaras Hindu University | BHU 770 |
| L. (L.) donovani 3 | Shyam Sundar, Banaras Hindu University | BHU 782 |
| L. (L.) donovani 4 | Shyam Sundar, Banaras Hindu University | BHU 796 |
| L. (L.) donovani 5 | Shyam Sundar, Banaras Hindu University | BHU 814 |
| L. (L.) donovani 6 | Shyam Sundar, Banaras Hindu University | BHU 921 |
| L. (L.) donovani 7 | Shyam Sundar, Banaras Hindu University | BHU 922 |
| L. (L.) major | M. Wilson, University of Iowa | CDCID:LW9 |
| L. (V.) braziliensis | R. Almeida and E. Carvalho, UFBA, Salvador, Brazil | CDCID:LW10 |
| L. (V.) braziliensis | A. Schriefer, UFBA, Salvador, Brazil | 13330 |
| L. (V.) braziliensis | A. Schriefer, UFBA, Salvador, Brazil | 13968 |
| L. (V.) guyanensis (French Guyana) | N. Aronson, WRAIR | WR 2853 |
| L. (V.) guyanensis (French Guyana) | N. Aronson, WRAIR | WR 2334 |
| L. (V.) panamensis (Panama) | N. Aronson, WRAIR | WR 2306 |
| L. (V.) panamensis (Panama) | N. Aronson, WRAIR | WR 2307 |
| L. (L.) mexicana | R. Almeida and E. Carvalho, UFBA, Salvador, Brazil | CDCID:LW5 |
| L. (L.) amazonensis | Diane McMahon-Pratt, Yale University | 4588 |
| Leptomonas sp. BHU | Shyam Sundar, Banaras Hindu University | BHU 151 (Leptomonas seymouri like) |
| Crithidia fasciculata | ATCC | LEM 138 (MHOM/IN/DEVI) |
qPCR primers and probe design.
Primers targeting kinetoplast minicircle, maxicircle, and nuclear genome DNA were designed to be used in a SYBR green qPCR assay for detection, species discrimination, and quantification (Table 2, forward and reverse primer sequences). In four cases, these assays were designed using primers from a previously reported TaqMan assay (marked as “adapted from” in Table 2). All other primer pairs used for SYBR green qPCR assays were designed from sequence databases to amplify either genes that had been targeted by other PCR diagnostic assays of leishmaniasis or genes targeted uniquely for the current study. In addition to SYBR green assays, we developed primers within a subset of these sequences as TaqMan assays. TaqMan probes were designed by J. L. Weirather, with the exception of the two sequences indicated in Table 2 (kDNA 5 and DNA polymerase 1).
Table 2.
Primers and probes used for Leishmania qPCR diagnosis and species identificationa
| Type of DNA targeted, primer set designation | SYBR green primer sequence |
TaqMan primer sequenceb | Sequence sourcec | Source or reference | |
|---|---|---|---|---|---|
| Forward | Reverse | ||||
| Minicircle | |||||
| kDNA 1 minicircle | GGGTAGGGGCGTTCTGC | TACACCAACCCCCAGTTTGC | M94088 | J.L.W.c | |
| kDNA 2 | AACTTTTCTGGTCCTCCGGGTAG | ACCCCCAGTTTCCCGCC | EU437406 | Leish1 and -2 (20) | |
| kDNA 3 minicircle | GGGTAGGGGCGTTCTGC | CCCGGCCTATTTTACACCAACC | M94088 | J.L.W. | |
| kDNA 4 minicircle | GGGTGCAGAAATCCCGTTCA | CCCGGCCCTATTTTACACCA | ACCCCCAGTTTCCCGCCCCG | Conserved kDNAd | J.L.W. |
| kDNA 5 minicircle | CTTTTCTGGTCCTCCGGGTAGG | CCACCCGGCCCTATTTTACACCAA | TTTTCGCAGAACGCCCCTACCCGC | L. infantum contig 1335 | TaqMan qPCR (40)e |
| kDNA 7 minicircle | AATGGGTGCAGAAATCCCGTTC | CCACCACCCGGCCCTATTTTAC | CCCCAGTTTCCCGCCCCGGA | Conserved kDNAd | J.L.W. |
| L. (L.) amazonensis kDNA 1 | GGTCCCGGCCCAAACTTTTC | CCGGGGTTTCGCACTCATTT | U19810 | J.L.W. | |
| L. (L.) amazonensis kDNA 2 | GGTAGGGGCGTTCTGCGAAT | CCCGGCCTATTTTACACCAACC | EU370875 | J.L.W. | |
| L. (L.) amazonensis kDNA 3 | GGGTAGGGGCGTTCTGC | TACACCAACCCCCAGTTTGC | M94089 | J.L.W. | |
| L. (L.) amazonensis kDNA 4 | TGAGTGCAGAAACCCCGTTCATA | ACACCAACCCCCAGTTGTGA | EU370875 | J.L.W. | |
| L (V.) braziliensis kDNA 1 | AATTTCGCAGAACGCCCCTAC | GTACTCCCCGACATGCCTCTG | U19807 | J.L.W. | |
| L (V.) braziliensis kDNA 3 | TGCTATAAAATCGTACCACCCGACA | GAACGGGGTTTCTGTATGCCATTT | TTGCAGAACGCCCCTACCCAGAGGC | AF231100 | Adapted from PCR (56)f |
| L (L.) infantum minicircle 1 | TCCGCAGGAGACTTCGTATG | CACGACTATCCACCCCATCC | CTGAGAGACCCGCCGGGGCG | AF032997 | Adapted from nested PCR (51) |
| L. (L.) major minicircle 1 | ACGGGGTTTCTGCACCCATT | GTAGGGGCGTTCTGCGAAAA | LM15_BIN_Contig406 | J.L.W. | |
| L. (L.) mexicana minicircle 1 | AATGCGAGTGTTGCCCTTTTG | GCCGAACAACGCCATATTAACC | AY145437 | J.L.W. | |
| L. (L.) tropica minicircle 1 | GGGGGTTGGTGTAAAATAGGG | ACCACCAGCAGAAGGTCAAAG | TCCTGGCGGGGGTTTTCGCT | AF308689 | J.L.W. |
| L. (L.) donovani minicircle 1 | GCGGTGGCTGGTTTTAGATG | TCCAATGAAGCCAAGCCAGT | CCCATACCACCAAACGCAGCCCA | FJ416603 | J.L.W. |
| Maxicircle | |||||
| Cytochrome B 1 | ATTTTAGTATGAGTGGTAGGTTTTGTT | CAATAACTGGGACGGTTGCT | CCATGTACGATGATGTCGTATTGAGGTCTAACA | AB095960 | J.L.W. |
| L. (L.) amazonensis cytochrome B 1 | GCGGAGAGGAAAGAAAAGGCTTA | AAAAGTCATGCTAAACACACACCACA | AB095964 | J.L.W. | |
| L. (L.) tropica cytochrome B 1 | CAGGTTGCTTACTACGTGTTTATGGTG | TCGTATTACAAACCCTAAATCAAAATCTCA | AB095960 | J.L.W. | |
| L. (L.) tropica cytochrome B 2 | TCAGGTTGCTTACTACGTGTTTATGGTG | TGCTAAACAAACACCACATATGATCTGC | AB095960 | J.L.W. | |
| L. (L.) tropica cytochrome B 3 | TGACACACATATTTTAGTGTGGGTGGTAGG | TCCCCAATAAGACATCATTGTACATGGTAA | EF579904 | J.L.W. | |
| L. (L.) tropica cytochrome B 4 | CACATATTTTAGTGTGGGTGGTAGGTTTTG | TCCCCAATAAGACATCATTGTACATGGTAA | EF579904 | J.L.W. | |
| Maxicircle 1 | GCTTGGTTGGATTATTTTTGCTG | AACAACATTTTAACTCTTGTAGGATTCG | CTTTAGGTAGGGAGTTGTACTACGTTTTTTGACCT | DQ492252 | J.L.W. |
| Genomic | |||||
| Alpha-tubulin 1 | GAGGTGTTTGCCCGCATC | CTCGCCCATGTCGTCG | TGAGGGCATGGAGGAGGGCG | XM_001681731 | J.L.W. |
| DNA polymerase 1 | TGTCGCTTGCAGACCAGATG | GCATCGCAGGTGTGAGCA | CAGCAACAACTTCGAGCCTGGCACC | AF009147 | TaqMan qPCR (8)e |
| DNA polymerase 2 | AGGAGGATGGCAAGCGGAAG | GCGACGGGTACAGGGAGTTG | TGGGGTCGAGCACCATGCCGCC | AF009136 | J.L.W. |
| Miniexon 1 | CGAAACTTCCGGAACCTGTCTT | CACCACACGCACGCACAC | CGGCAAGATTTTGGAAGCGCGCA | AL389894 | J.L.W. |
| Miniexon 2 | GTGTGGTGGCGGGTGTATGT | GCCCAGGTCGCTGTGAGG | LbrM02_V2.0710 | J.L.W. | |
| MAG 1 (MSP-associated gene 1) | AGAGCGTGCCTTGGATTGTG | CGCTGCGTTGATTGCGTTG | TGCGCACTGCACTGTCGCCCCC | AF058760 | J.L.W. |
| MAG 2 (MSP-associated gene 2) | AGTTTTGGTTGGCGCTCCTG | CCCACTCGCTTTCCTTGGTC | CGCTGAGAGCGAGGCAGGCACGC | AF058760 | J.L.W. |
| SIDER repeat 1 | CGACCCTGTCACCACCACAG | GAGGCCACCCTATCGCTGAC | AM937229 | J.L.W. | |
| L. (V.) braziliensis DNA polymerase 1 | TCGTTGAGGGAGGAGGTGTTTC | TCGGCTTTGAGGTTGGCTTC | XM_001563712 | J.L.W. | |
| L. (V.) braziliensis DNA polymerase 2 | ACGTCGCCAACTGCTTCACC | GTGTTCGCACCGCCTTGAC | XM_001563712 | J.L.W. | |
| L. major MAG 1 | GTCGTTGTCCGTGTCGCTGT | CGCTGTGTGTGTCCGTGTGT | XM_001681328 | J.L.W. | |
| L (L.) amazonensis DNA polymerase 1 | GACGACGACGAGGAGGATGG | GCGACGGGTACAGGGAGTTG | AF009136 | J.L.W. | |
| GPI | CCAGATGCCGACCAAAGC | CGCGCACGTGATGGATAAC | AM117195 | (66) | |
| HSP70-1 | GAAGGTGCAGTCCCTCGTGT | CCTCCGTCTGCTTGCTCTTG | FN395029 | J.L.W. | |
| HSP70-4 | TCGAGATCGACGCGTTGTT | CCGCACAGCTCCTCGAA | FN395037 | J.L.W. | |
| SLACS | GGAGAAACTCACGGCACAGG | GCGCCTCGTAGGTCACAGTT | XM_001562078 | J.L.W. | |
| Leptomonas DNA | |||||
| Leptomonas miniexon 1 | TGGAGCGGGTGCATTAACTC | GGTCTCGAGGTGCCCATGAC | S78663 | J.L.W. | |
| Leptomonas GAPDH 2 | AGAAGCCGGATGTGCTTGTG | GCCCTCAGCCTTCACCTTGT | AF053738 | J.L.W. | |
| Human DNA | |||||
| Human TNF-α 1 | GCCCTGTGAGGAGGACGAAC | AAGAGGTTGAGGGTGTCTGAAGGA | CCTTCCCAAACGCCTCCCCTGCCCC | NM_000594 | J.L.W. |
| Human TNF-α 2 | GCGCTCCCCAAGAAGACAGG | TGCCACGATCAGGAAGGAGAAG | CACCGCCTGGAGCCCTGGGGC | NM_000594 | J.L.W. |
| Human GAPDH 1 | GGGCTCTCCAGAACATCATCC | CCAGTGAGCTTCCCGTTCAG | NG_007073 | J.L.W. | |
| Human GAPDH 2 | CATCAAGAAGGTGGTGAAGCAG | CGTCAAAGGTGGAGGAGTGG | NG_007073 | J.L.W. | |
| Human GAPDH 3 | GCATGGCCTTCCGTGTCC | CGCCTGCTTCACCACCTTCT | NG_007073 | J.L.W. | |
All sequences were developed for use in SYBR green and/or TaqMan assays.
The TaqMan probe used for human DNA was 5′VIC/3′TAMRA. The TaqMan probe used for all other DNA was 5′FAM/3′TAMRA.
Primers and probes were designed by one of us (J. L. Weirather [J.L.W.]) from sequences available on the World Wide Web. GenBank accession numbers are listed.
The “conserved kDNA” sequences used multiple sources aligned with CLUSTALW.
For TaqMan qPCR, the primers and probes are the exact ones used for TaqMan but not SYBR green assay reported in the literature cited.
“Adapted from” indicates that the targeted gene in the literature cited was used by J. L. Weirather to redesign primers and probes.
Sequences from kinetoplast minicircles were derived from the NCBI Entrez nucleotide database (http://www.ncbi.nlm.nih.gov/sites/entrez?db=nuccore). Primers and probes suitable for qPCR were designed using the Primer3 website (http://frodo.wi.mit.edu/primer3/) (56). Product sizes were designed to be less than 150 bp.
Minicircle primers were designed against both conserved targets and species-specific sequences. Sequences from the kinetoplast maxicircles were derived from protein coding sequences that are not subject to the extensive RNA editing (24). The maxicircle 1 primer set was specifically designed against a variable region to maximize usefulness in species discrimination. Other maxicircle genes were designed to amplify cytochrome B sequences.
Within the nuclear genome, the assay for the DNA polymerase 1 gene was based upon a reported TaqMan assay (8). The flanking primer set for this TaqMan assay was found to be adequate for an independent SYBR green qPCR. The DNA polymerase gene is a single-copy gene, increasing its utility for quantitative assay of parasites. The miniexon 1 and 2, alpha-tubulin 1, HSP70, and SIDER repeat 1 primer sets were chosen because they amplify repetitive sequences and should therefore be sensitive. The MSP-associated gene (MAG) 1 and 2 primer sets were designed against mag gene sequences only known to be present in Leishmania (L.) infantum and L. (L.) chagasi (44), in anticipation that these sequences could be useful in species discrimination. L. (L.) major contains a hypothetical ortholog to mag (LmjF.10.0483), identified by a shared P-fam B domain (21814) identified by the ADDA algorithm (26). This prompted the design of L. (L.) major MAG 1 primers. Finally, the gene encoding glucose phosphate isomerase (GPI) was selected due to prior reports of its use in qPCR assays (66).
The genus Leishmania has two subgenera, designated L. Viannia spp. or L. Leishmania spp. Examination of genomes has revealed that L. Viannia braziliensis has retrotransposable elements not found in L. Leishmania species (49). We designed a primer set to target the splice-leader-associated (SLACS) retrotransposons of L. (V.) braziliensis. Although genome sequences are not available for the other members of the Viannia subgenus, L. (V.) guyanensis and L. (V.) panamensis, we also tested the ability of this primer set to differentiate between L. (V.) braziliensis and each of these other species.
Two non-Leishmania species members of the Trypanosomatidae (Crithidia fasciculata and Leptomonas) were studied for potential cross-reactivity of primers. The latter organism, Leptomonas sp., has been recovered from the spleens of several individuals with symptoms of visceral leishmaniasis (60). TNF-α 1 and 2, primers for human tumor necrosis factor alpha (TNF-α), target the single-copy human TNF-α gene (51). These and primers for human glyceraldehyde-3-phosphate dehydrogenase (GAPDH) were designed as positive controls for qPCR amplification.
DNA extraction.
Five- to 10-ml cultures were suspended in lysis buffer (150 mM NaCl, 4 mg/ml SDS, 10 mM EDTA, 10 mM Tris-HCl, pH 7.5) with 200 μg proteinase K/ml for 1 h at 56°C. DNA was extracted in Tris-equilibrated phenol and then in phenol-CHCl3-isoamyl alcohol (25:24:1), followed by CHCl3-isoamyl alcohol (49:1). Samples were ethanol precipitated and resuspended in water.
qPCR assay conditions.
All reactions were conducted on a 7900 fast real-time PCR system from Applied Biosystems (ABI) in 10-μl reaction mixture volumes. SYBR green reaction mixtures were composed of ABI Power SYBR green 2× master mix and 500 nM (each) forward and reverse primers. TaqMan reactions were performed with ABI TaqMan universal PCR 2× master mix, 375 nM (each) forward and reverse primers, and 250 nM probe (label, 5′FAM, and quench, 3′TAMRA). The thermocycling parameters consisted of a hold at 95° for 10 min followed by 40 temperature cycles of 95° for 15 s and 60 degrees for 1 min. A melt curve analysis was performed on all SYBR green reactions. The results were analyzed with SDS 2.4 software with an automatic baseline and a manual cycle threshold (CT) of 0.2 for all reactions. The R software package was used to generate melt curve plots for primer sets in which melting temperatures differed by at least 1 degree between species.
Multiplex TaqMan qPCR.
A multiplex TaqMan qPCR assay was designed to detect all tested Leishmania species and simultaneously differentiate between members of the visceralizing L. donovani complex and other species that cause primarily tegumentary disease. The DNA polymerase 2 (label, 5′TET, and quench, Iowa Black) and MAG 2 (label, 5′FAM, and quench, TAMRA) primers and probes were used together in a 10-μl reaction mixture with 125 nM each forward and reverse primers and 83 nM each probe. The thermocycling parameters were identical to the conditions described above.
RESULTS
Detection and differentiation between Leishmania species.
Based on the SYBR green primer sets and TaqMan probes designed as described above, we developed qPCR assays with the dual objectives of developing a sensitive assay that could detect all Leishmania species infections in clinical or experimental specimens and discriminate between the different Leishmania species. For these purposes, 41 Leishmania-specific primer pairs were tested in SYBR green assays. All were tested in the presence of a 10-fold excess of human DNA compared to the amount of DNA extracted from parasites. Seventeen targeted the kinetoplast minicircle, 7 targeted maxicircle sequences, and 17 targeted genes in the nuclear genome. Two primer pairs were developed to identify the related Leptomonas spp. protozoa, which have been reported as coisolates with L. donovani (60). Five primer pairs were also included as positive controls for human DNA in specimens. These could additionally be used for normalization to a baseline of human DNA in testing clinical samples.
For some but not all primer sets, differences in melt temperature curves were useful in distinguishing between Leishmania species (Fig. 1). For instance, kDNA 1 clearly distinguished between L. (L.) amazonensis and L. (L.) mexicana or L. (L.) tropica and L. (L.) major, whereas L. (L.) donovani, L. (L.) chagasi, and L. (L.) infantum exhibited very similar melting point (Tm) peaks. In contrast, the MAG 1 primer pair distinguished L. (L.) donovani from L. (L.) chagasi/L. (L.) infantum.
Fig. 1.

Melt curves of selected qPCR assays useful for species discrimination. The rate of change in the intensity of the fluorescent qPCR signal is plotted across a 10° temperature range for each species with each primer set. Data are only shown for sets that amplified with a CT of less than 30 and showed melt curve peaks that were more than 1° apart between species. Peaks of curves indicate the melting temperature of the amplicon. Each melt curve is color coded according to the parasite species it represents. Plots with a solid line indicate that the reaction mixture amplified with a CT of less than 25. Plots with a dotted line indicate that the reaction mixture amplified with a CT of more than or equal to 25 but less than 30.
TaqMan assays corresponding to a subset of the SYBR green primers that are listed in Table 2 were experimentally validated. The data in Table 3 show the relative efficiency of each SYBR green or TaqMan assay for detecting and differentiating between Leishmania spp. The CT values listed on this table reflect the cycle in which the intensity exceeded a threshold of 0.2 when amplifying 0.1 ng of total parasite DNA in the presence of 1 ng of human DNA. Products that amplified with a CT greater than 30 were excluded because these extreme values were often caused by primer-dimers or off-target amplification in SYBR green reaction assays. Such artifacts can be discerned using melt curve analyses (not shown) for some primer sets. The cutoff could be raised for TaqMan assays, as primer-dimers do not affect those results. The cutoff of 30 cycles is roughly equivalent to 0.005 parasites per well when amplifying minicircle sequences or 102 parasites when amplifying a genomic sequence with SYBR green reagents.
Table 3.
Relative efficiencies of SYBR green or TaqMan assays for detecting and differentiating Leishmania spp. or Leptomonas spp. or Crithidia fasciculata
| Type of PCR assay, primer set designation | Avg CT valuea |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| L. (L.) amazonensis | L. (V.) braziliensis | L. (L.) chagasi | L. (L.) donovani | L. (L.) infantum | L. (L.) major | L. (L.) mexicana | L. (L.) tropica | Leptomonas | Crithidia | |
| SYBR green | ||||||||||
| Alpha-tubulin | 21.4 | >30 | 20.7 | 20.3 | 21.1 | 20.8 | 21.4 | 23.4 | 27.8 | 28.3 |
| Cytochrome B 1 | >30 | >30 | 23.8 | 20.0 | 22.0 | >30 | >30 | >30 | >30 | >30 |
| DNA polymerase 1 | 25.1 | 29.0 | 23.5 | 23.0 | 24.7 | 24.5 | 25.6 | 27.5 | >30 | >30 |
| DNA polymerase 2 | 24.1 | 23.4 | 22.9 | 22.5 | 24.3 | 24.2 | 24.7 | 27.9 | 29.2 | 26.5 |
| GPI | 24.3 | 29.4 | 23.1 | 22.9 | 24.4 | 24.6 | 25.2 | 27.1 | >30 | >30 |
| HSP70-1 | 21.3 | 21.4 | 19.8 | 19.4 | 21.3 | 20.8 | 22.5 | 23.7 | >30 | >30 |
| HSP70-4 | 21.7 | 22.1 | 20.1 | 19.9 | 21.5 | 20.9 | 22.8 | 24.0 | 27.0 | 23.6 |
| kDNA 1 | 15.6 | 27.2 | 15.7 | 14.4 | 15.3 | 19.6 | 16.5 | 15.4 | 26.6 | 13.2 |
| kDNA 2 | >30 | 29.3 | 14.6 | 12.8 | 14.4 | 11.8 | 19.2 | 29.8 | 26.4 | 29.6 |
| kDNA 3 | 14.7 | 18.9 | 14.8 | 13.0 | 14.3 | 11.4 | 13.7 | 15.4 | 23.4 | 18.3 |
| kDNA 4 | 16.3 | 25.6 | 14.6 | 12.9 | 14.4 | 11.7 | 14.8 | 21.5 | 26.0 | 27.8 |
| kDNA 5 | >30 | 28.7 | 14.8 | 13.1 | 14.3 | 11.6 | 20.3 | 19.0 | 26.7 | 26.5 |
| kDNA 7 | 19.8 | 28.9 | 14.8 | 12.6 | 14.2 | 11.5 | 18.6 | 23.9 | 25.6 | 29.4 |
| L. (L.) amazonensis cytochrome B 1 | 28.4 | 25.6 | 29.1 | 29.8 | 27.4 | >30 | >30 | >30 | >30 | >30 |
| L. (L.) amazonensis DNA polymerase 1 | 24.4 | 23.2 | 23.4 | 22.9 | 24.8 | 25.4 | 24.7 | 28.0 | 29.1 | 25.5 |
| L. (L.) amazonensis kDNA 3 | 15.6 | 27.0 | 15.6 | 14.5 | 15.2 | 19.3 | 16.5 | 15.2 | 27.2 | 13.1 |
| L. (L.) amazonensis kDNA 4 | 15.9 | >30 | >30 | >30 | >30 | >30 | 19.0 | >30 | >30 | >30 |
| L. (L.) amazonensis kDNA 2 | 15.2 | >30 | >30 | 28.2 | 29.5 | 24.3 | 21.8 | >30 | >30 | >30 |
| L. (L.) amazonensis kDNA 1 | 15.0 | 29.8 | >30 | 27.9 | >30 | 23.8 | 21.6 | >30 | >30 | >30 |
| L. (V.) braziliensis DNA polymerase 1 | >30 | 22.9 | >30 | >30 | >30 | >30 | >30 | >30 | >30 | >30 |
| L. (V.) braziliensis DNA polymerase 2 | 28.1 | 23.1 | 24.3 | 23.7 | 25.5 | >30 | 28.6 | 28.3 | 26.6 | >30 |
| L. (V.) braziliensis kDNA 1 | >30 | 13.5 | >30 | >30 | >30 | >30 | >30 | >30 | >30 | >30 |
| L. (V.) braziliensis kDNA 2 | >30 | 23.9 | >30 | >30 | >30 | >30 | >30 | >30 | >30 | >30 |
| L. (V.) braziliensis kDNA 3 | >30 | 17.7 | >30 | >30 | >30 | >30 | >30 | >30 | >30 | >30 |
| L. (L.) chagasi SIDER 1 | >30 | >30 | 21.7 | 21.7 | 22.4 | >30 | >30 | 26.4 | >30 | >30 |
| L. (L.) infantum minicircle 1 | >30 | >30 | 23.8 | >30 | 26.6 | >30 | >30 | >30 | >30 | >30 |
| L. major MAG 1 | 23.2 | 27.6 | 22.4 | 21.3 | 24.5 | 24.1 | 25.1 | 23.2 | >30 | >30 |
| L. (L.) major minicircle 1 | 18.6 | 29.4 | 16.1 | 22.6 | 15.9 | 22.6 | 18.9 | >30 | 25.9 | 29.9 |
| L. (L.) mexicana minicircle 1 | >30 | >30 | >30 | >30 | >30 | >30 | 20.1 | >30 | >30 | >30 |
| L. (L.) tropica cytochrome B 1 | >30 | >30 | >30 | >30 | >30 | >30 | >30 | 25.1 | >30 | >30 |
| L. (L.) tropica cytochrome B 2 | >30 | >30 | 24.6 | 26.2 | 22.7 | >30 | >30 | 24.3 | >30 | >30 |
| L. (L.). tropica cytochrome B 3 | 28.4 | >30 | >30 | 29.4 | >30 | 23.2 | >30 | 26.2 | >30 | >30 |
| L. (L.) tropica cytochrome B 4 | 24.1 | >30 | 26.7 | 23.2 | 24.8 | 22.0 | 24.5 | 25.0 | >30 | >30 |
| L. (L.) tropica minicircle 1 | >30 | >30 | >30 | >30 | >30 | >30 | >30 | 19.8 | >30 | >30 |
| L. (L.) donovani minicircle 1 | >30 | >30 | 29.4 | 15.3 | 25.9 | >30 | >30 | >30 | >30 | >30 |
| Leptomonas GAPDH 2 | >30 | 24.1 | >30 | >30 | >30 | >30 | >30 | >30 | 26.6 | 23.5 |
| Leptomonas miniexon 1 | >30 | >30 | >30 | >30 | >30 | >30 | >30 | >30 | 20.7 | >30 |
| MAG 1 | >30 | >30 | 21.2 | 20.8 | 23.6 | >30 | >30 | 25.8 | >30 | >30 |
| MAG 2 | >30 | >30 | 21.0 | 20.8 | 23.4 | >30 | >30 | >30 | >30 | >30 |
| Maxicircle 1 | >30 | >30 | 22.3 | 19.6 | 19.2 | >30 | >30 | >30 | >30 | >30 |
| Miniexon 1 | 21.6 | 26.3 | 17.9 | 16.7 | 19.6 | 19.0 | 25.5 | 18.9 | 29.6 | 29.1 |
| Miniexon 2 | 29.9 | 16.9 | 29.0 | 29.9 | 29.3 | 29.3 | 30.0 | >30 | >30 | >30 |
| SLACS | 29.9 | 19.7 | 29.8 | 30.0 | 29.9 | >30 | 29.9 | >30 | >30 | 29.8 |
| TaqMan | ||||||||||
| TaqMan alpha-tubulin | 26.1 | >30 | 25.0 | 24.9 | 25.9 | 24.7 | 26.2 | 28.8 | >30 | >30 |
| TaqMan cytochrome B 1 | >30 | >30 | 27.7 | 24.5 | 26.1 | >30 | >30 | >30 | >30 | >30 |
| TaqMan DNA polymerase 1 | 27.1 | >30 | 25.8 | 25.1 | 27.3 | 26.7 | 27.8 | 29.4 | >30 | >30 |
| TaqMan DNA polymerase 2 | 27.0 | 26.7 | 26.5 | 26.0 | 27.9 | 26.9 | 27.9 | 28.8 | >30 | >30 |
| TaqMan kDNA 4 | >30 | >30 | 18.1 | 17.3 | 17.8 | >30 | >30 | >30 | >30 | >30 |
| TaqMan kDNA 5 | >30 | >30 | 18.1 | 16.1 | 18.0 | 16.1 | 25.5 | 25.3 | >30 | >30 |
| TaqMan kDNA 7 | >30 | >30 | 18.4 | 17.5 | 18.1 | 23.6 | >30 | >30 | >30 | >30 |
| TaqMan L. (V.) braziliensis kDNA 3 | >30 | 21.9 | >30 | >30 | >30 | >30 | >30 | >30 | >30 | >30 |
| TaqMan L. (L.) infantum minicircle 1 | >30 | >30 | 28.8 | >30 | >30 | >30 | >30 | >30 | >30 | >30 |
| TaqMan L. (L.) tropica minicircle 1 | >30 | >30 | >30 | >30 | >30 | >30 | >30 | 23.6 | >30 | >30 |
| TaqMan L. (L.) donovani minicircle 1 | >30 | >30 | >30 | 18.1 | >30 | >30 | >30 | >30 | >30 | >30 |
| TaqMan MAG 1 | >30 | >30 | 23.5 | 24.3 | 25.9 | >30 | >30 | 28.3 | >30 | >30 |
| TaqMan MAG 2 | >30 | >30 | 23.8 | 23.5 | 26.1 | >30 | >30 | >30 | >30 | >30 |
| TaqMan maxicircle 1 | >30 | >30 | 26.7 | 25.0 | 24.2 | >30 | >30 | >30 | >30 | >30 |
| TaqMan miniexon 1 | 23.8 | >30 | 26.5 | 24.0 | 26.8 | 22.0 | >30 | 22.9 | >30 | >30 |
| Multiplex TaqMan | ||||||||||
| TaqMan MAG 2; 5′FAM/3′TAMRA | >30 | >30 | 23.7 | 23.4 | 26.0 | >30 | >30 | >30 | >30 | >30 |
| TaqMan DNA polymerase 2; 5′TET /3′Iowa Black | 25.3 | 24.9 | 25.7 | 24.9 | 26.6 | 25.6 | 25.9 | 28.8 | >30 | >30 |
The data indicate the average CT values of 2 replicates for Leptomonas and L. amazonensis or of 4 or more replicates for all other species. CT values reflect the cycle in which fluorescence intensity reached 0.2 when amplifying 0.1 ng of total parasite DNA in the presence of 1 ng of human DNA. The cutoff of CT = 30 avoids ambiguities caused by primer-dimers in SYBR green assays. Please note that CT cutoffs could differ between different strains or when assays are performed in different laboratories.
Primer pairs targeting the multicopy minicircle kDNA were the most sensitive tests to detect any species of Leishmania. For many of the minicircle primers, exponential amplification occurred at a CT below 15 when amplifying 100 femtograms of parasite DNA using SYBR green. Maxicircle and nuclear genome targets were also effective in detecting parasites, although these primer sets were not as sensitive and required 25 or more cycles to reach detection (Table 3). Some of the late-amplifying markers demonstrated melt curves that were useful for distinguishing between species (Table 3). L. (L.) major MAG 1 is notable in that it amplifies all Leishmania species without amplifying the Leptomonas or Crithidia negative controls. The CT values in Table 3 can be used to assess the relative performance difference between species, but strains within species may differ slightly. Also, absolute CT values may vary somewhat between different laboratories.
Species in the subgenus Viannia were recognized by the primer set designated L. (V.) braziliensis kDNA 3, which was designed against the L. (V.) braziliensis genome [average CT values were 17.7, 13.5, or 13.3 when amplifying from L. (V.) braziliensis, L. (V.) guyanensis, or L. (V.) panamensis genomic DNA, respectively]. Therefore, we tested additional primers that might differentiate this species from the other Viannia subgenus members. The splice-leader-associated (SLACS) retrotransposons are found in the L. (V) braziliensis genome but not the subgenus Leishmania spp. analyzed to date (49). The primer pair SLACS amplified L. (V.) braziliensis DNA as predicted, although the primer set suffered from primer-dimer peaks at CTs greater than 28 in many species (Table 3). Because of a lack of genomic sequence information, it was unknown whether these primers would also amplify DNA from other Viannia subgenus members, L. (V.) guyanensis and L. (V.) panamensis. Analyses revealed that despite the background amplification at CT values of >28 due to primer-dimers, the SLACS primer set specifically amplified sequences in genomic DNA of L. (V.) braziliensis but not L. (V.) guyanensis or L. (V.) panamensis DNA [average CT values were 19.7. 30.2, or 32.5 using genomic DNA from L. (V.) braziliensis, L. (V.) guyanensis, or L. (V.) panamensis as the template]. SLACS, therefore, provided the only primer set tested that distinguished between species within the Viannia subgenus.
To quantitatively assess the relative sensitivities of SYBR green versus TaqMan assays of kDNA or genomic DNA sequences, we used kDNA 5 (minicircle) or DNA polymerase to amplify L. (L.) chagasi DNA. Standard curves were generated from DNA extracted from a known number of parasites. The approximate CT values for detection of a single parasite and the number of parasites detected at the CT cutoff point of 30 were determined (Table 4). The data in Table 4 demonstrate, first, increased sensitivity of the minicircle target sequence compared to the sensitivity of the single-copy gene target, as expected (20,400-fold enhanced sensitivity in the SYBR green assay). The data also show increased sensitivity of SYBR green compared to the sensitivity of TaqMan for detection of either sequence when using earliest amplification as a measure of performance. TaqMan is expected to have greater specificity than SYBR green, although when used in combination with melt curve analysis, SYBR green can approach the sensitivity of TaqMan assays for numbers of parasites exceeding the threshold.
Table 4.
Sensitivity of qPCR assays for parasite detectiona
| Type of DNA, type of assay | Approximate CT value for 1 promastigote | Detection threshold with CT cutoff of 30 |
|---|---|---|
| Minicircle, SYBR green | 24.2 | 0.005 |
| Minicircle, TaqMan | 27.7 | 0.18 |
| Genomic, SYBR green | 37.0 | 102 |
| Genomic, TaqMan | 38.5 | 318 |
DNA extracted from known numbers of L. infantum chagasi promastigotes was amplified with kDNA 5 minicircle primers or with primers hybridizing to the single-copy gene encoding DNA polymerase. The average CT values corresponding to numbers of promastigote genomes in a single well are indicated. Please note that this table should not be used for exact quantification, because the efficiency of the PCR is expected to vary between the Leishmania species.
Specificity of primer pairs.
The reactions whose results are shown in Table 3 were carried out using 0.1 ng of parasite DNA in the presence of 10 ng of human DNA. No primer sets amplified in human DNA alone at a CT lower than 30. Alpha-tubulin, DNA polymerase 2, HSP70-4, L. amazonensis DNA polymerase 1, L. (V.) braziliensis DNA polymerase 2, kDNA 1 to kDNA 7, L. (L.) amazonensis kDNA 3, L. (L.) major minicircle 1, and miniexon 1 showed cross-reactivity with Crithidia fasciculata or Leptomonas but not human DNA, indicating primer hybridization to sequences in other trypanosomatid protozoa. The CT can be used to differentiate species. A low CT serves as a criterion for choosing one marker over another with a higher expected CT for the species of interest. Examination of melt curves for some of these primer sets (e.g., miniexon 2 in Fig. 1) shows low amplification (high CT value, shown as dotted lines in the figure) for many different species, migrating at a lower melt temperature than the main peak amplifying at the lowest CT value [L. (V.) braziliensis in Fig. 1]. This low amplification could have been caused by the production of primer-dimers, although we cannot rule out a universally shared off-target DNA sequence. The final identification of Leishmania rather than a cross-reactive sequence can be made either by examining the melt temperature in SYBR green assays or by use of the TaqMan probe at the validation step.
Leishmania species identification.
The above-described qPCR assays can be used, first, to identify and quantify any organism belonging to the genus Leishmania, and second, to discriminate between Leishmania species. Species identification is accomplished either by observing the melt temperature of the amplicon or observing the presence or absence of some amplicons. Nine of the designed primer sets were found to be useful in discriminating between Leishmania species based upon the melt temperature of the amplicon (Fig. 1). L. (L.) infantum and L. (L.) chagasi could not be distinguished based on any melt curve analysis. This provides validity to the melt temperature approach. More distantly related but nonidentical Leishmania species could be readily distinguished depending on the primer set used. In some instances, species identification could be attained through the exclusive amplification of one or a few species by a primer set. For example, L. (L.) mexicana minicircle 1 uniquely amplifies L. (L.) mexicana DNA and not other species.
We used the above information to generate a flow chart of qPCR tests recommended to (i) determine whether any Leishmania spp. is present in a clinical or environmental sample and (ii) identify which species is present (Fig. 2). The example shows a sequence of tests appropriate for species found in Latin America. According to the flow chart, the DNA is first amplified using SYBR green qPCR assays for kDNA 1 and L. (V.) braziliensis kDNA 3, to detect all Leishmania species. The next step is examination of the melting temperatures of kDNA amplicons, a step that will differentiate false from true positives. Samples that test positive for Leishmania DNA would undergo a secondary set of SYBR green qPCRs to determine the species of infecting parasite and to validate the initial result. These include L. (L.) amazonensis kDNA 2 and MAG 1 for specimens in which L. (L.) amazonensis, L. (L.) chagasi/infantum, or L. (L.) donovani are suspected from the kDNA 1 melt curve and L. mexicana minicircle 1 when L. mexicana or L. major are suspected. The melting temperature differs based on size and GC composition and should be unique to each species. It is recommended to conduct the species test along with appropriate positive-control DNAs, as melt curve temperatures may vary slightly between experiments due to subtle differences in buffers or between machines. As a final verification, which could be considered optional, especially in settings where cost is a primary issue, a TaqMan assay specific for the infecting Leishmania species may be used. The inability of any primers to distinguish between L. chagasi and L. infantum is consistent with the current belief that these species seem to be genetically identical (16, 42).
Fig. 2.

Flow chart of the serial qPCR assay. The chart shows a minimal application of the serial diagnostic qPCR assay to determine the presence and species of Leishmania in a sample. In step 1 (detection), an unknown DNA sample can be tested for the presence of Leishmania spp. DNA using SYBR green primers. kDNA 1 amplifies most species. L. (V.) braziliensis minicircle 3 amplifies kDNA sequences within L. (V.) braziliensis, a primer set that would be included when testing samples from Latin America to provide the most sensitive detection across species. In step 2, a sample which has tested positive for the presence of parasite kDNA 1 but not L. braziliensis kDNA 3 can be classified according to its species. Melt curve analysis of kDNA 1 amplicons can distinguish between several Old World species [L. (L.) tropica, L. (L.) major, and L. (L.) infantum]. The application of SYBR green melt curve analysis using the MAG 1 (MSP-associated gene 1) primer set is capable of separating L. (L.) chagasi and L. (L.) infantum from L. (L.) donovani. The presence or absence of L. (L.) mexicana-specific minicircle amplicons is sufficient to differentiate between L. (L.) mexicana and L. (L.) major. Distinguishing L. (L.) amazonensis from the members of the L. (L.) donovani complex requires an additional primer set, such as L. (L.) amazonensis kDNA 2 (not shown in flowchart). The inability of any primer pairs tested to distinguish between L. (L.) chagasi and L. (L.) infantum is consistent with the current belief that these species are genetically identical (42). Note that the primer sets listed are minimal sets. It is advisable to select additional primers based upon the expected Leishmania species in the geographic region.
Other series of tests would be recommended for detection/species discrimination in other geographic regions. Table 5 lists markers to distinguish common species. The list is not exhaustive, and existing tests will need to be tested for their capacity to distinguish additional species as needed for some regions.
Table 5.
Serial qPCR studies recommended for detection and species discrimination of Leishmania spp. in clinical or environmental specimens based on geographic region
| Region | Clinical syndromea | Expected Leishmania species | 1st step: detection | 2nd step: species discrimination |
|---|---|---|---|---|
| North, Central, and South America | LCL | L. (L.) amazonensis | kDNA 1 | kDNA 1 melt curve |
| L. (L.) mexicana | L. braziliensis kDNA 3 | MAG 1 melt curve | ||
| L. (V.)b braziliensis, L. (V.) panamensis, L. (V.) guyanensis | L. braziliensis kDNA 3 | |||
| L. (L.) infantum chagasi (rare) | SLACS | |||
| North, Central, and South America | MCL, DL, DCL | L. (V.) braziliensis, L. (V.) panamensis, L. (V.) guyanensis | L. amazonensis kDNA 3 | L. amazonensis kDNA 3 melt curve |
| L. (L.) amazonensis | L. braziliensis kDNA 3 | L. braziliensis kDNA 3 | ||
| L. (L.) mexicana | SLACS | |||
| North, Central, and South America | VL | L. (L.) infantum | kDNA 2 | kDNA 2c |
| L. (L.) amazonensis (rare) | L. amazonensis kDNA 4 | L. amazonensis kDNA 4c | ||
| Europe | VL | L. (L.) infantum | kDNA 1 | MAG 1 melt curve |
| L. (L.) donovani | ||||
| Middle East and Northern and sub-Saharan Africa | CL, DCL, LR | L. (L.) major | kDNA 1 | kDNA 1 melt curve |
| L. (L.) tropica | L. tropica cytochrome B 1c | |||
| L. (L.) infantum (rare) | MAG 1 melt curve | |||
| L. (L.) donovani (rare) | ||||
| Middle East and Northern and sub-Saharan Africa | VL, PKDL | L. (L.) infantum | kDNA 1 | kDNA 1 melt curve |
| L. (L.) donovani | L. tropica cytochrome B 1c | |||
| L. (L.) tropica (rare) | MAG 1 melt curve | |||
| India/Bangladesh/Pakistan/Nepal | VL, PKDL | L. (L.) donovani | kDNA 1 to 7 (any) | |
| Asia | VL | L. (L.) donovani | kDNA 1 to 7 (any) |
Clinical syndromes are as follows: DCL, diffuse cutaneous leishmaniasis; DL, disseminated leishmaniasis; LCL, localized cutaneous leishmaniasis; LR, leishmaniasis recidivans; MCL, mucocutaneous leishmaniasis; VL, visceral leishmaniasis; CL, cutaneous leishmaniasis; and PKDL, post-kala-azar dermal leishmaniasis.
V. refers to parasites belonging to the subgenus Leishmania Viannia. All other species listed belong to the Leishmania Leishmania subgenus.
This marker amplifies only one species listed in the 3rd column of this row.
The combination of DNA polymerase 2 and MAG 2 TaqMan qPCR assays was developed into a multiplex reaction that distinguished between visceralizing and other Leishmania species. DNA polymerase 2 is capable of amplifying DNA from all species, albeit less efficiently in L. (L.) tropica. The latter required several more cycles of amplification than the other species tested. Among the visceralizing species, L. (L.) chagasi, L. (L.) donovani, and L. (L.) infantum, the MAG amplification occurred at earlier CTs than DNA polymerase, mitigating the possibility that the DNA polymerase 2 amplification could consume the reaction reagents and mask the results (Table 3). L. (L.) tropica did show some amplification at late CTs with the MAG 2 primer set. However, since this occurred late in the reaction, later than the DNA polymerase 2 amplification, it is easily discernible from the L. (L.) donovani complex members. This finding is of interest due to the ability of L. (L.) tropica to cause not only cutaneous ulcers but also disseminated disease (36), although one cannot draw conclusions without sequence and functional data.
kDNA copy numbers.
Real-time and TaqMan PCR are capable of quantifying parasite numbers in clinical or experimental specimens. Absolute quantification is determined by comparison to standard curves from parasite DNA. Since kDNA probes are most sensitive, it makes sense to perform quantification with these primers. However, the use of kDNA for quantification is complicated by the fact that Leishmania species contain multiple copies of both minicircle and maxicircle kDNA. It is not known whether copy numbers differ between the Leishmania species, between different isolates of the same species, or between parasite stages. We therefore employed minicircle and maxicircle kDNA qPCR assays to determine the relative difference in minicircle and maxicircle kDNA copy numbers between three different species of Leishmania (Table 6), between a recent and a multiply passaged line of the same L. (L.) chagasi isolate (Table 6, L. donovani complex), between eight strains of L. (L.) donovani derived from patients in the same region of endemicity of Bihar State, India (Table 6, L. donovani isolates), and between four L. (L.) tropica isolates recently derived from infected humans in the Middle East (Table 6, L. tropica isolates). L. tropica isolates were kindly provided by F. Steurer of the Centers for Disease Control. kDNA copy numbers were normalized to the results for the DNA polymerase I gene, which is a single-copy gene in the parasite genome. Relative copy numbers were calculated using the delta delta CT method.
Table 6.
Relative copy number difference in kinetoplast mini- and maxicircle sequences between the indicated Leishmania species
| Leishmania species | Relative copy no. |
|
|---|---|---|
| Maxicircle | Minicircle | |
| L. donovani complexa | ||
| L. (L.) chagasi | 6.75 | 4.21 |
| L. (L.) chagasi (attenuated) | 4.36 | 1.85 |
| L. (L.) donovani | 2.34 | 7.19 |
| L. (L.) infantum | 1.00 | 1.00 |
| Mean ± SE fold increase | 3.6 ± 1.45 | 3.5 ± 1.6 |
| L. donovani isolatesb | ||
| L. (L.) donovani (reference) | 1.00 | 1.00 |
| L. (L.) donovani 1 | 0.38 | 0.21 |
| L. (L.) donovani 2 | 0.23 | 0.16 |
| L. (L.) donovani 3 | 0.39 | 0.23 |
| L. (L.) donovani 4 | 1.72 | 0.66 |
| L. (L.) donovani 5 | 0.41 | 0.23 |
| L. (L.) donovani 6 | 0.99 | 1.80 |
| L. (L.) donovani 7 | 0.56 | 1.03 |
| Mean ± SE fold decrease/increase | 2.2 ± 0.4 | 3.1 ± 0.8 |
| L. tropica isolatesc | ||
| L. (L.) tropica 1 | 1.00 | 1.00 |
| L. (L.) tropica 2 | 1.99 | 1.53 |
| L. (L.) tropica 3 | 4.25 | 5.64 |
| L. (L.) tropica 4 | 3.92 | 2.76 |
| L. (L.) tropica 5 | 1.04 | 1.72 |
| Mean ± SE fold increase | 2.4 ± 0.8 | 2.5 ± 0.9 |
Relative copy number difference in kinetoplast mini- and maxicircle sequences between the indicated Leishmania donovani complex species. Expression changes are relative to the results for the L. (L.) infantum sample.
Relative copy number difference in maxi- or minicircle DNA between 8 isolates of L. donovani. Expression changes are relative to the results for the L. (L.) donovani reference sample.
Relative copy number difference in maxi- or minicircle DNA between 5 isolates of L. tropica. Expression changes are relative to the results for the L. (L.) tropica 1 sample.
These data showed that there are indeed differences between the copy numbers of maxi- or minicircle DNA both between species and between isolates within a single species. The minicircle or maxicircle copy numbers varied by 2.53 ± 0.92 or 2.44 ± 0.78 (fold, average ± standard error), respectively, between different isolates of L. tropica (Table 6, L. tropica isolates). Mini- or maxicircle copy numbers differed by 3.13 ± 0.77 or 2.18 ± 0.41, respectively, between different isolates of L. donovani (Table 6, L. donovani isolates). Although we did not compare all Leishmania species listed in Tables 1 and 3, there were differences in maxi- or minicircle copy numbers between the 3 species tested (3.61 ± 1.45 or 3.56 ± 1.60, respectively).
As a means of addressing changes in kDNA copy numbers during stage transition, we applied the same technique to an L. (L.) chagasi strain capable of transforming between promastigote and axenic amastigote in culture. Mini- and maxicircle copies were quantified weekly during the conversion from promastigote to amastigote or from amastigote to promastigote in response to culture conditions inducing stage transition (Fig. 3). At the week 1 time point, there was no significant difference between minicircle copy numbers as parasites converted either from amastigote to promastigote or from promastigote to amastigote (P > 0.1, n = 3, two-tailed t test assuming unequal variances). Maxicircle copy numbers underwent a greater fold change, although this did not reach significance. One week after the transition from amastigote to promastigote, newly transformed promastigotes contained 0.74-fold fewer copies of maxicircles than the amastigote baseline (P = 0.09, n = 3) (Fig. 3, week 1 solid bar). Conversely, 1 week after conversion from promastigotes, axenic amastigotes contained 1.50-fold-higher numbers of maxicircle copies than the baseline in promastigotes (P = 0.08, n = 3) (Fig. 3, week 1 striped bar). Although axenic amastigotes are not identical to tissue-derived amastigotes, these findings suggest that standard curves generated from promastigote-derived DNA, particularly using minicircle kDNA probes, can be validly applied to quantification of amastigotes of the same species/strain.
Fig. 3.

kDNA copy numbers during stage transition. Maxicircle (left) or minicircle (right) kDNA primer pairs were used to quantify copy numbers in the promastigote and amastigote life stages when converting in vitro between the two life stage forms. (Solid bars) Total parasite DNA was extracted from amastigotes to determine basal expression and then weekly after in vitro conversion to promastigotes for comparison. (Striped bars) Total parasite DNA was extracted from promastigotes to use as baseline and then weekly after in vitro conversion to amastigotes for comparison. kDNA abundance was normalized to the single-copy gene DNA polymerase I and expressed as fold change in promastigotes (solid bars) or amastigotes (striped bars) relative to the abundance at the preconversion stage.
Detection of L. donovani in clinical and other unknown isolates.
SYBR green-based qPCR assays were applied to serum samples from patients with visceral leishmaniasis (VL) caused by L. donovani and control patients undergoing treatment for other diseases (non-VL) (Fig. 4). A priori, serum specimens would be expected to be cell-free and, therefore, not contain DNA from intracellular Leishmania spp. parasites. Nonetheless, if there were inapparent lysis of leukocytes during blood collection, DNA could be present. Indeed, there was DNA extracted from serum specimens, and these were positive for Leishmania DNA in all five patients with VL, using the kDNA 3 SYBR green assay (Fig. 4). All negative controls were negative for parasite DNA, including five Bangladeshi control subjects, one individual from Iowa, and control human DNA from a North American subject. Parasites were quantified in all positive subjects, revealing 595 ± 178 (±standard error) leishmania parasites per ml of serum.
Fig. 4.
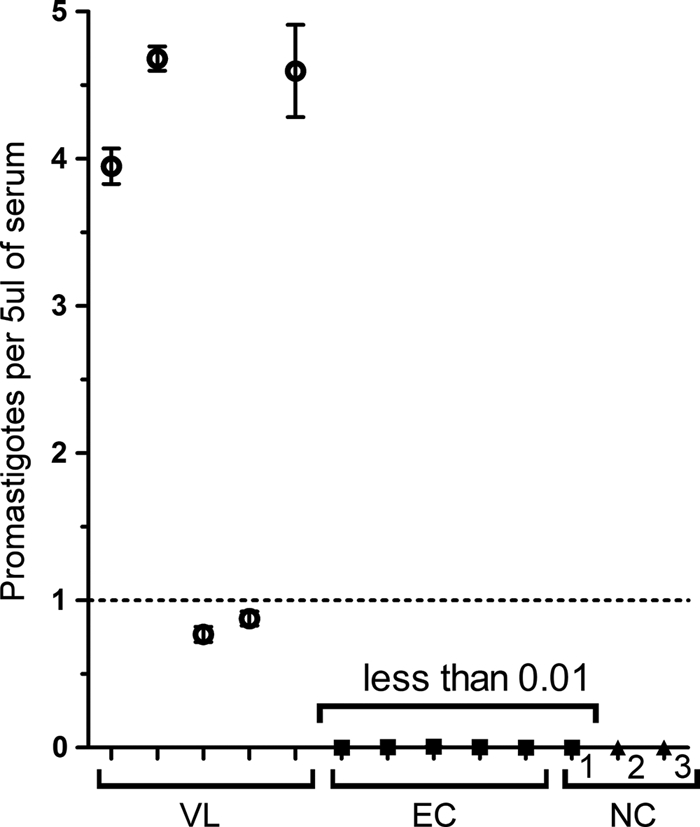
qPCR detection of L. (L.) donovani in human sera. DNA extracted from 200 μl of serum was used to detect and quantify Leishmania DNA in five VL patients from Bangladesh (VL). The low numbers of parasites detected may either be the result of whole parasites being sampled or of lysed parasite DNA in peripheral blood. Non-VL endemic control (EC) patients and serum from a donor without exposure to Leishmania (NC 1) showed no evidence of infection. Other negative controls (NC) were a human DNA template (NC 2) and water (NC 3).
Serial qPCR was used to determine the species of parasites from a different type of clinical specimen, i.e., cutaneous biopsy specimens for diagnosis of cutaneous leishmaniasis. Biopsy specimens are routinely obtained for diagnostic evaluation of cutaneous ulcers that are suspected to be cutaneous leishmaniasis in clinical settings in northeast Brazil. To evaluate the serial qPCR for diagnosis/species determination using these specimens, DNA was extracted from the biopsy specimens of three patients living in a region of the state of Bahia, Brazil, where L. (V.) braziliensis and L. (L.) amazonensis have been endemic (4, 58). Extracted DNA samples from all three patient biopsy specimens were tested by serial qPCR as described in Fig. 2, and all three were positive for the presence of L. Viannia spp. according to the L. (V.) braziliensis kDNA 3 probe pair. We tested an additional 11 DNA samples extracted from parasites isolated from cutaneous lesions of subjects living in Manaus, Amazonas, Brazil, as well as 19 DNA samples from different clinical forms of tegumentary leishmaniasis from Corte de Pedra, Bahia, Brazil (6 from cutaneous, 7 from disseminated, and 6 from mucosal leishmaniasis). L. (V.) braziliensis kDNA 3 amplified members of the L. Viannia spp. subgenus regardless of species. Furthermore, all 19 isolates from subjects in Corte de Pedra amplified with the primer set SLACS (29), which has specifically amplified L. (V.) braziliensis from among the Viannia subgenus, suggesting that these samples were L. (V.) braziliensis, as opposed to L. (V.) guyanensis or L. (V.) panamensis. These data also suggested that the SLACS genes were present regardless of the clinical form of the disease. None of the 11 samples from Manaus were amplified by the SLACS primer set, indicating that these 11 isolates were Leishmania Viannia subgenus but not L. (V.) braziliensis.
The presence of Leishmania RNA virus 1 (LRV1) viruses in L. (V.) guyanensis has recently been reported to be associated with distinct clinical presentations of disease (29). It was of interest to consider whether retrotransposons might also be a biomarker for the clinical presentation of symptomatic infections with Leishmania Viannia braziliensis infections. However, no significant differences in SLACS genomic copy numbers were observed between isolates from patients with different clinical presentations [tested via the delta-delta CT method relative to the results for the genomic L. (V.) braziliensis DNA polymerase 2 primer set, according to a Student's t tests at a P value of <0.05 between each clinical presentation].
Five DNA samples originally obtained from patients in different regions of the world were tested as unknowns using the serial qPCR assay. No information about geographic location or species was provided at the time of serial qPCR testing. Species identification of the isolates, determined at the Centers for Disease Control by isoenzyme testing, indicated that they were L. (V.) braziliensis, L. (L.) donovani, L. (L.) infantum, L. (L.) mexicana, and L. (L.) major. Using the qPCR primer pair selection strategy outlined in Table 5, the species identified through the qPCR serial testing corresponded to the species designation according to isoenzyme analysis for all 5 unknowns. The primer sets used in this analysis were kDNA 1, L. (V.) braziliensis kDNA 3, MAG 1, and L. (L.) mexicana minicircle 1.
DISCUSSION
The global burden of leishmaniasis is high (18). Visceral leishmaniasis has reached epidemic proportions in three regions (Bangladesh/Nepal/Bihar State, India; southern Sudan-Ethiopia/Afghanistan; and Brazil). Drug resistance has further augmented the disease burden in northern India (11), and the HIV epidemic has led to new patterns of visceral leishmaniasis in Spain and Portugal (2). Across the ocean, urbanization has caused the spread of visceral leishmaniasis in periurban settings of northeast and southern Brazil (30). Cutaneous leishmaniasis is highest in the Middle East, Syria, Brazil, and Peru (64), but imported cutaneous leishmaniasis is becoming problematic in the United States, particularly in military personnel (31, 57). Leishmaniasis in the blood supply is becoming a concern (10, 48), and an outbreak of Leishmania (L.) infantum infection affecting foxhounds has introduced a possible reservoir into the United States (52). As such, leishmaniasis is emerging as a disease of concern with both human and veterinary importance.
Diagnostic testing for leishmaniasis is less than optimal. The need for improved diagnostic procedures is prominent in the current setting of expanding disease burden. Clinical presentation and a positive test of immune response to the parasite (serology during visceral leishmaniasis or leishmania skin testing during cutaneous leishmaniasis) can be suggestive, but definitive diagnosis requires parasite identification. The latter can be in the form of microscopic examination of tissue biopsy specimens, bone marrow or splenic aspirates, and/or cultivation of live organisms from clinical tissue specimens. The sensitivity varies with the specimen source. Microscopic exam and culture from splenic aspirate have >95% and close to 100% sensitivity, respectively, for diagnosis of Indian kala azar, but the procedure is risky in all but a few clinicians' hands. The sensitivity of bone marrow aspirate is lower (52 to 69%) but the procedure is safer, albeit still invasive (7, 43, 67). Microscopic identification and/or culture from biopsied cutaneous lesions is less sensitive and very dependent on the disease form, with parasites isolated from only 30 to 50% of localized cutaneous ulcers and rarely isolated from lesions of mucosal or disseminated leishmaniasis. The species of Leishmania is most commonly determined by electrophoretic mobility assay of isoenzymes from cultured promastigotes (63), a lengthy method that is only feasible if parasites are isolated in culture.
Molecular methods to detect Leishmania spp. DNA include hybridization of infected tissues (17) and amplification using routine PCR, PCR-enzyme-linked immunosorbent assay (ELISA) (13, 35, 53, 55), or quantitative PCR (40, 46, 62). These methods have been successfully applied for detection of individual Leishmania species in clinical samples, including peripheral blood leukocytes of patients with visceral leishmaniasis or asymptomatic infection (39, 47, 59). Discrimination between the Leishmania species has been reported using RAPD (random amplification of polymorphic DNA) (25) and qPCR. Species-discriminating qPCR assays that can distinguish between the two Leishmania subgenera Viannia and Leishmania, between Leishmania complexes [L. (L.) mexicana, L. (L.) donovani/infantum, and L. (L.) major], or between L. (L.) donovani strains (54) have been reported. Despite many reports, a standardized rapid and sensitive test that distinguishes the spectrum of common Leishmania species is still needed (45).
The purpose of this study was to develop a comprehensive series of qPCR assays adequate for detection and species determination of Leishmania spp. in clinical, environmental, or experimental samples. To approach this goal, we chose qPCR targets that were multicopy genes or reported targets for conventional or quantitative PCR in the literature. Targets included kinetoplast minicircle DNA (46, 62), other repetitive sequences, such as rRNA coding or intergenic spacer regions, and single-copy genes, including those encoding DNA polymerase (8) or glucose phosphate isomerase (66). From these we designed a series of primers and probes that were then tested for their sensitivity and specificity to detect eight different Leishmania species. All assays retained specificity in the presence of a 10-fold excess of human DNA.
As expected, multicopy targets were the most sensitive assays for detection of Leishmania spp. DNA. The most efficient was minicircle kDNA, although the sensitivity of individual kDNA primer sets differed between Leishmania species. A combination of two kDNA primers was finally chosen [kDNA 1 and L. (V.) braziliensis kDNA 3] to detect all Leishmania spp., with the latter used to detect L. (V.) braziliensis alone. Species discrimination was approached starting with the melt curve information from the kDNA 1 primer set. The MAG 1 primer set could be used to differentiate L. (L.) donovani from L. (L.) infantum chagasi. Identification of L. (L.) amazonensis could be confirmed with the L. (L.) amazonensis kDNA 2 primer set. The L. mexicana minicircle 1 primer set was found to differentiate L. mexicana from L. major. If the above-described assays led to ambiguity regarding species identification, a number of other primer sets could be employed, with melt curve criteria used for species differentiation as shown in Fig. 1. Thus, detection and species determination can be achieved using SYBR green alone, without a need for expensive TaqMan probes, and the sets should provide enough flexibility to detect and differentiate infections with Leishmania isolates in many clinical specimens.
In addition to the above-described SYBR green assays, we also tested several TaqMan probes and primer sets. These could be used to provide additional validation of findings in problematic cases. Furthermore, at least some of these can be developed into multiplex assays for detection/species determination, illustrated by the ability of a multiplex assay containing MAG 2 and DNA polymerase 2 TaqMan primer pairs to distinguish visceralizing species from all other Leishmania species (Table 3).
There are a few reports documenting individuals with symptoms resembling visceral leishmaniasis from whom Leptomonas has been isolated alone or in combination with L. donovani (60). Whether these are primary causes of disease or coinfections is not yet clear. Therefore, we incorporated Leptomonas into our set of primers and probes. GAPDH and repetitive miniexon sequences were sufficient to identify and quantify Leptomonas species. Although Leptomonas GAPDH 2 primer sets also amplified L. braziliensis, Leptomonas miniexon 1 did not, making these primer sets useful even in the case of Leptomonas-Leishmania spp. coinfections.
Because of a need to quantify parasites in clinical specimens using the most sensitive primer sets, we investigated the validity of minicircle primers for comparison of clinical samples with standard curves of promastigote DNA. Therefore, we applied qPCR tests using primers for mini- or maxicircles, normalized to a single-copy gene (DNA polymerase I), to compare kDNA copy numbers between the promastigote and amastigote life stages, between different strains of individual Leishmania species, and between different species of Leishmania.
There were not significant differences between copy numbers of either mini- or maxicircle DNA between the promastigote and the amastigote stages, suggesting that the use of promastigote DNA to quantify amastigotes in mammalian specimens would be valid. However, copy numbers differed moderately between strains of the same species and even more between different species of the Leishmania donovani complex. The latter conclusions are complicated by the expected variability of primer hybridization to kDNA of different species. We conclude that the use of minicircle primer sets for quantification might be most accurate by comparison with a curve generated using DNA from the same species, but absolute numbers would not be as accurate as quantification based on a chromosomal gene target. Logically, it seems valid to use the kDNA primer set for relative quantification in a single patient to follow response to therapy or in a single household or family that might share parasite strains. However, caution must be used in comparing absolute parasite numbers between isolates or species using kDNA primers.
In conclusion, we report herein a series of qPCR assays that is sufficient to identify and make a species determination for parasite DNA in serum samples from patients with visceral leishmaniasis or lesion biopsy specimens of subjects with tegumentary leishmaniasis. The strategy using SYBR green makes this a rapid and relatively inexpensive means of detecting, quantifying, and determining the species of Leishmania in clinical or epidemiologic samples. In addition to diagnosis, additional useful applications could include quantitative analysis of parasites in buffy coat or serum specimens to document response to therapy, species determination in tissue biopsy specimens or tissues scraped from microscopic slides, and studies of parasite loads in sand flies from regions of endemicity. The qPCR serial testing strategy requires a reference laboratory with the technical capacity to perform qPCR. This technology is becoming available in many countries and could be developed in a central diagnostic laboratory in countries where leishmaniasis is endemic.
ACKNOWLEDGMENTS
This work was supported in part by a Merit Review grant, a Persian Gulf program grant, and an OEF-OIF program grant from the Department of Veterans' Affairs. It was supported in part by Tropical Diseases Medicine Center grants P50 AI-074321 (S.S., S.G., and M.E.W.) and P50 AI-30639 (S.M.B.J., A.S., E.M.C., and M.E.W.) and by NIH grants NIAID R01 AI045540 (M.E.W.), R01AI067874 (M.E.W.), R01 AI076233 (M.E.W.), and R01 AI059451 (J.E.D. and M.E.W.). It was performed in part during the tenure of J.L.W. on NIH training grants T32 GM008629 and T32 GM082729.
We thank Frank Steurer and Marcos de Almeida of the Centers for Diseases Control for providing parasite isolates and DNA and for validation of species identity.
Footnotes
Published ahead of print on 21 September 2011.
REFERENCES
- 1. Ahmed S., et al. 2003. Intradermal infection model for pathogenesis and vaccine studies of murine visceral leishmaniasis. Infect. Immun. 71:401–410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Alvar J., et al. 1997. Leishmania and human immunodeficiency virus coinfection: the first 10 years. Clin. Microbiol. Rev. 10:298–319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Aoun O., et al. 2009. Canine leishmaniasis in south-east of France: screening of Leishmania infantum antibodies (Western blotting, ELISA) and parasitaemia levels by PCR quantification. Vet. Parasitol. 166:27–31 [DOI] [PubMed] [Google Scholar]
- 4. Barral A., et al. 1991. Leishmaniasis in Bahia, Brazil: evidence that Leishmania amazonensis produces a wide spectrum of clinical disease. Am. J. Trop. Med. Hyg. 44:536–546 [DOI] [PubMed] [Google Scholar]
- 5. Berens R. L., Brun R., Krassner S. M. 1976. A simple monophasic medium for axenic culture of hemoflagellates. J. Parasitol. 62:360–365 [PubMed] [Google Scholar]
- 6. Berman J. 2005. Clinical status of agents being developed for leishmaniasis. Expert Opin. Invest. Drugs 14:1337–1346 [DOI] [PubMed] [Google Scholar]
- 7. Boelaert M., et al. 2008. Diagnostic tests for kala-azar: a multi-centre study of the freeze-dried DAT, rK39 strip test and KAtex in East Africa and the Indian subcontinent. Trans. R. Soc. Trop. Med. Hyg. 102:32–40 [DOI] [PubMed] [Google Scholar]
- 8. Bretagne S., et al. 2001. Real-time PCR as a new tool for quantifying Leishmania infantum in liver in infected mice. Clin. Diagn. Lab. Immunol. 8:828–831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bryceson A. D. M. 1970. Immunological aspects of clinical leishmaniasis. Proc. R. Soc. Med. 63:40–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cardo L. J. 2006. Leishmania: risk to the blood supply. Transfusion 46:1641–1645 [DOI] [PubMed] [Google Scholar]
- 11. Chappius F., et al. 2007. Visceral leishmaniasis: what are the needs for diagnosis, treatment and control? Nat. Rev. Microbiol. 5:S7–S16 [DOI] [PubMed] [Google Scholar]
- 12. Chen J., Rauch C. A., White J. H., Englund P. T., Cozzarelli N. R. 1995. The topology of the kinetoplast DNA network. Cell 80:61–69 [DOI] [PubMed] [Google Scholar]
- 13. Costa J. M., et al. 1996. PCR enzyme-linked immunosorbent assay for diagnosis of leishmaniasis in human immunodeficiency virus-infected patients. J. Clin. Microbiol. 34:1831–1833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Croft S. L., Sundar S., Fairlamb A. H. 2006. Drug resistance in leishmaniasis. Clin. Microbiol. Rev. 19:111–126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Davies C. R., et al. 1997. Cutaneous leishmaniasis in the Peruvian andes: factors associated with variability in clinical symptoms, response to treatment, and parasite isolation rate. Clin. Infect. Dis. 25:302–310 [DOI] [PubMed] [Google Scholar]
- 16. de Almeida M. E., et al. 2011. Identification of Leishmania spp. by molecular amplification and DNA sequencing analysis of a fragment of rRNA internal transcribed spacer 2. J. Clin. Microbiol. 49:3143–3149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. De Bruijn M. H. L., Barker D. C. 1992. Diagnosis of New World leishmaniasis: specific detection of species of the Leishmania braziliensis complex by amplification of kinetoplast DNA. Acta Trop. 52:45–58 [DOI] [PubMed] [Google Scholar]
- 18. Desjeux P. 1996. Leishmaniasis: public aspects and control. Clin. Dermatol. 14:417–423 [DOI] [PubMed] [Google Scholar]
- 19. Fillola G., et al. 1992. Peripheral intramonocytic leishmanias in an AIDS patient. J. Clin. Microbiol. 30:3284–3285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Francino O., et al. 2006. Advantages of real-time PCR assay for diagnosis and monitoring of canine leishmaniosis. Vet. Parasitol. 137:214–221 [DOI] [PubMed] [Google Scholar]
- 21. Gardener P. J. 1977. Taxonomy of the genus Leishmania: a review of nomenclature and classification. Trop. Dis. Bull. 74:1069–1088 [PubMed] [Google Scholar]
- 22. Gramiccia M., Smith D. F., Angelici M. C., Ready P. D., Gradoni L. 1992. A kinetoplast DNA probe diagnostic for Leishmania infantum. Parasitology 105:29–34 [DOI] [PubMed] [Google Scholar]
- 23. Guevara P., et al. 1993. Leishmania braziliensis in blood 30 years after cure. Lancet 341:1341. [DOI] [PubMed] [Google Scholar]
- 24. Hajduk S. L., Harris M. E., Pollard V. W. 1993. RNA editing in kinetoplastid mitochondria. FASEB J. 7:54–63 [DOI] [PubMed] [Google Scholar]
- 25. Hanafi R., Barhoumi M., Ali S. B., Guizani I. 2001. Molecular analyses of Old World Leishmania RAPD markers and development of a PCR assay selective for parasites of the L. donovani species complex. Exp. Parasitol. 98:90–99 [DOI] [PubMed] [Google Scholar]
- 26. Heger A., Wilton C. A., Sivakumar A., Holm L. 2005. ADDA: a domain database with global coverage of the protein universe. Nucleic Acids Res. 33:D133–D191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Henderson D. M., et al. 1992. Cloning of the gene encoding Leishmania donovani S-adenosylhomocysteine hydrolase, a potential target for antiparasitic chemotherapy. Mol. Biochem. Parasitol. 53:169–184 [DOI] [PubMed] [Google Scholar]
- 28. Herwaldt B. L. 1999. Leishmaniasis. Lancet 354:1191–1199 [DOI] [PubMed] [Google Scholar]
- 29. Ives A., et al. 2011. Leishmania RNA virus controls the severity of mucocutaneous leishmaniasis. Science 331:775–778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Jeronimo S. M. B., et al. 2004. An emerging peri-urban pattern of infection with Leishmania chagasi, the protozoan causing visceral leishmaniasis in northeast Brazil. Scand. J. Infect. Dis. 36:443–449 [DOI] [PubMed] [Google Scholar]
- 31. Johnson K. E. 2004. Leishmaniasis, US Armed Forces, 2003. MSMR 10:2–5 [Google Scholar]
- 32. Laskay T., Gemetchu T., Teferedegn H., Frommel D. 1991. The use of DNA hybridization for the detection of Leishmania aethiopica in naturally infected sandfly vectors. Trans. R. Soc. Trop. Med. Hyg. 85:599–602 [DOI] [PubMed] [Google Scholar]
- 33. Lee S. T., Chiang S. C., Singh A. K., Liu H. Y. 1995. Identification of Leishmania species by a specific DNA probe that is conserved only in the maxicircle DNA of human-infective Leishmania parasites. J. Infect. Dis. 172:891–894 [DOI] [PubMed] [Google Scholar]
- 34. le Fichoux Y., et al. 1999. Occurrence of Leishmania infantum parasitemia in asymptomatic blood donors living in an area of endemicity in Southern France. J. Clin. Microbiol. 37:1953–1957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lopez M., et al. 1993. Diagnosis of Leishmania using the polymerase chain reaction: a simplified procedure for field work. Am. J. Trop. Med. Hyg. 49:348–356 [DOI] [PubMed] [Google Scholar]
- 36. Magill A. J., Gasser R. A., Oster C. N., Grogl M. 1992. Viscerotropic leishmaniasis in persons returning from Operation Desert Storm—1990-1991. MMWR Morb. Mortal. Wkly. Rep. 41:131–134 [PubMed] [Google Scholar]
- 37. Magill A. J., Grogl M., Gasser R. A., Wellington S., Oster C. N. 1993. Visceral infection caused by Leishmania tropica in veterans of Operation Desert Storm. N. Engl. J. Med. 328:1383–1387 [DOI] [PubMed] [Google Scholar]
- 38. Martínez P., et al. 1993. Diagnosis of visceral leishmaniasis in HIV-infected individuals using peripheral blood smears. AIDS 7:227–230 [DOI] [PubMed] [Google Scholar]
- 39. Martin-Sanchez J., et al. 2004. Detection of Leishmania infantum kinetoplast DNA in peripheral blood from asymptomatic individuals at risk for parenterally transmitted infections: relationship between polymerase chain reaction results and other Leishmania infection markers. Am. J. Trop. Med. Hyg. 70:545–548 [PubMed] [Google Scholar]
- 40. Mary C., Faraut F., Lascombe L., Dumon H. 2004. Quantification of Leishmanai infantum DNA by a real-time PCR assay with high sensitivity. J. Clin. Microbiol. 42:5249–5255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Massamba N. N., Mutinga M. J. 1992. Recombinant kinetoplast DNA (kDNA) probe for identifying Leishmania tropica. Acta Trop. 52:1–15 [DOI] [PubMed] [Google Scholar]
- 42. Mauricio I. L., Stothard J. R., Miles M. A. 2000. The strange case of Leishmania chagasi. Parasitol. Today 16:188–189 [DOI] [PubMed] [Google Scholar]
- 43. Maurya R., et al. 2010. Evaluation of blood agar microtiter plates for culturing leishmania parasites to titrate parasite burden in spleen and peripheral blood of patients with visceral leishmaniasis. J. Clin. Microbiol. 48:1932–1934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. McCoy J. J., Beetham J. K., Myung K. S., Wilson M. E., Donelson J. E. 1998. Regulatory sequences and a novel gene in the MSP (GP63) gene cluster of Leishmania chagasi. Mol. Biochem. Parasitol. 95:251–265 [DOI] [PubMed] [Google Scholar]
- 45. Myjak P., et al. 2009. Usefulness of PCR method for detection of Leishmania in Poland. Pol. J. Microbiol. 58:219–222 [PubMed] [Google Scholar]
- 46. Nicolas L., Prina E., Lang T., Milon G. 2002. Real-time PCR for detection and quantitation of leishmania in mouse tissues. J. Clin. Microbiol. 40:1666–1669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Nuzum E., et al. 1995. Diagnosis of symptomatic visceral leishmaniasis by use of the polymerase chain reaction on patient blood. J. Infect. Dis. 171:751–754 [DOI] [PubMed] [Google Scholar]
- 48. Otero A. C. S., et al. 2000. Short report: occurrence of Leishmania donovani DNA in donated blood from seroreactive Brazilian blood donors. Am. J. Trop. Med. Hyg. 62:128–131 [DOI] [PubMed] [Google Scholar]
- 49. Peacock C. S., et al. 2007. Comparative genomic analysis of three Leishmania species that cause diverse human disease. Nat. Genet. 39:839–847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Pearson R. D., Sousa A. Q. 1996. Clinical spectrum of Leishmaniasis. Clin. Infect. Dis. 22:1–13 [DOI] [PubMed] [Google Scholar]
- 51. Pennica D., et al. 1984. Human tumour necrosis factor: precursor structure, expression and homology to lymphotoxin. Nature 312:724–729 [DOI] [PubMed] [Google Scholar]
- 52. Petersen C. A. 2009. Leishmaniasis, an emerging disease found. in companion animals in the United States. Top. Companion Anim. Med. 24:182–188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Piarroux R., et al. 1993. Isolation and characterization of a repetitive DNA sequence from Leishmania infantum: development of a visceral leishmaniasis polymerase chain reaction. Am. J. Trop. Med. Hyg. 49:364–369 [DOI] [PubMed] [Google Scholar]
- 54. Quispe-Tintaya K. W., et al. 2005. Fluorogenic assay for molecular typing of the Leishmania donovani complex: taxonomic and clinical applications. J. Infect. Dis. 192:685–692 [DOI] [PubMed] [Google Scholar]
- 55. Ramos A., Maslov D. A., Fernandes O., Campbell D. A., Simpson L. 1996. Detection and identification of human pathogenic Leishmania and Trypanosoma species by hybridization of PCR-amplified mini-exon repeats. Exp. Parasitol. 82:242–250 [DOI] [PubMed] [Google Scholar]
- 56. Rozen S., Skaletsky H. 2000. Primer3 on the WWW for general users and for biologist programmers. Methods Mol. Biol. 132:356–386 [DOI] [PubMed] [Google Scholar]
- 57. Sanders J. W., et al. 2005. Impact of illness and non-combat injury during Operations Iraqi Freedom and Enduring Freedom (Afghanistan). Am. J. Trop. Med. Hyg. 73:713–719 [PubMed] [Google Scholar]
- 58. Schriefer A., et al. 2009. Geographic clustering of leishmaniasis in northeastern Brazil. Emerg. Infect. Dis. 15:871–876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Smyth A. J., et al. 1992. Rapid and sensitive detection of Leishmania kinetoplast DNA from spleen and blood samples of kala-azar patients. Parasitology 105:183–192 [DOI] [PubMed] [Google Scholar]
- 60. Srivastava P., et al. 2010. Detection of Leptomonas sp. parasites in clinical isolates of kala-azar patients from India. Infect. Genet. Evol. 10:1145–1150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. van der Meide W., et al. 2008. Comparison between quantitative nucleic acid sequence-based amplification, real-time reverse transcriptase PCR, and real-time PCR for quantification of Leishmania parasites. J. Clin. Microbiol. 46:73–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Vitale F., et al. 2004. TaqMan-based detection of Leishmania infantum DNA using canine samples. Ann. N. Y. Acad. Sci. 1026:139–143 [DOI] [PubMed] [Google Scholar]
- 63. WHO. 1990. Control of leishmaniasis. Report of a WHO Expert Committee. WHO technical report series, no. 793. World Health Organization, Geneva, Switzerland. [PubMed] [Google Scholar]
- 64. WHO. 2011. Leishmaniasis: the vector. http://www.who.int/leishmaniasis/en/. World Health Organization, Geneva, Switzerland. [Google Scholar]
- 65. Wirth D. F., McMahon-Pratt D. 1982. Rapid identification of Leishmania species by specific hybridization of kinetoplast DNA in cutaneous lesions. Proc. Natl. Acad. Sci. U. S. A. 79:6999–7003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Wortmann G., et al. 2005. Rapid identification of Leishmania complexes by a real-time PCR assay. Am. J. Trop. Med. Hyg. 73:999–1004 [PubMed] [Google Scholar]
- 67. Zijlstra E. E., et al. 1992. Kala-azar: a comparative study of parasitological methods and the direct agglutination test in diagnosis. Trans. R. Soc. Trop. Med. Hyg. 86:505–507 [DOI] [PubMed] [Google Scholar]